

Abstract
Myotonic dystrophy type 1 (DM1) is caused by expansion of a CTG repeat in the gene DMPK. The expansion is highly unstable in somatic cells, a feature that may contribute to disease progression. The RNA expressed from the mutant allele exerts a toxic gain of function, due to the presence of an expanded CUG repeat (CUGexp). This RNA dominant mechanism is amenable to therapeutic intervention with antisense oligonucleotides (ASOs). For example, CAG-repeat ASOs that bind CUGexp RNA are beneficial in DM1 models by altering the protein interactions or metabolism of the toxic RNA. Because CUGexp RNA has been shown to aggravate instability of expanded CTG repeats, we studied whether CAG-repeat ASOs may also affect this aspect of DM1. In human cells the instability of (CTG)800 was suppressed by addition of CAG-repeat ASOs to the culture media. In mice that carry a DMPK transgene the somatic instability of (CTG)800 was suppressed by direct injection of CAG-repeat ASOs into muscle tissue. These results raise the possibility that early intervention with ASOs to reduce RNA or protein toxicity may have the additional benefit of stabilizing CTG:CAG repeats at subpathogenic lengths.
Introduction
Myotonic dystrophy type 1 (DM1) is a dominantly inherited neuromuscular disease caused by expansion of a CTG repeat in the 3′ UTR of DMPK. In addition to the skeletal myopathy, DM1 affects smooth muscle, cardiac conduction, the central nervous system, and ocular lens.1
A striking feature of DM1 is the marked instability of the expanded repeat. While instability is also characteristic of other repeat expansion disorders, in DM1 it is particularly extreme.2,3 In germline cells, the instability can lead to insertion of hundreds of additional CTG repeats in a single intergenerational transmission.4 In somatic cells the expansion process continues throughout life, at rates that are variable between tissues.5 This can lead to 10-fold variations of expansion length in different tissues of an individual, ultimately producing expansions of three to six thousand repeats in skeletal muscle and heart.6,7,8,9 The progression of DM1 may depend on the growth of the expanded repeat over time, suggesting that stabilization of the repeat is a means to postpone the onset or slow the progression.
Instability of expanded repeats is enhanced by transcription, and recent studies indicate that RNA repeats have a role in this process.10,11,12,13 RNAs comprised of expanded CUG, CAG, CCG, CGG, GAA, or CCUG repeats can bind to the template strand of DNA, forming RNA:DNA hybrids (R-loops).14,15,16,17 Whether these R-loops result from incomplete dissociation of nascent transcripts from template, or reassociation of transcripts with template, is unclear. Either way, the presence of R-loops may instigate the formation of extrahelical slipped strand structures on the nontemplate strand. Recognition of these structures by mismatch repair proteins may stimulate error-prone repair, leading to further expansion.
A second unusual feature of the DM1 mutation is that it leads to the production of a toxic RNA.18,19 Interactions of CUGexp RNA with proteins are thought to underlie many symptoms of the disease. One approach for therapy has focused on CAG-repeat antisense oligonucleotides (ASOs) designed to bind CUG repeat RNA, thereby blocking RNA-protein interactions by steric inhibition.20,21 By this strategy the activity of RNA binding proteins and regulation of alternative splicing are normalized in mouse models of DM1.20 Unexpectedly these “blocker ” ASOs, although not competent to activate RNase H, were shown to reduce the level of CUGexp transcripts.20,21 Although the mechanism for this effect has not been determined, evidence suggests that release of CUGexp transcripts from nuclear foci can facilitate their transport to the cytoplasm, where they may undergo more rapid decay.
While therapeutic efforts have focused on using ASOs to reverse RNA toxicity in DM1, the potential impact on DNA instability has not been addressed. If R-loops at the DM1 locus result partly from reassociation of CUG repeats with template, then increased nuclear export or knockdown of CUGexp transcripts may inhibit this process and stabilize the expanded repeat. By contrast, CAG-repeat ASOs designed to bind toxic RNA may also hybridize to the nontemplate strand of DNA (CTG-repeat strand), forming D-loops that could, in theory, increase the instability of expanded repeats. If this occurs, the possibility exists that continued growth of the expanded repeat may ultimately defeat the therapeutic effect, due to increased length and toxicity of the CUGexp RNA. Here we used human cells and transgenic mice to examine the effects of CAG-repeat ASOs on instability of expanded CTG•CAG repeats.
Results
Generation of CUG-expressing cells that have free uptake of ASOs
In previous studies, we found that CTG repeat expansions or contractions were frequent events in human cells, that could be evaluated over intervals as short as 4–8 weeks, provided that the CTG repeat was highly expanded and actively transcribed.13 To study ASO effects continuously over several weeks we took advantage of the capacity of HT1080 human fibrosarcoma cells for free uptake of oligonucleotides from the culture media.22 To insert an expanded repeat in HT1080 cells we used a plasmid (LC15-F, Figure 1a) that expresses a puromycin resistance marker (puro) from one transcription unit, and from a second transcription unit it expresses 800 CUG repeats in the DMPK 3′ UTR. The uninterrupted repeat tract was generated by rolling circle amplification and cell-free cloning as previously described.23 HT1080 cells were cotransfected with LC15-F-CTG800 and plasmid PhiC31o, expressing the phiC31 integrase, in order to obtain single-copy genomic integrations of the expanded repeat.13,24 Following puromycin selection, we obtained more than 15 stably transfected clones and then selected a clone with robust transgene expression, as determined by formation of nuclear foci of CUGexp RNA.
Figure 1.

Antisense oligonucleotides (ASOs) reduce RNA foci in HT1080 cells. (a) Diagram of pLC15-F construct for expression of expanded-CUG repeats in the DMPK 3′ UTR. The attB site supports genomic integration by PhiC31 integrase. CMV/CBA, CMV enhancer/chicken β-actin promoter; puro, puromycin resistance. (b) Sequence of LNA-ASOs. The position of locked nucleic acid (LNA)-modified subunits is shown by upper case letters, lower case letters are unmodified DNA. Gapmer and mixmer ASOs have full phosphorothioate backbones. (c) FISH showing foci of CUGexp RNA (red) in nuclei (blue) of stably transfected HT1080 cells. (d) Histogram showing the percentage of cells with nuclear foci of CUGexp RNA. The number of cells counted was 526 for no treatment, 300 for gapmer-treatment, and 398 for blocker-treatment. Mean ± SD, n = 3 or more. *P < 0.001. ASO, antisense oligonucleotide; CMV, cytomegalovirus.
Design of ASOs
To examine antisense effects on instability, we used oligonucleotides having a phosphorothioate backbone and locked nucleic acid (LNA) modification, a chemistry that increases hybridization affinity and nuclease resistance.25 LNA ASOs were previously shown to exhibit activity in HT1080 cells when added to the culture media without transfection reagents.22 We used two 18-mer ASOs having the identical (CAG)6 sequence but differing in the distribution of LNA-modified nucleotides. One design was a 4-10-4 gapmer in which a central stretch of 10 DNA monomers is flanked by four LNA nucleotides on either end (Figure 1b). The other design was a mixmer in which eight LNA nucleotides were interspersed throughout the oligonucleotide (Figure 1b). Both ASOs were designed for strong hybridization to CUGexp RNA (or CTGexp DNA) but only the gapmer is competent to activate RNase H.25
RNase H-active and inactive ASOs have similar capacity to reduce CUGexp transcripts
Next, we examined the effects of CAG-repeat ASOs on CUGexp transcripts in HT1080 cells. Previous studies demonstrated that transcripts with expanded-CUG repeats are retained in nuclear foci.26,27 Consistent with these observations, 49% of the stably transfected HT1080 cells showed nuclear foci of CUGexp RNA, a feature never observed in nontransfected cells (Figure 1c,d). One week after addition of ASOs to the culture media (final concentration 1 µmol/l) the frequency of cells showing nuclear foci was reduced to 6.0% or 6.5% by gapmer or mixmer ASOs, respectively (Figure 1c,d). We performed quantitative real-time reverse transcriptase-PCR (qRT-PCR) to determine the extent of target knockdown, using an assay that detects the combined output from transgene and endogenous DMPK alleles. The expression of DMPK 3′ UTR in stably transfected cells was 20-fold higher than in nontransfected cells (Figure 2a). One week after the addition of gapmer or mixmer to the culture media, the level of DMPK 3′ UTR was markedly reduced, reaching levels similar to nontransfected cells. In contrast, mRNA for CASK, a transcript with (CUG)16 in the 3′ UTR, was not affected by either ASO (Figure 2b). Furthermore, a different qRT-PCR assay, to quantify transgene output without detection of endogenous DMPK mRNA, showed that CUGexp transcripts in the cytoplasm were similarly reduced by both ASOs (Figure 2c). We also assessed puro, expressed from the contiguous promoter. The level of puro mRNA was not affected by either ASO (Figure 2d), indicating that reduction of CUGexp RNA did not result from general silencing of the transgene locus.
Figure 2.

RNA levels in CUGexp-expressing or nontransfected HT1080 cells, determined by quantitative real-time reverse transcriptase (qRT)-PCR and normalized to 18S rRNA. (a) The level of DMPK 3′ UTR RNA (transgene + endogenous) was reduced by both antisense oligonucleotides (ASOs), to levels similar to nontransfected cells. (b) No effect of ASOs on expression of CASK, an endogenous transcript with (CUG)16 in the 3′ UTR. (c) The level of DMPK 3′ UTR (transgene only) in cytoplasmic RNA was reduced by both ASOs. (d) No effect of ASOs on puro, expressed from the contiguous promoter. Data are the mean ± SD of triplicates. *P < 0.001.
LNA-ASOs stabilize an expanded CTG repeat in HT1080 cells
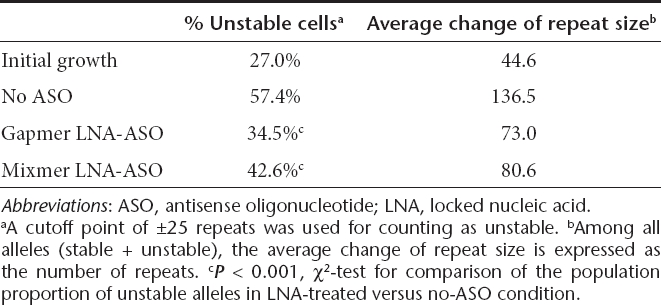
Having developed a system for sustained reduction of CUGexp RNA, we next examined the effects of LNA-ASOs on instability of expanded CTG repeats. We used small-pool PCR coupled with southern blot analysis, a method designed to resolve changes of repeat length for individual transgene alleles.13 During the initial derivation of stably transfected cells, the (CTG)800 tract incurred substantial instability, reflecting the expansion or contraction events that occurred during the first 19 days of clonal expansion (27% unstable alleles, Figure 3a,b). Similar results were obtained in a previous study using human fibroblasts.13 Cells were then grown in the presence or absence of gapmer ASO for an additional 4 weeks. The untreated cells continued to accrue variant alleles having altered CTG repeat length, including expansion and contraction events, reaching levels of 57% unstable alleles. In contrast, the instability in gapmer-treated cells was significantly reduced (35% unstable alleles, P < 0.001, Figure 3a,b and Table 1), and the average size of the length change was smaller than in untreated cells. The mixmer ASO also suppressed repeat instability (43% unstable alleles), with a smaller average size change (Figure 3a,b and Table 1). Neither ASO had a noticeable effect on cell morphology or proliferation.
Figure 3.

Effects of CAG-repeat antisense oligonucleotide (ASO) on CTG•CAG repeat instability in HT1080 cells. (a) Representative data showing small-pool PCR followed by Southern blot for analysis of CTG repeat length. The scale on the left shows molecular weight markers converted into repeat number for CTG-repeat fragments of equivalent size. (b) Histograms showing repeat length distributions in HT1080 cells. The frequency distribution of unstable alleles is shown by gray bars (left vertical axis). The frequency of stable alleles is shown by black bars (right vertical axis). Allele lengths are grouped in bins spanning 50 repeats. More than 110 alleles were sized for each group.
Table 1. Effect of LNA-ASO on repeat instability in HT1080 cells.
LNA-ASOs reduce the accessibility of the nontemplate DNA strand to bisulfite modification
Lin et al. recently found evidence that R-loops are formed at a transcribed (CAG:CTG)67 tract in human cells, leaving the nontemplate strand unpaired and more accessible to bisulfite modification.15 Following this method, we isolated genomic DNA and exposed it to bisulfite under nondenaturing conditions. We then PCR-amplified and cloned the repeat tract, followed by sequencing. In this experiment a modification of cytosine on the nontemplate (CTG) strand would appear as C to T transition, and a modification of the template (CAG) strand would appear as G to A transition.28 Reduced frequency of unpairing in the nontemplate strand would indicate that formation of R-loops was reduced, whereas ASO hybridization to the nontemplate strand (forming D-loops) would increase the frequency of unpairing on the template strand. Due to the tendency for contraction of CTG:CAG repeats in Escherichia coli,29 and the length of sequencing reads that we could obtain, we focused our analysis on the 5′ region of the CTG:CAG repeat. In accord with the previous study,15 the untreated CUGexp-expressing HT1080 cells showed an interspersed pattern of modified Cs (4.1% on average) on the nontemplate strand, consistent with formation of R-loops (Figure 4a,b). The frequency of converted C residues on the nontemplate strand was significantly reduced by ASO treatments (P < 0.01), whereas bisulfite modification of C residues in the template strand was not affected by either ASO (Figure 4b). These results suggest that ASOs have reduced the frequency of R-loops without causing the formation of D-loops.
Figure 4.

Reduced R-loop formation by antisense oligonucleotides (ASOs) in HT1080 cells. (a) Detection of unpaired DNA at the 5′ proximal region of the CTG•CAG repeat tract. Shown are the positions of bisulfite-modified unpaired Cs on the nontemplate strand of representative clones. Vertical marks indicate C residues that have been converted to U. Numbers at the top of figure represent increments of 10 CTG repeats. (b) Effects of antisense oligonucleotides (ASOs) treatment on the frequency of C to U conversion in the CTG•CAG repeat. A total of 36 clones (corresponding to 6,468 CTG·CAG repeats) from nontemplate (CTG) strand and 30 clones (corresponding to 4,717 CTG·CAG repeats) from template (CAG) strand were analyzed. Data are indicated as the mean ± SE, n = 10 or more. *P < 0.01.
LNA-ASOs stabilize CTG repeats in DM1 model mice
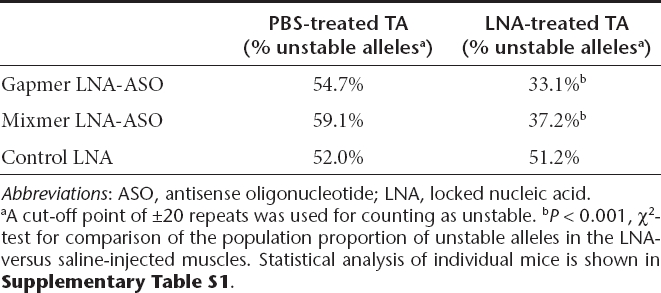
Next, we examined the effects of ASOs in vivo, using a disease-relevant tissue. We used transgenic mice that carried a 45-kb human genomic fragment that included the entire DMPK gene with an expanded CTG repeat.30 In this line of transgenic mice the expanded CTG repeat shows intergenerational and somatic instability, the latter becoming more pronounced with advancing age. In the DM300-328-XXL colony used for these experiments the basal length of the CTG repeat was around 800 repeats, and the DMPK transgene was expressed in skeletal muscle.31,32 We injected gapmer, mixmer, or control ASOs into hindlimb (tibialis anterior) muscle, followed by in vivo electroporation to load the oligonucleotide into muscle fibers. In previous studies, the duration of ASO action in muscle was very prolonged, up to 14 weeks.20,33 The opposite limb was injected with vehicle (saline) alone, followed by the same electroporation procedure. Muscle tissue was obtained 4 weeks later and CTG repeat length was determined by small-pool PCR and southern blot. The frequency of unstable alleles was consistently lower in gapmer- or mixmer-treated tissues (Figure 5a,b, Supplementary Figures S1 and S2, and Table 2). Also, although somatic instability varied among mice, a side-to-side comparison for each individual animal showed reduced instability on the gapmer- or mixmer-treated side (P < 0.05, Supplementary Table S1). In contrast, a control oligonucleotide, targeting β-globin but having a similar LNA gapmer design, did not suppress repeat instability (Table 2). The expression of the DMPK transgene showed a modest reduction at 4 weeks following ASO injection (Figure 5c), as compared to saline-injected muscle from the opposite hindlimb. The histologic appearance of ASO- and saline-injected muscle was similar.
Figure 5.

Effects of CAG-repeat antisense oligonucleotides (ASOs) on CTG•CAG repeat instability in XXL transgenic mice. (a) Small-pool PCR followed by Southern blot for analysis of CTG repeat length. The scale on the left shows molecular weight markers converted into numbers of CTG repeats. M indicates DIG-labeled molecular weight marker. (b) The percentage of unstable alleles in tibialis anterior muscle of XXL mice 4 weeks after the injection and electroporation of phosphate-buffered saline (PBS) or locked nucleic acid (LNA)-ASOs. Data from PBS vs. ASO-injected muscle from same mouse are connected by a solid or dotted line. (c) Level of human DMPK transgene mRNA determined by quantitative real-time reverse transcriptase (qRT)-PCR and normalized to 18S rRNA, expressed as the ratio of LNA-treated vs. PBS-treated TA muscles. Data are indicated as the mean ± SE of triplicates (n = 3 in each group). *P < 0.05.
Table 2. Repeat instability in XXL mice.
Discussion
CTG instability occurs throughout the life of an individual with DM1, apparently with no upper limit on expansion size.5,6,7,8,9 The ongoing CTG repeat expansion raises two concerns in connection with therapeutic CAG-repeat ASOs. First, that these agents may interact with the DM1 locus in a way that promotes somatic expansion, and second whether an initial beneficial effect on the toxic transcript will ultimately be lost due to ongoing expansion and increased production of CUGexp RNA.
The major finding of this study is that CAG-repeat ASOs have not exacerbated the instability of expanded CTG repeats in two different model systems. On the contrary, our results indicate that somatic instability is suppressed by ASOs in both models. Further studies are needed to determine the dose dependency of this effect, and whether it can be obtained in other tissues, with systemic administration, over longer periods of administration, and in other (CTG)•(CAG) expansion disorders. It will also be interesting to examine antisense effects on repeat instability in the germline.
Compounds that stabilize (CTG)•(CAG) repeats in cultured cells have been previously identified (reviewed in ref. 34). These include DNA intercalators, DNA alkylating agents, or drugs that affect DNA methylation or replication. Zinc finger nucleases were designed for cleavage of (CTG)•(CAG) repeats, and were shown to induce contractions of expanded repeats in cell culture.35 Although these studies are informative about the mechanisms of instability, the long-term safety of any approach that affects global DNA metabolism or causes DNA cleavage is unclear.
To our knowledge the present study is the first to show drug-induced reduction of CTG expansion instability in an affected tissue in vivo. Thus, if barriers to tissue delivery can be overcome, these ASOs may provide a targeted approach to modulate instability. However, the stabilizing effect that we observed in vivo was modest. In part this may reflect a limitation of our experimental system. The extent of somatic instability was variable between mice, and, in contrast to the cell culture experiments, we could not determine and subtract the instability that had already occurred before ASOs were administered. Accordingly, we could only suppress instability that occurred in the 4 weeks after ASO administration, which represented 30% of the post-conception life of the animal (4 weeks prenatal + 6 weeks postnatal + 4 weeks postadministration of ASO). Therefore, it is difficult from these data to determine the extent to which instability was suppressed, and any conclusion about the clinical utility of this approach would be premature.
The specificity of this method will require careful examination, because CAG-repeat oligonucleotides have potential to hybridize with short tracts of CUG repeats on several other transcripts. However, it is noteworthy that sensitivity to knockdown by CAG-repeat ASOs is greater for CUG-expanded than nonexpanded transcripts. Preferential reduction of expanded CUG transcripts was also observed for CAG-repeat oligonucleotides having the morpholino or 2-O-methyl chemistries.20,21 ASOs targeting CAG repeats in huntingtin or ATXN3 also showed selectivity for silencing the expanded versus nonexpanded alleles.36
In HT1080 cells the reduction of (CUG)800 transcripts by mixmer (RNase H-inactive) ASOs was equivalent to that induced by gapmer (RNase H-active) ASOs. The capacity for RNase H-inactive ASOs to reduce CUGexp transcripts was noted in previous studies.20,21 This effect has now been observed with CAG-repeat oligonucleotides of different chemistry (LNA, 2-O-methyl, morpholino) and length (18, 21, or 25 nt, respectively). Whereas CAG-repeat morpholinos caused an increase of nucleocytoplasmic transport and translation for (CUG)250 transcripts in a previous study,20 the present study did not show an increase of cytoplasmic CUGexp RNA in HT1080 cells. The mechanism for knockdown of the RNA target by these ASOs remains an open question.
The instability of expanded (CTG)•(CAG) repeats is increased by transcription across the repeat in one or both directions,13,37 R-loops being implicated in this process.15,17 Accordingly, the most straightforward mechanistic explanation for suppression of instability is that ASO knockdown of CUGexp RNA has decreased the formation of R-loops. The analysis of bisulfite sensitivity of the repeat tract is consistent with this interpretation. We cannot, however, exclude the possibility that ASOs have a direct interaction with the nontemplate strand, or that transcription of the expanded repeat has been silenced. Whichever mechanism applies, our findings suggest the intriguing possibility that intervention with CAG-repeat ASOs early in the disease process may have the dual benefits of preventing RNA toxicity while also stabilizing the repeat at a subpathogenic or minimally pathogenic length.
Materials and Methods
ASOs. LNA oligonucleotides having the following sequences were purchased from Exiqon (Woburn, MA): gapmer LNA-ASO, 5′-CAGCagca gcagcaGCAG-3′ mixmer LNA-ASO, 5′-CaGcaGcaGcaGcaGcAG-3′ control LNA-ASO, CCTCttacctcagtTACA. Upper case letters indicate LNA nucleotide. All ASOs had full phosphorothioate intersubunit linkage.
Stable transfection of HT1080 cells and introduction of LNA ASOs. HT1080 cells were obtained from ATCC (Manassas, VA). Cells were cultured in DMEM (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin. 5 × 105 cells were cotransfected with 200 ng of repeat-containing plasmid LC15-F13 and 1.8 µg of plasmid PhiC31o-encoding PhiC31 integrase.38 Transfection was performed using Nucleofector (Lonza, Basel, Switzerland) according the manufacturer's program L-005. Stably transfected clones were selected with puromycin (0.4 µg/ml). For delivery of LNA-ASOs, HT1080 cells were seeded at low plating density in 12-well plates. Eight hours after plating, oligonucleotides were added and mixed at a final concentration of 1 µmol/l. Cells were passaged twice weekly, and continuously exposed to 1 µmol/l ASO.
Intramuscular injection of LNA ASOs in XXL mice. Mouse handling and experimental procedures were conducted in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care. Six-week-old heterozygous XXL mice were pretreated with hyaluronidase (Sigma, St Louis, MO) and injected intramuscularly with 15 µg of LNA-oligonucleotides or phosphate-buffered saline alone, followed by electroporation. Mice were sacrificed 4 weeks later and the injected muscles were obtained for analysis of repeat instability and transgene expression.
Repeat length analysis. Genomic DNA was extracted from HT1080 cells and mouse muscles using the Gentra Puregene Kit (Qiagen, Valencia, CA). Expanded-CTG repeats were sized by small-pool PCR followed by Southern blot, as described previously.13 At least 110 alleles in HT1080 cells and 40 alleles in mouse muscle were analyzed for each group.
For statistical analysis, χ2-tests were performed to compare the frequency of unstable alleles for each set of experiments, as reported previously.13 The χ2-test was previously used to compare the population proportion of unstable repeat alleles in two experimental groups.12,39,40 In our studies, the number of nonvariant versus variant (expansion or contraction) alleles was compared between untreated and ASO-treated cells, or between phosphate-buffered saline- and ASO-treated muscles.
mRNA quantification. RNA extraction, DNase treatment, and reverse transcription were performed as described previously.13 Cytoplasmic RNA was prepared from HT1080 cells using RNeasy Mini Kit (Qiagen), according to the manufacturer's instruction. Quantitative reverse transcriptase-PCR was performed using TaqMan Gene Expression assays on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Carlsbad, CA). The level of endogenous DMPK plus transgene-derived mRNA, CASK mRNA, transgene mRNA, and puro transcript was normalized to18S rRNA. DMPK primers and probe sequences are described previously.13 Primer sequences for cytoplasmic transgene mRNA were 5′-GACTGACCGCGTTACTCC-3′ and 5′-AGAATAGGAACTTCGGAATAGGAAC-3′. The probe sequence was 5′-CACAATAACCAGCACGTTGCCCA-3′.
FISH. FISH was performed as previously described.13 At least 300 cells were counted to determine the frequency of nuclei having ribonuclear foci.
Detection of unpaired single-stranded DNA. Genomic DNA from HT1080 cells were subjected to bisulfite modification for 16 hours at 37 °C by using EpiTect Bisulfite Modification Kit (Qiagen). Two hundred nanogram of bisulfite-modified DNA was amplified for 36 cycles with the same primers as small-pool PCR, then tailed with 3′-terminal deoxyadenocine by Go Taq polymerase (Promega, Madison, WI). PCR products were electrophoresed on agarose gels, purified, and cloned into pGEM-T Easy vector (Promega). The DNA sequencing was performed with at least 10 individual clones by Big-Dye v.3.1 terminators (Applied Biosystems).
SUPPLEMENTARY MATERIAL Figure S1. Histograms of repeat length distributions in mouse muscle treated with the gapmer LNA-ASO or PBS. Figure S2. Histograms of repeat length distributions in mouse muscle treated with the mixmer LNA-ASO or PBS. Table S1. Repeat instability in individual XXL mice.
Acknowledgments
This work comes from the University of Rochester Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (NIH/NS048843) with support from the National Institute of Health (AR049077, AR48143); the Saunders Family Fund, the Muscular Dystrophy Association (M.N.); Run America Foundation, and postdoctoral fellowships (to M.N.) from the Cell Science Research Foundation and the Uehara Memorial Foundation. The authors thank Christopher Pearson for critical reading of the manuscript.
Supplementary Material
Histograms of repeat length distributions in mouse muscle treated with the gapmer LNA-ASO or PBS.
Histograms of repeat length distributions in mouse muscle treated with the mixmer LNA-ASO or PBS.
Repeat instability in individual XXL mice.
REFERENCES
- Harper PS. W.B. Saunders Company: London; 2001. Myotonic Dystrophy. [Google Scholar]
- López Castel A, Cleary JD., and, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. 2010;11:165–170. doi: 10.1038/nrm2854. [DOI] [PubMed] [Google Scholar]
- La Spada AR., and, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman JB, Fenwick RG, Jr, Fu YH, Pizzuti A., and, Caskey CT. Relationship between parental trinucleotide GCT repeat length and severity of myotonic dystrophy in offspring. JAMA. 1993;269:1960–1965. [PubMed] [Google Scholar]
- Martorell L, Monckton DG, Gamez J, Johnson KJ, Gich I, Lopez de Munain A.et al. (1998Progression of somatic CTG repeat length heterogeneity in the blood cells of myotonic dystrophy patients Hum Mol Genet 7307–312. [DOI] [PubMed] [Google Scholar]
- Ashizawa T, Dubel JR., and, Harati Y. Somatic instability of CTG repeat in myotonic dystrophy. Neurology. 1993;43:2674–2678. doi: 10.1212/wnl.43.12.2674. [DOI] [PubMed] [Google Scholar]
- Thornton CA, Johnson K., and, Moxley RT.3rd (1994Myotonic dystrophy patients have larger CTG expansions in skeletal muscle than in leukocytes Ann Neurol 35104–107. [DOI] [PubMed] [Google Scholar]
- Zatz M, Passos-Bueno MR, Cerqueira A, Marie SK, Vainzof M., and, Pavanello RC. Analysis of the CTG repeat in skeletal muscle of young and adult myotonic dystrophy patients: when does the expansion occur. Hum Mol Genet. 1995;4:401–406. doi: 10.1093/hmg/4.3.401. [DOI] [PubMed] [Google Scholar]
- López Castel A, Nakamori M, Tomé S, Chitayat D, Gourdon G, Thornton CA.et al. (2011Expanded CTG repeat demarcates a boundary for abnormal CpG methylation in myotonic dystrophy patient tissues Hum Mol Genet 201–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Dion V., and, Wilson JH. Transcription promotes contraction of CAG repeat tracts in human cells. Nat Struct Mol Biol. 2006;13:179–180. doi: 10.1038/nsmb1042. [DOI] [PubMed] [Google Scholar]
- Ditch S, Sammarco MC, Banerjee A., and, Grabczyk E. Progressive GAA.TTC repeat expansion in human cell lines. PLoS Genet. 2009;5:e1000704. doi: 10.1371/journal.pgen.1000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindler PM., and, Bidichandani SI. Role of transcript and interplay between transcription and replication in triplet-repeat instability in mammalian cells. Nucleic Acids Res. 2011;39:526–535. doi: 10.1093/nar/gkq788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M, Pearson CE., and, Thornton CA. Bidirectional transcription stimulates expansion and contraction of expanded (CTG)*(CAG) repeats. Hum Mol Genet. 2011;20:580–588. doi: 10.1093/hmg/ddq501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E, Mancuso M., and, Sammarco MC. A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 2007;35:5351–5359. doi: 10.1093/nar/gkm589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Dent SY, Wilson JH, Wells RD., and, Napierala M. R loops stimulate genetic instability of CTG.CAG repeats. Proc Natl Acad Sci USA. 2010;107:692–697. doi: 10.1073/pnas.0909740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIvor EI, Polak U., and, Napierala M. New insights into repeat instability: role of RNA•DNA hybrids. RNA Biol. 2010;7:551–558. doi: 10.4161/rna.7.5.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K.et al. (2011Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats Nucleic Acids Res 391749–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JR., and, Swanson MS. Mechanisms of RNA-mediated disease. J Biol Chem. 2009;284:7419–7423. doi: 10.1074/jbc.R800025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TA, Wan L., and, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT.et al. (2009Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA Science 325336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ.et al. (2009Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy Proc Natl Acad Sci USA 10613915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, Hansen JB, Lai J, Wu S, Voskresenskiy A, Høg A.et al. (2010Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents Nucleic Acids Res 38e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne RJ., and, Thornton CA. Cell-free cloning of highly expanded CTG repeats by amplification of dimerized expanded repeats. Nucleic Acids Res. 2008;36:e24. doi: 10.1093/nar/gkn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalberg TW, Portlock JL, Olivares EC, Thyagarajan B, Kirby PJ, Hillman RT.et al. (2006Integration specificity of phage phiC31 integrase in the human genome J Mol Biol 35728–48. [DOI] [PubMed] [Google Scholar]
- Vester B., and, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–13241. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- Davis BM, McCurrach ME, Taneja KL, Singer RH., and, Housman DE. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci USA. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamshere MG, Newman EE, Alwazzan M, Athwal BS., and, Brook JD. Transcriptional abnormality in myotonic dystrophy affects DMPK but not neighboring genes. Proc Natl Acad Sci USA. 1997;94:7394–7399. doi: 10.1073/pnas.94.14.7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Roy D, Huang FT., and, Lieber MR. Detection and structural analysis of R-loops. Meth Enzymol. 2006;409:316–329. doi: 10.1016/S0076-6879(05)09018-X. [DOI] [PubMed] [Google Scholar]
- Kang S, Jaworski A, Ohshima K., and, Wells RD. Expansion and deletion of CTG repeats from human disease genes are determined by the direction of replication in E. coli. Nat Genet. 1995;10:213–218. doi: 10.1038/ng0695-213. [DOI] [PubMed] [Google Scholar]
- Seznec H, Lia-Baldini AS, Duros C, Fouquet C, Lacroix C, Hofmann-Radvanyi H.et al. (2000Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability Hum Mol Genet 91185–1194. [DOI] [PubMed] [Google Scholar]
- Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N.et al. (2001Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities Hum Mol Genet 102717–2726. [DOI] [PubMed] [Google Scholar]
- Gomes-Pereira M, Foiry L, Nicole A, Huguet A, Junien C, Munnich A.et al. (2007CTG trinucleotide repeat “big jumps”: large expansions, small mice PLoS Genet 3e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Lueck JD, Swanson MS, Dirksen RT., and, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-Pereira M., and, Monckton DG. Chemical modifiers of unstable expanded simple sequence repeats: what goes up, could come down. Mutat Res. 2006;598:15–34. doi: 10.1016/j.mrfmmm.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Liu G, Chen X, Bissler JJ, Sinden RR., and, Leffak M. Replication-dependent instability at (CTG) x (CAG) repeat hairpins in human cells. Nat Chem Biol. 2010;6:652–659. doi: 10.1038/nchembio.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Matsui M, Gagnon KT, Schwartz JC, Gabillet S, Arar K.et al. (2009Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs Nat Biotechnol 27478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Leng M, Wan M., and, Wilson JH. Convergent transcription through a long CAG tract destabilizes repeats and induces apoptosis. Mol Cell Biol. 2010;30:4435–4451. doi: 10.1128/MCB.00332-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CS., and, Soriano P. High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE. 2007;2:e162. doi: 10.1371/journal.pone.0000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J., and, Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–1859. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- Shelbourne PF, Keller-McGandy C, Bi WL, Yoon SR, Dubeau L, Veitch NJ, US-Venezuela Collaborative Research Group et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. 2007;16:1133–1142. doi: 10.1093/hmg/ddm054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Histograms of repeat length distributions in mouse muscle treated with the gapmer LNA-ASO or PBS.
Histograms of repeat length distributions in mouse muscle treated with the mixmer LNA-ASO or PBS.
Repeat instability in individual XXL mice.