

Abstract
Abnormal methylation in gene promoters is a hallmark of the cancer genome; however, factors that may influence promoter methylation have not been well elucidated. As the one-carbon metabolism pathway provides the universal methyl donor for methylation reactions, perturbation of this pathway might influence DNA methylation and, ultimately, affect gene functions. Utilizing approximately 800 breast cancer tumor tissues from a large population-based study, we investigated the relationships between dietary and genetic factors involved in the one-carbon metabolism pathway and promoter methylation of a panel of 13 breast cancer-related genes. We found that CCND2, HIN1 and CHD1 were the most “dietary sensitive” genes, as methylation of their promoters was associated with intakes of at least two out of the eight dietary methyl factors examined. On the other hand, some micronutrients (i.e., B2 and B6) were more “epigenetically active” as their intake levels correlated with promoter methylation status in 3 out of the 13 breast cancer genes evaluated. Both positive (hypermethylation) and inverse (hypomethylation) associations with high micronutrient intake were observed. Unlike what we saw for dietary factors, we did not observe any clear patterns between one-carbon genetic polymorphisms and the promoter methylation status of the genes examined. Our results provide preliminary evidence that one-carbon metabolism may have the capacity to influence the breast cancer epigenome. Given that epigenetic alterations are thought to occur early in cancer development and are potentially reversible, dietary modifications may offer promising venues for cancer intervention and prevention.
Key words: epigenetics, DNA methylation, one-carbon, B vitamins, genetic polymorphism
Introduction
Promoter hypermethylation of cancer-related genes, accompanied by global hypomethylation, is a common feature of tumor cells.1 While large numbers of studies have been performed to investigate the functional consequence of aberrant DNA methylation, little is known about what might influence the DNA methylation status of the genome. Since epigenetic processes are dynamic, reversible and susceptible to exogenous factors, identifying environmental or lifestyle factors that influence the epigenome may offer a unique opportunity for chemoprevention or diet-based interventions that target epigenetic pathways.
Increasing evidence suggests that certain constituents in food and dietary supplements have the capacity to influence the epigenome and, ultimately, an individual's risk of developing cancer.2,3 Among such compounds, micronutrients in folate-mediated one-carbon metabolism are of particular interest.4 The universal methyl donor in the cellular reactions, S-adenosylmethionine (SAM), is generated in the one-carbon pathway. The key micronutrients in this pathway are folate, methionine and several B vitamins (i.e., B2, B6 and B12) that are essential co-factors for one-carbon transfer reactions. Other methyl donors, such as choline and betaine, can also affect SAM status, primarily through choline-mediated one-carbon metabolism, and ultimately impact DNA methylation. It has been shown that dietary methyl donors are capable of modulating methylation patterns in both animal models and humans.5–8 Furthermore, there are reports showing that functional polymorphisms in one-carbon-metabolizing genes can also influence DNA methylation status.4,9,10 Nevertheless, human data on whether one-carbon metabolism influences DNA methylation are limited and inconsistent. This issue has been explored primarily in colon cancer11,12 and to a much lesser extent in breast cancer.
In this study, we chose a panel of 13 genes that had been shown to play an important role in breast carcinogenesis and tumor progression. For example, steroid hormone genes (e.g., ESR1 and PGR) and tumor suppressors (e.g., BRCA1, APC and p16INK4a) are well-established breast cancer-related genes. More importantly, promoters of these genes are frequently methylated in breast tumor tissues and this aberrant methylation has been associated with malignant phenotypes and survival rates of breast cancer cases.13–22 Thus, promoter methylation was proposed to be the underlying mechanism for the loss of gene function.23,24
We previously reported that one-carbon metabolism influenced breast cancer risk and survival in the population-based Long Island Breast Cancer Study Project (LIBCSP).25–27 Herein, we investigate the influence of one-carbon metabolism (i.e., dietary intake of co-factors and genetic polymorphisms of genes) on promoter methylation status of a panel of breast cancer-related genes.
Results
Promoter methylation of 13 breast cancer-related genes was assessed in up to 851 breast tumor samples from the LIBCSP, including 104 in situ and 747 invasive tumors. Table 1 lists the genes examined in this study, with their established or proposed functions. Also shown in Table 1 are number of tumor samples examined for each gene's methylation status. The main reason for missing methylation data was an insufficient amount of DNA yielded from the tumor blocks.
Table 1.
Genes selected for methylation analysis and their putative functions
| Gene | Full name | Group | Function | Assayed sample no. | Methylated samples (%) |
| ESR1 | estrogen receptor 1 | steroid hormone receptor | hormone binding, DNA binding and activation of transcription | 851 | 44.8 |
| PGR | progesterone receptor | steroid hormone receptor | mediates the physiological effects of progesterone | 851 | 11.9 |
| BRCA1 | breast cancer 1, early onset | tumor suppressor | maintaining genomic stability | 851 | 59.0 |
| APC | adenomatous polyposis coli | tumor suppressor | an antagonist of the Wnt signaling; Defects cause familial adenomatous polyposis | 800 | 48.4 |
| p16/CDKN2A | cyclin-dependent kinase inhibitor 2A | tumor suppressor | cell cycle control | 777 | 3.6 |
| HIN1/SCGB3A1 | secretoglobin, family 3A, member 1 | tumor suppressor | growth-inhibitory cytokine, regulates epizthelial cell differentiation. | 765 | 62.9 |
| RASSF1A | Ras association domain family member 1 | tumor suppressor | involved in cell cycle control | 765 | 85.2 |
| DAPK1 | death-associated protein kinase 1 | tumor suppressor | a positive mediator of gamma-interferon induced programmed cell death | 765 | 14.1 |
| GSTP1 | glutathione S-transferase pi 1 | detoxification | xenobiotic metabolism | 765 | 27.8 |
| CCND2 | cyclin D2 | oncogene | regulators of CDK kinases | 765 | 19.6 |
| TWIST1 | twist homolog 1 | transcription factors | cell lineage determination and differentiation | 765 | 15.3 |
| CDH1 | E-cadherin | tumor suppressor | cell proliferation, invasion and/or metastasis | 765 | 5.8 |
| RARβ | retinoic acid receptor, beta | steroid hormone receptor | mediates cellular signalling in embryonic morphogenesis, cell growth and differentiation | 765 | 27.6 |
We have previously reported the relationship of promoter methylation of the same panel of genes with demographic and clinical-pathological characteristics in the population.28,29 This study focuses on the influence of diet on promoter methylation of the same panel of genes. Dietary methyl constituents examined in this study include folate, methionine, choline, betaine, B vitamins (B2, B6 and B12), and alcohol (a folate antagonist). Inter-relationships between dietary intake of these methyl constituents and 13 breast cancer genes are summarized in Table S1.
There are multiple gene-nutrient associations; however, the effects were moderate in size, 2-fold in general, when the high nutrient intake categories were compared to low ones (in tertiles). Although folate, methione and choline are considered key methyl donors in one-carbon metabolism, there is little evidence that these three micronutrients were associated with promoter methylation of any of the 13 breast cancer genes; the only exception is the inverse relationship between folate and CCND2 methylation (Table S1). Interesting patterns emerge from the gene-nutrient matrix (Table 2). Two co-factors for one-carbon metabolizing enzymes, B2 (for MTHFR) and B6 (for cSHMT), were associated with 3 of the 13 breast cancer genes (Fig. 1) and two of these genes, HIN1 and CDH1, were both associated with B2 and B6. Furthermore, both positive and inverse associations between dietary intake and promoter methylation were observed (Fig. 1). Another interesting observation from the gene-nutrient matrix is that promoter methylation of several genes (CCND2, HIN1 and CHD1) appeared to be associated with multiple (at least two) dietary micronutrients (Fig. 2). While high intake of methyl constituents positively associated with HIN1 promoter methylation (with B2 and B6), inverse associations were observed for CCND2 (with folate and B6) and CDH1 (with B2, B6 and B12).
Table 2.
Summary of significant trends between one-carbon micronutrient intake and gene promoter methylation status
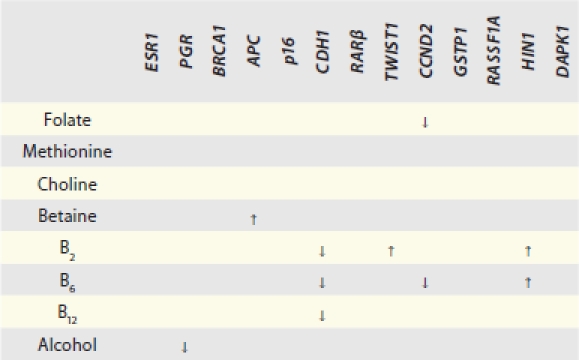 |
↑ indicates positive association, i.e., higher intake is associated with higher likelihood for a case to possess a methylated promoter for the gene of interest; ↓ indicates inverse association, i.e., higher intake is associated with lower likelihood for a case to possess a methylated promoter for the gene of interest. Cells without an arrow are those with insignificant trend test results.
Figure 1.

Example of epigenetically active micro-nutrients for association between gene promoter methylation status and dietary intake of one-carbon related nutrients. Odd ratios (ORs) and 95% confidence intervals are shown on the y-axis, comparing medium and high intake to the low intake group. p values are of the test for liner trend of ORs.
Figure 2.

Example of dietary sensitive genes for association between gene promoter methylation status and dietary intake one-carbon related nutrients. Odds ratios and 95% confidence intervals were shown on the y-axis, comparing medium and high intake to the low intake group. p values are of the test for liner trend of ORs.
We then explored associations between 13 functional polymorphisms in one-carbon metabolizing genes and gene promoter methylation status (Table S2). Several suggestive dose-dependent relations were observed (in other words, if there was a corresponding increase in the magnitude of the odd ratios (ORs) with an increase in the number of variant alleles for a particular polymorphism, then that association was considered a positive dose-dependent relationship and, conversely, if the magnitude of the ORs decreased with the number of alleles, then that association was considered to be an inverse dose-dependent relationship): the TYMS 2R allele, ESR1 hypermethylation; the DHFR 19 bp deletion allele, CDH1 hypomethylation; CHDH rs12676 T allele, GSTP1 hypermethylation; and CHDH rs9001 C allele, GSTP1 hypomethylation. However, unlike the gene-nutrient matrix above, we did not observe any clear patterns between one-carbon polymorphism and promoter methylation.
Discussion
Changes in DNA methylation have been shown to play important roles in breast cancer development; these alterations are potentially reversible.30 The panel of genes chosen in this study was based on their important role in breast carcinogenesis and tumor progression. These genes are frequently methylated in breast cancer and promoter methylation was proposed to be the underlying mechanism for the loss of function.23,24
To date, few studies have identified factors, especially modifiable lifestyle factors, that are capable of influencing the epigenome.3 In this study, we focus on the one-carbon metabolism, in which potentially modifiable factors are involved. The goal of this study was to examine the potential influence of nutrient intake and genetic polymorphisms in the key enzymes involved in the one-carbon metabolic pathway on promoter methylation of breast cancer-related genes. Our study, based on a large population-based case-control study, is the first to systematically examine lifestyle factors that may influence the breast cancer epigenome, and may lead to the development of better preventive strategies for the disease.
Several interesting findings emerged from our study. First, from a panel of 13 breast cancer related genes, promoter methylation of CCND2, HIN1 and CHD1 appeared to be more “sensitive” to dietary modulation, as they were correlated with at least 2 out of 8 micronutrients examined in the study. In the meantime, two one-carbon micronutrients, vitamin B2 and B6, appeared to be more “epigenetically active” as they were associated with promoter methylation of 3 out of 13 genes examined. While vitamin B2 is co-factor for the rate-limiting enzyme in folate-mediated one-carbon metabolism, MTHFR, vitamin B6 is the co-factor of another critical enzyme, cSHMT, involved in regenerating the methyl-carrying folate in the pathway. These results suggest that intake of certain B vitamins may affect the supply of key co-factors required for maintaining proper functions of one-carbon metabolism in the cell, thus influencing the metabolic output of methyl donors for methylation reactions.
Second, associations between high intake of one-carbon micronutrients and promoter methylation are not unidirectional; both positive (hypermethylation) and inverse (hypomethylation) relationships were observed. For example, high B2 intake was associated with lower likelihood of having a methylated promoter for CDH1 but with higher likelihood of having a methylated promoter for TWIST1 and HIN1. These results may reflect the complex regulatory mechanisms in the cell. One-carbon metabolism provides the methyl-donor (i.e., SAM) for the methylation reaction; interruption of this pathway by suboptimal nutrients intake or by suboptimal enzyme activity caused by genetic polymorphism may influence methyl supply. However, the regulation of the methyltransferases (e.g., DNMT3) is not known, especially for gene specific methylation. Genomic structure, sequence dependence and genetic mutation could all play a role.
Third, we found suggestive evidence that functional polymorphisms in one-carbon metabolizing genes could influence promoter methylation. However, we did not pursue the analyses of gene-diet interactions due to limited power. There are few other reports studying gene promoter methylation status in relation to one-carbon metabolism. One study examined seven genes (RARβ2, CDH1, ESR1, BRCA1, CCND2, p16 and TWIST) among ∼200 tumors.31 They found that patients who were homozygous for the TYMS double tandem repeat (2R/2R) showed a trend for more frequent promoter methylation (by methylated gene counts) in their breast cancers, and patients who were homozygous for the MTHFD1 G1958A polymorphism showed a significantly higher frequency of methylation. However, none of the individual methylated CpG sites showed a significant association with any of the polymorphisms. We observed an ESR1-TYMS association with the same trend in our study (Table S2). Another recent study examined promoter methylation of three genes (E-cadherin, p16 and RARβ2) among ∼800 breast tumors and found no evidence that MTHFR C677T, A1298C and MTR A2756G polymorphisms were associated methylation status.32 We did not observe significant association for these three polymorphisms in our study, either.
There are other studies on the influence of one-carbon metabolism on gene promoter methylation with limited sample size. The majority of these published papers are on colorectal cancer, focusing on one or few of the nutrients (e.g., folate), SNPs (MTHFR C677T) and/or gene methylation status (p16, hMLH).9,33–35 These results are far from conclusive. Variations in sample sizes, and the use of different assays to detect methylation status, could have contributed to the inconsistency.
Given the number or micronutrients and genetic polymorphisms examined in this study, one important issue that warrants careful consideration is that of “multiple comparison,” which results in increased likelihood of false positive findings. We examined eight micronutrients and 13 genetic polymorphisms in relation to the promoter methylation status of 13 genes. At the p < 0.05 level, there could be five significant results for micronutrients and eight for polymorphisms by chance alone. There is continuing debate on whether/when/how multiple comparisons should be taken into account.36 When evaluating results of molecular epidemiology studies, in addition to the magnitude of the p value, statistical power and the priority of the tested hypothesis, are also need to take into account.37 In this study, we opt to report the crude p values without adjusting for multiple comparisons for the following reasons. First, instead of randomly considering multiple genes without regarding of the biological relevance but only relying on statistical significance to interpret the study findings, we chose to focus only on genes and dietary/genetic factors that have functional relevance to breast cancer. Second, using the nutrient-gene matrix, our study focuses on the patterns of micronutrients-methylation associations, rather than of the magnitude of specific associations. Lastly, multiple comparison adjustment, such as Bonferroni method, may safeguard false positive findings, but it may increase Type II error (false negative) and reduce sensitivity,38 thus being too conservative for exploratory purpose. Nevertheless, we need to be cautious and results from our study warrant replication in other population studies.
Several limitations of our study are discussed below. (1) In our study, tumor DNA was not available for all cases of the LIBCSP participants. Although there were some differences between those with and without tumor DNA available for our analyses (as described in the Methods section), the benefit of utilizing our population-based sample was that we were able to quantify the differences between the two groups. This valuable contrast aided in our interpretation of the generalizability of our study results to the general population. It is this type of information that is often unavailable from other study populations, such as those derived from a hospital-based case series. (2) The association between one-carbon metabolism and gene promoter methylation was examined only in tumor tissues; whether the same association exists in tissues that precede malignancy (i.e., normal adjacent tissues) is not known. This limits our ability to draw definitive causal-inference. Nevertheless, our results add knowledge in the breast cancer research field and provide a rationale for future mechanistic research. (3) The dietary factors may affect methylation of multiple CpG sites in the gene promoter region. The assays used in this study examined a limited number of CpG sites in the region, which may limit the sensitivity of detecting a diet-methylation relationship. (4) In our study, dietary intake values for one-carbon related micronutrients and compounds were assessed using a modified Block food frequency questionnaire (FFQ). There is continuing debate over the accuracy of this method to assess usual adult diet. Despite these potential limitations, the ranking of intakes (which was the approach used in our analyses) was unlikely to be invalid, given that the Block FFQ has been shown to be a valid and reliable dietary assessment tool for estimating usual food intake and ranking individuals into categories of intake of micronutrients.39,40
In summary, we examined the influence of one-carbon metabolism on gene promoter methylation in 13 breast-cancer related genes in a relatively large epidemiologic study. Our results provide preliminary evidence that dietary nutrients might influence the breast cancer epigenome. Given that epigenetic alterations are reversible and occur early in cancer development, dietary intervention may offer promising venues for cancer prevention.
Materials and Methods
Study population.
We utilized the resources of the Long Island Breast Cancer Study Project, a population-based study. The study participants included 1,504 women newly diagnosed with a first primary breast cancer in 1996–1997 and who participated in the original, parent case-control study. Details of the study design have been described in detail previously in references 41 and 42. Participant information used in this ancillary study was obtained as part of in-person interviews and medical record abstraction that were collected as part of the case-control parent study.41 Questionnaires were administrated to assess the demographic characteristics and breast cancer-related factors. Tumor characteristics such as ER/PR status were extracted from the medical records. Among cases with this information available, 583 (58.9%) were ER+/PR+; 143 (14.4%) were ER+/PR−; 52 (5.3%) were ER−/PR+; and 212 (21.4%) were ER−/PR−.
Archived pathology blocks for the first primary breast cancer of the LIBCSP cases were requested from the 33 hospitals in the Long Island study area and successfully retrieved for 962 women.43 After review and processing in the laboratory by a trained breast cancer pathologist (H. Hibshoosh), tumor tissues from 859 subjects (89.3%) were available for our study analyses. We compared the demographic and clinico/pathological features between cases with or without tumor blocks available for methylation analysis in our study. Although most characteristics are similar between these two groups, some factors were different. Case women who had tumor samples available for methylation analysis tended to be older (mean age 59.6 vs. 57.9 years; p = 0.005), to have an invasive tumor (87.8% vs. 80.1%; p < 0.001), and to be post-menopausal (70.7% vs. 64.6%; p = 0.01). The study protocol was approved by the Institutional Review Boards of the collaborating institutions.
Determining gene promoter methylation in tumor DNA.
Tumor block retrieval and DNA extraction were performed as previously described in reference 43 and 44. Tumor DNA first underwent bisulfite modification using the CpGnome DNA Modification Kit (Chemicon International, cat. no. S7820) following the protocol from the manufacturer. We selected a panel of 13 genes that have been implicated in breast carcinogenesis (Table 1) for methylation analysis. Promoter methylation of ER, PR and BRCA1 was determined by methylation-specific PCR (MSP) as described previously in reference 44 and 45. The MethyLight assay was used for determining the methylation status of the rest of genes as described in reference 46 and 47. The percentage of methylation was calculated by the 2−ΔΔCT method, where ΔΔCT = (CT,Target − CT,Actin)sample − (CT,Target − CT,Actin)fully methylated DNA48 and multiplying by 100. Samples containing ≥4% fully methylated molecules were designated as methylated, whereas samples containing <4% were designated as unmethylated. The main reason for some missing methylation data was that there was not enough DNA as template for PCR reaction regardless of methods for assessing methylation status. The limiting amount of DNA is primarily due to the small size of the original tumor.
Genotyping and dietary assessment.
Genotyping was conducted on 13 functional polymorphisms in the one-carbon metabolism pathway using methods described in reference 25, 26 and 49. Dietary intake values for one-carbon related micronutrients and compounds were calculated based on data collected as part of the in-person case-control interview assessed using a modified Block food frequency questionnaire (FFQ), which was self-completed by the study participants.50,51
Statistical analysis.
The MSP-PCR assay for ER, PR and BRCA1 promoter methylation generated dichotomized outcomes, i.e., methylated and unmethylated status. MethyLight assay, on the other hand, yielded percentage of methylation for gene promoters that were then dichotomized into methylated or unmethylated cases using a cut-off of 4%, according to previous reports in reference 47, 52 and 53. Relationships of gene promoter methylation status with demographic and clinical-pathological characteristics were examined using chi-square test. Associations between methylation status and diet or genetic factors were analyzed in a case-case setting using unconditional logistic regression.54 With this approach, the odds ratios (ORs) and 95% confidence interval (CI) represent the likelihood of a case possessing a methylated promoter for the gene of interest given certain nutrient intake levels or genotypes. Nutrient intakes in the year prior to the case interviews were divided into tertiles based on the distributions in cases with methylation data. Tests for linear trend of ORs were calculated by regressing on the median values of each tertile.54 For genetic polymorphisms, an ordered categorical variable by assigning a score to each genotype [i.e., 0 (no variant allele), 1 (carrying one variant allele) and 2 (carrying two variant alleles)] was used as the variable to test for trend.
Potential confounders included age at diagnosis (entered as 5-year age groups), ER/PR IHC status [positive group: ER and PR both positive (ER+/PR+) and negative group: all others (ER+/PR−, ER−/PR+, ER−/PR−)], cancer type (in situ vs. invasive), menopausal status (pre- vs. post-), race and family history of breast cancer and benign breast. If eliminating a covariate from the full regression model changed the effect estimate by 10% or more, the covariate was considered a confounder and kept in the model.54 None of the covariates tested met this criterion and, thus, only results from the age-adjusted model are presented. Because of the exploratory nature of this study, we choose to report the crude p values without adjusting for multiple comparison; the implications of this approach are discussed in the ‘Discussion’ section. All statistical analyses were performed using SAS statistical software version 9.1(SAS Institute, Cary, NC).
Acknowledgments
This work was supported by grants from the National Cancer Institutes (R01CA109753 and 3R01CA109753-04S1) and in part by grants from Department of Defense (BC031746 and W81XWH-06-1-0298) and National Cancer Institute and National Institutes of Environmental Health and Sciences (UO1CA/ES66572, UO1CA66572, P30CA013696, P30ES009089 and P30ES10126).
Supplementary Material
References
- 1.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Xu X. Diet, epigenetic and cancer prevention. Adv Genet. 2010;71:237–255. doi: 10.1016/B978-0-12-380864-6.00008-0. [DOI] [PubMed] [Google Scholar]
- 3.Ulrich CM, Grady WM. Linking epidemiology to epigenomics—where are we today? Cancer Prev Res. 2010;3:1505–1508. doi: 10.1158/1940-6207.CAPR-10-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern LL, Mason JB, Selhub J, Choi SW. Genomic DNA hypomethylation, a characteristic of most cancers, is present in peripheral leukocytes of individuals who are homozygous for the C677T polymorphism in the methylenetetrahydrofolate reductase gene. Cancer Epidemiol Biomarkers Prev. 2000;9:849–853. [PubMed] [Google Scholar]
- 5.Christman JK, Sheikhnejad G, Dizik M, Abileah S, Wainfan E. Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis. 1993;14:551–557. doi: 10.1093/carcin/14.4.551. [DOI] [PubMed] [Google Scholar]
- 6.Fowler BM, Giuliano AR, Piyathilake C, Nour M, Hatch K. Hypomethylation in cervical tissue: Is there a correlation with folate status? Cancer Epidemiol Biomarkers Prev. 1998;7:901–906. [PubMed] [Google Scholar]
- 7.Rampersaud GC, Kauwell GP, Hutson AD, Cerda JJ, Bailey LB. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am J Clin Nutr. 2000;72:998–1003. doi: 10.1093/ajcn/72.4.998. [DOI] [PubMed] [Google Scholar]
- 8.Jacob RA, Gretz DM, Taylor PC, James SJ, Pogribny IP, Miller BJ, et al. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J Nutr. 1998;128:1204–1212. doi: 10.1093/jn/128.7.1204. [DOI] [PubMed] [Google Scholar]
- 9.Paz MF, Avila S, Fraga MF, Pollan M, Capella G, Peinado MA, et al. Germ-line variants in methyl-group metabolism genes and susceptibility to DNA methylation in normal tissues and human primary tumors. Cancer Res. 2002;62:4519–4524. [PubMed] [Google Scholar]
- 10.Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, Bagley PJ, et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci USA. 2002;99:5606–5611. doi: 10.1073/pnas.062066299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YI. Folate and DNA methylation: A mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol Biomarkers Prev. 2004;13:511–519. [PubMed] [Google Scholar]
- 12.Estécio MRH. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PloS one. 2007;2:399. doi: 10.1371/journal.pone.0000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jin Z, Tamura G, Tsuchiya T, Sakata K, Kashiwaba M, Osakabe M, et al. Adenomatous polyposis coli (APC) gene promoter hypermethylation in primary breast cancers. Br J Cancer. 2001;85:69–73. doi: 10.1054/bjoc.2001.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silva JM, Dominguez G, Villanueva MJ, Gonzalez R, Garcia JM, Corbacho C, et al. Aberrant DNA methylation of the p16INK4a gene in plasma DNA of breast cancer patients. Br J Cancer. 1999;80:1262–1264. doi: 10.1038/sj.bjc.6690495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agathanggelou A, Honorio S, Macartney DP, Martinez A, Dallol A, Rader J, et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumors. Oncogene. 2001;20:1509–1518. doi: 10.1038/sj.onc.1204175. [DOI] [PubMed] [Google Scholar]
- 16.Dulaimi E, Hillinck J, Ibanez de Caceres I, Al-Saleem T, Cairns P. Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin Cancer Res. 2004;10:6189–6193. doi: 10.1158/1078-0432.CCR-04-0597. [DOI] [PubMed] [Google Scholar]
- 17.Nass SJ, Herman JG, Gabrielson E, Iversen PW, Parl FF, Davidson NE, et al. Aberrant methylation of the estrogen receptor and E-cadherin 5′ CpG islands increases with malignant progression in human breast cancer. Cancer Res. 2000;60:4346–4348. [PubMed] [Google Scholar]
- 18.Sirchia SM, Ferguson AT, Sironi E, Subramanyan S, Orlandi R, Sukumar S, et al. Evidence of epigenetic changes affecting the chromatin state of the retinoic acid receptor beta2 promoter in breast cancer cells. Oncogene. 2000;19:1556–1563. doi: 10.1038/sj.onc.1203456. [DOI] [PubMed] [Google Scholar]
- 19.Esteller M, Corn PG, Urena JM, Gabrielson E, Baylin SB, Herman JG. Inactivation of glutathione S-transferase P1 gene by promoter hypermethylation in human neoplasia. Cancer Res. 1998;58:4515–4518. [PubMed] [Google Scholar]
- 20.Evron E, Umbricht CB, Korz D, Raman V, Loeb DM, Niranjan B, et al. Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res. 2001;61:2782–2787. [PubMed] [Google Scholar]
- 21.Fackler MJ, McVeigh M, Mehrotra J, Blum MA, Lange J, Lapides A, et al. Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res. 2004;64:4442–4452. doi: 10.1158/0008-5472.CAN-03-3341. [DOI] [PubMed] [Google Scholar]
- 22.Gort EH, Suijkerbuijk KP, Roothaan SM, Raman V, Vooijs M, van der Wall E, et al. Methylation of the TWIST1 promoter, TWIST1 mRNA levels and immunohistochemical expression of TWIST1 in breast cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:3325–3330. doi: 10.1158/1055-9965.EPI-08-0472. [DOI] [PubMed] [Google Scholar]
- 23.Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21:5462–5482. doi: 10.1038/sj.onc.1205606. [DOI] [PubMed] [Google Scholar]
- 24.Yang X, Yan L, Davidson NE. DNA methylation in breast cancer. Endocr Relat Cancer. 2001;8:115–127. doi: 10.1677/erc.0.0080115. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Gammon MD, Chan W, Palomeque C, Wetmur JG, Kabat GC, et al. One-carbon metabolism, MTHFR polymorphisms and risk of breast cancer. Cancer Res. 2005;65:1606–1614. doi: 10.1158/0008-5472.CAN-04-2630. [DOI] [PubMed] [Google Scholar]
- 26.Xu X, Gammon MD, Zhang H, Wetmur JG, Rao M, Teitelbaum SL, et al. Polymorphisms of one-carbon metabolizing genes and risk of breast cancer in a population-based study. Carcinogenesis. 2007;28:1504–1509. doi: 10.1093/carcin/bgm061. [DOI] [PubMed] [Google Scholar]
- 27.Xu X, Gammon MD, Wetmur JG, Bradshaw PT, Teitelbaum SL, Neugut AI, et al. B-vitamin intake, one-carbon metabolism and survival in a population-based study of women with breast cancer. Cancer Epidemiol Biomarkers Prev. 2008;17:2109–2116. doi: 10.1158/1055-9965.EPI-07-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu X, Gammon MD, Zhang Y, Cho YH, Wetmur JG, Bradshaw PT, et al. Gene promoter methylation is associated with increased mortality among women with breast cancer. Breast Cancer Res Treat. 2010;121:685–692. doi: 10.1007/s10549-009-0628-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho YH, Shen J, Gammon MD, Zhang YJ, Wang Q, Gonzalez K, et al. Prognostic significance of gene-specific promoter hypermethylation in breast cancer patients. Breast Cancer Res Treat. doi: 10.1007/s10549-011-1712-y. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10:687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 31.Li SY, Rong M, Iacopetta B. Germ-line variants in methyl-group metabolism genes and susceptibility to DNA methylation in human breast cancer. Oncol Rep. 2006;15:221–225. [PubMed] [Google Scholar]
- 32.Tao MH, Shields PG, Nie J, Marian C, Ambrosone CB, McCann SE, et al. DNA promoter methylation in breast tumors: No association with genetic polymorphisms in MTHFR and MTR. Cancer Epidemiol Biomarkers Prev. 2009;18:998–1002. doi: 10.1158/1055-9965.EPI-08-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Engeland M, Weijenberg MP, Roemen GM, Brink M, de Bruine AP, Goldbohm RA, et al. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: The netherlands cohort study on diet and cancer. Cancer Res. 2003;63:3133–3137. [PubMed] [Google Scholar]
- 34.Mokarram P, Naghibalhossaini F, Saberi Firoozi M, Hosseini SV, Izadpanah A, Salahi H, et al. Methylenetetrahydrofolate reductase C677T genotype affects promoter methylation of tumor-specific genes in sporadic colorectal cancer through an interaction with folate/vitamin B12 status. World J Gastroenterol. 2008;14:3662–3671. doi: 10.3748/wjg.14.3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stidley CA, Picchi MA, Leng S, Willink R, Crowell RE, Flores KG, et al. Multivitamins, folate and green vegetables protect against gene promoter methylation in the aerodigestive tract of smokers. Cancer Research. 2010;70:568–574. doi: 10.1158/0008-5472.CAN-09-3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michels KB, Rosner BA. Data trawling: To fish or not to fish. The Lancet. 1996;348:1152–1153. doi: 10.1016/S0140-6736(96)05418-9. [DOI] [PubMed] [Google Scholar]
- 37.Wacholder S, Chanock S, Garcia-Closas M, El ghormli L, Rothman N. Assessing the probability that a positive report is false: An approach for molecular epidemiology studies. Journal of the National Cancer Institute. 2004;96:434–442. doi: 10.1093/jnci/djh075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1:43–46. [PubMed] [Google Scholar]
- 39.Block G, Hartman AM, Dresser CM, Carroll MD, Gannon J, Gardner L. A data-based approach to diet questionnaire design and testing. Am J Epidemiol. 1986;124:453–469. doi: 10.1093/oxfordjournals.aje.a114416. [DOI] [PubMed] [Google Scholar]
- 40.Potischman N, Swanson CA, Coates RJ, Weiss HA, Brogan DR, Stanford JL, et al. Dietary relationships with early onset (under age 45) breast cancer in a case-control study in the united states: Influence of chemotherapy treatment. Cancer Causes Control. 1997;8:713–721. doi: 10.1023/a:1018475203820. [DOI] [PubMed] [Google Scholar]
- 41.Gammon MD, Neugut AI, Santella RM, Teitelbaum SL, Britton JA, Terry MB, et al. The long island breast cancer study project: Description of a multi-institutional collaboration to identify environmental risk factors for breast cancer. Breast Cancer Res Treat. 2002;74:235–254. doi: 10.1023/a:1016387020854. [DOI] [PubMed] [Google Scholar]
- 42.Gammon MD, Santella RM, Neugut AI, Eng SM, Teitelbaum SL, Paykin A, et al. Environmental toxins and breast cancer on long island. I. polycyclic aromatic hydrocarbon DNA adducts. Cancer Epidemiol Biomarkers Prev. 2002;11:677–685. [PubMed] [Google Scholar]
- 43.Rossner P, Jr, Gammon MD, Zhang YJ, Terry MB, Hibshoosh H, Memeo L, et al. Mutations in p53, p53 protein overexpression and breast cancer survival. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu X, Gammon MD, Zhang Y, Bestor TH, Zeisel SH, Wetmur JG, et al. BRCA1 promoter methylation is associated with increased mortality among women with breast cancer. Breast Cancer Res Treat. 2009;115:397–404. doi: 10.1007/s10549-008-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z, Maekawa M, Horii T, Morita M. The multiple promoter methylation profile of PR gene and ERα gene in tumor cell lines. Life Sci. 2003;73:1963–1972. doi: 10.1016/S0024-3205(03)00544-7. [DOI] [PubMed] [Google Scholar]
- 46.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Danenberg PV, Laird PW. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Res. 1999;59:2302–2306. [PubMed] [Google Scholar]
- 47.Eads CA, Lord RV, Kurumboor SK, Wickramasinghe K, Skinner ML, Long TI, et al. Fields of aberrant CpG island hypermethylation in barrett's esophagus and associated adenocarcinoma. Cancer Res. 2000;60:5021–5026. [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 49.Xu X, Gammon MD, Zeisel SH, Lee YL, Wetmur JG, Teitelbaum SL, et al. Choline metabolism and risk of breast cancer in a population-based study. FASEB J. 2008;22:2045–2052. doi: 10.1096/fj.07-101279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaudet MM, Britton JA, Kabat GC, Steck-Scott S, Eng SM, Teitelbaum SL, et al. Fruits, vegetables and micronutrients in relation to breast cancer modified by menopause and hormone receptor status. Cancer Epidemiol Biomarkers Prev. 2004;13:1485–1494. DOI:13/9/1485. [PubMed] [Google Scholar]
- 51.Zeisel SH, Mar MH, Howe JC, Holden JM. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. 2003;133:1302–1307. doi: 10.1093/jn/133.5.1302. [DOI] [PubMed] [Google Scholar]
- 52.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, et al. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006;8:209–217. doi: 10.2353/jmoldx.2006.050135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rothman KJ, Greenland S. Modern Epidemiology. Ed. Philadelphia, Pa: Lippincott-Raven Publishers; 1998. p. 2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.