

Abstract
Genomic alterations of the epidermal growth factor receptor (EGFR) gene play a crucial role in pathogenesis of glioblastoma multiforme (GBM). By systematic analysis of GBM genomic data, we have identified and characterized a novel exon 27 deletion mutation occurring within the EGFR carboxyl-terminus domain (CTD) in addition to identifying additional examples of previously reported deletion mutations in this region. We show that the GBM-derived EGFR CTD deletion mutants are able to induce cellular transformation in vitro and in vivo in the absence of ligand and receptor autophosphorylation. Treatment with the EGFR-targeted monoclonal antibody, cetuximab, or the small molecule EGFR inhibitor, erlotinib, effectively impaired tumorigenicity of oncogenic EGFR CTD deletion mutants. Cetuximab in particular prolonged the survival of intracranially xenografted mice with oncogenic EGFR CTD deletion mutants, compared to untreated control mice. Therefore, we propose that erlotinib and especially cetuximab treatment may be a promising therapeutic strategy in GBM patients harboring EGFR CTD deletion mutants.
Keywords: glioblastoma, epidermal growth factor receptor, cetuximab, EGFR mutation, targeted therapy
Introduction
Glioblastoma multiforme (GBM) is the most common type of malignant brain tumors. There are approximately 9,000 new cases diagnosed every year in the United States (1, 2). Due to the resistant nature of GBM to available therapeutic modalities, such as surgery, radiation, and chemotherapy, the median survival of patients diagnosed with GBM is only about 14 months (3, 4). Thus, it is critical to develop new effective therapeutic approaches to treat patients with this devastating disease.
Numerous studies, including recent large-scale genomic analyses, have identified epidermal growth factor receptor (EGFR), a member of the ErbB family of receptor tyrosine kinases, as a common genetically altered gene in primary GBM (5-10). Different classes of EGFR somatic mutations have been identified in GBM. These include the exon 2-7 deletion, which is known as variant III (EGFRvIII), and point mutations within the extracellular domain of EGFR (11-14). These genetic alterations have been shown to lead to oncogenic activation of the mutant receptor independent of ligand stimulation and, consequently, induce cellular transformation (14-16). In addition, various exon deletion mutations including exon 25–27 and exon 25–28 deletion mutations, which result in the truncation of the C-terminal domain of EGFR, have been identified in GBM patients although their oncogenic potential has not yet been characterized (17, 18). Furthermore, EGFR gene amplification and/or EGFR protein overexpression commonly occur in approximately 50% of GBM patients, suggesting that an increased abundance of the EGFR may also be responsible for tumorigenesis in primary GBM (7, 19). Interestingly, somatic mutations within the EGFR kinase domain, which are frequently identified in non-small cell lung cancer, have only rarely been identified in GBM (8, 14, 20).
Given that abnormal regulation of downstream signaling pathways such as PI3K/Akt, Ras/Erk and/or STAT5 originating from mutant EGFR appear to play a crucial role in pathogenesis of GBM, targeting oncogenic EGFR with small molecule kinase inhibitors or monoclonal antibodies has been tested as a therapeutic approach (21-23). Clinical trials with either erlotinib or gefitinib as a single agent therapy reveal that these drugs do not have additional clinical benefit over standard treatment regimens in unselected patients who have not been characterized for genomic alterations of EGFR (24, 25). Interestingly, a retrospective genetic analysis study with GBM patient tumor samples indicates that the clinical response to erlotinib is closely associated with co-expression of EGFRvIII and PTEN (26). This is consistent with the consensus that genetic factors in tumors may determine their clinical response, and identifying these genetic biomarkers is the key for successful targeted therapy with EGFR small molecule inhibitors. Cetuximab, a humanized monoclonal antibody, has been shown to be effective against GBM cell lines and in vivo xenograft mouse model as monotherapy or in combination with radiation or chemotherapy (27-29). However, only a single case study has reported the clinical effectiveness of cetuximab among GBM patients (30).
In this study, through genomic analysis of primary GBM patient samples collected under The Cancer Genome Atlas (TCGA), we have confirmed deletion mutations within the C-terminal domain of EGFR and have further identified novel C-terminal deletion mutations. In addition, we showed that the resulting C-terminal deletion mutants of EGFR are oncogenic in vitro and in vivo. Finally, we demonstrated that erlotinib and cetuximab inhibited the growth of tumors driven by C-terminal deletion EGFR mutants, indicating that both small molecule inhibitors and anti-EGFR monoclonal antibodies may be promising therapeutic approaches in treating GBM patients with tumors harboring such deletions.
Materials and Methods
Exon and copy number array analysis
RMA data from Affymetrix Human Exon 1.0 ST arrays and segmented and level 2 copy number data from SNP 6.0 arrays were batch-downloaded from the TCGA data portal (tcga-data.nci.nih.gov/tcga/tcgaCancerDetails.jsp?diseaseType=GBM&diseaseName=Glioblastoma multiforme). Exon expression data from tumor-derived RNA were normalized to exon expression data from total brain control RNA, and probes interrogating expression of EGFR exons 25 through 28 were compared to the average of those for EGFR exons 17 through 20. Segmented data were searched for tumor DNA copy number profiles with EGFR amplifications that contain segmentation breaks between exons 24 and 27 (chromosome 7 55269049 to 55270209) where the copy number of the 3’segment was lower than that of the 5’ segment. With level 2 copy number data, the copy number probe closest to exon 27 (CN_1227312) was compared to probes both 5’ and 3’ of exons 17 and exon 20. For more details, see SI Materials and Methods.
Expression Constructs
pBabe-puro plasmids encoding CT982NT, CT1054NT, and CT Del1 EGFR mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene) with wild-type EGFR as a template (31). The expression construct for the EGFR vIII mutant was previously described (32).
Cell culture and generation of cell lines by viral transduction
All EGFR-mutant expressing cell lines (Ba/F3, NIH-3T3 and LN443 cells) used in the study were established by retroviral infections, pooled and maintained as described previously (31, 33, 34). EGFR CT Del1 mutant were identified in the wild-type EGFR expressing Ba/F3 cell clone that grew after IL-3 withdrawal (see text for more detail).
Cell growth inhibition assay
For growth inhibition assays, Ba/F3 cells (10,000 cells) were plated in 180 μL media in 96-well flat-bottom plates (Corning). 24 hrs after plating, cell culture media was replaced with medium with and without either erlotinib or cetuximab. The concentrations of erlotinib and cetuximab used for the assay ranged from 3.3 μM to 10 μM or from 33 ng/mL to 100 μg/mL, respectively. The cells were incubated for another 72 hrs and the viable cell numbers were measured using Cell Counting Kit-8 solution (Dojindo, Kumamoto, Japan). Absorbance was measured at 450 nm after 3 hrs. Data are expressed as percentage of growth relative to that of untreated control cells.
Immunoblotting and antibodies
Cells were lysed in RIPA buffer supplemented with protease inhibitors (Roche) and phosphatase inhibitors (Calbiochem) and subjected to immunoblotting. Anti-EGFR (Ab-5) antibody was purchased from NeoMarker (Fremont, CA). Anti-phospho-tyrosine antibody (4G10) and anti-actin were from Millipore and Santa Cruz Biotechnology, respectively. Ab against phosho-Stat5 (Y705) was from Cell Signaling Biotechnology.
Generation of xenografted mice, erlotinib and cetuximab treatment
Animal (SCID mice) studies done in accordance with UCSD IACUC protocols. For subcutaneous studies, cells were resuspended in PBS and 1-2 million cells were injected in the flanks of mice and both cetuximab and erlotinib administration was initiated approximately twenty days after tumor cell inoculation, when the tumors had reached a diameter of 50 to 70 mm3 For intracranial studies, cells were resuspended in PBS at concentration of 2×105 viable cells/5 μl, and implanted into right striatum as previously described using stereotactic apparatus and the drug administration was initiated after 1 week (35). Cetuximab and erlotinib were obtained from Dana-Farber Cancer Institute pharmacy. For the cetuximab treated mice, we administered 1 mg per mouse of cetuximab by Intra-peritoneal (IP) injection to the mice 3 times per week, and for the erlotinib treated mice, we orally administered 1 mg per mouse of erlotinib 3 times per week. Mice were sacrificed when morbid and the date was recorded for survival analysis.
Pathology and immunohistochemistry analysis
All orthotopic xenograft tissue was fixed by host perfusion with phosphate buffered 4% formalin, followed by additional fixation in formalin after brain removal, dehydration by graded ethanols and embedding in wax (Paraplast Plus, McCormick Scientific) using routine techniques. All sections were 5μ on Superfrost /Plus slides (Fisher Scientific) and stained with hematoxylin and eosin by standard techniques. For immunohistochemistry, the following antibodies were used at the dilutions and incubation times/temperatures: 1) anti-EGFR ab (clone 5B7) (Ventana Medical Systems, predilute – diluted 1:4 in Dako diluent, 60 min/37°; 2) anti-phospho-EGFR (Tyr1173)(Cell Signaling #4407, 1:250 in Dako diluent, 60 min/37°. Tissue conditioning for both epitopes was performed using Tris buffer pH 8 with the following time/temperature intervals: 8 min/ 95°C; 4min/100°C; and 8 min cooling to 37°C. All immunohistochemistry was performed on the Ventana Medical Systems Discovery XT automated slide preparation system using the Ultraview (multimer) detection system for 12 min at 37°C.
Results
Identification and validation of EGFR C-terminal deletion mutants by exon array analysis in human GBM patients
Based on reports of somatic genome rearrangements in the region encoding the EGFR C-terminal domain (CTD) in GBM (17, 18), we examined the region of chromosome 7 from 55.268 MB to 55.276 MB (hg19) systematically by analyzing SNP 6.0 array copy number and exon array expression data (tcga-data.nci.nih.gov/tcga/tcgaCancerDetails.jsp?diseaseType=GBM&diseaseName=Glioblastoma multiforme) from GBM patient samples in the TCGA database (5, 8). We identified candidate deletions by the presence of either a) copy number segment boundaries following EGFR exon 24 where the segmented copy number of the 3’ segment was at least 1.5 normalized copies lower than that of the 5’ segment or b) expression values for one or more of EGFR exons 25 through 28 that was less than one-half the average expression value of EGFR exons 17 through 20. Where this approach identified deletions based on expression but not segmented copy number data, we analyzed raw SNP array data for the presence of a reduction in copy number of at least 1.5 normalized copies on probe CN_1227312, the copy number probe closest to exon 27 (see Supplementary Materials and Methods).
By these analyses, we found a total of 8 samples of 469 analyzed (7 out of 435 samples from exon array analysis and 6 out of 447 samples from SNP array analysis) showing evidence of EGFR C-terminal exonic deletions (Supplementary Table 1). The 8 samples with candidate EGFR C-terminal deletions can be divided into 4 categories, including previously reported exon 25-27 deletions, previously reported exon 25-28 deletions (18), hitherto unreported exon 27 deletions, and hitherto unreported exon 27-28 deletions.
To confirm candidate events, we performed qPCR analysis of genomic DNA using specific primers targeting exon 23 to 28 on 5 of the 8 samples for which sufficient DNA was available. These assays confirmed exon 25-28 deletions in three samples (TCGA 06-0216, TCGA 08-0511 and TCGA 08-0356), an exon 25-27 deletion in one sample (TCGA 02-0043), and an exon 27 deletion in one sample (TCGA 02-0102) (Fig. 1A). By PCR amplification with intronic primers from genomic DNA followed by amplicon sequencing, we further validated the exon 27 and exon 25-27 deletions in the relevant samples and mapped the breakpoints of these deletions (Fig. 1B). Since the deletion of exon 27 has not been previously reported, we sequenced cDNA from sample TCGA 02-0102 and confirmed that the EGFR exon 27 deletion results in the transcription of an aberrant mRNA consistent with a splice between exon 26 and exon 28 (Fig. 1B). However, we could not validate Ex25-28 deletion mutants using the same approach, because we were unable to determine the 3’ breakpoint of these deletion mutants. Instead, we mapped the 5’ breakpoint of Ex25-28 deletion mutants (TCGA 08-0511 and TCGA 08-0356) approximately within intron 24 using qPCR based analysis (Supplementary Fig. S1A). Thus, we believe that mutants harboring deletions within intron 24 likely result in aberrant transcripts missing Ex25-28 and containing a newly generated stop codon within exon 24, leading to an EGFR CTD deletion protein (Fig 1C and Supplementary Fig. S1C).
Figure 1. Identification and functional characterization of GBM-derived CT deletion mutants.
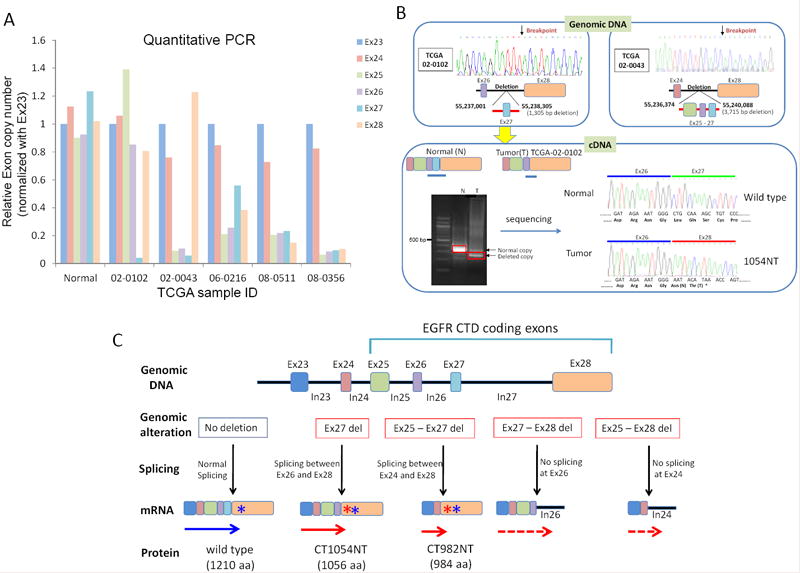
(A) Quantitative PCR analysis using primer sets for exons 23 through 28 of EGFR, marked according to the color legend on the right, performed on skin DNA from TCGA-08-0359 as a normal control and five samples harboring candidate EGFR CTD deletions based on exon array analysis shown in Table S1. The relative copy number for each exon is normalized to EGFR exon 23. (B) Direct sequencing of EGFR PCR fragments from TCGA-02-0102 and TCGA-02-0043 DNA revealed the location of intragenic deletions of exon 27 and exon 25-27 in these samples, respectively. (C) Schematic showing the proposed splicing and resulting protein products of GBM-derived mutants as well as wild-type EGFR. Deletion of exon 27 generates a frame-shifted exon 28, with the addition of Asn (N) and Thr (T) after amino acid 1054 followed by early termination (red solid arrow). Deletion of exons 25-27 also resulted in the addition of N and T after amino acid 982 followed by a stop codon (red solid arrow). Given that the 3’ end of the exon 27-28 and exon 25-28 deletions were not determined, the detailed transcripts are not defined (red dashed arrows). The blue asterisks indicate the position of the stop codon of the wild-type EGFR transcript (blue arrow). Red asterisks indicate the predicted stop codons generated by frame-shift of the indicated aberrant RNA transcripts.
In summary, we validated C-terminal EGFR deletions in the 5 tested GBM samples including a novel exon 27 deletion as well as the previously identified exon 25-27 and exon 25-28 deletions (Fig. 1C).
GBM-derived CT982NT and CT1054NT mutants are oncogenic
In order to explore the functional significance of GBM-derived EGFR CTD deletion mutants, we first generated retroviral vectors to express cDNA encoding the protein products of three EGFR mutants, CT982NT, CT1054NT, and Ex25-28 deletion. These mutants werestably introduced into Ba/F3 cells by retroviral infection along with the highly prevalent oncogenic EGFRvIII mutant as a positive control. We used the Ba/F3 pro-B cell line-as a model system for two reasons. First, the survival and proliferation of Ba/F3 cells are strictly dependent on IL-3 but ectopic oncogene expression relieves this dependency; thus we can not only determine the oncogenic activity of the EGFR CTD deletion mutants by assessing whether the forced expression of these mutants in Ba/F3 cells can functionally replace IL-3 dependency, but can also use these cells as an efficient system to test drug sensitivity, which has been highly predictive for clinical efficacy of specific EGFR mutants (36). Second, as Ba/F3 cells are known to express low to undetectable levels of endogenous ErbB proteins (37), the biological consequences of EGFR expression in the engineered Ba/F3 cells are likely due to the isogenically expressed EGFR mutants.
Ba/F3 cells expressing the CT982NT, CT1054NT, vIII mutants, or Ex25-28 deletion of EGFR were able to grow in the absence of IL-3, while the control parental Ba/F3 cell line failed to grow under the same conditions (Fig. 2A and data not shown). The IL-3 independent cell growth ability of Ba/F3 cells expressing the CT982NT mutant is comparable to that induced by oncogenic EGFRvIII mutant and higher than that of CT1054NT. Furthermore, while the EGFRvIII mutant underwent constitutive phosphorylation, we did not observe any detectable level of phosphorylation for the C-terminal deletion mutants, which is unsurprising as these mutants have deleted the major tyrosine phosphorylation sites of EGFR (Fig. 2B and Supplementary Fig. S1B). Consistent with the results in Ba/F3 cells, NIH-3T3 cells stably expressing either CT982NT or CT1054NT mutants, but not vector-infected cells, were able to grow in an anchorage independent manner in soft agar, a hallmark of cellular transformation, in the absence of ligands (Supplementary Fig. S2A).
Figure 2. GBM-derived CT982NT and CT1054NT EGFR CTD deletion mutants are oncogenic in the absence of tyrosine phosphorylation.
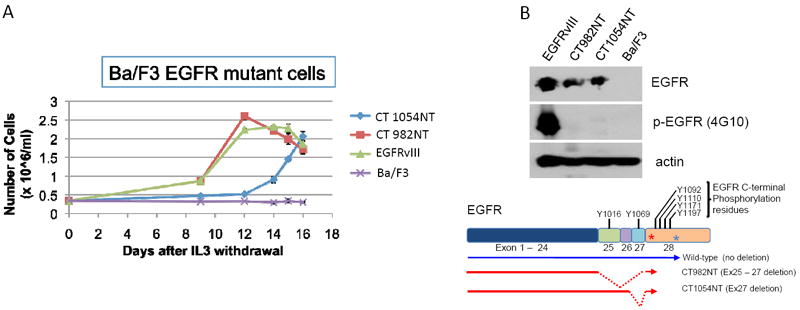
(A) CT982NT and CT1054NT mutants confer IL-3 independency to Ba/F3 cells. IL-3 independent cell proliferation ability of Ba/F3 cell lines stably expressing CT982NT, CT1054NT, vIII mutant EGFR as well as parental Ba/F3 cells, was assayed by counting cell numbers on 9, 12, 14, 15 and 16 days after IL-3 withdrawal. The results are indicated as means +/- SD of three independent counts. (B) Tyrosine phosphorylation of EGFR is dispensable for oncogenic activity of CT982NT and CT1054NT mutants. Whole cell lysates prepared from Ba/F3 cells analyzed in (A) were subjected to immunoblotting with antibodies against phospho-tyrosine (4G10), EGFR and actin. Schematic cartoon shows the location of tyrosine residues for phosphorylation within exons consisting of C-terminal domain of wild-type EGFR and CT deletion mutants.
Taken together, we concluded that GBM patient-derived CT982NT and CT1054NT EGFR mutants are oncogenic in vitro and are able to induce cellular transformation.
Tumors induced by EGFR CTD deletion mutants are sensitive to EGFR inhibitors
The clinical effectiveness of anti-EGFR targeted therapy with small molecule EGFR kinase inhibitors such as erlotinib or monoclonal antibodies such as cetuximab have been successfully proven in a subset of cancer types including non-small cell lung cancer and colorectal cancer (38, 39). Therefore, we sought to determine the efficacy of these drugs against the oncogenic EGFR CTD deletion mutants. Both erlotinib and cetuximab were able to effectively suppress the growth of Ba/F3 cells transformed by either the CT982NT or CT1054NT or Ex25-28 deletion EGFR mutants in a dose-dependent manner (Fig. 3A, 3B and Supplementary Fig. S1C). In contrast, the same drugs showed no inhibitory effects on the parental Ba/F3 cells in the presence of IL-3. Consistent with this result, erlotinib and cetuximab decreased the constitutive phosphorylation of Stat5 in both CT982NT and CT1054NT expressing Ba/F3 cell lines, but not in the parental cells, suggesting that these drugs specifically target the EGFR CTD deletion mutants and effectively inhibit their oncogenic activity (Supplementary Fig. S2B and data not shown).
Figure 3. CT982NT and CT1054NT oncogenic EGFR mutants are sensitive to erlotinib or cetuximab in vivo and in vitro.
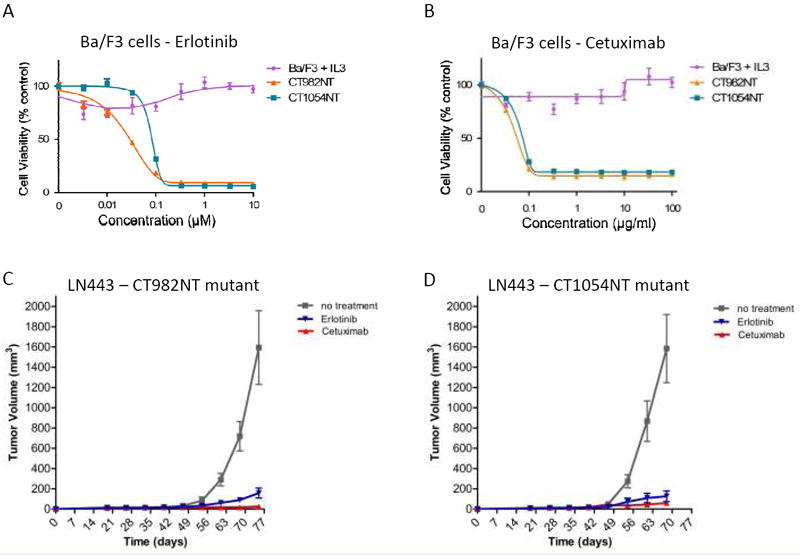
(A and B) Growth of Ba/F3 cells transformed with either CT982NT or CT1054NT EGFR mutant, but not the parental line with IL-3, was suppressed by either erlotinib (A) or cetuximab (B). Cells were treated with either erlotinib or cetuximab at the concentrations indicated for 72 hrs and assayed for cell viability. The results are indicated as mean +/- SD of three independent experiments. (C and D) Growth of mouse tumors driven by CT982NT and CT1054NT EGFR mutants are significantly suppressed by either erlotinib or cetuximab. LN443 cells expressing CT982NT (C) and CT1054NT (D) mutants were subcutaneously injected in the flanks of SCID mice (5 mice per group and 3 sites per each mouse). 20 days after cell injection, when tumors researched a size around 50-70 mm3, either erlotinib (50 mg/kg, gavage) or cetuximab (50 mg/kg, IP) was administered 3 times per week for 13 weeks. Tumor size was measured once a week, and volume was determined according to the formula (W2 × L)/2.
To further investigate the in vivo efficacy of erlotinib and cetuximab, we extended our study to xenografted mice generated by subcutaneous injection of non-tumorigenic LN443 glioma cell lines engineered to stably express either wild-type EGFR, or CT982NT or CT1054NT mutants by retroviral infection (34). Consistent with the results of the in vitro studies, we found that tumors were formed in xenografted mice harboring LN443 cells expressing either CT982NT or CT1054NT mutants. The sizes of the resulting tumors were significantly reduced in mice treated with either erlotinib or cetuximab, compared to tumor size in untreated mice (Fig. 3C and 3D). No tumors were observed in the mice injected with wild-type EGFR expressing cells (data not shown). These results demonstrate that GBM-derived EGFR CTD deletion mutants are tumorigenic and that tumors driven by these mutants exhibit a significant response to erlotinib and cetuximab treatment.
Deletion of the region containing amino acid residues 1010 through 1152 within the EGFR CTD is sufficient to confer oncogenic activation
In addition to the CT982NT and CT1054NT EGFR mutants found in GBM patients, we have identified an additional EGFR CTD deletion mutant harboring an intragenic deletion of amino acids 1010-1152. This mutation was discovered in an IL-3 independent clone that grew from a pool of Ba/F3 cells in which wild-type EGFR, which normally does not transform Ba/F3 cell, was introduced by retroviral infection (Supplementary Fig. S3A)(33). EGFR was sequenced from the transformed Ba/F3 clone and found to have undergone an intragenic deletion. Re-expression of the cDNA encoding this mutant EGFR (CT Del1) transformed the parental Ba/F3 cell line as well as NIH-3T3 cells in the absence of ligand (Supplementary Fig. S3B). These results confirmed that the oncogenic activation of EGFR is most likely induced by a deletion occurring within the CTD of the ectopically introduced EGFR in the cells and may be a direct cause of clonal selection from IL-3 withdrawal. As for the CT982NT and CT1054NT EGFR mutants, both IL-3 independent cell proliferation and the growth of mouse tumors driven by the oncogenic CT Del1 EGFR mutant are significantly suppressed by cetuximab or erlotinib (Supplementary Fig. S3C and S3D). Taken together, these results demonstrate that similar to GBM-derived CT982NT and CT1054NT mutants, the CT Del1 mutant is oncogenically activated by EGFR CTD deletion and its oncogenic activity is effectively suppressed by either cetuximab or erlotinib.
Antitumor effect of cetuximab and erlotinib against orthotopic xenografted brain tumors driven by EGFR CTD deletion mutants
Next, we wanted to further investigate the efficacy of cetuximab and erlotinib treatment as therapeutic approaches in treating GBM patients harboring EGFR CTD deletion mutants. To this end, we first generated orthotopic mouse models by intracranially implanting LN443 cells stably expressing oncogenic CT Del1, EGFRvIII mutants or wild-type EGFR. We chose the CT Del1 mutant as a representative of various oncogenic EGFR CTD deletion mutants for in vivo mouse study because unlike the CT982NT or CT1054NT mutants, CT Del1 mutant undergoes detectable constitutive phosphorylation at two tyrosine residues, Tyr1172 and Tyr1196, within the residual C-terminal segment of the mutant protein (Supplementary Fig. S4). Therefore, we can determine whether the enzymatic activation of the ectopically introduced EGFR CT Del1 mutant is responsible for tumorigenesis by monitoring its phosphorylation in tumors of the xenografted mice.
One week after tumor cell implantation, we assigned the xenografted mice to three different groups – no treatment, cetuximab treatment, and erlotinib treatment (Fig. 4A). For the cetuximab treated mice, we administered 1 mg per mouse of cetuximab by intraperitoneal (IP) injection to the mice 3 times per week, and for the erlotinib treated mice, we administered 1 mg per mouse of erlotinib by gavage three times per week. Consistent with the results from the subcutaneously xenografted mouse model and the in vitro study, while all mice subject to intracranial xenografts of LN443 cells expressing wild-type EGFR survived, the mice xenografted with LN443 cells expressing EGFRvIII or CT Del1 died early due to tumor formation in the brain (Fig. 4B). Immunohistochemical analysis revealed that both total and phosphorylated EGFR were specifically detected in brain tumors from EGFRvIII and CT Del1 mutant mice, demonstrating that these tumors were indeed induced by constitutive enzymatic activation of ectopically expressed EGFRvIII or CT Del1 mutants (Fig. 4C).
Figure 4. Anti-EGFR therapy is effective against brain tumors induced by oncogenic EGFR CTD deletion mutants.
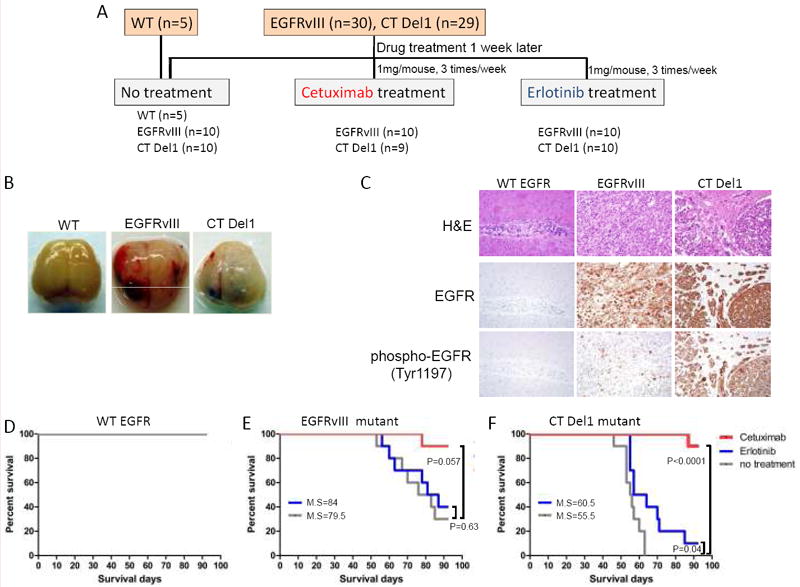
(A) Schematic to show the generation of the intracranial xenograft mouse model and the timing of anti-EGFR drug treatment. The indicated numbers of SCID mice were implanted intracranially with LN443 cells stably expressing either wild-type EGFR, EGFRvIII or CT Del1 mutants. After 1 week, each mutant group of xenografted mice was assigned to receive either no treatment or erlotinib or cetuximab three times per week with the dose indicated in the schematic. (B) Representative images of excised brains from the untreated group of mice xenografted with LN443 cells expressing wild-type EGFR or the EGFRvIII or CT Del1 mutants, respectively. (C) EGFRvIII and CT Del1 mutants are expressed and active in brain tumors originated from xenografted mice whereas wild-type EGFR is neither detected nor active. Brain tumors sections prepared from no treatment group of xenograft mice of wild-type EGFR or EGFRvIII or CT Del1 mutants were stained with H&E (top) or antibodies to total EGFR (middle) or phospho-EGFR (Tyr1197)(bottom). (D-F) Cetuximab treatment prolongs survival of xenografted mice with oncogenic EGFR CT Del1 mutant as well as EGFRvIII mutant. The survival of treated or non-treated groups of xenografted mice described in (A) were monitored and depicted as a Kaplan-Meier curve (M.S = median survival). P values were calculated between non-treated and drug-treated mouse groups.
We then tested the effects of cetuximab and erlotinib on survival of mice xenografted with LN443 cells expressing the various forms of EGFR. Given the lack of tumorigenicity by LN443 cells expressing wild-type EGFR, there was no decrease in survival for mice xenografted with these cells (Fig. 4D), while the mice xenografted with LN443 cells expressing the EGFR mutants exhibited diminished survival, with a median of 79.5 days for the EGFR vIII and 55.5 days for the EGFR CT Del1 deletion (Fig. 4E and 4F).
With the tested dose of cetuximab, 90% (17 out of 19) of the cetuximab-treated xenograft mice with oncogenic EGFR mutants survived for the 92 day duration of the treatment (Fig. 4E and 4F). These results show that cetuximab exerts strong pharmacological effects against brain tumors driven by oncogenic CT Del1 mutant as well as EGFRvIII mutant. In contrast to cetuximab, we observed that erlotinib had no statistically significant effect on survival of mice engrafted with LN443 cells expressing the EGFRvIII mutant, with median survival of 84 days for erlotinib treatment vs. 79.5 days for untreated control mice engrafted with the same cells, respectively (p=0.63) (Fig. 4E) and only modestly improved the survival of the mice xenografted with LN443 cells expressing the EGFR CT Del1 mutant (55.5 days vs. 60.5 days, p=0.04) (Fig. 4F). These results demonstrate that cetuximab is more potent in vivo than erlotinib at the tested concentrations for preventing growth of tumors driven by EGFR CTD deletions.
Given that there was a significant anti-tumor effect in the cetuximab treated xenograft mice with the EGFR CTD deletion mutants, we wanted to further study the long-term anti-tumor response of cetuximab in these mice. To this end, we divided the cetuximab-treated xenograft mice harboring CT Del1 mutant into two groups – in one group (five mice) we discontinued cetuximab treatment for sixty days after the initial cetuximab treatment of 92 days, while in the other group (five mice) we continued to administer the same dose of cetuximab as described above. Interestingly, the cetuximab-treated mice continued to survive for more than 60 days after the discontinuation of cetuximab treatment. Upon further investigation, we observed that none of the mice in either group had any sign of a brain tumor, suggesting that cetuximab may eradicate the tumors or prevent initial tumor formation after xenografting (data not shown).
Discussion
We here report both previously described (17, 18) and novel C-terminal deletion mutations of EGFR in GBM specimens. Furthermore, we showed that LN443 cells expressing an EGFR C-terminal deletion mutant form brain tumors in an intracranial mouse model. The survival of these xenografted mice was significantly prolonged by treatment with erlotinib or more potently by cetuximab. Based on our preclinical studies, we concluded that therapeutic targeting of EGFR with cetuximab may be a promising clinical approach to treat GBM patients harboring tumors with EGFR C-terminal deletion mutants. In addition, identification of EGFR C-terminal deletions is likely to be an important biomarker for selection of targeted therapy for GBM patients.
Compared to the previous reports (17, 18), a relatively lower frequency of EGFR CTD deletion mutations was detected among GBM tumors in the current study. One possible reason for this discrepancy is the use of distinct experimental approaches to achieve different study goals. We sought to identify any potential novel CT deletion mutation in addition to previous known mutations by systematic analysis of the pre-existing TCGA genomic data sets. Thus, we applied stringent analytic parameters for selecting the potential CTD deletion mutations. Expanding upon previous findings, we were able to identify the novel Ex27 deletions among GBM tumors and pursued further functional characterization of the mutants using in vivo and in vitro models. It is noted that 4 out of 5 tumors (TCGA 02-0102, 02-0043, 06-02511 and 08-0356) with mutant EGFR genes harboring CTD deletion exhibit high amplification at EGFR and do not harbor any other EGFRmutation (data not shown).
The detailed molecular mechanism underlying cellular transformation by EGFR C-terminal deletion mutants is currently unknown. Ligand-independent heterodimerization of EGFR CTD deletion mutants with other endogenous ErbB family members and/or Met receptors is unlikely to contribute to oncogenic activity of these mutants, as we did not observe detectable levels of total or phosphorylated forms of ErbB2, ErbB3, ErbB4 or Met in cells transformed by EGFR CTD deletion mutants (data not shown). In contrast to wild-type EGFR, we found that degradation of EGFRvIII mutant and EGFR CTD deletion mutants in response to EGF stimulation was diminished (Supplementary Fig. S5). As suggested in previous studies, this observation could be due to a low level of receptor internalization of EGFR CTD deletion mutant and/or lack of Y1069 phosphorylation on the mutant EGFR, which is required for Cbl-mediated receptor proteolysis (40, 41). Therefore, dysregulation of receptor degradation could be one of the possible oncogenic mechanisms of EGFR CTD deletion mutant, which is consistent with previous reports (42, 43). In addition, the CTD deletion mutants may have altered EGFR substrate specificity/receptor degradation or no longer bind regulatory proteins including Mig6 which has been found to negatively regulate EGFR kinase activity by blocking the activating dimer interface (44-46). Further experiments are needed to test these possibilities and to characterize a detailed mechanism of oncogenic activation of EGFR CTD deletion mutants.
We found that the levels of phosphorylation of the major EGFR downstream signaling mediators including STAT3, STAT5, Shc and Akt were increased in the cells expressing EGF-stimulated EGFR CTD deletion mutants, suggesting that the enzymatic activity of these mutants are able to be further enhanced by ligand unlike EGFRvIII mutant in the absence of autophosphorylation (Supplementary Fig. S6). Interestingly, the robust induction of phosphorylation of STAT3, STAT5 and Shc were observed specifically among EGFR CTD deletion mutants whereas high levels of Src phosphorylation were detected on the EGFRvIII mutant (Supplementary Fig. S6), suggesting that the EGFR CTD deletion mutants may activate different downstream signaling pathways compared to EGFRvIII and wild-type EGFR. Further investigation will be needed to validate this possibility. Given that both CT1054NT and CT Del1 mutants are oncogenic, we hypothesized that the region between amino acids 1055 and 1152 may play an important role in inhibiting ligand-independent activation of EGFR. This hypothesis is further supported by recent reports that EGFR CTD modulates kinase-domain dimer formation required for enzymatic activation of EGFR (47, 48).
Our in vivo studies showed that erlotinib and, more potently cetuximab, affected the tumorigenicity of the oncogenic EGFR CTD deletion mutants in mouse xenograft models. However, it is not clear whether the prolonged survival of mice observed in the intracranial mouse model is derived from drug’s direct antitumor effect against established mouse tumors or blockage of initial tumor formation. Given that the results using our subcutaneous mouse model clearly showed that both drugs effectively diminished growth of established tumors, we believe that eradication of mouse brain tumors in cetuximab-treated intracranial mice is likely due to drug’s direct killing effect on tumor cells formed by oncogenic EGFR mutants. Considering the translational impact of this distinction for potential clinical utility, it should be more clearly addressed by additional in vivo experiments in future.
Previous studies have suggested that cetuximab may exert antitumor effects via immune responses, promoting receptor degradation and antibody-dependent cellular cytotoxicity (ADCC) (49). In addition, prevention of receptor activation by directly blocking ligand binding and/or indirectly blocking the receptor dimerization was proposed as a possible pharmacological mechanism of cetuximab (50). Given that the degradation of CTD deletion mutants is impaired (42, 43), we believe that receptor degradation may not be a major mechanistic role of dramatic antitumor effects of cetuximab against EGFR CT deletion mutants in our model systems. The relative contribution of each mechanism is currently under investigation.
Supplementary Material
Acknowledgments
We thank Joshua Francis and Peter Hammerman for careful reading of the manuscript. This work was supported in part by National Cancer Institute grant R01CA116020 to M.M., NIH K08CA124804, NIH 3P30CA023100-25S8, Sontag Foundation Distinguished Scientist Award, James S. McDonnell Foundation to S.K. and Samsung Cancer Research Institute intramural grant to J.C.
Footnotes
Disclosure of potential conflicts of interest: No conflicts of interest were disclosed.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.DeAngelis LM. Brain tumors. N Engl J Med. 2001;344:114–23. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- 3.Gurney JG, Kadan-Lottick N. Brain and other central nervous system tumors: rates, trends, and epidemiology. Curr Opin Oncol. 2001;13:160–6. doi: 10.1097/00001622-200105000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Dietrich J, Diamond EL, Kesari S. Glioma stem cell signaling: therapeutic opportunities and challenges. Expert Rev Anticancer Ther. 2010;10:709–22. doi: 10.1586/era.09.190. [DOI] [PubMed] [Google Scholar]
- 5.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rao SK, Edwards J, Joshi AD, Siu IM, Riggins GJ. A survey of glioblastoma genomic amplifications and deletions. J Neurooncol. 2010;96:169–79. doi: 10.1007/s11060-009-9959-4. [DOI] [PubMed] [Google Scholar]
- 7.Toth J, Egervari K, Klekner A, Bognar L, Szanto J, Nemes Z, et al. Analysis of EGFR gene amplification, protein over-expression and tyrosine kinase domain mutation in recurrent glioblastoma. Pathol Oncol Res. 2009;15:225–9. doi: 10.1007/s12253-008-9082-4. [DOI] [PubMed] [Google Scholar]
- 8.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–7. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 11.Schwechheimer K, Huang S, Cavenee WK. EGFR gene amplification--rearrangement in human glioblastomas. Int J Cancer. 1995;62:145–8. doi: 10.1002/ijc.2910620206. [DOI] [PubMed] [Google Scholar]
- 12.Malden LT, Novak U, Kaye AH, Burgess AW. Selective amplification of the cytoplasmic domain of the epidermal growth factor receptor gene in glioblastoma multiforme. Cancer Res. 1988;48:2711–4. [PubMed] [Google Scholar]
- 13.Yamazaki H, Fukui Y, Ueyama Y, Tamaoki N, Kawamoto T, Taniguchi S, et al. Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-erbB) in human brain tumors. Mol Cell Biol. 1988;8:1816–20. doi: 10.1128/mcb.8.4.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006;3:e485. doi: 10.1371/journal.pmed.0030485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–31. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996;56:5079–86. [PubMed] [Google Scholar]
- 17.Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89:4309–13. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–7. [PubMed] [Google Scholar]
- 19.Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–70. [PubMed] [Google Scholar]
- 20.Lassman AB, Rossi MR, Raizer JJ, Abrey LE, Lieberman FS, Grefe CN, et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res. 2005;11:7841–50. doi: 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- 21.Narita Y, Nagane M, Mishima K, Huang HJ, Furnari FB, Cavenee WK. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 2002;62:6764–9. [PubMed] [Google Scholar]
- 22.Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Sci Signal. 2009;2:re6. doi: 10.1126/scisignal.287re6. [DOI] [PubMed] [Google Scholar]
- 23.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 24.van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–74. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. (2) 2010;96:219–30. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 27.Yang SH, Hong YK, Jeun SS, Kim IS, Hong JT, Sung JH, et al. Assessment of cetuximab efficacy by bioluminescence monitoring of intracranial glioblastoma xenograft in mouse. J Neurooncol. 2009;95:23–8. doi: 10.1007/s11060-009-9895-3. [DOI] [PubMed] [Google Scholar]
- 28.Diaz Miqueli A, Rolff J, Lemm M, Fichtner I, Perez R, Montero E. Radiosensitisation of U87MG brain tumours by anti-epidermal growth factor receptor monoclonal antibodies. Br J Cancer. 2009;100:950–8. doi: 10.1038/sj.bjc.6604943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eller JL, Longo SL, Hicklin DJ, Canute GW. Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery. 2002;51:1005–13. doi: 10.1097/00006123-200210000-00028. discussion 13-4. [DOI] [PubMed] [Google Scholar]
- 30.Belda-Iniesta C, Carpeno Jde C, Saenz EC, Gutierrez M, Perona R, Baron MG. Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol Ther. 2006;5:912–4. doi: 10.4161/cbt.5.8.3118. [DOI] [PubMed] [Google Scholar]
- 31.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji H, Zhao X, Yuza Y, Shimamura T, Li D, Protopopov A, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci U S A. 2006;103:7817–22. doi: 10.1073/pnas.0510284103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang J, Greulich H, Janne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65:8968–74. doi: 10.1158/0008-5472.CAN-05-1829. [DOI] [PubMed] [Google Scholar]
- 34.Stegh AH, Kesari S, Mahoney JE, Jenq HT, Forloney KL, Protopopov A, et al. Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via distinct mechanisms in glioblastoma. Proc Natl Acad Sci U S A. 2008;105:10703–8. doi: 10.1073/pnas.0712034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ligon KL, Huillard E, Mehta S, Kesari S, Liu H, Alberta JA, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–17. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:3076–83. doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 37.Walker F, Hibbs ML, Zhang HH, Gonez LJ, Burgess AW. Biochemical characterization of mutant EGF receptors expressed in the hemopoietic cell line BaF/3. Growth Factors. 1998;16:53–67. doi: 10.3109/08977199809017491. [DOI] [PubMed] [Google Scholar]
- 38.Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 39.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 40.Chen WS, Lazar CS, Lund KA, Welsh JB, Chang CP, Walton GM, et al. Functional independence of the epidermal growth factor receptor from a domain required for ligand-induced internalization and calcium regulation. Cell. 1989;59:33–43. doi: 10.1016/0092-8674(89)90867-2. [DOI] [PubMed] [Google Scholar]
- 41.Grandal MV, Zandi R, Pedersen MW, Willumsen BM, van Deurs B, Poulsen HS. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis. 2007;28:1408–17. doi: 10.1093/carcin/bgm058. [DOI] [PubMed] [Google Scholar]
- 42.Masui H, Wells A, Lazar CS, Rosenfeld MG, Gill GN. Enhanced tumorigenesis of NR6 cells which express non-down-regulating epidermal growth factor receptors. Cancer Res. 1991;51:6170–5. [PubMed] [Google Scholar]
- 43.Wells A, Welsh JB, Lazar CS, Wiley HS, Gill GN, Rosenfeld MG. Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science. 1990;247:962–4. doi: 10.1126/science.2305263. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J. Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature. 2007;450:741–4. doi: 10.1038/nature05998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ying H, Zheng H, Scott K, Wiedemeyer R, Yan H, Lim C, et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc Natl Acad Sci U S A. 2010;107:6912–7. doi: 10.1073/pnas.0914930107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beebe JA, Wiepz GJ, Guadarrama AG, Bertics PJ, Burke TJ. A carboxyl-terminal mutation of the epidermal growth factor receptor alters tyrosine kinase activity and substrate specificity as measured by a fluorescence polarization assay. J Biol Chem. 2003;278:26810–6. doi: 10.1074/jbc.M301397200. [DOI] [PubMed] [Google Scholar]
- 47.Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pines G, Huang PH, Zwang Y, White FM, Yarden Y. EGFRvIV: a previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene. 2010;29:5850–60. doi: 10.1038/onc.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harding J, Burtness B. Cetuximab: an epidermal growth factor receptor chemeric human-murine monoclonal antibody. Drugs Today (Barc) 2005;41:107–27. doi: 10.1358/dot.2005.41.2.882662. [DOI] [PubMed] [Google Scholar]
- 50.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.