

Abstract
Prenylated indole alkaloids are a diverse group of fungal secondary metabolites and represent an important biosynthetic class. In this study we have identified new halogenated prenyl-indole alkaloids from an invertebrate-derived Malbranchea graminicola strain. Using Direct Analysis in Real Time (DART) Mass Spectrometry, these compounds were initially detected from spores of the fungus grown on agar plates, without the need for any organic extraction. Subsequently, the metabolites were isolated from liquid culture in artificial seawater. The structures of two novel chlorinated metabolites, named (−)-spiromalbramide and (+)-isomalbrancheamide B, provide additional insights into the assembly of the malbrancheamide compound family. Remarkably, two new brominated analogs, (+)-malbrancheamide C and (+)-isomalbrancheamide C, were produced by enriching the growth medium with bromine salts.
Introduction
The ever-growing group of prenylated indole alkaloid natural products is a ubiquitous biosynthetic class for fundamental investigations on many fronts. This collection contains both therapeutic and toxic compounds, such as the antitumor molecule tryprostatin A and the ergot alkaloid ergotamine. The structures, bioactivities, chemical syntheses and biosyntheses of this family have been the subject of research for several decades.1 The high degree of structural diversity is derived from different biosynthetic pathways to produce numerous scaffolds including dipeptides, terpenes, and aromatic polyketides. Within this group, the pattern observed for indole prenylation, such as the degree, site, and direction, adds an extra element of variety to the resulting molecules. While these compounds are commonly isolated from plants and bacteria, the majority of them originate from terrestrial and marine-derived fungi.1
Some noteworthy examples from fungi are those of non-ribosomal peptide synthase (NRPS) biosynthetic origin including the tryprostatins,2 fumitremorgins,3,4 and verruculogen5 (see Figure S1, Supporting Information). The majority of these structures are made up of tryptophan and another cyclic amino acid, condensed into a diketopiperazine (DKP) ring, and are further modified by prenyltransferase and oxidative tailoring enzymes.6 An important subgroup of these polycyclic structures contains a bicyclo[2.2.2]diazaoctane ring system, postulated to arise from an intramolecular Diels Alder reaction (IMDA).7,8 Two fungal genera, Aspergillus sp. and Pencillium sp., are the source of at least 50 metabolites with this core structure, including the brevianamides,9–11 stephacidins,12 notoamides,13–15 and paraherquamides16–18 (Figure S1). The myriad of bioactivities displayed by these compounds, including antitumor, insecticidal, antibacterial, anthelmintic, and antinematodal, sustains great interest in the class.
Another subset of these structures are members of the malbrancheamide family, including the halogenated compounds (+)-malbrancheamide (1)19 and (+)-malbrancheamide B (2), 20 and the non-halogenated precursor, (+)-premalbrancheamide (3).21 These were first isolated from a strain of Malbranchea aurantiaca, cultured from bat guano collected in a Mexican cave. The compounds 1 and 2 are the only prenylated indole alkaloids containing a halogenated indole ring and the bicyclo[2.2.2]diazaoctane core. Another unique characteristic is their inhibitory activity against calmodulin (CaM)-dependent phosphodiesterase (PDE1),19,20,22 which has important implications in cancer, neurodegenerative and vascular diseases due to its effect on intracellular cAMP and cGMP concentrations.23–26
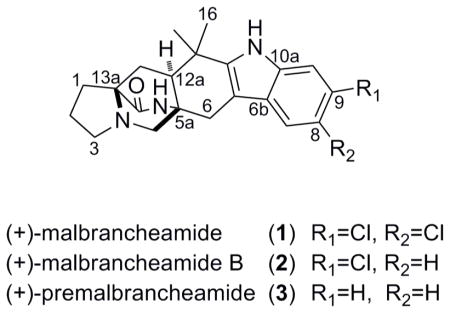
Since their discovery in 2006, these features have inspired several total syntheses of malbrancheamide and its analogs,21,22,27–30 and two biochemical studies have emerged regarding their biosynthesis. In the first investigation researchers identified a 4-dimethylallyltryptophan synthase from the genomic DNA of Malbranchea aurantiaca, named MaPT. It was found that this reverse prenyltransferase (PTase) enzyme was lacking the typical isoprene binding motif found in the majority of bacterial PTase enzymes, and displays high catalytic efficiency making it an attractive enzyme for synthetic biology and combinatorial biosynthesis.31 Similarly, the reverse PTase NotF from Aspergillus fumigatus, involved in notoamide biosynthesis, shares these characteristics.6 Another noteworthy study focused on the first chlorination step and timing of the reduction of the tryptophan carbonyl residue in malbrancheamide biosynthesis.21 A key insight was gained from the transformation of the radiolabeled precursor premalbrancheamide (3) into malbrancheamide B (2) during culture of Malbranchea aurantiaca. It was pointed out that the protein involved in this halogenation event must bind 3 in an “exquisitely defined orientation,”21 selectively adding a chlorine atom only to C-9, the less activated position of the indole ring. It was also postulated through the outcomes of feeding studies that reduction of the tryptophan carbonyl must take place before the Diels Alder cyclization and formation of premalbrancheamide (3). This result implies that this small class of monoketopiperazine compounds is biogenetically distinct from the related diketopiperazine compounds, including the brevianamides, stephacidins, and notoamides.21
The unique qualities summarized above of this fungal genus motivated our chemical exploration of a marine invertebrate-derived strain of Malbranchea graminicola (collection number 086937A). Additional justification for this project was the solid tumor selectivity observed for the crude extract of 086937A in a disk diffusion assay.32 As this work progressed, the known compounds of the malbrancheamide series (1, 2, 3) were encountered, along with four new structures in the same class. The rapid dereplication of these three known compounds and detection of new compounds was made possible by using an emerging mass spectrometry approach. We have been intrigued by the possibility of employing an open-air ion source to detect metabolites directly from a whole organism without the need for organic extraction. It seemed this would be achievable with direct analysis in real time (DART) mass spectrometry, an ion source for analysis of materials with little or no sample preparation.33,34 DART is a versatile device that has provided ion detection of polar and nonpolar analytes directly from a variety of substances, including currency, foods, body fluids, explosives and pharmaceuticals.33
It seems that this method has been overlooked by the natural products community, as the literature precedence for employing DART to profile natural metabolites is limited. Current awareness searches provided only a few examples, including detection of fatty acid methyl esters in whole cell bacteria,35 analysis of hydrocarbon pheromones from a live fly,36 and examination of marker compounds in herbal drugs.37,38 Herein we show that it is also possible to detect secondary metabolites from fungal spores taken directly from an agar culture plate, using a DART ion source coupled to ion trap mass spectrometer. Subsequently, milligram quantities of these metabolites were purified from the extracts of large scale liquid cultures, through selective monitoring of their molecular ions with traditional liquid chromatography-mass spectrometry (LCMS). Together these two mass spectrometry methods provided a rapid and facile route to pinpoint molecules of interest and streamline their isolation. These methods, results and implications are discussed below.
Results and Discussion
The study commenced with the goal of isolating and identifying the bioactive components from culture extracts of Malbranchea graminicola. A pertinent first step was to culture M. graminicola on a solid agar medium with artificial seawater, for use in the dereplication process and for inoculation of liquid cultures. To gain an overview of the secondary metabolites produced in this organism, spores were removed from the agar plate using a sterile platinum inoculation loop and positioned in front of the DART source. The resulting mass spectrum from the coupled ion trap mass spectrometer is shown in Figure 1. The molecular ions for malbrancheamide (1) and malbrancheamide B (2) were easily pinpointed by their isotope patterns indicative of mono- (m/z 404.6/406.6, 1:0.6) and di-chlorination (m/z 370.6/372.6, 1:0.4) patterns, shown in Figure 1a. The [M+H]+ molecular ion for premalbrancheamide (3) was also visible (m/z 336.6) in low relative intensity. A fourth ion, observed at 420.6/422.6 (1:0.6), suggested the presence of a new di-chlorinated metabolite, as no compounds with chlorine-containing molecular formulas were identified in mass-based searches (input 419.0–419.9) in relevant databases.
Figure 1.

DART spectra of Malbranchea graminicola spores grown on two different solid agar medium types, with labeled pseudomolecular ions [M+H]+ for 1–4 and 7–9. a) Agar medium containing chlorine salts; b) Agar medium containing bromine salts.
In order to identify the structure of this new metabolite and to obtain the malbrancheamide compounds for biological testing, a 20 L liquid culture of M. graminicola was prepared. Utilizing solid phase extraction (SPE) and HPLC techniques, each of these compounds were promptly obtained from the culture extract by tracking their respective pseudomolecular ions in each purification step using ESITOFMS. Concentrating first on the subfraction (coded H3H6) containing the 420.2/422.2 ion, the molecular formula C21H24N3O2Cl2 was established and 1H and 13C NMR spectra were obtained. In comparison to malbrancheamide (C21H24N3OCl2), one additional O atom was present in the new metabolite, and while the same proton types were present (1 aliphatic methine, 2 aromatic methines, 6 methylenes and 2 methyls), there were clear downfield shifts of several signals in the 1H NMR spectrum.
The 13C chemical shifts of the new structure 4 aligned well with that of 1, except for a carbonyl resonance at 184.3 ppm (1, C-11a: 145.2 ppm19) and a quaternary carbon at 63.9 ppm (1, C-6a: 104.8 ppm19). This data and the HMBC correlations shown in Table 1 led us to the hexacyclic structure 4, named spiromalbramide, similar to that of (−)-paraherquamide B (5), with a spiro junction at C-6a. The relative configuration for 4 about the bicyclo[2.2.2]diazaoctane ring system was expected to be syn based on biosynthetic analogy to 1,19 and was confirmed by NOE experiments. Using the study of NOE relationships in notoamide B (6) as a guide,13 the relative configuration of 5aR*,6aS*12aS*,13aS* was established for 4 by the correlations shown in Figure 2 (also see Figure S2, Supporting Information). The absolute configuration of 4 was determined by comparing its circular dichroism spectra and Cotton effects to that reported for (+)-5 and (−)-5, prepared synthetically by Cushing et al.39 The spectra for 4 coincided with (−)-5 (Figure 3), as was anticipated based on the negative optical rotation value obtained in MeOH ([α]23D −5.2), therefore supporting the assignment of 5aR,6aS,12aS,13aS configuration.
Table 1.
1H and 13C NMR data (CD3OD, 600 and 150 MHz, respectively) for (−)-spiromalbramide (4).
| δC type | δH (J[Hz) | COSY | HMBC | |
|---|---|---|---|---|
| 1 | 28.3 CH2 | 2.46 ddd (12.8, 9.4, 5.8) | 1, 2 | 14 |
| 1.45 ddd (12.2, 11.6, 5.0) | ||||
| 2 | 22.9 CH2 | 1.91 m | 1, 3 | 1, 13a |
| 3 | 54.5 CH2 | 3.11 td (8.7, 2.9) | 2 | |
| 2.25 dd (9.0, 8.8) | ||||
| 5 | 59.8 CH2 | 3.71 d (11.4) | 5 | 3, 5a, 13a |
| 2.65 dd (11.3, 1.5) | ||||
| 5a | 68.9 C | |||
| 6 | 39.3 CH2 | 2.39 d (15.1) | 6 | 5, 6a, 6b, 11a, 12a |
| 2.07 d (15.2) | ||||
| 6a | 63.9 C | |||
| 6b | 132.3 C | |||
| 7 | 128.9 CH | 7.52, s | 6a, 8, 9, 10a | |
| 8 | 125.9 C | |||
| 9 | 133.0 C | |||
| 10 | 112.1 CH | 7.02 s | 6b, 8 | |
| 10a | 143.7 C | |||
| 11a | 184.3 C | |||
| 12 | 47.1 C | |||
| 12a | 55.1 CH | 3.05 dd (11.0, 10.1) | 13a | |
| 13 | 28.7 CH2 | 1.86 dd (12.6, 11.2) | 12a, 13 | 5a, 12a, 13a, 14 |
| 1.71 dd (12.6, 10.0) | ||||
| 13a | 62.8 C | |||
| 14 | 175.9 C | |||
| 16 | 24.4 CH3 | 0.83 s | 6a, 12, 12a, 17 | |
| 17 | 20.9 CH3 | 1.12 s | 6a, 12, 12a, 16 |
Figure 2.

Significant NOE correlations, observed at 600MHz in CD3OD, establishing the relative configuration for (−)-spiromalbramide (4).
Figure 3.

a) Circular dichroism spectrum for (−)-spiromalbramide (4) and b) comparison of the Cotton effects of 4 to (±)-paraherquamide B (5).39

The isolation of compounds 1–3 was also straightforward using low resolution selective ion monitoring, and the purity of the resulting subfractions was confirmed by 1H NMR. Surprisingly, two subfractions (H5H2 and H5H3) contained only the molecular ion m/z 370.2/372.2 [M+H]+. While all of the 1H NMR signals for fraction H5H3 matched with the literature data for compound 2,20 there were three extra aromatic signals in the spectrum for H5H2 along with all of the same resonances as H5H3 (Figure 4a, 4b). The integration ratio of 0.5:1 for each of the six aromatic signals in H5H2 relative to an upfield methine signal (H-12a) proved that we had a 1:1 mixture of 2 and an isomer with a distinct aromatic substitution pattern, but otherwise identical scaffold. Through analysis of the aromatic chemical shifts, coupling constants, and HMBC correlations (Figure 5, Table 2) we proposed C-8 chlorination for the analogue 7 as opposed to C-9 in compound 2. The 13C chemical shifts for the analogue matched the reported data for a synthetic sample of isomalbrancheamide B (7), prepared by Miller et al, supporting this assignment.28 Separation of these two close isomers was not possible, but we propose the same configuration for our natural sample of 7 as the rest of the malbrancheamide series, based on the positive optical rotation value for the 1:1 mixture of 2 and 7 (H5H2, [α]24D +12, MeOH).40
Figure 4.

Overlaid 1H NMR spectra (600 MHz, CD3OD) of a) 2; b) mixture of 2 and 7 (1:1); c) 8; d) mixture of 8 and 9 (0.3:1), displaying the varying indole mono-halogenation patterns of the malbrancheamide family.
Figure 5.

Significant gHMBC correlations and δ13C values (C-6b, C-8 or C-9 and C-10a) for determination of indole halogenation patterns in 2, 7, 8, and 9.
Table 2.
1H and 13C NMR data (CD3OD, 600 and 150 MHz, respectively) for (+)-isomalbrancheamide B (7), (+)-malbrancheamide C (8), and isomalbrancheamide C (9).
| 7 | 8 | 9 | ||||
|---|---|---|---|---|---|---|
| δC type | δH (J[Hz) | δC type | δH (J[Hz) | δC type | δH (J[Hz) | |
| 1 | 28.1 CH2 | 2.54 ddd (14.3, 13.0, 6.2) | 28.3 CH2 | 2.55 ddd (12.6,9.0,5.6) | 28.3 CH2 | 2.55 ddd (12.6,9.0,5.6) |
| 1.47 ddd (12.6, 10.7, 7.0) | 1.48 ddd (12.6,11.0,7.0) | 1.48 ddd (12.6,11.0,7.0) | ||||
| 2 | 23.5 CH2 | 1.89 m | 23.7 CH2 | 1.89 m | 23.7 CH2 | 1.89 m |
| 3 | 55.3 CH2 | 3.08 ddd (9.7, 7.4, 3.7) | 55.5 CH2 | 3.09 ddd (9.2,7.3,3.6)) | 55.5 CH2 | 3.09 ddd (9.2,7.3,3.6)) |
| 2.19 m | 2.20 m | 2.20 m | ||||
| 5 | 59.4 CH2 | 3.47 d (10.3) | 59.6 CH2 | 3.47 d (10.3) | 59.6 CH2 | 3.47 d (10.3) |
| 2.29 dd (10.3, 1.7) | 2.29 dd (10.3,1.7) | 2.29 dd (10.3,1.7) | ||||
| 5a | 56.0 C | 57.7 C | 57.7 C | |||
| 6 | 30.2 CH2 | 2.87 d (1.1) | 30.3 CH2 | 2.88 d (3.5) | 30.3 CH2 | 2.86 d (1.6) |
| 6a | 104.5 C | 105.1 C | 105.1 C | |||
| 6b | 129.5 C | 127.3 C | 130.1 C | |||
| 7 | 118.0 CH | 7.34 d (1.9) | 119.9 CH | 7.27 d (8.3) | 121.1 CH | 7.49 dd (1.9,0.5) |
| 8 | 125.4 C | 122.8 CH | 7.08 dd (8.3,1.7) | 112.7 C | ||
| 9 | 122.4 CH | 7.01 d (8.6, 2.1) | 115.4 C | 113.4 CH | 7.14 dd (8.5, 1.9) | |
| 10 | 112.9 CH | 7.23 d (8.6) | 114.6 CH | 7.42 d (1.7) | 124.8 CH | 7.20 dd (8.5,0.4) |
| 10a | 136.6 C | 139.4 C | 137.2 C | |||
| 11a | 144.4 C | 143.5 C | 143.5 C | |||
| 12 | 35.4 C | 35.6 C | 35.6 C | |||
| 12a | 48.6 CH | 2.18 m | 49.0 CH | 2.17 m | 49.0 CH | 2.17 m |
| 13 | 32.4 CH2 | 2.02 dd (13.2, 11.1) | 32.6 CH2 | 2.02 dd (13.2,11.4) | 32.6 CH2 | 2.02 dd (13.2,11.4) |
| 1.97 dd (13.2, 5.0) | 1.97 dd (13.2,5.0) | 1.97 dd (13.2,5.0) | ||||
| 13a | 66.4 C | 66.3 C | 66.3 C | |||
| 14 | 176.8 C | 176.7 C | 176.7 C | |||
| 16 | 30.7 CH3 | 1.36 s | 30.9 CH3 | 1.34 s | 30.9 CH3 | 1.34 s |
| 17 | 24.2 CH3 | 1.45 s | 24.4 CH3 | 1.44 s | 24.4 CH3 | 1.44 s |

The isolation of 7, with C-8 chlorination, from our strain of M. graminicola was somewhat surprising, based on the previous proposal that the halogenase in M. aurantiaca displays complete selectivity for delivering a chlorine atom to C-9 in compound 3.21 Our result suggests that the halogenase in M. graminicola may exhibit lower specificity. This indication provided the basis for a proposal that our strain may have the ability to incorporate halogen atoms other than chlorine. To test this hypothesis, agar plates were prepared using artificial seawater enriched with bromine salts as a growth medium for M. graminicola. To our delight, the mass of a brominated metabolite was detected in the DART spectra from these spores, evident from the 1:1 isotopic ratio (m/z 414.2/416.2), as shown in Figure 1b.
A culture of M. graminicola was then prepared in 10 L of artificial seawater using bromine salts, with the goal of isolating the brominated analogue. Similar to the purification outcome of compound 2, two HPLC subfractions contained the molecular mass m/z 414.2/416.2 (coded P3H3 and P3H4), and it was clear from the 1H NMR spectra that both fractions contained a brominated metabolite nearly identical to compounds 1–3. It was also easily established from 1H NMR that P3H4 contained compound 8, but a 0.3:1 mixture of 8 and 9 was present in P3H3, as shown in Figure 4c and 4d. The molecular formula C21H24N3OBr was deduced from accurate mass measurements on both fractions, and using the same NMR guidelines as mentioned above (Figure 5), the bromine placements were established in these new compounds as C-9 in malbrancheamide C (8, P3H4), and C-8 in isomalbrancheamide C (9, major isomer, P3H3). The absolute configuration shown for 8 and 9 is supported by biosynthetic analogy to compounds 1–3 and 7, and by their positive optical rotation values (8, [α]24D +12.5; 9,8 (1:0.3), [α]24D +13.6, MeOH).
Our proposed biosynthesis of the malbrancheamide family of molecules is shown in Figure 6, which is based on previous biosynthetic proposals of compounds 1–3,6,21 a biochemical study of M. aurantiaca,31 and the additional insights gained from the isolation of compounds 4, 7, 8, and 9. This assembly line incorporates a diverse set of putative biosynthetic enzymes, and to date only the prenyltransferase enzyme (PTase), MaPT, has been characterized from M. aurantiaca.31 Other postulated enzymes in the construction of malbrancheamide (1) include an intramolecular Diels-Alderase (IMDA), two halogenases, oxidative and reductive proteins, and a deoxybrevianamide E (10) synthetase.
Figure 6.

Proposed biosynthesis of the malbrancheamide class6,21,31 in Malbranchea graminicola, including new insights from the isolation of compounds 4, 7, 8, and 9.a
a Solid arrows represent reactions that have been confirmed with biochemical analysis or precursor incorporation experiments in Malbranchea aurantiaca,21,31 and dashed arrows represent proposed biosynthetic steps. The symbol X indicates this step is not supported by the current study, and the symbol * indicates that this step is supported, but not proven, by this study. PTase = prenyltranferase; IMDA = intramolecular Diels Alderase.
The isolation of compounds 7, 8, and 9 provides evidence that the first halogenase enzyme in M. graminicola can incorporate more diverse substrates than previously hypothesized for M. aurantiaca. The integration of bromine from growth medium is documented in fungi41,42 and other microorganisms,43,44 and three purified tryptophan halogenases, PyrH (Streptomyces rugosporus), Thal (Streptomyces albogriseolus), and RebH (Lechevalieria aerocolonigenes), have been shown to incorporate both chlorine and bromine atoms.45 However, each of these characterized proteins showed excellent regioselectivity in the monohalogenation event, unlike the results obtained here for M. graminicola. It seems that the second halogenase in M. graminicola is incapable of incorporating substrates 8 and 9, as we did not observe a dibrominated analog (11), nor a mixed chlorine and bromine congener (12a, 12b). It is thus far unclear whether 7 is a substrate for the second halogenase; this matter could be resolved by feeding a radiolabeled analog of 7 to cultures of M. graminicola.
The co-occurrence of compounds 1 and 4 in our culture extract suggests that a tailoring event follows the construction of 1, thereby producing the spirooxindole functionality in 4. Researchers have proposed that a similar transformation takes place in the notoamide-producing strain of Aspergillus fumigatus, based on the isolation of an analogous compound set, (+)-stephacidin A (13) and (−)-notoamide B (6).13 The late stage conversion of 13 to 6 was postulated from a two-step sequence of oxidation followed by rearrangement.6 Similarly we propose that the hypothetical, and undoubtedly unstable, compound 14 (Figure 6) could be formed from a stereoselective oxidation of 1. One of two oxidative enzymes in notoamide biosynthesis in Aspergillus fumigatus, NotB or NotI, could be responsible for this type of oxidation.6 Compound 14 may be the relay substrate for a complex rearrangement to 4, and a “pinacol-type” mechanism has been hypothesized (see Supporting Information, ref. 2). The elucidation of these biosynthetic enzymes would be an important advance in understanding the assembly of this intriguing class.

Conclusions
There are several outcomes from this study that we consider significant. Firstly, we have shown that DART mass spectrometry is a useful tool for detection of secondary metabolites directly from M. graminicola fungal spores, thereby circumventing the need for organic extraction of the organism and mass analysis by coupled liquid chromatography-mass spectrometry. Secondly, this method enabled the facile detection and rapid isolation of the new compound (−)-spiromalbramide (4), in conjunction with selective ion monitoring in the purification process. Additionally, by modifying the solid growth medium, the biosynthesis of novel brominated metabolites (+)-malbrancheamide C (8) and (9) was induced in M. graminicola, and these compounds were easily pinpointed by using DART-MS. This study represents one of the most useful scenarios for DART methodology, because the isotopic patterns were easily identifiable (compound 4) or masses of interest were known before analysis (compound 8). Mass information obtained from DART-MS could also be important for verification of metabolite production in other microorganisms before large-scale culturing and for differentiation of seemingly identical strains. We anticipate that DART-MS will become an increasingly important dereplication tool for natural products chemists. However, utility of this method will be limited without taxonomy information that would provide insights for database searching along with the key m/z data from DART-MS. Finally, the identification of spiromalbramide (4), and the monohalogenated analogues 7, 8, and 9 provide chemical insight into the biosynthesis of the malbrancheamide class. We believe the genome of the marine invertebrate-derived M. graminicola holds important biological clues regarding the assembly of this group of prenylated indole alkaloids.
Experimental Section
General Experimental Procedures
The 1D and 2D NMR spectra were obtained using a 600 MHz spectrometer, outfitted with a cryoprobe, in CD3OD at 600 MHz for 1H and 150 MHz for 13C. Chemical shifts are reported in ppm relative to CD3OD (δH 3.31 and δC 49.0). High-resolution and low-resolution mass spectra were acquired with a bench top ESI-TOF-MS. Semi-preparative reverse-phase (RP) HPLC was performed using a C18 5 μm column, 10 × 250 mm, with UV peak detection and 2 mL/min flow rate. Analytical separation was achieved with a C18 5 μm column, 4.6 × 250 mm and 1 mL/min flow rate. A reverse-phase 10 μm column, 21.2 × 250 mm, was utilized for preparative RP HPLC with a 10 mL/min flow rate. The optical rotations were determined on a digital polarimeter, and the UV data were obtained on a UV-vis photodiode array spectrophotometer. The circular dichroism spectrum is reported as an average of three spectra, each measured with an integration time of 8s per step in a 10 mm quartz cuvette. Compound purity was determined to be >95% by analytical HPLC and 1H NMR, except when noted otherwise.
Biological Materials
Malbranchea graminicola (086937A) was isolated from an unidentified invertebrate collected by SCUBA in Kona, Hawaii, in December of 2008. The strain was taxonomically identified by molecular (ITS and D1/D2 regions of rDNA) and morphological methods at the University of Texas Fungus Testing Laboratory.46 It is maintained as a cryopreserved glycerol stock at UCSC.
Culture Conditions
086937A was grown in 20 L of artificial seawater medium (salt content per liter: 27 g NaCl, 0.6 g KCl, 0.3 g CaCl2, 2 g Tris Base, 7 g MgSO4•7H2O) containing 3.5% Czapek-Dox (086937AZa) at pH 7. A second culture of 086937A was grown in 10 L of brominated artificial seawater medium (salt content per liter: 26 g NaBr, 0.6 g KBr, 0.3 g CaBr2, 2 g Tris Base, 7 g MgSO4•7H2O) containing 3.5% Czapek-Dox at pH 7 (086937AZaBr). pH adjustments for both liquid cultures were made with 12N HCl. Cultures were shaken at 150 rpm for 21 days at room temperature.
Extraction and Isolation
086937AZa
On the 21st day, ~50 g (2% volume) of pre-washed XAD-16 resin was added to each flask and shaken for 12 hrs. The resin and mycelia were then separated from the culture broth by vacuum filtration with glass microfiber filters and a Buchner funnel. The mycelium and resin were extracted 3X with 1 L of 50% MeOH/50% DCM. The resulting crude extract (32 g) was desalted by first dissolving in 10% MeOH and adsorbing onto pre-washed HP-20 resin. The resin was flushed with 1 L of 5% MeOH and then 3 L of 100% MeOH. The MeOH-soluble extract was dried, providing 2 g of de-salted material. This extract was then subjected to flash chromatography using reversed-phase SPE cartridges. Eight fractions (A–H) were collected using the following solvents: A-20% aqueous MeOH (264.3 mg), B-40% aqueous MeOH (130.7 mg), C-60% aqueous MeOH (128.9 mg); D-80% aqueous MeOH (474.2 mg), E-100% MeOH (750.2 mg), F-70% MeOH/30% DCM (442.8 mg), G-30% MeOH/70% DCM (54.5 mg), H-100% DCM (7.8 mg). Fractions A, B, and C were combined based on similar LCMS profiles, and the resulting mixture was subjected to semi-preparative RP HPLC. A linear gradient (10% to 40% aqueous CH3CN over 30 min) provided 7 fractions (H1–H7). Fraction H1 was further purified by analytical HPLC using isocratic conditions (15% aqueous CH3CN), giving 3 (H1H2, 0.7 mg). Similarly, fractions H3 and H7 were each subjected to isocratic analytical RP HPLC (20% aqueous CH3CN) resulting in 4 (H3H6, 0.5 mg), and 1 (H7H1, 10.3 mg). Fraction H5 was also separated with the same conditions, giving 2 (H5H3, 1.0 mg) and an inseparable mixture (1:1) of 2 and 7 (H5H2, 0.9 mg).
086937AZaBr
The 10 L culture was extracted in an identical fashion to 086937AZa. The crude extract (14.6 g) was desalted, providing 2.7 g that was fractionated using the same reversed phase SPE scheme, producing 8 fractions (A, 960.8 mg; B, 447.0 mg; C, 177.7 mg; D, 216.4 mg; E, 448.4 mg; F, 264.6 mg; G, 75.6 mg; H, 2.4 mg). Fractions C and D were combined based on similar LCMS profiles and then subjected to a linear gradient with preparative reversed phase HPLC (20–40% CH3CN, 40 min). Three fractions were collected, containing semi-pure 3 (P1, 3.5 mg), a mixture of 2 and 7 (P2, 3.0 mg), and a semi-pure mixture (1.0:0.2) of 8 and 9 (P3, 4.8 mg). Fraction P3 was further purified by isocratic semi-preparative RP HPLC (48% CH3CN, 3 mL/min) giving pure 8 (P3H4, 1.9 mg) and an inseparable mixture (0.3:1) of 8 and 9 (P3H3, 0.6 mg).
(+)-Malbrancheamide (1): white solid (10.3 mg); [α]24D +34 (c 0.5, MeOH). Other physical properties are in accordance with published data.19
(+)-Malbrancheamide B (2): white solid (1.0 mg); [α]24D +28 (c 0.5, MeOH). Other physical properties are in accordance with published data. 20
(+)-Premalbrancheamide (3): white solid (0.7 mg); [α]24D +15 (c 0.5, MeOH). Other physical properties are in accordance with published data.21
(−)-Spiromalbramide (4): white solid (0.5 mg); [α]23D −5.2 (c 0.36, MeOH). UV (MeOH) λmax (log ε): 203 (3.43), 215 (3.43), 260 (2.78) nm; CD (MeOH) λmax (Δ ε): 200 (+1.7), 216 (−7.2), 237 (+6.9), 265 (−3.0), 302 (−1.5) nm. 1H and 13C NMR (see Table 1); HRESITOFMS [M+H]+ m/z 420.12427 (calcd for C21H24N3O2Cl2, 420.12401).
Inseparable mixture (1:1 ratio) of (+)-Malbrancheamide B (2) and (+)-Isomalbrancheamide B (7): yellow solid (0.9 mg); [α]24D +12 (c 0.5, MeOH). HRESITOFMS [M+H]+ m/z 370.16536 (calcd for C21H25N3OCl, 370.16807). 1H and 13C NMR for 7 (see Table 2). Other physical properties are in accordance with published data.28
(+)-Malbrancheamide C (8): white solid (1.9 mg); [α]24D +12.5 (c 0.5, MeOH). 1H and 13C NMR (see Table 2); HRESITOFMS [M+H]+ m/z 414.11821 (calcd for C21H25N3OBr, 414.11744).
Inseparable mixture (0.3:1 ratio) of (+)-Malbrancheamide C (8) and (+)-Isomalbrancheamide B (9): yellow solid (0.6 mg); [α]24D +13.6 (c 0.5, MeOH). 1H and 13C NMR (see Table 2); HRESITOFMS [M+H]+ m/z 414.11948 (calcd for C21H25N3OBr, 414.11744).
In vitro disk diffusion assay
This screen was performed as previously described32 on the crude extract of 086937AZa. Selectivity (19.5 mm) was defined between a human prostate adenocarcinoma cell line (LNCaP47) versus murine macrophage cells (CFU-GM).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH) (R01CA047135) and the United States State Department (IDBI-210030JA-08). Thank you to Captain Dustin Bumgardner and Crew of the Kona Aggressor II for field assistance in Hawaii. The staff of the UT San Antonio Health Sciences Center Fungus Testing Laboratory, E. Thompson, D. A. Sutton and B. Wickes, provided fungal ID’s. Eefei Chen at UCSC supplied the CD data.
Footnotes
Supporting Information Available. NMR spectra for compounds 4, 7, 8 and 9, isolation schemes, and structures of important prenylated indole alkaloids. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Li SM. Nat Prod Rep. 2010;27:57–78. doi: 10.1039/b909987p. [DOI] [PubMed] [Google Scholar]
- 2.Cui CB, Kakeya H, Okada G, Onose R, Osada H. J Antibiot. 1996;49:527–533. doi: 10.7164/antibiotics.49.527. [DOI] [PubMed] [Google Scholar]
- 3.Abraham WR, Arfmann HA. Phytochemistry. 1990;29:1025–1026. [Google Scholar]
- 4.Yamazaki M, Suzuki S, Miyaki K. Chem Pharm Bull. 1971;19:1739–40. doi: 10.1248/cpb.19.1739. [DOI] [PubMed] [Google Scholar]
- 5.Fayos J, Lokensgard D, Clardy J, Cole RJ, Kirksey JW. J Am Chem Soc. 1974;96:6785–6787. doi: 10.1021/ja00828a054. [DOI] [PubMed] [Google Scholar]
- 6.Ding Y, de WJR, Cavalcoli J, Li S, Greshock TJ, Miller KA, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, et al. J Am Chem Soc. 2010;132:12733–12740. doi: 10.1021/ja1049302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams RM, Stocking EM, Sanz-Cervera JF. Top Curr Chem. 2000;209:97–173. [Google Scholar]
- 8.Williams RM, Cox RJ. Acc Chem Res. 2003;36:127–139. doi: 10.1021/ar020229e. [DOI] [PubMed] [Google Scholar]
- 9.Birch AJ, Wright JJ. J Chem Soc D. 1969:644–5. [Google Scholar]
- 10.Birch AJ, Wright JJ. Tetrahedron. 1970;26:2329–44. doi: 10.1016/s0040-4020(01)92812-1. [DOI] [PubMed] [Google Scholar]
- 11.Birch AJ, Russell RA. Tetrahedron. 1972;28:2999–3008. [Google Scholar]
- 12.Qian-Cutrone J, Huang S, Shu YZ, Vyas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q. J Am Chem Soc. 2002;124:14556–14557. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]
- 13.Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S. Angew Chem, Int Ed. 2007;46:2254–2256. doi: 10.1002/anie.200604381. [DOI] [PubMed] [Google Scholar]
- 14.Tsukamoto S, Kato H, Samizo M, Nojiri Y, Onuki H, Hirota H, Ohta T. J Nat Prod. 2008;71:2064–2067. doi: 10.1021/np800471y. [DOI] [PubMed] [Google Scholar]
- 15.Tsukamoto S, Kawabata T, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. Org Lett. 2009;11:1297–1300. doi: 10.1021/ol900071c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ondeyka JG, Goegelman RT, Schaeffer JM, Kelemen L, Zitano L. J Antibiot. 1990;43:1375–9. doi: 10.7164/antibiotics.43.1375. [DOI] [PubMed] [Google Scholar]
- 17.Blanchflower SE, Banks RM, Everett JR, Manger BR, Reading C. J Antibiot. 1991;44:492–7. doi: 10.7164/antibiotics.44.492. [DOI] [PubMed] [Google Scholar]
- 18.Blanchflower SE, Banks RM, Everett JR, Reading C. J Antibiot. 1993;46:1355–63. doi: 10.7164/antibiotics.46.1355. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Luis S, Rodriguez R, Acevedo L, Gonzalez MC, Lira-Rocha A, Mata R. Tetrahedron. 2006;62:1817–1822. [Google Scholar]
- 20.Figueroa M, Del CGM, Mata R. Nat Prod Res. 2008;22:709–714. doi: 10.1080/14786410802012361. [DOI] [PubMed] [Google Scholar]
- 21.Ding Y, Greshock TJ, Miller KA, Sherman DH, Williams RM. Org Lett. 2008;10:4863–4866. doi: 10.1021/ol8019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller KA, Figueroa M, Valente MWN, Greshock TJ, Mata R, Williams RM. Bioorg Med Chem Lett. 2008;18:6479–6481. doi: 10.1016/j.bmcl.2008.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeon YH, Heo YS, Kim CM, Hyun YL, Lee TG, Ro S, Cho JM. Cell Mol Life Sci. 2005;62:1198–1220. doi: 10.1007/s00018-005-4533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menniti FS, Faraci WS, Schmidt CJ. Nat Rev Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 25.Bender AT, Beavo JA. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 26.Sharma RK, Das SB, Lakshmikuttyamma A, Selvakumar P, Shrivastav A. Int J Mol Med. 2006;18:95–105. [PubMed] [Google Scholar]
- 27.Valente MWN, Williams RM. Heterocycles. 2006;70:249–259. [Google Scholar]
- 28.Miller KA, Welch TR, Greshock TJ, Ding Y, Sherman DH, Williams RM. J Org Chem. 2008;73:3116–3119. doi: 10.1021/jo800116y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frebault F, Simpkins NS, Fenwick A. J Am Chem Soc. 2009;131:4214–4215. doi: 10.1021/ja900688y. [DOI] [PubMed] [Google Scholar]
- 30.Frebault FC, Simpkins NS. Tetrahedron. 2010;66:6585–6596. [Google Scholar]
- 31.Ding Y, Williams RM, Sherman DH. J Biol Chem. 2008;283:16068–16076. doi: 10.1074/jbc.M801991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valeriote F, Grieshaber CK, Media J, Pietraszkewicz H, Hoffmann J, Pan M, McLaughlin S. J Exp Ther Oncol. 2002;2:228–236. doi: 10.1046/j.1359-4117.2002.01038.x. [DOI] [PubMed] [Google Scholar]
- 33.Cody RB, Laramee JA, Durst HD. Anal Chem. 2005;77:2297–2302. doi: 10.1021/ac050162j. [DOI] [PubMed] [Google Scholar]
- 34.Cody RB. Anal Chem. 2009;81:1101–1107. doi: 10.1021/ac8022108. [DOI] [PubMed] [Google Scholar]
- 35.Pierce CY, Barr JR, Cody RB, Massung RF, Woolfitt AR, Moura H, Thompson HA, Fernandez FM. Chem Comm. 2007:807–809. doi: 10.1039/b613200f. [DOI] [PubMed] [Google Scholar]
- 36.Yew JY, Cody RB, Kravitz EA. Proc Natl Acad Sci U S A. 2008;105:7135–7140. doi: 10.1073/pnas.0802692105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HJ, Jee EH, Ahn KS, Choi HS, Jang YP. Arch Pharmacal Res. 2010;33:1355–1359. doi: 10.1007/s12272-010-0909-7. [DOI] [PubMed] [Google Scholar]
- 38.Kim HJ, Jang YP. Phytochem Anal. 2009;20:372–377. doi: 10.1002/pca.1136. [DOI] [PubMed] [Google Scholar]
- 39.Cushing TD, Sanz-Cervera JF, Williams RM. J Am Chem Soc. 1996;118:557–79. [Google Scholar]
- 40.After our work was completed, a new current awareness search uncovered a publication of this compound from Malbranchea aurantiaca. See: Figueroa M, Gonzalez-Andrade M, Sosa-Peinado A, Madariaga-Mazon A, Del R-PF, Gonzalez MDC, Mata R. J Enzyme Inhib Med Chem. 2011;26:378–385. doi: 10.3109/14756366.2010.518964.
- 41.Stander MA, Steyn PS, Lubben A, Miljkovic A, Mantle PG, Marais GJ. J Agricul Food Chem. 2000;48:1865–1871. doi: 10.1021/jf9912708. [DOI] [PubMed] [Google Scholar]
- 42.Amagata T, Takigawa K, Minoura K, Numata A. Heterocycles. 2010;81:897–907. [Google Scholar]
- 43.Ezaki N, Koyama M, Kodama Y, Shomura T, Tashiro K, Tsuruoka T, Inouye S, Sakai S. J Antibiot. 1983;36:1431–8. doi: 10.7164/antibiotics.36.1431. [DOI] [PubMed] [Google Scholar]
- 44.Bister B, Bischoff D, Nicholson GJ, Stockert S, Wink J, Brunati C, Donadio S, Pelzer S, Wohlleben W, Süssmuth RD. ChemBioChem. 2003;4:658–662. doi: 10.1002/cbic.200300619. [DOI] [PubMed] [Google Scholar]
- 45.Wagner C, El OM, Koenig GM. J Nat Prod. 2009;72:540–553. doi: 10.1021/np800651m. [DOI] [PubMed] [Google Scholar]
- 46.Sigler L, Lacey J, Carmichael JW. Mycotaxon. 1982;15:465–471. [Google Scholar]
- 47.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP. Cancer Res. 1983;43:1809–18. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.