

Abstract
Microwave-assisted optimized transglycosylation reactions were used to prepare eleven modified l-3′-azido-2′,3′-dideoxypurine nucleosides. These l-nucleoside analogs were evaluated against HIV and hepatitis B virus. The l-3′-azido-2′,3′-dideoxypurines nucleosides were metabolized to nucleoside 5′-triphosphates in primary human lymphocytes, but exhibited weak or no antiviral activity against HIV-1. The nucleosides were also inactive against HBV in HepG2 cells. Pre-steady-state kinetic experiments demonstrated that the l-3′-azido-2′,3′-dideoxypurine triphosphates could be incorporated by purified HIV-1 reverse transcriptase, although their catalytic efficiency (kpol/Kd) of incorporation was low. Interestingly, a phosphoramidate prodrug of l-3′-azido-2′,3′-dideoxyadenosine exhibited anti-HIV-1 activity without significant toxicity.
Keywords: l-nucleoside analogues, antiviral agents, HIV, HBV, 3′-azidopurine nucleosides
1. Introduction
Nucleoside analogs continue to be the cornerstone of anti-HIV-1 therapy [1], but the emergence of drug-resistant mutants that render current therapies ineffective [2], drives interest in the development of new and novel nucleoside analogs with high potency, low toxicity, and favorable resistance profiles. The first synthesis of an l-nucleoside was reported in 1964[3]; however, for decades it was assumed that only natural d-configuration nucleosides could exhibit biological activity due to the stereospecificity of enzymes in living systems [4,5]. As a consequence, little attention was paid to l-nucleoside analogs until the 1990's with the discovery of lamivudine (3TC, Fig. 1) that exhibited potent antiviral activity against HIV-1 and HBV [6,7]. Following this discovery, a large number of l-nucleoside analogs were synthesized and their antiviral activities evaluated [8,9,10,11,12,13,14,15]. In addition to lamivudine (3TC, Epivir®) [16], emtricitabine (FTC, Emtriva®) [17] exhibited antiviral activity toward HIV-1 and HBV infection with mild or no toxicity (Fig. 1). Further, l-2′-deoxy-thymidine (LdT, Telbivudine, Tyzeka®) is a specific and selective anti-HBV agent [18]. Lamivudine, emtricitabine, and telbivudine are FDA-approved for clinical use. This experience has revealed that l-nucleoside analogs may display antiviral activity comparable to, and in some cases greater than their d-counterparts. Moreover, l-nucleoside analogs may provide a more favorable toxicological profile along with greater stability against cellular enzymes such as deaminases [19].
Fig. 1.

Clinically approved antiviral l-nucleoside analogues.
We recently identified 3′-azido-2′,3′-dideoxyguanosine (3′-azido-ddG) as a lead compound [20] that exhibits potent activity against HIV-1 variants containing either discrimination mutations such as K65R, L74V or M184V or multiple thymidine analog mutations (TAMs) that enhance nucleotide analog excision. Furthermore, 3′-azido-ddG does not exhibit cytotoxicity in primary lymphocytes or epithelial and T-cell lines, and does not decrease the mitochondrial DNA content of HepG2 cells. We have refined and improved the synthetic methodology to prepare purine modified d-3′-azido nucleosides [21] and subsequently prepared a number of analogs which were studied extensively at both the cellular and HIV-1 RT level [22]. The low toxicity and broad antiviral activity against clinically relevant mutation profiles discovered with the d-3′-azido purine series lead us to investigate the synthesis and antiviral activity of the corresponding l-nucleosides.
2. Results and discussion
2.1.1. Chemistry
Our previous work [21] identified transglycosylation as the most efficient method by which the desired 3′-azido purine analogs could be prepared. Problems of trace AZT leading to false positives was identified as an issue in the d-series and ultimately led us to use β -d-3′-azido-2′,3′-dideoxyuridine (AZU, CS-87) as the source of the 3′-azido sugar in the transglycosylation reaction. Our best conditions led to yields of 30-45% as a 1.1:1 mixture of α:β anomers when 1.7 equivalents of silylated base was reacted with 1 equivalent of 5′-protected AZU and 5.2 equivalents of TMSOTf at 85 °C under positive nitrogen pressure for 18 h. Our observations of extreme water sensitivity and negative effects of extended reaction time were most likely related to the stability of the azide group to these conditions and ultimately led us to examine the use of microwave irradiation as a method to reduce the overall reaction time and improve the yield of the desired β anomer. Microwave heating has the potential to significantly shorten reaction time, increase product yields, and enhance product purities by reducing unwanted side reactions compared to conventional heating methods. The short reaction times provided by microwave synthesis make it an ideal tool for rapid reaction scouting and optimization of reaction conditions [23]. Herein, we report our optimization of the microwave assisted transglycosylation methodology, its application to the synthesis of l-3′-azido purine analogues, and evaluation of their antiviral activity, cytotoxicity and incorporation by HIV-1 RT.
2.1.2. Microwave transglycosylation optimization
To rapidly optimize the transglycosylation conditions, an HPLC analysis of the crude reaction was used to determine the amount of co-eluting α:β anomers based on a calibration curve (r2of 0.9998) with 2′-methyl-2′,3′,5′-tribenzoyl-6-chloropurine-furanosyribose as the internal standard. The ratio of α and β anomers was estimated by analysis of the 1H-NMR of the crude reaction mixture. Because AZU is readily available in three steps from dUrd, we chose its transglycosylation to compound 2 as the model reaction to optimize the microwave-assisted transglycosylation reaction (Scheme 1).
Scheme 1.

Model reaction for the optimization of the transglycosylation reaction with a 3′-azido nucleoside.
We examined SnCl4, TMSI, TMSOTf, and Ti(OiPr)4 as potential Lewis acids to affect the transglycosylation of 1 to 2. SnCl4 was found to be incompatible with 1 and led to complex mixtures. On the other end of the reactivity spectrum, the use of Ti(OiPr)4 resulted in no reaction. When TMSI was utilized a low yield of 2 was formed that favored the α-isomer as a 1.2:1 mixture. TMSOTf provided a 1:1 anomer mixture and the yield of 2 was higher than any other Lewis acid tested.
With knowledge of the reported microwave assisted Vorbrüggen glycosylation reaction which was optimized to a 5 min 130 °C ribosylation reaction [24], we began our study with variation of the reaction temperature. The stability of the azido group to TMSOTf at elevated temperature drove us to study temperatures up to but not higher than 130 °C. The best yields of 2 were obtained at 105 °C and 120 °C with 10 minutes of heating and 2.7 equivalents of TMSOTf. At 90 °C the incomplete reaction gave a 32% yield while at 130 °C some decomposition was noted. All temperatures studied had no effect on the 1:1 anomer ratio. We next studied the effect of reaction time on the yield of 2. We chose to reduce the amount of TMSOTf to 1 equivalent and use 130 °C in an effort to reduce the reaction time to 5 min or less. Starting with a 10-min reaction time the yield was increased to 41% of 2versus25% obtained with 2.7 equivalents of TMSOTf. Reducing the reaction time to 5 min had little effect on the reaction outcome while a reaction time of 3 min increased the yield to 61% of 2. Further reductions in reaction time resulted in incomplete reactions. Sub-stoichiometric amounts of TMSOTf gave reduced yields of 2; however, it is worth noting that even 0.1 equivalents of TMSOTf gave a 47% yield of 2, again with no effect on anomer ratio.
With partially optimized conditions in hand we evaluated several solvents for their effect on the outcome of the microwave assisted transglycosylation reaction. Surprisingly the choice of solvent had a large effect on anomer ratio. We evaluated dichloromethane, chloroform, dichloroethane, THF, and acetonitrile for yield and anomer ratio under a standard set of transglycosylation conditions. The yields and α:β anomer ratios were determined to be as follows: dichloromethane (80%, 1.6:1); chloroform (85%, 1.9:1); dichloroethane (70%, 1.5:1); THF (50%, 1.9:1); and acetonitrile (61%, 1:1). Cleaner and higher yielding reactions were noted in the chlorinated solvents; however, all solvents except acetonitrile favored or strongly favored the undesired α anomer.
To test the generality of the optimized transglycosylation conditions we evaluated several different bases and obtained the following yields and α:β anomer ratios: 2-amino-6-chloropurine (40%, 1:1), 6-chloropurine (82%, 1:2), 2,6-dichloropurine (34%, 1:1.4), 2-fluoro-6-aminopurine (56%, 1:1). The consistently acceptable yields obtained from these optimized microwave assisted transglycosylation conditions confirmed their generality and broad synthetic usefulness. It is quite interesting that the ratio of α and β anomers shows a clear dependence on the purine base employed and warrants further investigation.
2.1.3 l-Nucleoside Synthesis
We next turned our attention to the synthesis of l-3′-azido purine nucleoside analogues. Although l-AZA [25,26] and l-AZG [27] have both been studied previously and found to have little antiviral activity against HIV-1 and HBV, we questioned if that was due to lack of binding or other factors. Our plan included cellular pharmacology in human PBM cells, preparation of the corresponding 5′-triphosphate (TP) forms, incorporation studies with purified HIV-1 RT, preparation and evaluation of purine analogs and monophosphate prodrugs. To take advantage of the transglycosylation conditions developed above, we identified LdT as an inexpensive readily available source to prepare the 3′-azido sugar for the transglycosylation. While trace LdT could lead to false positives when screening for HBV activity, at least five chemical steps and as many as five purification steps separated the desired l-3′-azido purine nucleoside analogue from the starting LdT.
As illustrated in Scheme 2, 5′-TBS protected l-AZT, 5 was synthesized with 40-50% overall yield in three steps through a straightforward procedure. In brief, compound 3, which was synthesized from LdT by reaction with TBSCl, was treated with a mixture of triphenylphosphine and diisopropyl azodicarboxylate at 80 °C for 30 min to form the anhydride 4. Treatment of compound 4 with lithium azide in DMF at 130 °C for 2 days furnished compound 5 in 82% yield, which is the key transglycosylation synthon for the synthesis of various l-3′-azido purine nucleosides.
Scheme 2.

Transglycosylation to l-3-azido purines starting from LdT. Reagents and conditions: (a) 1 eq LdT, 1.02 eq TBSCl, pyridine, CH2Cl2, rt, 18 h, 92%; (b) DIAD, tpp, tol, 80 °C, 30 min, 79%; (c) LiN3, dmf, 130 °C, 2 days, 67%; (d) BSA, TMSOTf, acetonitrile, 85 °c, 18 h, 81%; (e) TBAF, rt, 16 h, 35%; (f) t-BuMgCl, phenyl ethoxyalaninyl phosphorochloridate, THF, rt, 18 h, 27%.
Scheme 5.

Synthesis of some l-2-amino 6-substituted 3′-azido purines. Reagents and conditions: (a) i) 2-amino-6-chloropurine, BSA, TMSOTf, acetonitrile, 85 °C, 18 h, 54%; ii) TBAF, THF, 5 h, rt, 27%; (b) t-BuMgCl, phenyl ethoxyalaninyl phosphorochloridate, THF, rt, 18 h, 40%; (c) NaOCH3, CH3OH, 80 °C, 16 h, 71%; (d) NH3/CH3OH, CH3OH, 110 °C, 18 h, 87%; (e) t-BuMgCl, phenyl ethoxyalaninyl phosphorochloridate, THF, rt, 18 h, 60%; (f) methylamine, THF, 80 °C, 18 h, 71%.
With 3′-azido 5 in hand, the 3′-azido 6-chloropurine analogue, 6 was prepared by the above microwave transglycosylation reaction conditions in a very satisfying 81% yield as a 1:2 mixture of α:β anomers. Deprotection by TBAF/acetic acid and separation of the two anomers delivered 3′-azido-6-chloropurine 7, which was further converted to its prodrug 8 with t-BuMgCl and phenyl ethoxyalaninyl phosphorochloridate in 27% isolated yield as a 1:1 mixture of phosphorous diastereomers [28].
From the 6-Cl purine nucleoside 7, 6-substituted-2-H-purine compounds 9, 10,29 and 12, were prepared by reaction with sodium methoxide, ammonia, and methyl amine, respectively, with yields ranging from 40 to 80% (Scheme 3). The phosphoramidate 11 was prepared in 35% yield, as described above.
Scheme 3.

Synthesis of some l-3′-azido adenosine analogs from 6-chloro Purine 7. Reagents and conditions: (a) NaOCH3, CH3OH, rt, 18 h, 51%; (b) NH3/CH3OH, CH3OH, 110 °C, 12 h, 72%; (c) t-BuMgCl, phenyl ethoxyalaninyl phosphorochloridate, THF, rt, 18 h, 35%; (d) methylamine, THF, 80 °C, 61%.
From 5′-TBS-l-AZT, 5, protected l-AZG 13 was obtained using the developed transglycosylation conditions in 37% yield (Scheme 4). Sequential deprotection of the 2-amino group with ammonia in methanol followed by 5′-hydroxyl group unmasking with TBAF afforded l-AZG, 15, which was further converted to the phosphoramidate 16 in 46% yield.
Scheme 4.

Synthesis of l-AZG and its phosphoramidate 16. Reagents and conditions: (a) 2-isopropylguanine, BSA, TMSOTf, acetonitrile, 85 °C, 18 h, 37%; (b) NH3/CH3OH, CH3OH, 110 °C, overnight, 85%; (c) TBAF, rt, 16 h, 30%; (d) t-BuMgCl, phenyl ethoxyalaninyl phosphorochloridate, THF, rt, 18 h, 46%.
For the synthesis of l-AZG analogs the 2-amino-6-chlororpurine nucleoside 17 as the pure β-anomer was prepared in 15% yield in two steps from 2-amino-6-chloro purine as described above (Scheme 5). Reaction with different nucleophilic reagents afforded the 6-methoxypurine, 19, 2,6-diaminopurine, 20 and 6-N-methylaminopurine, 21 in 71%, 87%, 71% yield, respectively. The 2,6-diaminopurine analogue, 20, was transformed to its phosphoramidate prodrug in 60% yield.
The synthesis of the 2,6-dichloropurine nucleoside 24 [30], and the 2-fluoro-6-aminopurine nucleoside, 26, was efficiently carried out in two steps shown as Scheme 6. In the case of 26, it proved to be quite difficult to separate the mixture of α and β anomers. Ultimately, repeated development of a preparative TLC plate with dichloromethane : methanol : ammonium hydroxide in a ratio of 10 : 1 : 0.1 provide sufficient resolution to obtain pure β anomer 26.
Scheme 6.

Synthesis of some l-2-halo 6-substituted 3′-azido purines. Reagents and conditions: (a) 2,6-dichloropurine, BSA, TMSOTf, acetonitrile, 85 °C, 18 h, 26%; (b) TBAF, rt, 16 h, 41%; (c) 2-fluoro-6-aminopurine, BSA, TMSOTf, acetonitrile, 85 °C, 18 h, 46%; (d) TBAF, rt, 16 h, 34%.
2.2. Pharmacological profiling
2.2.1. Anti-HIV Activity
In total, eleven l-3′-azido purine nucleosides were prepared and five were also further converted to their corresponding phosphoramidate prodrug forms. All were evaluated against HIV-1 in human PBM cells and cytotoxicity was determined in PBM, CEM and Vero cells [31]. The EC50, EC90, and cytotoxicity are listed in Table 1. The 6-Cl, 7, and 2,6-dichloro, 24, purine analogs showed weak anti-HIV activity that may result from the cytotoxicity displayed toward PBM cells. The phosphoramidate prodrug of 7 (compound 8) had little impact on antiviral potency or cytotoxicity. The same antiviral activity and cytotoxicity relationship was observed for the 6-Cl, 2-amino purine analogue 17 and its prodrug 18. The 6-OMe, 9, 6-N-Me-amino, 12, 2-amino, 6-OMe, 19, 2-amino, 6-N-Me amino, 21, and 2,6-diamino, 20 analogs as well as l-AZG, 15, and l-AZT, 27, and their phosphoramidates, 16 and 28, respectively, were all inactive against HIV-1 and also did not display cytotoxicity up to 100 μM in PBM, CEM, or Vero cells. However, conversion of the 2,6-diamino analogue, 20 to the phosphoramidate, 22 resulted in an EC50value of 16 μM with no observed toxicity up to 100 μM. l-AZA, 10, displayed very weak anti-HIV-1 activity with an EC50 value of 62 μM with no observed toxicity up to 100 μM; fluorine substitution at the 2-position gave nucleoside analogue 26 with no effect on HIV-1 activity or cytotoxicity. The most potent compound in this series was realized by conversion of l-AZA, 10, to its phosphoramidate, 11, resulting in an EC50 value of 1.4 μM with no observed toxicity up to 100 μM in human PBM cells.
Table 1.
Antiviral activity and cytotoxicity of l-3′-azidopurine nucleosides.
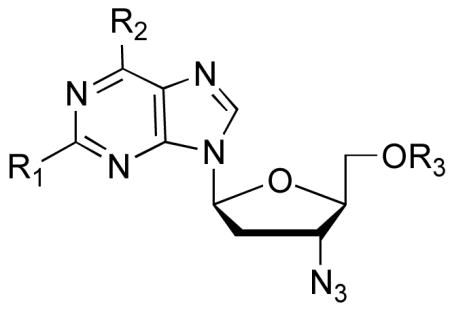 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | Anti-HIV activity (μM) | Anti-HBVactivity (μM) | Cytotoxicity (IC50, μM) |
|||
| EC50 | EC90 | EC50 | PBM | CEM | Vero | ||||
| 7 | H | Cl | H | 18.1 | 46.3 | 98 | 23.8 | 5.3 | 36.5 |
|
| |||||||||
| 8 | H | Cl |
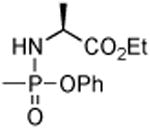
|
16.5 | 83.1 | > 100 | 69.2 | 5.8 | > 100 |
|
| |||||||||
| 9 | H | OCH3 | H | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 10 | H | NH2 | H | 62.3 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 11 | H | NH2 |
|
1.4 | 15.6 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 12 | H | NHCH3 | H | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 15 (l-AZG) | NH2 | OH | H | >100 | >100 | > 100 | >100 | >100 | >100 |
|
| |||||||||
| 16 (l-AZG-PD) | NH2 | OH |
|
>100 | >100 | > 100 | >100 | >100 | >100 |
|
| |||||||||
| 17 | NH2 | Cl | H | 13.2 | 78.3 | > 100 | 59.1 | 46.2 | >100 |
|
| |||||||||
| 18 | NH2 | Cl |
|
16.5 | 55.9 | > 100 | 71 | 23.1 | >100 |
|
| |||||||||
| 19 | NH2 | OCH3 | H | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 20 | NH2 | NH2 | H | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 21 | NH2 | NHCH3 | H | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 22 | NH2 | NH2 |
|
15.8 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 24 | Cl | Cl | H | 11.3 | 26.7 | 51 | 10.7 | 1.4 | 19 |
|
| |||||||||
| 26 | NH2 | F | H | 55.4 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 27 (l-AZT) | — | — | — | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
|
| |||||||||
| 28 (l-AZT-PD) | — | — |
|
> 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| AZT | — | — | — | 0.005 | 0.02 | > 10 | 100 | 14.3 | 50.6 |
| Lamivudine | — | — | — | 0.067 | 0.32 | 0.06 | > 100 | > 100 | > 100 |
2.2.2. Anti-HBV Activity
All compounds were evaluated against wild-type HBV using the HepG2 AD38 system [32,33,30b]. Unfortunately, none demonstrated significant anti-HBV activity (EC50> 50 μM, Table 1) or cytotoxicity toward HepG2 cells (data not shown). Lamivudine, used as a positive control, had an EC50 of 0.067 μM.
2.2.3. Cellular Pharmacology in human PBM Cells
As a first step towards understanding the antiviral data obtained with these compounds we undertook a study of their cellular pharmacology in PBM cells. Briefly, the l-3′-azidopurine nucleoside analogs were incubated for 4 h at 50 μM in PHA activated human PBM cells at 37 °C. After incubation, cells were washed with PBS, intracellular nucleoside and nucleotide metabolites were then extracted with 60% ice-cold MeOH, samples were dried, re-suspended in HPLC mobile phase, and analyzed by LC-MS/MS. Quantification of intracellular nucleoside and nucleotide metabolites was based on calibration curves generated from the l-nucleoside or from the mono-, di-, or tri-phosphate nucleotides prepared from the corresponding d-nucleoside series.
We studied l-AZA, 10, l-AZG, 15, along with three compounds: 9, 12, and 20, which showed no antiviral activity in our assays. In the d-series, compounds 9, 12, and 20 were found to possess anti-HIV activity, and were efficiently converted to their corresponding 6-hydroxy purines in PBM cells [21]. We also studied three additional compounds: 7, 17, and 18, which displayed weak anti-HIV activity. For each nucleoside analogue we looked for intracellular levels of the nucleoside, nucleotides, and possible metabolism to hypoxanthine or guanine nucleoside analogues.
In contrast to the broad 6-position metabolism observed in the d-series, the seven analogs that could potentially undergo 6-position metabolism remained intact under these conditions. Five of the eight nucleosides/prodrugs tested were successfully phosphorylated to the NTP (Table 2). However, intracellular NTP levels were much lower (fmol) than was previously observed with d-nucleosides (pmol) (except for 10 and 11). The detection of intracellular NTPs correlated fairly well with antiviral activity. Specifically, compounds 10, 11, and 18 had detectable NTP levels and also displayed anti-HIV-1 activity while compounds 9 and 12 had nucleoside TP levels below the level of detection and were devoid of anti-HIV-1 activity. l-AZG, 15, and the 2,6-diaminopurine nucleoside 20 had detectable TP levels, but perhaps are not ideal substrates for HIV-1 RT as they did not show any anti-HIV-1 activity in our assay. Compounds 7 and 17 displayed anti-HIV-1 activity with no detectable TPs indicating that the activity may be more related to cytotoxicity toward PBM cells. The most potent compound in the series, phosphoramidate 11, displayed significantly higher levels of nucleoside 10 and its phosphates. At the triphosphate level, prodrug 11 gave 11 times higher levels of triphosphate relative to its parent nucleoside 10. This correlates relatively well with the observed 45-fold increase in anti-HIV activity seen with 11 versus 10 (Table 1).
Table 2.
Cellular pharmacology of l-3′-azidopurine nucleosides.
| |||||
|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | Cellular pharmacology in human PBM Cells | |
| Metabolites detected | Amount fmol/106 cells | ||||
| 7 | H | Cl | H | 7 | 7.8 |
|
| |||||
| 9 | H | OCH3 | H | None | BLDa |
|
| |||||
| 10 | H | NH2 | H | 10 | 4400 |
| 10-MP | 1400 | ||||
| 10-DP | 76 | ||||
| 10-TP | 670 | ||||
|
| |||||
| 11 | H | NH2 |
|
10 | 7500 |
| 10-MP | 4200 | ||||
| 10-DP | 1200 | ||||
| 10-TP | 7400 | ||||
|
| |||||
| 12 | H | NHCH3 | H | 12-MP | 17 |
|
| |||||
| 15 | NH2 | OH | H | 15 | 3200 |
| 15-MP | 46 | ||||
| 15-TP | 54 | ||||
|
| |||||
| 17 | NH2 | Cl | H | 17 | 7.6 |
| 17-DP | 38.3 | ||||
| 17-TP | BLD | ||||
|
| |||||
| 18 | NH2 | Cl |
|
18 | 12 |
| 17 | 49 | ||||
| 17-MP | 430 | ||||
| 17-DP | 190 | ||||
| 17-TP | 10 | ||||
|
| |||||
| 20 | NH2 | NH2 | H | 20-MP | 3.5 |
| 20-TP | 7.6 | ||||
BLD = below the limit of detection.
2.2.4. HIV-1 RT Incorporation Studies
Pre-steady-state kinetic analyses were performed to elucidate, in detail, the interactions between the l-3′-azido-2′,3′-dideoxypurine-5-triphosphates and the polymerase active site of HIV-1 RT. The TP forms of l-AZA, l-AZG, l-AZT, and the 2,6-diamino analogue, 20, were synthesized and purified using the methods of Ludwig and Eckstein [34]. The results (Table 3) demonstrated that each of the l-nucleoside triphosphate analogs was incorporated into the nascent DNA by purified wild-type HIV-1 RT. Interestingly the l-2,6-diamino analog (20-TP) was incorporated opposite thymine indicating that the molecule acts as an adenosine mimetic. We previously reported that the d-analog of this nucleotide also acted as an adenosine mimetic [35]. In general, the catalytic efficiencies of incorporation (kpol/Kd) of each of the l-nucleotide analogs were markedly lower than the kpol/Kd values calculated for the natural dNTPs. The low catalytic efficiencies of l-3′-azido-ddNTP analogs suggests selective incorporation of natural dNTPs, which is consistent with a lack of l-nucleotide triphosphate incorporation observed under steady state kinetic conditions in the presence of natural dNTP (data not shown). The decreased kpol/Kd values for the l-nucleoside analogs was driven primarily by exceptionally slow rates of incorporation (kpol). The nucleotide incorporation rates of l-AZA-TP, l-AZT-TP, and l-AZG-TP were also 2,307-, 915-, and 2,740-fold lower than values previously reported for their d-enantiomer 3′-azido-2′,3′-ddNTP counterparts [36]. By contrast 3TC-TP was incorporated 12-500-fold better than the l-3′-azido-ddNTPs. This suggests that the 3′-azido group in the l-configuration may be the dominant structural characteristic decreasing incorporation. Interestingly, the M184V mutation that confers high levels of resistance to 3TC and (-)-FTC also conferred resistance to the l-3′-azido-2′,3′-dideoxypurine-5′-triphosphates, albeit at lower levels of resistance. Similar to 3TC-TP, the M184V mutation in HIV-1 RT discriminates against the l-3′-azido-nucleotides by decreasing their affinity for the enzyme's active site without significantly impacting their rates of incorporation.
Table 3.
Pre-steady state kinetic constants of l-AZA-TP, l-AZG-TP, l-AZT-TP, and 20-TP.
| dNTP | l-3′-azido-ddNTP | |||||||
|---|---|---|---|---|---|---|---|---|
| kpol(min-1)a | K (μM)a | kpol/Kd(μM-1min-1) | kpol(min-1) | Kd(μM)a | kpol/Kd(μM-1min-1)a | Selectivityb | Resistancec | |
| dATPd | l-AZA-TP | |||||||
| WT | 1464 ± 234 | 3.2 ± 1.2 | 455 | 0.34 ± 0.05 | 15 ± 1.8 | 0.023 | 19783 | -- |
| M184V | 888 ± 384 | 2.5 ± 0.8 | 355 | 0.19 ± 0.04 | 31 ± 3.7 | 0.006 | 59167 | 3 |
| dATPd | 20-TP | |||||||
| WT | 1464 ± 234 | 3.2 ± 1.2 | 455 | 0.14 ± 0.01 | 2.3 ± 1.9 | 0.062 | 7339 | -- |
| M184V | 888 ± 384 | 2.5 ± 0.8 | 355 | 0.18 ± 0.02 | 20 ± 15 | 0.009 | 39444 | 5.4 |
| TTPd | l-AZT-TP | |||||||
| WT | 485 ± 59 | 2.1 ± 0.7 | 231 | 0.58 ± 0.11 | 55 ± 6.6 | 0.011 | 21000 | -- |
| M184V | 394 ± 53 | 2.9 ± 0.8 | 138 | ndg | ≫60 | -- | -- | -- |
| dGTPd | l-AZG-TP | |||||||
| WT | 452 ± 65 | 4.2 ± 1.4 | 108 | 0.10 ± 0.02 | 0.23 ± 0.17 | 0.458 | 235.8 | -- |
| M184V | nd | nd | -- | nd | ≫60 | -- | -- | -- |
| dCTPd | 3TC-TP | |||||||
| WT | 72 ± 42 | 1.1 ± 0.4 | 65 | 0.71 ± 0.06 | 0.13 ± 0.05 | 5.46 | 11.9 | -- |
| M184Ve | 78 ± 24 | 3.0 ± 1.5 | 26 | 0.3 ± 0.06 | 18 ± 4.7 | 0.017 | 1529 | 128 |
Values are the mean ± S.D. of three independent experiments.
Selectivity is defined as (kpol/Kd)dNTP/(kpol/Kd)analog.
Resistance (n-fold) is defined as selectivityM184V/selectivityWT.
Data are adapted from reference 20. The experimental conditions and T/P sequences were identical to those used in this study.
Data are adapted from reference 47. The experimental conditions and T/P sequences were identical to those used in this study.
2.4. Modeling Studies
Model structures were generated as described in the experimental section. Surprisingly, examination of the nucleotide binding site revealed pockets that allow for binding of either d- or l-3′-azido substituted analogs in different modes (Fig. 2). While binding to the active site is necessary, full activity of chain terminators depends upon bond formation between nucleotide α-phosphate and 3′-oxygen of the primer. The d-sugars guide the 3′-azido moiety into a complimentary region of the receptor pocket formed by the phenylalanine side chain of F116 and backbone interaction of Y115 similar to that shown in crystallographic studies [37]. This positions the α-phosphate about 3.4 Å from the primer 3′-oxygen. Tight association between the positive residues R72 and K65, the Mg++, and the α-phosphate are thought to mediate the phosphotransfer reaction. However, the 3′-azido of l-nucleotides, such as l-AZG-TP (Fig. 2b), best fill a receptor cavity flanked by methylene substituents of M184 and D185 above the F160 pi system. While providing relatively high affinity binding for certain base pairs (l-AZG-TP Kd = 0.23 μM), the 3′-azido group impairs the 3′-OH/α-phosphate bond formation (Kpol) for all l-3′ analogs tested (Table 2). The models suggest the l-3′-azido substitution sterically inhibits movement to the expected transition state geometry where the reacting α-phosphate is 2.9 Å between the primer 3′-oxygen and (β-phosphate. The difference in binding mode sheds light on the observed effect of the M184V mutation in reducing l-AZG-TP binding. A comparison between the mutant binding site for d- and l-3′-azido nucleotides suggests that the d analogs have room to bind and react with the primer 3′-oxygen, while the l-3′-azido version would have a van der Waals (VDW) steric collision with the most favored rotamer of V184 (Fig. 2c, 2d). A less favored rotomer (< 11%) of V184 may allow incorporation to occur at low rates similar to 2b, and consistent with experimental results (Table 2).
Fig. 2.
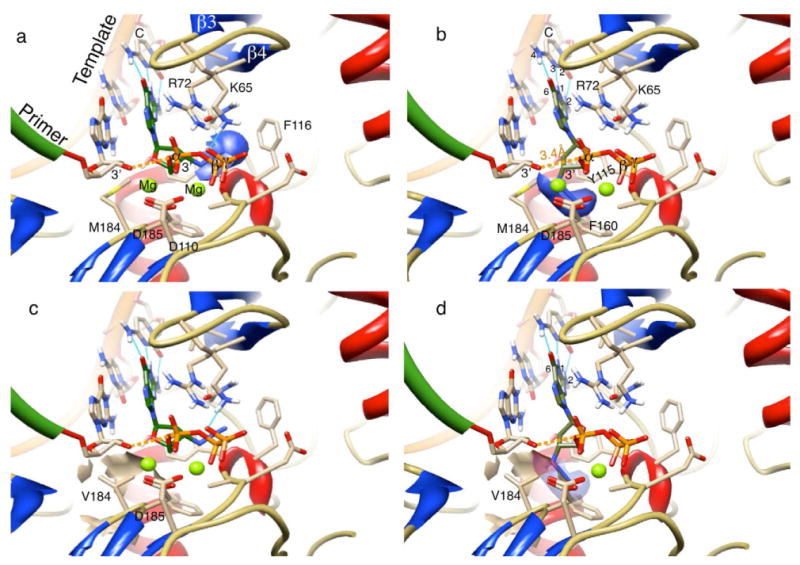
Proposed models of d- and l-AZG-TP interacting with HIV-RT WT and M184V active sites, a) d-AZG-TP (green carbons) bound to HIV-1 WT RT. The 3′-azido Van der Waals (VDW) surface (blue) is interacting with the receptor at Y115 and F116 as described in the text. A space of 3.4 Å between the primer 3′-oxygen and α-phosphate (dashed orange) is unencumbered for bond formation; b) l-AZG-TP (green carbons) bound to HIV-1 WT RT. The 3′-azido binds to the receptor above F160 in the space between the primer 3′-oxygen and the α-phosphate in a way that enhances affinity, but inhibits incorporation. The formation of three hydrogen bonds (Light Blue) between bases (G-C) of the NTP and the template act through the l-sugar to stabilize a planar alignment of the 3′-azido within the pocket; c) d-AZG-TP is bound to the M184V mutant and has no conflict with the VDW steric surface of the dominant V rotomer; d) l-AZG-TP binding to the M184V mutant does show a severe clash between the 3′-azido group and the steric surface (beige) of valine consistent with the relatively high calculated Kd value (Table 2). Mg++atom is hidden for clarity.
Base pair hydrogen bonding also appears to be critical for binding of the l-3′-azido nucleotides to HIV-1 RT. The three hydrogen bonds forming between G-C pairs stabilize a planar relationship between substrate/template bases and the 3′-azido. This stable relationship precisely in the binding pocket (Fig. 2b) must be associated with the observed strong binding affinity (Kd). Conversely, the out of plane wobble between A-T due to loss of the 2 position H-bond is transferred through the l-sugar to the 3′-azido group causing a collision with either M184, D184, or the pi system of F160. The negative effects of this collision can be overcome by restoring the 2-2 position hydrogen bonding interaction as demonstrated by the l-2,6-diamino analog (20-TP) binding with a Kd of 2.26 μM compared to 14.8 μM for l-AZA-TP. The l-analog 20-TP, incorporated across from T like its d counterpart (not shown).
3. Conclusion
We have described a rapid, simple, and general microwave-assisted transglycosylation reaction for the synthesis of 3′-azidopurine nucleosides of both d- and l-configuration. The synthesized compounds were evaluated for antiviral activity against both HIV and HBV and in addition cytotoxicity was studied in five different cell lines. The cellular pharmacology in PBM cells was studied for a representative subset of the synthesized compounds. We also examined incorporation of l-3′-azido purine nucleoside-triphosphates by WT and M184V RT. Our data and modeling suggest that l-3′-azido substituted nucleotides having C, G, or diamino bases can bind to WT HIV-RT with high affinity, however, the bulky l-3′ group interferes with incorporation due to steric conflict at residue 184. Increased affinity does not correlate with efficacy for this class of inhibitors. The l-AZA prodrug, 11, demonstrated significant anti-HIV activity with an EC50 value of 1.4 μM, but incorporation studies with HIV-RT show a lack of significant incorporation of l-AZA. Cellular pharmacology studies with 11 in PBM cells resulted in a significant enhancement in the delivery of nucleoside 10 and of its phosphates that may explain the observed anti-HIV activity. However, these l-nucleosides may be acting by a mechanism other than chain termination of HIV-RT. Further exploration of 3′-azido nucleosides including modification of 3′-azido sugar continues in our laboratories.
4. Experimental
4.1. Chemistry
Unless otherwise stated, all reactions were carried out under an atmosphere of dry argon or nitrogen in oven-dried glassware. Anhydrous solvents were purchased from EMD Chemicals Inc (E. Merck, Darmstadt, Germany). Unless noted otherwise, the reagents and materials used were obtained from commercial suppliers. Thin layer chromatography (TLC) was carried out on GHLF 250 μm silica gel plates item 21521 from Analtech, Inc. (Newark, DE, USA). Plate layer chromatography (PLC) was employed for purification of some products, item 02013 from Analtech, Inc. 1H NMR spectra were taken on a Varian Unity Plus 400 spectrometer (Varian, Inc., Polo Alto, CA, USA) at room temperature and reported in parts per million downfield from internal tetramethylsilane. Signal multiplicities are represented by s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quadruplet), br (broad), bs (broad singlet), m (multiplet). All J values are in hertz. Mass spectral analyses were performed on a Micromass TOF instrument (Hewlett-Packard HPLC driven electrospray MS instrument). Purity of final compounds was determined to be > 95%, using an analytical HPLC analyses performed on a Hewlett-Packard 1100 HPLC with a Phenomenex Gemini-NX column (2 mm × 50 mm, 3 μm, C18, 110 Å) and further supported by clean NMR spectra. Mobile phase flow was 0.5 mL/min with a 3.5 min initial hold, a 6.5 min gradient from 96% aqueous media (0.05% formic acid) to 96% CH3CN (0.05% formic acid), and a 15 min total acquisition time. Photo diode array detection was from 190 to 360 nm. Microwave reactions were carried out in a Biotage Initiator EXPUS.
4.1.1. General procedure A for the microwave-assisted transglycosylation reaction
To a solution of 5 (0.47 g, 1.24 mmol) and an appropriate purine base (1.48 mmol) in acetonitrile (3 mL) in an open air undried microwave reaction vessel was added bis(trimethylsilyl) acetamide (BSA) (1.4 mL, 5.7 mmol) the resulting heterogeneous mixture was sealed with a crimp cap and heated near reflux with a heat gun until a clear solution resulted (∼2-5 min). The microwave reaction vessel was placed in an ice bath for 5 min before TMSOTf (ranged from 0.14 mL, 0.77 mmol to 0.24 mL, 1.24 mmol) was added dropwise with gentle agitation while remaining in the ice bath. The resulting reaction mixture was heated to 130 °C under microwave irradiation for 3 min, cooled to room temperature, poured into saturated sodium bicarbonate (5 mL), extracted with ethyl acetate (3 × 5 mL), dried, purified by silica gel chromatography obtain the desired product as an oil.
4.1.2. General procedure B for the preparation of phosphoramidate prodrugs
To a solution of the appropriate nucleoside analogue (50 mg, 0.17 mmol) in THF (10 mL) was added t- BuMgCl (1 M, 0.6 mL) dropwise. The reaction mixture was stirred for 30 min at rt, then phenyl ethoxyalaninyl phosphorochloridate (0.6 mL, 0.6 mmol) was added. The reaction mixture was stirred for 3 h, then ethanol (1 mL) and ammonium chloride (0.2 mL) were added to quench the reaction. The reaction mixture was purified by column chromatography on silica gel to afford pure phosphoramidate as an approximate 1:1 mixture of phosphorous diastereomers.
4.1.1.1. 5′-O-tert-Butyldimethylsilyl-2′-deoxy-β-L-thymidine (3)
To a solution of LdT (1 g, 4.12 mmol) in DMF (1 mL) at 0 °C was added imidazole (0.56 g, 8.24 mmol) followed by dropwise addition of TBSCl (0.62 g, 4.12 mmol) over a period of 10 min. The cooling bath was removed and the reaction was stirred toward room temperature overnight. The solvent was evaporated; the residue was diluted with ethyl acetate (20 mL), washed with water, then saturated sodium bicarbonate, and dried over sodium sulfate. The solvent was removed and the resulting solid was purified by column silica gel chromatography. Elution with ethyl acetate:hexane (1:1) gave 3 as a white solid; yield (1.25 g, 85%). 1H NMR (CDCl3): δ 8.49 (bs, 1H, NH), 7.47 (d, J = 1.2 Hz, 1H, H-6), 6.32 (dd, J = 5.6 Hz, J = 8.0 Hz, 1H, H-1′), 4.44-4.45 (m, 1H, H-3′), 4.00 (q, J = 2.4 Hz, 1H, H-4′), 3.85 (dd, J = 2.8 Hz, J = 11.2 Hz, 1H, H-5′), 3.80 (dd, J = 2.4 Hz, J = 11.2 Hz, 1H, H-5′), 2.34 (dd, J = 2.8 Hz, J = 5.6 Hz, 0.5H, H-5′), 2.31 (dd, J = 2.8 Hz, J = 6.0 Hz, 0.5H, H-5′), 2.20 (bs, 1H, OH), 2.05 (m, 1H, H-2′), 1.89 (d, J = 1.2 Hz, 3H, CH3), 0.90 (s, 9H, 3X CH3), 0.099 (s, 3H, CH3), 0.094 (s, 3H, CH3). LRMS (ESI): m/z calcd for C16H29N2O5Si [M+H]+: 357.2, observed 357.1.
4.1.1.2. 5′-O-tert-Butyldimethylsilyl-2′,3′-dideoxy-2,3′-anhydo-β-L-thymidine (4)
To a solution of 3 (0.8 g, 2.24 mmol) and triphenylphosphine (0.73 g, 2.78 mmol) at 80 °C in toluene (20 mL) was added diisopropyl azodicarboxylate (0.83 mL, 4.21 mmol). After 30 min, the reaction mixture was concentrated, dissolved in ethyl acetate (10 mL), filtered, and again concentrated to a white solid, which was purified by column silica gel chromatography. Elution with ehyl acetate:hexane (1:1 to 1:0) gave 4 as a white solid; yield (200 mg, 79%). 1H NMR (CDCl3): δ 6.92 (d, J = 1.2 Hz, 1H, H-6), 5.46 (d, J = 4.0 Hz, 1H, H-1′), 5.15 (s, 1H, H-3′), 4.21 (dt, J = 2.4 Hz, J = 7.6 Hz, 1H, H-4′), 3.69 (m, 2H, H-5′), 2.66 (d, J = 13.2 Hz, 1H, H-2′), 2.38 (dt, J = 3.6 Hz, J = 12.8 Hz, 1H, H-2′), 1.89 (s, 3H, CH3), 0.84 (s, 9H, 3X CH3), 0.03 (s, 3H, CH3), 0.02 (s, 3H, CH3). LRMS (ESI): m/z calcd for C16H27N2O4Si [M+H]+: 339.2, observed 339.1.
4.1.1.3. 5′-O-tert-Butyldiemthylsilyl-3′-azido-2′,3′-dideoxy-β-L-thymidine (5)
A solution of 4 (2 g, 5.90 mmol) and lithium azide (0.5 g, 10.21 mmol) in DMF (10 mL) at 120 °C was stirred for 2 days, then the solvent was removed and the resulting residue was dissolved in ethyl acetate (30 mL). The organic layer was washed with water, dried over sodium sulfate and purified by column silica gel chromatography. Elution with ethyl acetate:hexane (1:1) gave 5 as a white solid; yield (1.5 g, 66.6%). 1H NMR (CDCl3): δ 8.68 (s, 1H, NH), 7.40 (d, J = 1.2 Hz, 1H, H-6), 6.18 (t, J = 6.0 Hz, 1H, H-1′), 4.19-4.23 (m, 1H, H-3′), 3.90-3.95 (m, 2H, H-4′ and H-5′), 3.76 (dd, J = 2.0 Hz, J = 11.2 Hz, 1H, H-5′), 2.38-2.44 (m, 1H, H-2′), 2.17-2.23 (m, 1H, H-2′), 1.90 (d, J = 1.2 Hz, 3H, CH3), 0.91 (s, 9H, 3X CH3), 0.11 (s, 6H, 2X CH3). LRMS (ESI): m/z calcd for C16H28N5O4Si [M+H]+: 382.2, observed 382.1.
4.1.1.4. 9-(3-Azido-5′-O-tert-butyldimethylsilyl-2,3-dideoxy-α,β-L-ribofuranosyl)-6-chloro-9H-purine (6)
General procedure A was employed, purified by silica gel chromatography. Elution with ethyl acetate:hexane (1:3); yield (410 mg, 81%). β-isomer/α-isomer = 2:1. β-isomer: 1H NMR (CDCl3): δ 8.72 (s, 1H, H-8), 8.41 (s, 1H, H-2), 6.47 (dd, J = 2.4 Hz, J = 6.8 Hz, 1H, H-1′), 4.39-4.41 (m, 1H, H-3′), 4.37 (q, J = 2.8 Hz, 1H, H-4′), 3.72-3.80 (m, 2H, H-5′), 2.90 (m, 1H, H-2′), 2.63-2.66 (m, 1H, H-2′), 0.87-0.90 (m, 9H, 3X CH3), 0.084 (s, 6H, 2X CH3). α-isomer: 1H NMR (CDCl3): δ 8.71 (s, 1H, H-8′), 8.46 (s, 1H, H-2′), 6.41 (t, J = 6.0 Hz, 1H, H-1′), 4.45 (q, J = 6.8 Hz, 1H, H-4′), 4.06-4.08 (m, 1H, H-3′), 3.93 (dd, J = 3.2 Hz, J = 11.2 Hz, 1H, H-5′), 3.80 (dd, J = 2.4 Hz, J = 10.8 Hz, 1H, H-5′), 2.80-2.83 (m, 1H, H-2′), 2.57-2.64 (m, 1H, H-2′), 0.87 (s, 9H, 3X CH3), 0.08 (s, 6H, 2X CH3). LCMS (ESI): m/z calcd for C16H25ClN7O2Si [M+H]+: 410.1 observed 410.1.
4.1.1.5. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-6-chloro-9H-purine (7)
A mixture of TBAF (1 mL, 1 M in THF) and acetic acid (0.2 mL) was added to a solution of 6 (150 mg, 0.36 mmol) in THF (3 mL) and the resulting solution was stirred for 5 h. After removal of the solvent the resulting solid was purified by column silica gel chromatography. Elution first with ethyl acetate:hexane (1:1) then with methanol:dichloromethane (1:50) gave the β-isomer 7 as a white solid; yield (18.8 mg, 34.8%). 1H NMR (CDCl3): δ 8.79 (s, 1H, H-8), 8.72 (s, 1H, H-2), 6.47 (dd, J = 5.2 Hz, J = 6.4 Hz, 1H, H-1′), 4.60 (q, J = 6.0 Hz, 1H, H-4′), 4.02-4.05 (m, 1H, H-3′), 3.81 (dd, J = 3.6 Hz, J = 12.4 Hz, 1H, H-5′), 3.72 (dd, J = 3.2 Hz, J = 12.0 Hz, 1H, H-5′), 2.98 (m, 1H, H-2′), 2.59 (m, 1H, H-2′). 13C-NMR (CDCl3): 151.7, 151.2, 150.2, 145.7, 131.7, 85.7, 85.1, 61.3, 60.6, 37.2. HRMS (EI): m/z calcd for C10H10ClN7O2 [M+H]+: 296.0622, found 296.0657.
4.1.1.6. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-6-chloro-9H-purine-5′-O-[phenyl-(ethoxy-L-alaninyl)]-phosphate (8)
General procedure B was employed and purified with silica gel chromatography. Elution with ethyl acetate:hexane (1:1); yield (20 mg, 27.0%.). 1H NMR (CD3OD): δ 8.72, 8.69 (ds, 1H, H-8), 8.71, 8.65 (ds, 1H, H-2), 7.25 (t, J = 8.0 Hz, 2H, phenyl-H), 7.08-7.16 (m, 3H, phenyl-H), 6.45-6.51 (m, 1H, H-1′), 4.74 (q, J = 6.0 Hz, 0.4 Hz, H-4′), 4.63 (q, J = 0.6 Hz, 0.6 Hz, H-4′), 4.26-4.39 (m, 2H, H-3′ and H-5′), 4.16-4.23 (m, 1H, H-5′), 3.99-4.11 (m, 2H, CH2), 3.71-3.88 (m, 1H, H-2′), 3.01-3.12 (m, 1H, H-2′), 2.60-2.72 (m, 1H, CH), 1.13-1.34 (m, 6H, 2X CH3). HRMS (EI): m/z calcd for C21H24ClN8O6P [M+H]+: 551.1323, found 551.1316.
4.1.1.7. 3′-Azido-2′,3′-dideoxy-β-L-adensoine (10)
A solution of 7 (100 mg, 0.33 mmol) in saturated ammonia in methanol (10 mL) was heated to 110 °C in steel bomb. The reaction mixture was heated overnight then concentrated and purified by column chromatography on silica gel. Gradient elution with dichloromethane:methanol (20:1) to (10:1) gave 10 as a white solid; yield (67 mg, 72%). 1H NMR (DMSO-d6): δ 8.34 (s, 1H, H-8), 8.13 (s, 1H, H-2), 7.34 (bs, 2H, NH2), 6.29 (t, J = 6.4 Hz, 1H, H-1′), 5.34 (t, J = 6.0 Hz, 1H, OH), 4.61-4.65 (m, 1H, H-3′), 3.92 (q, J = 4.0 Hz, 1H, H-4′), 3.54-3.64 (m, 2H, H-5′), 2.92-2.99 (m, 1H, H-2′), 2.45-2.51 (m, 1H, H-2′). HRMS (EI): m/z calcd for C10H12N8O2 [M+H]+: 277.1161, found 277.1155.
4.1.1.8. 3′-Azido-2′, 3′-dideoxy-β-L-adenosine-5′-O-[phenyl-(ethoxy-L-alaninyl)]-phosphate (11)
General procedure B was employed and purified by silica gel chromatography. Elution with dichloromethane:methanol (10:1); yield (20 mg, 35%). 1H NMR (CD3OD): δ 8.28, 8.22 (ds, 1H, H-8), 8.19, 8.18 (ds, 1H, H-2), 7.27 (t, J = 8.4 Hz, 2H, phenyl-H), 7.12-7.20 (m, 3H, phenyl-H), 6.33-6.39 (m, 1H, H-1′), 4.57-4.71 (m, 1H, H-3′), 4.32-4.37 (m, 1H, H-5′), 4.24-4.29 (m, 1H, H-5′), 4.11-4.18 (m, 1H, H-4′), 3.99-4.10 (m, 2H, CH2), 3.77-3.90 (m, 1H, CH), 2.89-3.01 (m, 1H, H-2′), 2.53-2.64 (m, 1H, H-2′), 1.22-1.26 (m, 3H, CH3), 1.12-1.19 (m, 3H, CH3). HRMS (EI): m/z calcd for C21H26N9O6P [M+H]+: 532.1821, found 532.1814.
4.1.1.9. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-6-methoxyl-9H-purine (9)
A solution of 7 (30 mg, 0.11 mmol) in methanol (2 mL) was added sodium methoxide (25%, 1 mL). The reaction mixture was stirred overnight, carefully neutralized with Dowex 25 resin, filtered. The solvent was concentrated and the resulting residue was purified by column silica gel chromatography. Elution with dichloromethane:methanol (10:1) gave 9 as a clear oil; yield, (15 mg, 51%). 1H NMR (CD3OD): δ 8.50 (s, 1H, H-8), 8.49 (s, 1H, H-2), 6.41 (t, J = 6.4 Hz, 1H, H-1′), 4.58-4.62 (m, 1H, H-3′), 4.15 (s, 3H, OCH3), 4.03 (q, J = 3.2 Hz, 1H, H-4′), 3.81 (dd, J = 3.6 Hz, J = 12.4 Hz, 1H, H-5′), 3.71 (dd, J = 4 Hz, J = 12.4 Hz, 1H, H-5′), 2.94-3.01 (m, 1H, H-2′), 2.54-2.61 (m, 1H, H-2′). HRMS (EI): m/z calcd for C11H13N7O3 [M+H]+: 292.1158, found 292.1152.
4.1.1.10. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-6-methylamino-9H-purine (12)
A solution of 7 (50 mg, 0.16 mmol) in methylamine (1 M in THF, 3 mL) was heated at 80 °C in a steel bomb with stirring for 24 h. The reaction mixture was concentrated to a residue and purified by column chromatography on silica gel. Elution with dichloromethane:methanol (10:1) gave 12 as a oily product; yield (30 mg, 61.2%). 1H NMR (CD3OD): δ 8.22 (s, 1H, H-8), 8.19 (s, 1H, H-2), 6.30 (t, J = 6.8 Hz, 1H, H-1′), 4.54-4.57 (m, 1H, H-3′), 4.03 (q, J = 2.8 Hz, 1H, H-4′), 3.81 (dd, J = 3.2 Hz, J = 12.8 Hz, 1H, H-5′), 3.70 (dd, J = 3.2 Hz, J = 12.4 Hz, 1H, H-5′), 3.05 (bs, 3H, CH3), 2.88-2.95 (m, 1H, H-2′), 2.47-2.53 (m, 1H, H-2′). HRMS (EI): m/z calcd for C11H12N8O2 [M+H]+: 291.1318, found 291.1312.
4.1.1.11. N2-Isobutyryl-5′-O-tert-butyldimethylsilyl-3′-azido-2′, 3′-dideoxy-α,β-L-guanosine (13)
General procedure B was employed and purified by silica gel chromatography, Elution with dichlromethane:methanol (30:1) yield (700 mg, 37%). 1H NMR (CDCl3): δ 12.14 (s, 1H, NH-3), 9.02 (s, 1H, NH), 7.94 (s, 1H, H-8), 6.09 (t, J = 6.4 Hz, 1H, H-1′), 4.37 (q, J = 4.8 Hz, 1H, H-3′), 3.99 (q, J = 4 Hz, 1H, H-4′), 3.82 (dd, J = 3.6 Hz, J = 11.6 Hz, 1H, H-5′), 3.77 (dd, J = 2.8 Hz, J = 11.2 Hz, 1H, H-5′), 2.60-2.75 (m, 2H, CH and H-2′), 2.42-2.48 (m, 1H, H-2′), 1.25 (d, J = 3.6 Hz, 3H, CH3), 1.23 (d, J = 3.6 Hz, 3H, CH3), 0.87 (s, 9H, 3X CH3), 0.067 (s, 3H, CH3), 0.060 (s, 3H, CH3). Alpha/beta = 1/4. LCMS (ESI): m/z calcd for C20H33N8O4Si [M+H]+: 477.2, observed 477.0.
4.1.1.12. 5′-O-tert-Butyldimenthylsilyl-3′-azido-2′,3′-dideoxyguanosine (14)
A solution of 13 (140 mg, 0.29 mmol) in saturated NH3/CH3OH (5 mL) was heated at 110 °C overnight with stirring in steel bomb. The reaction mixture was concentrated to a residue and purified column chromatography on silica gel. Elution with dichloromethane:methanol (10:1) gave 14 as a white solid; yield (0.10 g, 85%). Alpha/beta = 1/4. 1H NMR (CD3OD): δ 7.90 (s, 1H, H-8), 6.14 (dd, J = 5.2 Hz, J = 6.8 Hz, 1H, H-1′), 4.50 (q, J = 6.4 Hz, 1H, H-3′), 3.94 (q, J = 4.4 Hz, 1H, H-4′), 3.83-3.85 (m, 2H, H-5′), 2.77-2.84 (m, 1H, H-2′), 2.46-2.53 (m, 1H, H-2′), 0.88 (s, 9H, 3X CH3), 0.05 (s, 3H, CH3), 0.06 (s, 3H, CH3). LCMS (ESI) Calcd for C16H27N8O3Si [M+H]+: 407.2, observed 407.1.
4.1.1.13. 3′-Azido-2′,3′-dideoxy-β-L-guanosine (15)
To a solution of 14 (200 mg, 0.49 mmol) in THF (5 mL) at 0 °C was added TBAF/AcOH (2 mL, 1 M in THF/0.4 mL). The reaction mixture was stirred for 6 h at 0 °C. The reaction mixture was concentrated to a residue and purified gradient column chromatography on silica gel. Elution with dichloromethane:methanol (50:1) to (50:5) gave β isomer 15 as a white solid; yield (43 mg, 30%). 1H NMR (CD3OD): δ 7.93 (s, 1H, H-8), 6.16 (t, J = 6.4 Hz, 1H, H-1′), 4.52-4.56 (m, 1H, H-3′), 3.96 (q, J = 4.4 Hz, 1H, H-4′), 3.77 (dd, J = 3.2 Hz, J = 11.6 Hz, 1H, H-5′), 3.70 (dd, J = 4.0 Hz, J = 12.0 Hz, 1H, H-5′), 2.80-2.87 (m, 1H, H-2′), 2.44-2.50 (m, 1H, H-5′). HRMS (EI): m/z calcd for C10H12N8O3 [M+H]+: 293.1110, found 293.1105.
4.1.1.14. 3′-Azido-2′, 3′-dideoxy-β-L-guanosine-5′-O-[phenyl-(ethoxy-L-alaninyl)]-phosphate (16)
General procedure B was employed and purified with column chromatography on silica gel. Elution with dichloromethane:methanol (6:1), yield (26 mg, 46%). The faster moving compound: 1H NMR (CD3OD): δ 7.82 (s, 1H, H-8), 7.14-7.34 (m, 5H, phenyl-H), 6.15 (t, J = 6.4 Hz, 1H, H-1′), 4.55-4.59 (m, 1H, H-3′), 4.33-4.38 (m, 1H, H-4′), 4.25-4.30 (m, 1H, H-5′), 4.03-4.21 (m, 3H, CH2 and H-5′), 3.82-3.88 (m, 1H, CH), 2.77-2.84 (m, 1H, H-2′), 2.44-2.51 (m, 1H, H-2′), 1.15-1.26 (m, 6H, CH3 and CH3). HRMS (EI): m/z calcd for C21H26N9O7P [M+H]+: 548.1771, found 548.1765.
The slower moving compound: 1H NMR (CD3OD): δ 7.86 (s, 1H, H-8), 7.29-7.34 (m, 2H, phenyl-H), 7.14-7.20 (m, 3H, phenyl-H), 6.17-6.21 (t, J = 4.8 Hz, 1H, H-1′), 4.65-4.70 (m, 1H, H-3′), 4.28-4.41 (m, 2H, H-4′ and H-5′), 4.05-4.14 (m, 3H, CH2 and H-5′), 3.88-3.92 (m, 1H, CH), 2.90-2.96 (m, 1H, H-2′), 2.48-2.54 (m, 1H, H-2′), 1.24 (dd, J = 1.2 Hz, J = 7.2 Hz, 3H, CH3), 1.15 (t, J = 7.2 Hz, 3H, CH3). LRMS (ESI): m/z calcd for C21H27N9O7P [M+H]+: 548.2, observed 548.0.
4.1.1.15. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-2-amino-6-chloropurine (17)
General procedure A was employed and purified by column chromatography on silica gel. Elution with ethyl acetate:hexane (1:1); yield (300 mg, 54% as an 1/1 α/β mixture). β-isomer: 1H NMR (CDCl3): δ 8.05 (s, 1H, H-8), 6.21 (t, J = 6.0 Hz,1H, H-1′), 5.12 (s, 2H, NH2), 4.38-4.42 (m, 1H, H-3′), 4.02 (q, J = 4.0 Hz, 1H, H-4′), 3.86 (dd, J = 4.0 Hz, J = 11.2 Hz, 1H, H-5′), 3.79 (dd, J = 3.2 Hz, J = 8.4 Hz, 1H, H-5′), 2.70-2.77 (m, 1H, H-2′), 2.47-2.54 (m, 1H, H-2′), 0.89 (s, 9H, 3X CH3), 0.08 (s, 6H, 2X CH3).
α-isomer: 1H NMR (CDCl3): δ 8.04 (s, 1H, H-8), 6.24 (dd, J = 3.2 Hz, J = 7.2 Hz, 1H, H-1′), 5.12 (s, 2H, NH2), 4.29-4.32 (m, 2H, H-3′ and H-4′), 3.76 (dd, J = 3.2 Hz, J = 12.4 Hz, 1H, H-5′), 3.71 (dd, J = 4.4 Hz, J = 11.2 Hz, 1H, H-5′), 2.81-2.88 (m, 1H, H-2′), 2.61 (dt, J = 3.6 Hz, J = 14.0 Hz, 1H, H-2′), 0.90 (s, 9H, 3X CH3), 0.08 (s, 6H, 2X CH3). LRMS (ESI): m/z calcd for C16H26ClN8O2Si [M+H]+: 425.2. observed 425.0.
To a solution of 9-(5-O-tert-butyldimethylsilyl-3-azido-2,3-dideoxy-α,β-L-ribofuranosyl)-2-amino-6-chloropurine (100 mg, 0.23 mmol) from above (as the α/β mixture) in THF (2 mL) was added a mixture of TBAF (1 mL, 1 M in THF) and acetic acid (0.2 mL). The reaction mixture was stirred for 4 h, concentrated to a residue, and purified by column chromatography on silica gel. Elution with ethyl acetate:hexane = 1:1 to 5:1 gave the β isomer 17 as a white solid; yield (20 mg, 27%).
1H NMR (CD3OD): δ 8.27 (s, 1H, H-8), 6.24 (t, J = 6.4 Hz, 1H, H-1′), 4.57-4.61 (m, 1H, H-3′), 3.98 (q, J = 3.6 Hz, 1H, H-4′), 3.79 (dd, J = 4.0 Hz, J = 12.4 Hz, 1H, H-5′), 3.71 (dd, J = 4.0 Hz, J = 12.0 Hz, 1H, H-5′), 2.89-2.95 (m, 1H, H-2′), 2.48 (m, 1H, H-2′). HRMS (EI): m/z calcd for C10H11ClN8O2 [M+H]+: 311.0771, found 311.0766.
4.1.1.16. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-2-amino-6-chloropurine-5-O-[phenyl-(ethoxy-L-alaninyl)]-phosphate (18)
General procedure B was employed and purified by column chromatography on silica gel. Elution with ethyl acetate:hexane (1:1 to 5:1), (10 mg, 40%). 1H NMR (CD3OD): δ 8.18 and 8.14 (ds, 1 H), 7.25-7.32 (m, 2 H), 7.12-7.18 (m, 3 H), 6.23-6.28 (m, 1 H), 4.61-4.75 (m, 1 H), 4.25-4.43 (m, 2 H), 4.02-4.17 (m, 3 H), 3.79-3.90 (m, 1 H), 2.91-3.06 (m, 1 H), 2.49-2.58 (m, 1 H), 1.22-1.26 (m, 3 H), 1.14-1.18 (m, 3 H). HRMS (ESI) Calcd for C21H25ClN9O6P: 565.14; observed (M + 1): 566.33. HRMS (EI): m/z calcd for C21H25ClN9O6P [M+H]+: 566.1432, found 566.1425.
4.1.1.17. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-2-amino-6-methoxypurine (19)
To a solution of 17 (50 mg, 0.16 mmol) in methanol (5 mL) was added sodium methoxide in methanol (4.6 M, 0.2 mL) at rt. The reaction mixture was stirred overnight, concentrated to a residue and purified by column chromatography on silica gel. Elution with ethyl acetate:hexane (1:1) gave 19 as a oily product; yield (35 mg, 71%). 1H NMR (CD3OD): δ 8.01 (s, 1H, H-8), 6.20 (t, J = 6.8 Hz, 1H, H-1′), 4.55-4.58 (m, 1H, H-3′), 4.02 (s, 4H, CH3and H-4′), 3.80 (dd, J = 3.6 Hz, J = 12.4 Hz, 1H, H-5′), 3.70 (dd, J = 3.6 Hz, J = 12.4 Hz, 1H, H-5′), 2.87-2.94 (m, 1H, H-2′), 2.43-2.49 (m, 1H, H-2′). HRMS (EI): m/z calcd for C11H14N8O3 [M+H]+: 307.1267, found 307.1261.
4.1.1.18. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl) -2,6-diaminopurine (20)
A solution of 17 (25 mg, 0.08 mmol) in saturated ammonia in methanol (3 mL) was heated with stirring in a pressure tube at 120 °C for 14 h. The reaction mixture was cooled to room temperature, concentrated to a residue and purified by column chromatography on silica gel. Elution with dichloromethane:methanol (10:1) gave 20 as a white solid; yield (20 mg, 87%). 1H NMR (CD3OD): δ 7.91 (s, 1H, H-8), 6.17 (t, J = 6.8 Hz, 1H, H-1′), 4.53-4.57 (m, 1H, H-3′), 4.03 (q, J = 2.8 Hz, 1H, H-5′), 3.82 (dd, J = 3.2 Hz, J = 12.4 Hz, 1H, H-5′), 3.70 (dd, J = 3.2 Hz, J = 12.4 Hz, 1H, H-5′), 2.87-2.94 (m, 1H, H-2′), 2.41 (dq, J = 3.6 Hz, J = 14 Hz, 1H, H-2′). HRMS (EI): m/z calcd for C10H13N9O2 [M+H]+: 292.1270, found 292.1263.
4.1.1.19. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl) -2,6-diaminopurine-5-O-(phenyl-[ethoxyl-L-alaninyl])-phosphate (22)
General procedure B was employed and purified with silica gel chromatography. Elution with dichloromethane:methanol (10:1) to afford 22 as a white solid (20 mg, 60%). 1H NMR (CDCl3): δ 7.55 and 7.57 (s, 1H, H-8), 7.10-7.33 (m, 5H, phenyl-H), 6.06 (q, J = 6.8 Hz, 1H, H-1′), 5.51 (bs, 2H, NH2), 5.12 (bs, 1H, H-3′), 5.01 (bs, 1H, H-4′), 4.52-4.74 (m, 3H, NH and H-5′), 3.93-4.27 (m, 5H, CH2, NH2, and CH), 2.89-3.17 (m, 1H, H-2′), 2.34-2.41 (m, 1H, H-2′), 1.29-1.33 (m, 3 H), 1.18-1.22 (m, 3 H). HRMS (EI): m/z calcd for C21H27N10O6P [M+H]+: 547.1930, found 547.1923.
4.1.1.20. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-2-amino-6-methylaminopurine (21)
A solution of 17 (50 mg, 0.16 mmol) in methylamine (1M in THF, 3 mL) was heated with stirring in a steel bomb at 110 °C overnight. The reaction mixture was concentrated to a residue and purified by column chromatography on silica gel. Elution with dichloromethane:methanol (10:1) gave 21 as a white solid; yield (35 mg, 71%). 1H NMR (CD3OD): δ 7.85 (s, 1H, H-8), 6.15 (t, J = 7.2 Hz, 1H, H-1′), 4.52-4.56 (m, 1H, H-3′), 4.03 (q, J = 2.8 Hz, 1H, H-4′), 3.82 (dd, J = 3.2 Hz, J = 12.4 Hz, 1H, H-5′), 3.70 (dd, J = 2.8 Hz, J = 12.0 Hz, 1H, H-5′), 3.00 (bs, 3H, CH3), 2.87-2.94 (m, 1H, H-2′), 2.39 (dq, J = 3.6 Hz, J = 14 Hz, 1H, H-2′). HRMS (EI): m/z calcd for C11H15N9O2 [M+H]+: 306.1427, found 306.1420.
4.1.1.21. 9-(5-tert-Butyldimethylsilyl-3-azido-2,3-dideoxy-α, β-L-ribofuranosyl)-2,6-dichloropurine (23)
General procedure A was employed and purified by column chromatography on silica gel. Elution with ethyl acetate:hexane (1:2); yield (400 mg, 26%). α isomer/β isomer = 1/1.4. β-isomer: 1H NMR (CDCl3): δ 8.40 (s, 1H, H-8), 6.42 (dd, J = 2.0 Hz, J = 7.2 Hz, 1H, H-1′), 4.38-4.43 (m, 2H, H-3′ and H-4′), 3.76 (dd, J = 3.2 Hz, J = 11.6 Hz, 1H, H-5′), 3.70 (dd, J = 4.4 Hz, J = 11.6 Hz, 1H, H-5′), 2.88-2.95 (m, 1H, H-2′), 2.54-2.59 (m, 1H, H-2′), 0.88 (s, 9H, 3X CH3), 0.07 (s, 6H, 2X CH3). α-isomer: 1H NMR (CDCl3): δ 8.48 (s, 1H, H-8), 6.37 (t, J = 5.6 Hz, 1H, H-1′), 4.37 (q, J = 2.4 Hz, 1H, H-4′), 4.06-4.09 (m, 1H, H-3′), 3.95 (dd, J = 3.2 Hz, J = 11.2 Hz, 1H, H-5′), 3.80 (dd, J = 2.8 Hz, J = 11.6 Hz, 1H, H-5′), 2.72 (m, 1H, H-2′), 2.61-2.64 (m, 1H, H-2′), 0.86 (s, 9H, 3X CH3), 0.084 (s, 3H, CH3), 0.080 (s, 3H, CH3). LRMS (ESI): m/z calcd for C16H24Cl2N7O2Si [M+H]+: 444.1; observed 444.1.
4.1.1.22. 9-(3′-Azido-2′,3′-dideoxy-β-L-ribofuranosyl) -2,6-dichloropurine (24)
To a solution of 23 (200 mg, 0.45 mmol) in THF (10 mL) was added TBAF/AcOH (2 mL, 1 M in THF/0.4 mL) at rt. The resulting solution was stirred for 10 h, then concentrated to a residue and purified by column chromatography on silica gel. Elution with ethyl acetate:hexane (1:1) gave the β-isomer 24 as a white solid; yield (60 mg, 40.5%). 1H NMR (CD3OD): δ 8.79 (s, 1H, H-8), 6.40 (dd, J = 5.2 Hz, J = 6.8 Hz, 1H, H-1′), 4.55 (q, J = 6.4 Hz, 1H, H-4′), 4.01-4.04 (m, 1H, H-3′), 3.81 (dd, J = 3.6 Hz, J = 12.4 Hz, 1H, H-5′), 3.72 (dd, J = 3.2 Hz, J = 12 Hz, 1H, H-5′), 2.92-2.99 (m, 1 H, H-2′), 2.59-2.66 (m, 1H, H-2′). HRMS (EI): m/z calcd for C10H9Cl2N7O2 [M+H]+: 330.0273, found 330.0267.
4.1.1.23. 9-(5-O-tert-Butyldimethylsilyl-3-azido-2,3-dideoxy-α, β-L-ribofuranosyl)-2-fluoro-6-aminopurine (25)
General procedure A was employed purified by column chromatography on silica gel. Elution with dichloromethane:methanol (10:1); yield (500 mg, 46%). α/β isomer = 1/1. β isomer: 1H NMR (CDCl3): δ 8.07 (s, 1H, H-8), 6.52 (bs, 2H, NH2), 6.25 (t, J = 6.0 Hz, 1H, H-1′), 4.34 (m, 1H, H-3′), 4.00 (q, J = 4.0 Hz, 1H, H-4′), 3.79 (dd, J = 3.2 Hz, J = 11.2 Hz, 1H, H-5′), 3.72 (dd, J = 4.4 Hz, J = 13.2 Hz, 1H, H-5′), 2.74-2.79 (m, 1H, H-2′), 2.50-2.55 (m, 1H, H-2′), 0.89 (s, 9H, 3X CH3), 0.07 (s, 6H, 2X CH3). α-isomer: 1H NMR (CDCl3): δ 8.06 (s, 1H, H-8), 6.55 (s, 2H, NH2), 6.31 (dd, J = 2.8 Hz, J = 7.2 Hz, H-1′), 4.40 (q, J = 5.6 Hz, 1H, H-4′), 4.34 (m, 1H, H-3′), 3.92 (dd, J = 4.0 Hz, J = 11.2 Hz, 1H, H-5′), 3.69 (dd, J = 4.4 Hz, J = 11.2 Hz, 1H, H-5′), 2.83 (m, 1H, H-2′), 2.56 (m, 1H, H-2′), 0.89 (s. 9H, 3X CH3), 0.08 (s, 6H, 2X CH3). LRMS (ESI): m/z calcd for C16H26FN8O2Si [M+H]+: 409.2, observed 409.0.
4.1.1.24. 9-(3-Azido-2,3-dideoxy-β-L-ribofuranosyl)-2-fluoro-6-aminopurine (26)
To a stirred solution of 25 (120 mg, 0.29 mmol) in THF (4 mL) was added a mixture of TBAF/AcOH (1 mL, 1 M in THF/0.1 mL). The resulting solution was stirred at 0 °C for 30 min then the cooling bath was removed and stirred toward room temperature for 4 h. The reaction was concentrated to a residue and purified by column chromatography on silica gel. Elution with ethyl acetate:hexane:methanol (60:20:3.5) with trace ammonium hydroxide gave 26 as a white solid; yield (30 mg, 34%). 1H NMR (CD3OD): δ 8.24 (s, 1H, H-8), 6.24 (t, J = 6.4 Hz, 1H, H-1′), 4.52-4.55 (m, 1H, H-3′), 4.00 (q, J = 3.6 Hz, 1H, H-4′), 3.80 (dd, J = 3.2 Hz, J = 12 Hz, 1H, H-5′), 3.70 (dd, J = 4.0 Hz, J = 12.4 Hz, 1H, H-5′), 2.87-2.93 (m, 1H, H-2′), 2.49-2.55 (m, 1H, H-2′). HRMS (EI): m/z calcd for C10H11FN8O2 [M+H]+: 295.1067, found 295.1060.
4.1.1.25. 3′-Azido-2′-3′-dideoxy-β-L-thymidine (27)
To a mixture of TBAF (1 mL, 1 M in THF) and acetic acid (0.2 mL) was added to a solution of 5 (200 mg, 0.52 mmol) in THF (10 mL). The reaction mixture was stirred at rt for 5 h, evaporated the solvent, purified by column chromatography on silica gel, eluting with ethyl acetate:hexane (1:1 to 2:1) gave 27 as a white solid (100 mg, 71%). 1H NMR (CD3OD): δ 7.78 (d, J = 1.2 Hz, 1 H, H-6), 6.13 (t, J = 6.4 Hz, 1 H, H-1′), 4.31-4.36 (m, 1 H, H-3′), 3.87-3.90 (m, 1 H, H-4′), 3.79 (dd, J = 2.8 Hz, J = 12.4 Hz, 1 H, H-5′), 3.70 (dd, J = 2.8 Hz, J = 11.6 Hz, 1 H, H-5′), 2.31-2.43 (m, 2 H, H-2′), 1.85 (d, J = 1.2 Hz, 3 H, CH3). LRMS (ESI): m/z calcd for C10H14N5O4 [M+H]+: 268.1, observed 268.0.
4.1.1.26. 1-(3′-Azido-2′, 3′-dideoxy-β-L-thymidine)-O-(phenyl-[ethoxyl-L-alaniny])-phosphate (28)
To a solution of 27 (40 mg, 0.15 mmol) in THF (2 mL) at 0 °C was added N-methylimidazole (0.1 mL, 1.2 mmol), then slowly added phenyl ethoxyalaninyl phosphorochloridate (0.45 mL, 0.45 mmol). The reaction mixture was stirred at rt overnight, diluted with ethyl acetate (10 mL), washed with Sat. sodium bicarbonate, dried over sodium sulfate, concentrated, purified by column chromatography on silica gel, eluting with ethyl acetate : hexane (1 : 1), to get 28 as oily product (34 mg, 43%). 1H NMR (CD3OD): δ 7.55 (d, J = 1.2 Hz, 0.5 H, H-6), 7.47 (d, J = 1.2 Hz, 0.5 H, H-6), 7.32-7.36 (m, 2 H, phenyl-H), 7.16-7.24 (m, 3 H, phenyl-H), 6.13 (q, J = 6.8 Hz, 1 H, H-1′), 4.25-4.44 (m, 3 H, H-3′, H-5′), 4.04-4.16 (m, 3 H, H-4′, CH2, H-4′), 3.89-3.95 (m, 1 H, CH), 2.22-2.39 (m, 2 H, H-2′), 1.85 and 1.81 (d, J = 1.2 Hz, 3 H, CH3), 1.18-1.35 (m. 6 H, 2 × CH3). LRMS (ESI): m/z calcd for C21H28N6O8P [M+H]+: 523.1, observed 523.06.
4.2 Pharmacological profiling
4.2.1 HIV-1 RT inhibition assays
The WT HIV-1 RT (EC2.7.7.49) and M184V RT enzymes were purified as described previously [38,39]. The protein concentration of these enzymes was determined spectrophotometrically at 280 nm using an extinction coefficient (ε280) of 260450 M−1cm−1, and by Bradford protein assays (Sigma-Aldrich, St. Louis, MO). The RNA- and DNA-dependent DNA polymerase activities of the purified WT and mutant enzymes were essentially identical (data not shown). Deoxynucleotide triphosphates were purchased from GE Healthcare (Piscataway, NJ), and [γ-32P] ATP was acquired from PerkinElmer Life Sciences (Boston, MA). DNA heteropolymeric oligonucleotides were synthesized by IDT (Coralville, IA). A 20-nucleotide (nt) DNA primer (5′-TCGGGCGCCACTGCTAGAGA-3′) and a 57-nt DNA template derived from the HIV-1 PBS region (5′-CTCAGACCCTTTTAGTCAGAATGGAAANTCTCTAGCAGTGGCGCCCGAACAGGGACA-3′) were used in steady-state and pre-steady state DNA synthesis experiments. Four DNA templates were synthesized, each of which contained a different nucleotide at position 30 (indicated by N in the sequence). This strategy allowed us to evaluate the kinetics of single nucleotide incorporation for each L-3′-azidopurine nucleotide triphosphate (L-3′-azido-NTP) in essentially the same sequence context and using the same 20-nt primer.
4.2.2 Pre-steady state kinetic analyses
The typical experiment was performed at 37 °C in 50 mM Tris-HCl (pH 7.5) containing 50 mM KCl, 10 mM MgCl2, and varying concentrations of nucleotide. All concentrations reported refer to the final concentrations after mixing. HIV-1 RT (200 nM) was pre-incubated with 20 nM T/P substrate prior to mixing with nucleotide and divalent metal ions to initiate the reaction. At various timepoints (1-60 minutes) 5 μL aliquots were removed and quenched with 10 μL of gel loading buffer. Products were separated from substrates as described above. The disappearance of substrate (20mer) and the formation of product (21mer) were quantified using Quantity One Software. Data were fitted by nonlinear regression with Sigma Plot software (Systat Software, Inc., San Jose, CA) using the appropriate equations (20) [40]. The apparent burst rate constant (kobs) for each particular concentration of nucleotide was determined by fitting the time courses for the formation of product (21mer) using the following equation: [21mer] = A[1-exp (-kobst)], where A represents the burst amplitude. The turnover number (kpol) and apparent dissociation constant for nucleotide (Kd) were then obtained by plotting the apparent catalytic rates (kobs) against nucleotide concentration and fitting the data with the following hyperbolic equation: kobs = (kpol [dNTP])/([dNTP] + Kd). Catalytic efficiency was calculated as the ratio of turnover number over dissociation constant ([kpol/Kd]). Selectivity for natural dNTP versusL-3′-azido-NTP was calculated as the ratio of catalytic efficiency of dNTP over that of L-3′-azido-NTP.
4.3 Computational Modeling
4.3.1 Preparation of reference systems
Starting crystal structure coordinates of HIV-1 RT/DNA duplex complexed with different ligands, (1RTD, 1TO5, 1N6Q) [37,41,42] were downloaded from the RCSBProtein Data Bank at www.pdb.org. Structure 1RTD, with T-TP bound was chosen as the reference receptor system for fitting all others using the Matchmaker function in Chimera [43]. An oxygen atom was added to the 3′-position of the primer and hydrogen atoms were added using the “Protonate 3D” function in MOE 2009.1 with default settings [44]. Residues within 8 Å of the active site were minimized within the MMFF94x force field to a shallow 1.0 Kcal gradient using stepwise decreasing tether strengths. A 3′-azido group was added to T-TP, typed for MMFF45 and similarly optimized to produce a “pre-incorporation” model of d-AZT-TP liganded in relation to the primer 3′-OH and the 2 catalytic Mg atoms. Catalytic site interactions were compared to the experimental structure of “post-incorporation”/pre-translocation AZT-MP-terminated DNA (1N6Q) [37]. The bases of the bound TP and its' complementary base of the template, were pair-wise modified from T-A to form, C-G, A-T, and G-C models using “Nucleic Acid Explorer “ in MOE.
4.3.2 Analysis of D/L substrate interactions
Sugar geometries of all d-3′-azido ligand interaction models described were modified to the L-stereochemistry in Sybyl 8.1 while keeping base and triphosphate positions fixed [46]. 3′-Azido substitution was fit into the receptor pocket and the entire ligand geometry was minimized to a 1 kcal gradient with MMFF95 atom types and charges. Steric surfaces, atomic distances, H-bond interactions, and 184 mutant rotamers were analyzed for all models using Chimera 1.4.1 and Discovery Studio 3.0. [43,47]
Highlights.
Microwave transglycosylation prepared l-3′-azido-2′,3′-dideoxypurine nucleosides.
The l-nucleosides had weak or no antiviral activity against HIV-1 and HBV.
Pre-steady-state kinetics and cellular pharmacology clarified the results.
An l-nucleoside phosphoramidate had anti-HIV-1 activity without significant toxicity.
Acknowledgments
This work was supported primarily by NIH grant 5R01-AI-071846, and in part by 5R37-AI-025899, 2P30-AI-050409, 5R37-AI-041980, and the Department of Veterans Affairs (to RFS). RFS is the founder and a major shareholder in RFS Pharma, LLC. JWM is a consultant to RFS Pharma, LLC, and holds share options in the company. Computational modeling was performed with assistance of the Emory School of Medicine Biomolecular Computing Resource (BIMCORE). Hardware and software was supported in part by Academic Excellence Grants from Sun Microsystems and Accelrys Corporation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.De Clercq E. Curr Opin Microbiol. 2005;8:552–560. doi: 10.1016/j.mib.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richman DD. Antiviral Research. 2006;71:117–121. doi: 10.1016/j.antiviral.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Smejkal J, Sorm F. Collect Czech Chem Commun. 1964;29:2809–2813. [Google Scholar]
- 4.Maury G. Chemother Antiviral Chem. 2000;11:165–190. doi: 10.1177/095632020001100301. [DOI] [PubMed] [Google Scholar]
- 5.Focher F, Spadari S, Maga G. Curr Drug Targets Infect Disord. 2003;3:41–53. doi: 10.2174/1568005033342163. [DOI] [PubMed] [Google Scholar]
- 6.(a) Beach JW, Jeong LS, Alves AJ, Pohl D, Kim HO, Chang CN, Doong SL, Schinazi RF, Cheng YC, Chu CK. J Org Chem. 1992;57:2217–2219. [Google Scholar]; (b) Cameron JM, Collins P, Daniel M, Storer R. Drugs Fut. 1993;18:319–323. [Google Scholar]
- 7.Schinazi RF, Lloyd RM, Jr, Nguyen MH, Cannon DL, McMillan A, Ilksoy N, Chu CK, Liotta DC, Bazmi HZ, Mellors JW. Antimicrob Agents Chemother. 1993;37:875–881. doi: 10.1128/aac.37.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Choi WB, Yeola S, Liotta DC, Schinazi RF, Painter GR, Davis M, Clair St, Furman M, Bioorg PA. Med Chem Lett. 1993;3:693–696. [Google Scholar]; (b) Furman PA, Painter GR. Int Antiviral News. 1995;3:74–77. [Google Scholar]
- 9.Nair V, Jahnke TS. Antimicrob Agents Chemother. 1995;39:1017–1029. doi: 10.1128/aac.39.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Xiang YJ, Cavalcanti S, Chu CK, Schinazi RF, Pai SB, Zhu YL, Cheng YC. Bioorg Med Chem Lett. 1995;5:877–880. [Google Scholar]; (b) Wang P, Hong JH, Cooperwood JS, Chu CK. Antiviral Res. 1998;40:19–44. doi: 10.1016/s0166-3542(98)00041-2. [DOI] [PubMed] [Google Scholar]
- 11.Graciet JC, Schinazi RF. Adv Antiviral Drug Des. 1999;3:1–68. [Google Scholar]
- 12.Kotra LP, Xiang YJ, Newton MG, Schinazi RF, Cheng YC, Chu CK. J Med Chem. 1997;40:3635–3644. doi: 10.1021/jm970275y. [DOI] [PubMed] [Google Scholar]
- 13.Chun BK, Schinazi RF, Cheng YC, Chu CK. Carbohydrate Research. 2000;328:49–59. doi: 10.1016/s0008-6215(99)00312-2. [DOI] [PubMed] [Google Scholar]
- 14.Lee K, Choi Y, Hong JH, Schinazi RF, Chu CK. Nucleosides Nucleotides. 1999;18:537–540. doi: 10.1080/15257779908041489. [DOI] [PubMed] [Google Scholar]
- 15.Lee K, Choi Y, Gullen E, Schlueter-Wirtz S, Schinazi RF, Cheng YC, Chu CK. J Med Chem. 1999;42:1320–1328. doi: 10.1021/jm980651u. [DOI] [PubMed] [Google Scholar]
- (a) Schinazi RF, Lloyd RM, Jr, Nguyen MH, Cannon DL, McMillan A, Ilksoy N, Chu CK, Liotta DC, Bazmi HZ, Mellors JW. Antimicrob Agents Chemother. 1993;37:875–881. doi: 10.1128/aac.37.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jarvis B, Faulds D. Drugs. 1999;58:101–141. doi: 10.2165/00003495-199958010-00015. [DOI] [PubMed] [Google Scholar]
- 17.Schinazi RF, McMillan A, Cannon D, Mathis R, Lioyd RM, Jr, Peck A, Sommadossi JP, Clair M, Wilson St J, Furman PA. Antimicrob Agents Chemother. 1992;36:2423–2431. doi: 10.1128/aac.36.11.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Cretton-Scott E, Zhou XJ, Bridges EG, Tennant B, Juodawlkis A, Gosselin G, Imbach JL, Pierra C, Dukhan D, Schinazi RF, Sommadossi JP, Bryant M. Antivir Ther. 1999;4:A124. [Google Scholar]; (b) Kim JW, Park SH, Louie SG. Ann Pharmacother. 2006;40:472–478. doi: 10.1345/aph.1G027. [DOI] [PubMed] [Google Scholar]
- 19.Mathé C, Gosselin G. Antiviral Research. 2006;71:276–281. doi: 10.1016/j.antiviral.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 20.Sluis-Cremer N, Koontz D, Bassit L, Hernez-Santiago BI, Detorio M, Rapp KL, Amblard F, Bondada L, Grier J, Coats SJ, Schinazi RF, Mellors JW. Antimicrob Agents Chemother. 2009;53:3715–3719. doi: 10.1128/AAC.00392-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang HW, Coats SJ, Bondada L, Amblard F, Detorio M, Asif G, Fromentin E, Solomon S, Obikhod A, Whitaker T, Sluis-Cremer N, Mellors JW, Schinazi RF. Bioorg Med Chem Lett. 2010;20:60–64. doi: 10.1016/j.bmcl.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman BD, Obikhod A, Detorio M, Zhang HW, Nettles J, Coats SJ, Schinazi RF, Mellors JW, Sluis-Cremer N. XVIII International HIV Drug Resistance Workshop; June 9-13; Fort Myers, Florida. 2009. Abstract 123. [Google Scholar]
- 23.Kappe CO, Dallinger D, Murphree SS. Practical microwave synthesis for organic chemists: strategies, instruments, and protocols. WILEY·VCH Verlag GmbH & Co. KGaA; Weinheim: 2009. [Google Scholar]
- 24.Bookser BC, Raffaele NB. J Org Chem. 2007;72:173–179. doi: 10.1021/jo061885l. [DOI] [PubMed] [Google Scholar]
- 25.Bryant ML, Bridges EG, Placidi L, Faraj A, Loi AG, Pierra C, Dukhan D, Gosselin G, Imbach JL, Hernez B, Juodawlkis A, Tennant B, Korba B, Cote P, Marion P, Cretton-Scott E, Schinazi RF, Sommadossi JP. Antimicrob Agents Chemother. 2001;45:229–235. doi: 10.1128/AAC.45.1.229-235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.String DN, Bridges EG, Placidi L, Faraj A, Loi AG, Pierra C, Dukhan D, Gosselin G, Imbach JL, Hernez B, Juodawlkis A, Tennant B, Korba B, Cote P, Cretton-Scott E, Schinazi RF, Myers M, Bryant ML, Sommadossi JP. Antivir Chem Chemother. 2001;12:119–129. [PubMed] [Google Scholar]
- 27.Pathak T. Chem Rev. 2002;102:1623–1667. doi: 10.1021/cr0104532. [DOI] [PubMed] [Google Scholar]
- 28.Perrone P, Daverio F, Valente R, Rajyaguru S, Martin JA, Leveque V, Pogam SL, Najera I, Klumpp K, Smith DB, McGuigan CJ. Med Chem. 2007;50:5463–5470. doi: 10.1021/jm070362i. [DOI] [PubMed] [Google Scholar]
- 29.(a) Bryant ML, Bridges EG, Placidi L, Faraj A, Loi AG, Pierra C, Benzaria S, Dukhan D, Gosselin G, Imbach JL, Hernez B, Juodawlkis A, Tennant B, Korba B, Cote P, Cretton-Scott E, Schinazi RF, Myers M. In: Frontiers in viral hepatitis. Schinazi RF, Sommadossi JP, Rice C, editors. Elsevier; 2003. pp. 245–261. [Google Scholar]; (b) Bryant ML, Bridges EG, Placidi L, Faraj A, Loi AG, Pierra C, Benzaria S, Dukhan D, Gosselin G, Imbach JL, Hernez B, Juodawlkis A, Tennant B, Korba B, Cote P, Cretton-Scott E, Schinazi RF, Myers M, Sommadossi JP. Nucleosides, Nucleotides Nucleic acids. 2001;20:597–607. doi: 10.1081/NCN-100002336. [DOI] [PubMed] [Google Scholar]
- 30.March A, Lioux T, Mathé C, Imbach JL, Gosselin GJ. Chem Soc Perkin Trans. 1999;1:2249–2254. [Google Scholar]
- 31.(a) Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie MW, Hart GC, Smith GA, Hahn EF. Antimicrob Agents Chemother. 1990;34:1061–1067. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stuyver LJ, Lostia S, Adams M, Mathew JS, Pai BS, Grier S, Tharnish PM, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob Agents Chemother. 2002;46:3854–3860. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Antimicrob Agents Chemother. 1997;41:1715–1720. doi: 10.1128/aac.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.HBV Quantitative-PCR Assay. The HBV 7-day assay was performed in HepAD38 wild type cells as previously described (reference 31b). Compounds controls were prepared in medium without tetracycline added in duplicate at various concentrations. On day seven, total DNA was extracted using DNeasy 96 Tissue kit (Qiagen), HBV DNA was amplified by real-time PCR.
- 34.Ludwig J, Eckstein F. J Org Chem. 1989;54:631–635. [Google Scholar]
- 35.Herman BD, Obikhod A, Detorio M, Zhang HW, Nettles J, Coats SJ, Schinazi RF, Mellors JW, Sluis-Cremer N. Antiviral Therapy. 2009;14(Supp 1):A144. [Google Scholar]
- 36.Sluis-Cremer N, Arion D, Parikh U, Koontz D, Schinazi RF, Mellors JW, Parniak MA. J Biol Chem. 2005;280:29047–29052. doi: 10.1074/jbc.M503166200. [DOI] [PubMed] [Google Scholar]
- 37.Sarafianos SG, Clark AD, Jr, Das K, Tuske S, Birktoft JJ, Ilankumaran P, Ramesha AR, Sayer JM, Jerina DM, Boyer PL, Hughes SH, Arnold E. EMBO J. 2002;21:6614–6624. doi: 10.1093/emboj/cdf637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sluis-Cremer N, Sheen CW, Zelina S, Argoti Torres PS, Parikh UM, Mellors JW. Antimicrob Agents Chemother. 2007;51:48–53. doi: 10.1128/AAC.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parikh UM, Zelina S, Sluis-Cremer N, Mellors JW. AIDS. 2007;21:1405–1414. doi: 10.1097/QAD.0b013e3281ac229b. [DOI] [PubMed] [Google Scholar]
- 40.Herman BD, Votruba I, Holý A, Sluis-Cremer N, Balzarini J. J Biol Chem. 2010;285:12101–12108. doi: 10.1074/jbc.M109.096529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang H, Chopra R, Verdine GL, Harrison SC. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 42.Tuske S, Sarafianos SG, Clark AD, Jr, Ding J, Naeger LK, White KL, Miller MD, Gibbs CS, Boyer PL, Clark P, Wang G, Gaffney BL, Jones RA, Jerina DM, Hughes SH, Arnold E. Nat Struct Mol Biol. 2004;11:469–474. doi: 10.1038/nsmb760. [DOI] [PubMed] [Google Scholar]
- 43.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 44.Molecular Operating Environment (MOE) Chemical Computing Group; Montreal: 2009. [Google Scholar]
- 45.Chen FF, Wang F. Molecules. 2009;14:2656–2668. doi: 10.3390/molecules14072656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sybyl 8.1. Tripos; St. Louis: 2008. [Google Scholar]
- 47.Discovery Studio. Accelrys, Inc.; San Diego: 2010. [Google Scholar]