

Abstract
Myoclonus-dystonia (M-D) is a movement disorder characterized by myoclonic jerks with dystonia. DYT11 M-D is caused by mutations in SGCE which codes for ε-sarcoglycan. SGCE is maternally imprinted and paternally expressed. Abnormal nuclear envelope has been reported in mouse models of DYT1 generalized torsion dystonia. However, it is not known whether similar alterations occur in DYT11 M-D. We developed a mouse model of DYT11 M-D using paternally-inherited Sgce heterozygous knockout (Sgce KO) mice and reported that they had myoclonus and motor coordination and learning deficits in the beam-walking test. However, the specific brain regions that contribute to these phenotypes have not been identified. Since ε-sarcoglycan is highly expressed in the cerebellar Purkinje cells, here we examined the nuclear envelope in these cells using a transmission electron microscope and found that they are abnormal in Sgce KO mice. Our results put DYT11 M-D in a growing family of nuclear envelopathies. To analyze the effect of loss of ε-sarcoglycan function in the cerebellar Purkinje cells, we produced paternally-inherited cerebellar Purkinje cell-specific Sgce conditional knockout (Sgce pKO) mice. Sgce pKO mice showed motor learning deficits, while they did not show abnormal nuclear envelope in the cerebellar Purkinje cells, robust motor deficits, or myoclonus. The results suggest that ε-sarcoglycan in the cerebellar Purkinje cells contributes to the motor learning, while loss of ε-sarcoglycan in other brain regions may contribute to nuclear envelope abnormality, myoclonus and motor coordination deficits.
Keywords: ε-sarcoglycan, motor learning, myoclonus-dystonia, nuclear envelope, Purkinje cell, Sgce
1. Introduction
Myoclonus-dystonia (M-D) is a movement disorder characterized by myoclonic jerks with dystonic symptoms. Myoclonus is a sudden brief jerk caused by involuntary muscle activity [1]. Dystonia is a movement disorder characterized by involuntary, repetitive, sustained muscle contractions or postures [2–4]. DYT11 M-D is a major type of genetic M-D caused by mutations in SGCE which codes for ε-sarcoglycan [5], while M-D itself is genetically heterogeneous [6]. Sarcoglycans are transmembrane glycoproteins with six different isoforms, α, β, γ, δ, ε and ζ [7]. The cDNA clones coding for ε-sarcoglycan have been reported in mouse [8–11], human [12, 13] and rat [14]. This gene is maternally-imprinted and paternally-expressed in both humans and mice [11, 13, 15–17], and widely expressed in the body, including the brain, lungs and smooth muscles [8, 12]. Therefore, DYT11 M-D is caused by paternally-inherited SGCE mutation in most cases [18], while de novo SGCE mutations were also reported [19]. Many mutations in SGCE have been reported in M-D patients and it is believed that loss of ε-sarcoglycan function causes this disease [6]. We previously reported the generation of Sgce knockout (KO) mice lacking exon 4 and demonstrated that paternally-inherited Sgce heterozygous KO mice do not express maternally-inherited wild-type (WT) Sgce in the brain [11, 20]. Exclusive paternal expression of ε-sarcoglycan has also been confirmed in human brains [13]. Therefore we use the paternally-inherited Sgce heterozygous KO mice as Sgce KO mouse in this paper and in our earlier study [21]. Sgce KO mice exhibit increased total numbers of slips and impaired motor learning in the beam-walking test, numbers of spontaneous myoclonus, vertical activity in the open-field test that is similar to obsessive-compulsive disorder (OCD), and levels of striatal dopamine and its metabolites [21].
We previously showed that Dyt1 ΔGAG heterozygous knock-in (KI) mice exhibit motor deficits and pontine inclusion bodies [22]. Recent studies suggested that there are functional microstructural brain alterations in DYT1 generalized torsion dystonia patients [23] and a DYT1 dystonia mouse model [24, 25], although there is no overt neurodegeneration. Abnormal nuclear envelope has been reported in transfected cells over-expressing the mutant forms of torsinA [26, 27]. Abnormal nuclear envelope was also reported in Tor1a−/− (Dyt1 KO) mice and Tor1a Δgag/Δgag (Dyt1 ΔGAG homozygous KI) mice exhibiting neonatal lethality [28]. It was suggested that neuron-specific nuclear envelope abnormality in Dyt1 ΔGAG homozygous KI mice is caused by malfunction of torsinA with incomplete compensation by torsinB which is weakly expressed in neurons [29]. However both transgenic mice overexpressing human WT torsinA and mutant torsinA using murine prion promoter exhibit abnormal nuclear envelope and impaired motor performance [30]. Both cerebral cortex-specific Dyt1 conditional knockout mice and striatum-specific Dyt1 conditional knockout mice exhibit motor deficits without abnormal nuclear envelope in the cerebral cortex and striatum, respectively [31, 32]. Moreover, nuclear envelope abnormality has not been found in Dyt1 ΔGAG heterozygous KI mouse [20, 28] or DYT1 dystonia patient brains. Therefore, it is not clear whether abnormal nuclear envelope is a pathological biomarker for DYT1 dystonia. Such a nuclear envelope abnormality has not been examined in DYT11 M-D patients because of the difficulty of obtaining brain samples of deceased patient.
The cerebellum contributes to motor deficits in several genetic rodent models. Cerebellectomy eliminates the motor syndrome of a genetically dystonic rat [33]. Bilateral electrolytic and/or excitatory amino acid lesions of the medial cerebellar nucleus, nucleus interpositus, lateral cerebellar nucleus and lateral vestibular nuclei produce significant improvements in motor function and decreases in the frequency of abnormal motor signs in a genetically dystonic rat [34]. The lesions of the cerebellum improve the motor performance of Weaver mutant mice [35]. Moreover, motor dysfunction in tottering mouse is abolished when crossed into a cerebellar Purkinje cell degeneration (pcd) mutant background [36]. Since ε-sarcoglycan is highly expressed in the cerebellar Purkinje cells of mice [37] and humans [13], loss of ε-sarcoglycan may affect cerebellar Purkinje cell function. In the present study, we examined the nuclear envelopes of the cerebellar Purkinje cells in Sgce KO mice using a transmission electron microscope.
DYT1 dystonia patients exhibit not only dystonic symptoms but also impaired sequence learning [38]. Consistent with the learning deficits in the patients, impaired motor learning was reported in a DYT1 dystonia mouse model [39]. Some DYT11 M-D patients exhibit impaired verbal learning and memory [40]. Sgce KO mice exhibit not only motor coordination and balance deficits but also impaired motor learning [21]. However the brain regions that contribute to the impaired motor learning have not been identified. It is known that the cerebellar Purkinje cells play principal a role in motor coordination and motor learning [41]. To analyze the in vivo function of ε-sarcoglycan in the cerebellar Purkinje cells, we produced paternally-inherited cerebellar Purkinje cell-specific Sgce conditional knockout (Sgce pKO) mice and analyzed their nuclear envelopes in the cerebellar Purkinje cells, locomotion, motor performance, and myoclonus.
2. Materials and Methods
2.1. Animals
Sgce KO mice and Sgce loxP neo mice were prepared and genotyped as described previously [11]. Sgce loxP neo mice were crossed with FLP mice (Jackson Laboratory, Stock No. 003946) to remove PGKneo cassette flanked by FRT sites [42]. FLP was removed by backcrossing with C57BL/6 mice to produce Sgce loxP mice. We used the cre-loxP system [43] applied to mouse gene recombination [44] to selectively inactivate Sgce in the cerebellar Purkinje cells. Pcp2-cre mice were purchased from the Jackson laboratory [45]. Sgce loxP male mice were crossed with Pcp2-cre female mice to produce Sgce pKO mice. The genotyping of Sgce pKO mice and their littermates was performed by multiplex PCR using tail DNA with a combination of SgceloxP5/SgceloxP3 [11] and creA/cre6 primers [46]. A group of 16 Sgce pKO (8 males and 8 females) and 17 control littermate mice (CT mice; 7 males and 10 females) was used for the behavioral semi-quantitative assessments of motor disorders, open-field test, beam-walking test, and accelerated rotarod test in this order. All behavior tests were performed by investigators blind to the genotypes. All experiments were carried out in compliance with the USPHS Guide for Care and Use of Laboratory Animals and approved by the IACUCs of University of Illinois at Urbana-Champaign (UIUC) and University of Alabama at Birmingham (UAB).
2.2. Transmission electron and light microscopy analysis
Brain sections for transmission electron microscope were prepared as described earlier [20]. Sgce KO mice and their WT littermates (n = 3 each, 2 to 4 months of age) were perfused with chilled 0.1M phosphate-buffered saline (pH7.4) followed by Karnovsky’s Fixative in phosphate buffered 2% glutaraldeyde and 2.5 % paraformaldehyde. The brains were dissected out and left in Karnovsky’s Fixative overnight. The tissue was then trimmed and washed in cacodylate buffer with no further additives. Microwave fixation was used with the secondary 2% osmium tetroxide fixative, followed by the addition of 3% potassium ferricyanide for 30 minutes. After washing with water, saturated uranyl acetate was added for en bloc staining. The tissue was dehydrated in a series of increasing concentrations of ethanol starting at 50%. Acetonitrile was used as the transition fluid between ethanol and the epoxy. Infiltration series was done with an epoxy mixture using the epon substitute Lx112. The resulting blocks were polymerized at 90 °C overnight, trimmed with a razor blade, and ultrathin sectioned with diamond knives. Sections were then stained with uranyl acetate and lead citrate, and examined or photographed with a Hitachi H600 transmission electron microscope.
Sgce pKO mice and CT adult mice (n = 3 each) were also perfused and processed. Semi-thin sections (0.5 μm) were cut and stained with 1% Toluidine Blue O (Fisher scientific) in 1% Sodium borate (EMS) for a few minutes followed by washing with distilled water and acetone, and then dried. The sections were examined by light microscopy. Images were captured using ZEISS Axiophot RZGF-1 microscope with 20× Plan-NEOFLUAR objective lens and MBF Bioscience Neurolucida 7 software (MicroBrightField, Inc). The semi-thin sections were further ultrathin sectioned with diamond knives. Sections were then stained with uranyl acetate and lead citrate, and examined or photographed with a Hitachi 7600 transmission electron microscope with digital camera. The nuclear envelopes in the cerebellar Purkinje cells were examined by investigators blind to the genotypes.
2.3. Immunohistochemistry
Generation of a novel mouse monoclonal antibody (mSE 3A9) against mouse ε-sarcoglycan has been described earlier [20]. In Western blot analysis, this antibody detects a single band at 52 kDa in the striatal extract from a WT mouse while no bands at the position are detected from a Sgce KO mouse. Sgce pKO and CT mice were anesthetized and perfused with ice-cold 0.1 M phosphate-buffered saline (pH7.4; PBS) followed by 4% paraformaldehyde in 0.1 M phosphate-buffer (pH 7.4; PB). The brains were soaked in paraformaldehyde-PB at 4°C overnight, and then incubated in 30% sucrose in 0.1 M PBS at 4°C overnight until the brain sank. The brains were frozen with dry-ice powder and cut sagittally into 40 μm sections with a Histoslide 2000 sliding microtome (Reichert-Jung). Sections were collected in 0.1 M PB and treated with water for 5 min, then incubated with 0.3% H2O2 for 30 min. After washing with 10 mM PBS for 5 min, the sections were blocked with 10 mM PBS containing goat normal serum for 20 min, and then incubated with 1 μg/ml mouse monoclonal ε-sarcoglycan antibody (mSE 3A9) in the same buffer. Immunohistochemistry was performed with Vectastain ABC kit for peroxidase mouse IgG and DAB peroxidase substrate kit with nickel solution (Vector Lab) according to the manufacture’s protocols. Images were captured using a Nikon ECLIPSE E800M microscope with 40× Plan Fluor objective lens.
2.4. Behavioral semi-quantitative assessments of motor disorders
Behavioral semi-quantitative assessments of motor disorders were performed for Sgce pKO and CT mice from 183 to 221-days old as described earlier [22, 47]. Each mouse was placed on a table and assessments of hind paw clasping, hind paw dystonia, truncal dystonia and balance adjustments to a postural challenge were made. The hind paw clasping was assessed as hind paw movements for postural adjustment and attempt to straighten up while the mouse was suspended by the mid-tail. The hind paw dystonia was assessed as the increased spacing between the limbs, poor limb coordination, crouching posture and impairment of gait. Truncal dystonia was assessed as the flexed posture. Postural challenge was performed by flipping the mouse onto its back and the ease of righting was noted.
2.5. Open-field test
Locomotion of Sgce pKO and CT mice from 190 to 227-days old was assessed with the open-field apparatus (AccuScan Instruments) as described earlier [21, 48]. Spontaneous movements of individual mice were recorded by infrared light beam sensors in a 41 × 41 × 31 cm acrylic case for 15 min at 1 min intervals using DigiPro software. The test was conducted under light condition in the light period.
2.6. Beam-walking test
The beam-walking test for Sgce pKO and CT mice from 201 to 239-days old was performed as described earlier [21, 22, 32, 49], using a slightly modified method of the standard protocol [50] to avoid unnecessary overtraining and tests. Mice were trained to transverse a medium square beam (14 mm wide) in three consecutive trials each day for two days. The trained mice were tested twice on the medium square beam and medium round beam (17 mm diameter) on the third day and small round beam (10 mm diameter) and small square beam (7 mm wide) on the fourth day. The hind paw slips on each side while walking the 80 cm beams were recorded.
2.7. Accelerated rotarod test
The motor performance was assessed for Sgce pKO and CT mice from 210 to 248-days old with Economex accelerating rotarod (Columbus Instruments) as described earlier [22]. The apparatus started at an initial speed of 4 rpm and gradually accelerated at a rate of 0.2 rpm/s. The latency to fall was measured with a cutoff time of 2 min. Mice were tested for three trials on each day for 4 days. The trials within the same day were performed at approximately 1 hour intervals.
2.8. Spontaneous myoclonus test
Sgce pKO and CT mice from 264 to 302-days old were placed in transparent flat bottom rodent restrainers (model 541-RR; Plas-labs, Inc. MI) to minimize voluntary movements and videotaped for an hour. The mice were habituated for approximately 30 min in the restrainers and the numbers of spontaneous myoclonus jerks of the whole body were counted during the subsequent 30 min as described earlier [21].
2.9. Statistics
The numbers of cerebellar Purkinje cell sections with abnormal nuclear envelopes and those with normal nuclear envelopes between Sgce KO and WT littermate mice were analyzed by chi square test for independence with Yates’s correction. Locomotion in the open-field test, latency to fall in the accelerated rotarod test, and the numbers of myoclonus for Sgce pKO and CT mice were analyzed by using ANOVA mixed model in SAS/STAT Analyst software (Version 9.1.3; SAS Institute Inc.) as described earlier [22]. The data of slip numbers in the beam-walking test for all four beams were analyzed together by logistic regression (GENMOD) with negative binominal distribution using GEE model in the software [21, 22, 32]. Sex, age, and body weight were used as variables. CT mice were normalized to zero. To compare the effect of motor learning during the beam-walking test, a dummy variable that combined genotype and trial number was used and contrasted to derive the p-value between trials 1 and 2 for medium round, small round and small square beams. Data in the accelerated rotarod, beam-walking, and myoclonus tests were analyzed after natural log transformation to obtain a normal distribution. Significance was assigned at p < 0.05.
3. Results
3.1. Abnormal nuclear envelopes of the cerebellar Purkinje cells in Sgce KO mice
Sgce KO mice and WT littermates were produced as described earlier [11]. Nuclear envelopes in the cerebellar Purkinje cells were examined using a transmission electron microscope. Although we examined 178 cerebellar Purkinje cell sections, no abnormal nuclear envelope was detected in WT littermates (Fig. 1A). However, we found 5 cerebellar Purkinje cell sections with abnormal nuclear envelopes among the examined 96 sections in Sgce KO mice (Fig. 1B). All three Sgce KO mice showed abnormal nuclear envelopes. Numbers of cerebellar Purkinje cell sections with abnormal nuclear envelopes were significantly higher in Sgce KO mice than those in WT littermates (χ2 = 6.453, p = 0.011). Detailed analysis with high magnification revealed that the nuclear envelope blebbing and other nuclear envelope abnormalities in Sgce KO mice (Fig. 1C) were similar to those reported in DYT1 dystonia models [28]. The appearance of abnormal envelopes in the cerebellar Purkinje cells suggests a functional alteration of the cerebellar Purkinje cells in Sgce KO mice.
Fig. 1.
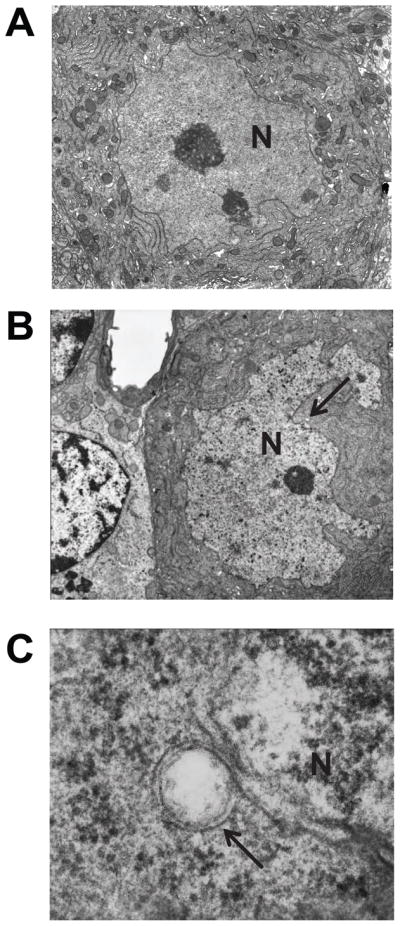
Nuclear envelope structures of the cerebellar Purkinje cells in WT and Sgce KO mice. Although no abnormal nuclei were detected in the cerebellar Purkinje cells of WT mice (A), abnormal nuclei were detected in those of Sgce KO mice (B). Enlarged and inverted images of B in Sgce KO mice (C) revealed that the nuclear envelope blebbing and other nuclear envelope abnormalities were similar to those reported in DYT1 dystonia models. Magnifications; A, B: 5,000 ×; C: 100,000 ×. Nuclei (N) are marked in the images. Arrows in B and C indicate the abnormal nuclear envelopes. Representative electron microscope images are shown.
3.2. Generation of Sgce pKO mice
To analyze the effect of loss of ε-sarcoglycan function in the cerebellar Purkinje cells, Sgce pKO mice and CT mice were produced by crossing Sgce loxP male mice with Pcp2-cre female mice (Fig. 2A). Genotyping was performed by multiplex PCR with their tail DNA (Fig. 2B). The cerebellar Purkinje cells were clearly present in both CT and Sgce pKO mice in the cerebellar sections stained with Toluidine Blue O (Fig. 3), suggesting loss of ε-sarcoglycan in the cerebellar Purkinje cells alone did not affect robust development and surviving of the cerebellar Purkinje cells. The cerebellar Purkinje-cell specific reduction of ε-sarcoglycan was confirmed by immunohistochemistry with the mouse monoclonal ε-sarcoglycan antibody (mSE 3A9). Molecular layer (ML), cerebellar Purkinje cell layer (PCL) and Granule cell layer (GCL) are clearly identified as shown in in a CT mouse (Fig. 4A). Both the soma and dendrite of the cerebellar Purkinje cells in the CT mouse were strongly stained. However, staining for the cerebellar Purkinje cells were significantly decreased in a Sgce pKO mouse (Fig. 4B), while staining for the ML and GCL were similar to those in the CT mouse, suggesting Sgce was knocked out in the cerebellar Purkinje cells as predicted. The sections were not stained in the negative control without the primary antibody and the boundary between the layers were not clear (Fig. 4C), suggesting that the staining shown in Fig. 4A and 4B depended on the ε-sarcoglycan antibody. Since the cerebellar Purkinje cells were intact in Sgce pKO mice as shown in Fig. 3B, the absence of staining in the immunohistochemistry shown in Fig. 4B was not due to cell loss.
Fig. 2.

Generation of Sgce pKO mice. (A) Strategies to generate Sgce pKO mice. Sgce loxP neo mice were crossed with FLP mice to remove PGKneo cassette flanked by FRT sites. FLP was removed by backcrossing with CB57BL/6 mice to make Sgce loxP mice. Sgce loxP male mice were crossed with Pcp2-cre female mice to produce Sgce pKO mice. (B) A representative PCR-based genotyping for Sgce pKO mice. Top bands were PCR products of cre. Middle bands were those of Sgce loxP locus. The bottom bands were those of Sgce WT locus. Lane 1: Pcp2-cre mouse; Lane 2: Sgce loxP heterozygous mouse; Lane 3: WT mouse; Lane 4: Sgce pKO mouse.
Fig. 3.

The cerebellar Purkinje cells stained with Toluidine Blue O in CT and Sgce pKO mice. (A) A representative sagittal section of the cerebellum in a CT mouse. (B) A representative sagittal section of the cerebellum in a Sgce pKO mouse. Granule cell layer (GCL), cerebellar Purkinje cell layer (PCL) and Molecular layer (ML) are clearly identified. Arrows indicate representative cerebellar Purkinje cells. Inset scale bars indicate 50 μm.
Fig. 4.

Immunohistochemistry of the cerebellum in CT and Sgce pKO mice. The sagittal sections were stained with mouse monoclonal antibody (mSE 3A9) against mouse ε-sarcoglycan. (A) A sagittal section of the cerebellum in a CT mouse stained with the ε-sarcoglycan antibody. (B) A sagittal section of the cerebellum in a Sgce pKO mouse stained with the ε-sarcoglycan antibody. (C) A sagittal section of the cerebellum in the same Sgce pKO mouse without the primary antibody as a negative control. Molecular layer (ML), cerebellar Purkinje cell layer (PCL) and Granule cell layer (GCL) are clearly identified in A and B. Both the soma and dendrite of the cerebellar Purkinje cells were strongly stained in CT mouse (A), while those were significantly diminished in Sgce pKO mice as predicted (B). Arrows indicate representative cerebellar Purkinje cells. Inset scale bars indicate 50 μm.
3.3. Normal nuclear envelopes of the cerebellar Purkinje cells in Sgce pKO mice
Since Sgce KO mice showed nuclear envelope abnormality in cerebellar Purkinje cells, we further examined the nuclear envelopes of the cerebellar Purkinje cells in Sgce pKO mice using a transmission electron microscope to determine whether abnormal nuclear envelopes were caused by cell-autonomous phenomenon in the absence of ε-sarcoglycan in the cerebellar Purkinje cells alone. There was no blebbing or other nuclear envelope abnormalities in the cerebellar Purkinje cells in the examined 77 sections of CT mice (n = 3) as predicted (Fig. 5A, C). We could not find any blebbing or other nuclear envelope abnormalities either in the cerebellar Purkinje cells in the examined 82 sections of Sgce pKO mice (n = 3; Fig. 5B, D). The results suggest that loss of ε-sarcoglycan in the cerebellar Purkinje cells alone did not produce the nuclear envelope abnormalities.
Fig. 5.

Normal nuclear envelope structures of the cerebellar Purkinje cells in CT and Sgce pKO mice. No abnormal nuclei were detected in the cerebellar Purkinje cells of both CT (A, C) and Sgce pKO (B, D) mice. Magnifications of A and B: 8,000 ×. Enlarged images of A and B are also shown in C and D, respectively. Nucleus (N) is marked in the images. Arrows indicate nuclear envelopes. Inset scale bars indicate 2 μm. Representative electron microscope images are shown.
3.4. No overt abnormal postures in Sgce pKO mice
When suspended from the tail, both Sgce pKO and CT mice had normal splaying of hind paws. Sgce pKO mice had no observable hind paw extension or truncal arching in comparison to CT mice. All mice exhibited strong righting reflexes when tipped on their side. The results suggest that similar to previously reported in Sgce KO mice [21], Sgce pKO mice had no overt abnormal postures.
3.5. Decreased stereotypic behaviors in Sgce pKO mice
Locomotion of Sgce pKO mice were assessed in the open-field test (Table 1). Sgce pKO mice did not exhibit significant differences in comparison to CT mice in horizontal activity, total distance, movement number, movement time, rest time, and clockwise or anti-clockwise revolutions. Furthermore, Sgce pKO mice did not exhibit significant differences in vertical activity, vertical movement number, or vertical time, suggesting no significant OCD-like behaviors [21]. Sgce pKO mice showed no significant alteration either in the central time or the central distance ratio, suggesting no significant anxiety-like behaviors. However, Sgce pKO mice exhibited decreased stereotypic activity, decreased stereotypic movement numbers, and decreased stereotypic movement time.
Table 1.
Open-field behavior in CT and Sgce pKO mice.
| Open-field parameters | CT | Sgce pKO | p |
|---|---|---|---|
| Horizontal activity (beam breaks) | 4471 ± 200 | 3877 ± 230 | 0.066 |
| Total distance (cm) | 2635 ± 181 | 2276 ± 208 | 0.213 |
| Horizontal movement number | 207 ± 6 | 194 ± 7 | 0.161 |
| Movement time (sec) | 264 ± 13 | 226 ± 15 | 0.068 |
| Rest time (sec) | 636 ± 13 | 674 ± 15 | 0.070 |
| Clockwise revolutions (count) | 10 ± 1 | 8 ± 1 | 0.175 |
| Anticlockwise revolutions (count) | 9 ± 1 | 8 ± 1 | 0.339 |
| Vertical activity (beam breaks) | 231 ± 19 | 203 ± 21 | 0.342 |
| Vertical movement number | 87 ± 6 | 74 ± 7 | 0.199 |
| Vertical movement time (sec) | 90 ± 7 | 81 ± 8 | 0.377 |
| Central time (sec) | 165 ± 17 | 130 ± 20 | 0.208 |
| Central distance ratio | 0.27 ± 0.02 | 0.24 ± 0.02 | 0.353 |
| Stereotypic activity (beam breaks) | 2390 ± 131 | 1937 ± 151 | 0.034* |
| Stereotypic movement number | 161 ± 3 | 150 ± 4 | 0.030* |
| Stereotypic movement time (sec) | 242 ± 11 | 202 ± 13 | 0.028* |
The values of each parameter in open-field test are shown as means ± standard errors.
p < 0.05.
3.6. Motor learning deficits in Sgce pKO mice
Motor performance was assessed in the beam-walking and accelerated rotarod tests. Although Sgce KO mice exhibit increased total slip numbers in beam-walking test as reported earlier [21], Sgce pKO mice in the present study did not exhibit significant difference in the total numbers of slips on the four beams in the same beam-walking test (Fig 6A, P = 0.28), suggesting the motor coordination and balance were not largely affected. Since beam-walking test data showed a significant interaction between genotype and trial (p = 0.003), we further analyzed the motor learning performance in beam-walking test by comparing the slip numbers on the last three beams at the first and second trials. Although CT mice improved significantly in trial 2 over trial 1 (Fig 6B, p = 0.008), there was no significant difference between trials 1 and 2 in Sgce pKO mice (Fig 6B, p = 0.50). The results suggest impaired motor learning for the hind paws in Sgce pKO mice. The present results suggest that the impaired motor learning in Sgce KO mice is produced by the loss of ε-sarcoglycan function in the cerebellar Purkinje cells.
Fig. 6.

Impaired motor learning of Sgce pKO mice in the beam-walking and accelerated tests. (A) No significant difference in total slips numbers in the beam-walking test. The data in CT mice were normalized to zero. (B) Impaired motor learning of Sgce pKO mice in beam-walking test. CT mice improved significantly in trial 2 over trial 1. However, there was no significant difference between trials 1 and 2 in Sgce pKO mice. Data were normalized to trial 2 of CT mice. (C) Latency to fall in each trial of the accelerated rotarod test. Sgce pKO mice exhibited significantly shorter latency to fall only in the fourth trial and did not exhibit significant difference in other trials. (D) No significant difference in overall latency to fall in the accelerated rotarod test. The data in CT mice were normalized to zero. (E) No significant myoclonus in Sgce pKO mice. The total spontaneous myoclonus numbers in 30 min were compared with those of CT mice. The numbers in CT mice were normalized to zero. The data are analyzed after natural log transformation to obtain a normal distribution. Vertical bars represent means ± standard errors. *p < 0.05.
In the accelerated rotarod test, Sgce pKO mice exhibited significantly shorter latency to fall only in the fourth trial which was examined at the first trial on the second day (Fig 6C, p = 0.040) and did not exhibit significant difference in other trials (Fig 6C, p > 0.05). Therefore, the motor performance of Sgce pKO seems to be recovered in the following trials after the fourth trial, suggesting subtle motor learning deficit in Sgce pKO mice. Sgce pKO mice did not exhibit significant difference in overall latency to fall (Fig 6D, p = 0.273) as reported in Sgce KO mice [21].
3.7. No significant myoclonus in Sgce pKO mice
To analyze whether myoclonus observed in Sgce KO mice [21] are caused by the loss of ε-sarcoglycan function in the cerebellar Purkinje cells, the spontaneous myoclonus numbers of individual Sgce pKO and CT mice were counted for 30 min. Sgce pKO mice did not exhibit significant myoclonus (Fig. 6E, p = 0.252), suggesting that the loss of ε-sarcoglycan in the cerebellar Purkinje cells alone doesn’t contribute to myoclonus. Since Sgce KO mice exhibit significant myoclonus, the present results suggest that loss of ε-sarcoglycan in other brain regions may contribute to myoclonus.
4. Discussion
DYT11 M-D is a complex disease with myoclonus, dystonia and psychiatric symptoms [5]. Consistent with the symptoms, Sgce KO mice exhibit spontaneous myoclonus, impaired motor coordination and motor learning in the beam-walking test, and OCD- and anxiety-like behaviors [21]. While ε-sarcoglycan is expressed in multiple brain regions [13, 37], the regions that specifically contribute to the symptoms have not been identified. The cerebellar Purkinje cells are known to function not only in motor coordination and balance but also in motor learning [41]. It was also suggested that functional alterations of the cerebellar Purkinje cells contribute to dystonic symptoms in another mouse model [36]. Since ε-sarcoglycan is highly expressed in the cerebellar Purkinje cells, we examined the nuclear envelopes of the cerebellar Purkinje cells in Sgce KO mice and found abnormal nuclear envelopes. The results suggest that DYT11 myoclonus-dystonia can be categorized as a nuclear envelopathy [51]. To analyze the in vivo function of ε-sarcoglycan in the cerebellar Purkinje cells, we produced Sgce pKO mice. The loss of ε-sarcoglycan in cerebellar Purkinje cells did not affect robust development or survival of the cerebellar Purkinje cells. Moreover we could not find any blebbing or other nuclear envelope abnormalities in the cerebellar Purkinje cells of Sgce pKO mice, suggesting that loss of ε-sarcoglycan in the cerebellar Purkinje cells alone may not produce nuclear envelope abnormalities. Sgce pKO mice did not exhibit robust motor deficits, alteration in most parameters in the open-field test, or significant myoclonus. However, Sgce pKO mice exhibited decreased stereotypic behaviors, significant impaired motor learning in the beam-walking test, and subtle motor learning deficits in the accelerated rotarod test. The results suggest that the loss of ε-sarcoglycan in the cerebellar Purkinje cells contributes to the impaired motor learning, while loss of ε-sarcoglycan in other brain regions may contribute to the nuclear envelope abnormality, myoclonus and motor coordination deficits. This is the first report that ε-sarcoglycan in the cerebellar Purkinje cells specifically contributes to the motor learning in vivo.
Functional alterations in the cerebellum have been reported in both dystonia patients and rodent models. Since cerebellum controls balance and motor leaning, functional damage of the cerebellum often causes motor deficits relating to dystonia. Trauma to the cerebellum or cerebellar atrophy causes dystonia in humans [52, 53]. Pharmacological perturbation of the cerebellar signaling induces dystonia in mice [54]. Structural grey matter alterations in the cerebellum were observed in upper limb dystonia [55], cervical dystonia [56], and focal dystonia [57]. Abnormal activities of the cerebellum in DYT1 mutation carriers and alterations in the olivo-cerebellar pathway of patients with primary focal dystonia were also reported [58]. Moreover, a pharmacological perturbation using ouabain, a sodium pump blocker, in the cerebellum induces dystonia-like symptoms in a rapid-onset dystonia Parkinsonism mouse model [59]. These studies suggest the contribution of cerebellum in dystonia. However, here Sgce pKO mice showed no overall motor deficit in beam walking test, an indirect test for dystonia [4, 60]. And the present results suggest that the contribution of loss of ε-sarcoglycan function in the cerebellar Purkinje cells is limited to the motor leaning deficits in DYT11 M-D. Generation of other Sgce conditional KO mouse models targeting the basal ganglia circuits, such as a striatum-specific conditional KO mouse, will elucidate the brain regions which may contribute to dystonic symptom.
The abnormal nuclear envelopes were detected in 5 of the 96 sections from Sgce KO mice and none of 178 sections from CT mice. Although statistical analysis suggested that the appearance of the abnormal nuclear envelopes was significantly high in Sgce KO mice, the phenomenon may seem relatively rare. To determine whether the appearance of the abnormal nuclear envelopes is really rare in the examined sections, we made a simple mathematical model to calculate the probability of the appearance of the nuclear blebbing in the sections as shown in the Supplementary Material. The mathematical model also suggests that the appearance of the nuclear blebbing in Sgce KO mice is significantly high. In order to have 5% of probability of the blebbing detection, 231 of blebbings should exist on average per nucleus in Sgce KO mice.
Although the nuclear envelopes of cerebellar Purkinje cells were examined in Sgce pKO mice, no blebbing or other nuclear envelope abnormalities were found. Therefore loss of ε-sarcoglycan in the cerebellar Purkinje cells alone may not produce abnormal nuclear envelope, while it causes motor learning deficits. The results suggest that abnormal nuclear envelope do not correlate with motor learning deficits. Furthermore, the present results suggest that the abnormal nuclear envelopes in Sgce KO mice are caused by non-cell autonomous mechanism. This phenomenon is unique in comparison to other nuclear envelopathies. For examples, emerinopathy and laminopathy are known to exhibit abnormal nuclear envelopes. The former is caused by mutations in STA (EMD) coding for emerin and the latter is caused by mutations in LMNA coding for lamins A/C [61]. Emerin deficiency causes X-linked Emery-Dreifuss muscular dystrophy. Lamins A/C deficiency causes the autosomal dominant form of Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy with atrioventricular conduction disturbances, hypertrophic cardiomyopathy and Dunnigan-type familial partial lipodystrophy [51]. Both emerin and lamins A/C are nuclear envelope proteins and their loss of function causes abnormal nuclear envelopes. Moreover, abnormal nuclear envelopes have been reported in transfected cells over-expressing the mutant forms of torsinA [26, 27] and in Dyt1 KO mice and Dyt1 ΔGAG homozygous KI mice exhibiting neonatal lethality [28, 31]. TorsinA is located within the lumen of the nuclear envelope and endoplasmic reticulum, and interacts with nesprins which span the outer nuclear membrane and may contribute to the abnormal nuclear envelope [62]. TorsinA also interacts with LAP1 which concentrates in the inner nuclear membrane and contributes to maintain normal nuclear membrane [29, 63]. These nuclear envelopathies associated with functional loss of nuclear envelope proteins are believed to be caused by cell-autonomous mechanism. On the other hand, ε-sarcoglycan exists in the synapse as shown in the previous biochemical fractionation studies [10, 20]. The present immunohistochemical data also suggest that ε-sarcoglycans exist in the dendrites of the cerebellar Purkinje cells as well as the soma. Therefore loss of ε-sarcoglycan in the cerebellar Purkinje cells may not directly cause abnormal nuclear envelope in the cells.
The cellular mechanism underlying the motor learning is also unclear. The ε-sarcoglycan exist both in pre- and post-synaptic neurons of the mouse brain [8]. The existence of ε-sarcoglycan in the synaptosomal fraction was also confirmed in the mouse cerebral cortex [38]. Since ε-sarcoglycan has a cadherin-like motif in the N-terminal region [44], it may function in the synapse for linking the post-synaptic neurons and the pre-synaptic neurons during synaptogenesis and remodeling. A synaptic function of ε-sarcoglycan is further supported by our finding that alternatively spliced ε-sarcoglycans possess PDZ binding motifs that could potentially associate with synaptic PDZ domain containing proteins [11]. The cerebellar Purkinje cells receive input signals from the parallel fibers and climbing fibers, and send the major inhibitory signal from the cerebellum to the deep cerebellar nuclei and there are direct connections going from deep cerebellar nuclei to striatum by a disynaptic pathway and to globus pallidus externa by a tri-synaptic pathway [64]. Future experiments characterizing the synaptic dysfunction of cerebellar circuits in Sgce pKO mice will provide insight of in vivo function of ε-sarcoglycan. Electrophysiological approach, such as LTD measurement in the cerebellar Purkinje cells [65], will elucidate whether the cerebellar Purkinje cells receive some altered signals produced in other brain regions of Sgce KO mice.
The pathophysiology of myoclonus is complex and can be conceptualized at many levels [66]. The motor cortex is the most common source of myoclonus, while the subcortical areas, brainstem, spinal, and peripheral nervous system can be involved as well [1]. Identification of the affected brain regions or other nerves is important to elucidate the mechanism of myoclonus. In the previous study [21], we reported that Sgce KO mice exhibit significant myoclonus, while the present results showed no significant myoclonus in Sgce pKO mice. Our results suggest that the loss of ε-sarcoglycan function in other brain regions may contribute to myoclonus. Generation of brain regions- or cell type-specific Sgce conditional KO mice other than cerebellar Purkinje cells may identify brain regions or cell types that contribute to myoclonus in DYT11 M-D.
5. Conclusions
We found abnormal nuclear envelopes in the cerebellar Purkinje cells of Sgce KO mice. Sgce pKO mice exhibited impaired motor learning and decreased stereotypic behaviors, while they did not exhibit abnormal nuclear envelope in their cerebellar Purkinje cells, overt dystonic postures, myoclonus, OCD-like behaviors or anxiety-like behaviors. The results suggest that ε-sarcoglycan in the cerebellar Purkinje cells contributes to the motor learning, while the loss of ε-sarcoglycan in other brain regions may contribute to myoclonus, motor coordination deficits, and emotional alterations.
Supplementary Material
Highlights.
Impaired motor learning in Purkinje cell-specific Sgce knockout mice.
Purkinje cells may need ε-sarcoglycan to process proper signals for motor learning.
Sgce KO mice exhibited abnormal nuclear envelopes in the cerebellar Purkinje cells.
DYT11 myoclonus-dystonia can be categorized as a nuclear envelopathy.
Acknowledgments
We thank Lisa Foster, Andrea McCullough and their staff for animal care, Lou Ann Miller, Miki Jinno, Veena Ganesh, Mark P. DeAndrade, George Kasnic, Drs. Jill Verlander Reed, Sharon Matthews and Huan-Xin Chen for their technical assistance. We also thank Dr. William T. Dauer for his help to identify the abnormal nuclear envelopes. This work was supported by National Institutes of Health grants (NS37409, NS47466, NS47692, NS54246, NS57098, NS65273, NS72876, and NS74423) and the startup funds from the Lucille P. Markey Charitable Trust (UIUC), Department of Neurology (UAB), and Tyler’s Hope for a Dystonia Cure, Inc. (UF).
Abbreviations
- CT mouse
control littermate mouse
- GCL
Granule cell layer
- KI mouse
knock-in mouse
- M-D
Myoclonus-dystonia
- ML
Molecular layer
- OCD
obsessive-compulsive disorder
- PB
phosphate buffer
- PBS
phosphate-buffered saline
- PCL
cerebellar Purkinje cell layer
- Sgce KO mouse
paternally-inherited Sgce heterozygous knockout mouse
- Sgce pKO mouse
paternally-inherited cerebellar Purkinje cell-specific Sgce conditional knockout mouse
- WT mouse
wild-type mouse
Footnotes
Conflicts of interest
The authors state no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Caviness JN, Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol. 2004;3:598–607. doi: 10.1016/S1474-4422(04)00880-4. [DOI] [PubMed] [Google Scholar]
- 2.Fahn S. Concept and classification of dystonia. Adv Neurol. 1988;50:1–8. [PubMed] [Google Scholar]
- 3.Muller U. The monogenic primary dystonias. Brain. 2009;132:2005–25. doi: 10.1093/brain/awp172. [DOI] [PubMed] [Google Scholar]
- 4.Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–34. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- 5.Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. 2001;29:66–9. doi: 10.1038/ng709. [DOI] [PubMed] [Google Scholar]
- 6.Kinugawa K, Vidailhet M, Clot F, Apartis E, Grabli D, Roze E. Myoclonus-dystonia: an update. Mov Disord. 2009;24:479–89. doi: 10.1002/mds.22425. [DOI] [PubMed] [Google Scholar]
- 7.Sandona D, Betto R. Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects. Expert Rev Mol Med. 2009;11:e28. doi: 10.1017/S1462399409001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ettinger AJ, Feng G, Sanes JR. epsilon-Sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J Biol Chem. 1997;272:32534–8. doi: 10.1074/jbc.272.51.32534. [DOI] [PubMed] [Google Scholar]
- 9.Ettinger AJ, Feng G, Sanes JR. Additions and Corrections to epsilon-Sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J Biol Chem. 1998;273:19922. doi: 10.1074/jbc.272.51.32534. [DOI] [PubMed] [Google Scholar]
- 10.Nishiyama A, Endo T, Takeda S, Imamura M. Identification and characterization of epsilon-sarcoglycans in the central nervous system. Brain Res Mol Brain Res. 2004;125:1–12. doi: 10.1016/j.molbrainres.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Yokoi F, Dang MT, Mitsui S, Li Y. Exclusive paternal expression and novel alternatively spliced variants of epsilon-sarcoglycan mRNA in mouse brain. FEBS Lett. 2005;579:4822–8. doi: 10.1016/j.febslet.2005.07.065. [DOI] [PubMed] [Google Scholar]
- 12.McNally EM, Ly CT, Kunkel LM. Human epsilon-sarcoglycan is highly related to alpha-sarcoglycan (adhalin), the limb girdle muscular dystrophy 2D gene. FEBS Lett. 1998;422:27–32. doi: 10.1016/s0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- 13.Ritz K, van Schaik BD, Jakobs ME, van Kampen AH, Aronica E, Tijssen MA, et al. SGCE isoform characterization and expression in human brain: implications for myoclonus-dystonia pathogenesis? Eur J Hum Genet. 2011;19:438–44. doi: 10.1038/ejhg.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao J, LeDoux MS. Cloning, developmental regulation and neural localization of rat epsilon-sarcoglycan. Brain Res Mol Brain Res. 2003;119:132–43. doi: 10.1016/j.molbrainres.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Piras G, El Kharroubi A, Kozlov S, Escalante-Alcalde D, Hernandez L, Copeland NG, et al. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: identification by a subtractive screen of novel uniparental fibroblast lines. Mol Cell Biol. 2000;20:3308–15. doi: 10.1128/mcb.20.9.3308-3315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grabowski M, Zimprich A, Lorenz-Depiereux B, Kalscheuer V, Asmus F, Gasser T, et al. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur J Hum Genet. 2003;11:138–44. doi: 10.1038/sj.ejhg.5200938. [DOI] [PubMed] [Google Scholar]
- 17.Lancioni A, Luisa Rotundo I, Monique Kobayashi Y, D’Orsi L, Aurino S, Nigro G, et al. Combined deficiency of alpha and epsilon sarcoglycan disrupts the cardiac dystrophin complex. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roze E, Apartis E, Clot F, Dorison N, Thobois S, Guyant-Marechal L, et al. Myoclonus-dystonia: clinical and electrophysiologic pattern related to SGCE mutations. Neurology. 2008;70:1010–6. doi: 10.1212/01.wnl.0000297516.98574.c0. [DOI] [PubMed] [Google Scholar]
- 19.Hedrich K, Meyer EM, Schule B, Kock N, de Carvalho Aguiar P, Wiegers K, et al. Myoclonus-dystonia: detection of novel, recurrent, and de novo SGCE mutations. Neurology. 2004;62:1229–31. doi: 10.1212/01.wnl.0000118286.75059.35. [DOI] [PubMed] [Google Scholar]
- 20.Yokoi F, Yang G, Li J, Deandrade MP, Zhou T, Li Y. Earlier onset of motor deficits in mice with double mutations in Dyt1 and Sgce. J Biochem. 2010;148:459–66. doi: 10.1093/jb/mvq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yokoi F, Dang MT, Li J, Li Y. Myoclonus, motor deficits, alterations in emotional responses and monoamine metabolism in epsilon-sarcoglycan deficient mice. J Biochem. 2006;140:141–6. doi: 10.1093/jb/mvj138. [DOI] [PubMed] [Google Scholar]
- 22.Dang MT, Yokoi F, McNaught KS, Jengelley TA, Jackson T, Li J, et al. Generation and Characterization of Dyt1 deltaGAG Knock-in Mouse as a Model for Early-Onset Dystonia. Exp Neurol. 2005;196:452–63. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 23.Carbon M, Kingsley PB, Su S, Smith GS, Spetsieris P, Bressman S, et al. Microstructural white matter changes in carriers of the DYT1 gene mutation. Ann Neurol. 2004;56:283–6. doi: 10.1002/ana.20177. [DOI] [PubMed] [Google Scholar]
- 24.Yokoi F, Dang MT, Miller CA, Marshall AG, Campbell SL, Sweatt JD, et al. Increased c-fos expression in the central nucleus of the amygdala and enhancement of cued fear memory in Dyt1 DeltaGAG knock-in mice. Neurosci Res. 2009;65:228–35. doi: 10.1016/j.neures.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Yokoi F, Jin YH, Deandrade MP, Hashimoto K, Standaert DG, et al. Altered Dendritic Morphology of Purkinje cells in Dyt1 DeltaGAG Knock-In and Purkinje Cell-Specific Dyt1 Conditional Knockout Mice. PLoS One. 2011;6:e18357. doi: 10.1371/journal.pone.0018357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naismith TV, Heuser JE, Breakefield XO, Hanson PI. TorsinA in the nuclear envelope. Proc Natl Acad Sci U S A. 2004;101:7612–7. doi: 10.1073/pnas.0308760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez-Alegre P, Paulson HL. Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J Neurosci. 2004;24:2593–601. doi: 10.1523/JNEUROSCI.4461-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–32. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Kim CE, Perez A, Perkins G, Ellisman MH, Dauer WT. A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc Natl Acad Sci U S A. 2010;107:9861–6. doi: 10.1073/pnas.0912877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grundmann K, Reischmann B, Vanhoutte G, Hubener J, Teismann P, Hauser TK, et al. Overexpression of human wildtype torsinA and human DeltaGAG torsinA in a transgenic mouse model causes phenotypic abnormalities. Neurobiol Dis. 2007;27:190–206. doi: 10.1016/j.nbd.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 31.Yokoi F, Dang MT, Li J, Standaert DG, Li Y. Motor Deficits and Decreased Striatal Dopamine Receptor 2 Binding Activity in the Striatum-Specific Dyt1 Conditional Knockout Mice. PLoS One. 2011;6:e24539. doi: 10.1371/journal.pone.0024539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokoi F, Dang MT, Mitsui S, Li J, Li Y. Motor deficits and hyperactivity in cerebral cortex-specific Dyt1 conditional knockout mice. J Biochem. 2008;143:39–47. doi: 10.1093/jb/mvm191. [DOI] [PubMed] [Google Scholar]
- 33.LeDoux MS, Lorden JF, Ervin JM. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol. 1993;120:302–10. doi: 10.1006/exnr.1993.1064. [DOI] [PubMed] [Google Scholar]
- 34.LeDoux MS, Lorden JF, Meinzen-Derr J. Selective elimination of cerebellar output in the genetically dystonic rat. Brain Res. 1995;697:91–103. doi: 10.1016/0006-8993(95)00792-o. [DOI] [PubMed] [Google Scholar]
- 35.Grusser C, Grusser-Cornehls U. Improvement in motor performance of Weaver mutant mice following lesions of the cerebellum. Behav Brain Res. 1998;97:189–94. doi: 10.1016/s0166-4328(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 36.Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–78. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- 37.Chan P, Gonzalez-Maeso J, Ruf F, Bishop DF, Hof PR, Sealfon SC. Epsilon-sarcoglycan immunoreactivity and mRNA expression in mouse brain. J Comp Neurol. 2005;482:50–73. doi: 10.1002/cne.20377. [DOI] [PubMed] [Google Scholar]
- 38.Ghilardi MF, Carbon M, Silvestri G, Dhawan V, Tagliati M, Bressman S, et al. Impaired sequence learning in carriers of the DYT1 dystonia mutation. Ann Neurol. 2003;54:102–9. doi: 10.1002/ana.10610. [DOI] [PubMed] [Google Scholar]
- 39.Sharma N, Baxter MG, Petravicz J, Bragg DC, Schienda A, Standaert DG, et al. Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J Neurosci. 2005;25:5351–5. doi: 10.1523/JNEUROSCI.0855-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doheny DO, Brin MF, Morrison CE, Smith CJ, Walker RH, Abbasi S, et al. Phenotypic features of myoclonus-dystonia in three kindreds. Neurology. 2002;59:1187–96. doi: 10.1212/wnl.59.8.1187. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe M. Molecular mechanisms governing competitive synaptic wiring in cerebellar Purkinje cells. Tohoku J Exp Med. 2008;214:175–90. doi: 10.1620/tjem.214.175. [DOI] [PubMed] [Google Scholar]
- 42.Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28:106–10. [PubMed] [Google Scholar]
- 43.Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci U S A. 1988;85:5166–70. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–1. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barski JJ, Dethleffsen K, Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–8. [PubMed] [Google Scholar]
- 46.Campos VE, Du M, Li Y. Increased seizure susceptibility and cortical malformation in beta-catenin mutant mice. Biochem Biophys Res Commun. 2004;320:606–14. doi: 10.1016/j.bbrc.2004.05.204. [DOI] [PubMed] [Google Scholar]
- 47.Fernagut PO, Diguet E, Stefanova N, Biran M, Wenning GK, Canioni P, et al. Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterisation. Neuroscience. 2002;114:1005–17. doi: 10.1016/s0306-4522(02)00205-1. [DOI] [PubMed] [Google Scholar]
- 48.Cao BJ, Li Y. Reduced anxiety- and depression-like behaviors in Emx1 homozygous mutant mice. Brain Res. 2002;937:32–40. doi: 10.1016/s0006-8993(02)02461-7. [DOI] [PubMed] [Google Scholar]
- 49.Dang MT, Yokoi F, Pence MA, Li Y. Motor deficits and hyperactivity in Dyt1 knockdown mice. Neurosci Res. 2006;56:470–4. doi: 10.1016/j.neures.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carter RJ, Morton AJ, Dunnett SB. Motor Coordination and Balance in Rodents. In: Crawley J, editor. Current Protocols in Neuroscience. John Wiley & Sons, Inc; 2001. pp. 8.12.1–8..4. [DOI] [PubMed] [Google Scholar]
- 51.Nagano A, Arahata K. Nuclear envelope proteins and associated diseases. Curr Opin Neurol. 2000;13:533–9. doi: 10.1097/00019052-200010000-00005. [DOI] [PubMed] [Google Scholar]
- 52.Rumbach L, Barth P, Costaz A, Mas J. Hemidystonia consequent upon ipsilateral vertebral artery occlusion and cerebellar infarction. Mov Disord. 1995;10:522–5. doi: 10.1002/mds.870100424. [DOI] [PubMed] [Google Scholar]
- 53.Le Ber I, Clot F, Vercueil L, Camuzat A, Viemont M, Benamar N, et al. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology. 2006;67:1769–73. doi: 10.1212/01.wnl.0000244484.60489.50. [DOI] [PubMed] [Google Scholar]
- 54.Pizoli CE, Jinnah HA, Billingsley ML, Hess EJ. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci. 2002;22:7825–33. doi: 10.1523/JNEUROSCI.22-17-07825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Delmaire C, Vidailhet M, Elbaz A, Bourdain F, Bleton JP, Sangla S, et al. Structural abnormalities in the cerebellum and sensorimotor circuit in writer’s cramp. Neurology. 2007;69:376–80. doi: 10.1212/01.wnl.0000266591.49624.1a. [DOI] [PubMed] [Google Scholar]
- 56.Draganski B, Thun-Hohenstein C, Bogdahn U, Winkler J, May A. “Motor circuit” gray matter changes in idiopathic cervical dystonia. Neurology. 2003;61:1228–31. doi: 10.1212/01.wnl.0000094240.93745.83. [DOI] [PubMed] [Google Scholar]
- 57.Obermann M, Yaldizli O, De Greiff A, Lachenmayer ML, Buhl AR, Tumczak F, et al. Morphometric changes of sensorimotor structures in focal dystonia. Mov Disord. 2007;22:1117–23. doi: 10.1002/mds.21495. [DOI] [PubMed] [Google Scholar]
- 58.Carbon M, Ghilardi MF, Argyelan M, Dhawan V, Bressman SB, Eidelberg D. Increased cerebellar activation during sequence learning in DYT1 carriers: an equiperformance study. Brain. 2008;131:146–54. doi: 10.1093/brain/awm243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Calderon DP, Fremont R, Kraenzlin F, Khodakhah K. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci. 2011;14:357–65. doi: 10.1038/nn.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeAndrade MP, Yokoi F, van Groen T, Lingrel JB, Li Y. Characterization of Atp1a3 mutant mice as a model of rapid-onset dystonia with parkinsonism. Behav Brain Res. 2011;216:659–65. doi: 10.1016/j.bbr.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emery AE. Emery-Dreifuss muscular dystrophy - a 40 year retrospective. Neuromuscul Disord. 2000;10:228–32. doi: 10.1016/s0960-8966(00)00105-x. [DOI] [PubMed] [Google Scholar]
- 62.Nery FC, Zeng J, Niland BP, Hewett J, Farley J, Irimia D, et al. TorsinA binds the KASH domain of nesprins and participates in linkage between nuclear envelope and cytoskeleton. J Cell Sci. 2008;121:3476–86. doi: 10.1242/jcs.029454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goodchild RE, Dauer WT. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol. 2005;168:855–62. doi: 10.1083/jcb.200411026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoshi E, Tremblay L, Féger J, Carras PL, Strick PL. The cerebellum communicates with the basal ganglia. Nat Neurosci. 2005;8:1491–3. doi: 10.1038/nn1544. [DOI] [PubMed] [Google Scholar]
- 65.Ito M. Cerebellar circuitry as a neuronal machine. Prog Neurobiol. 2006;78:272–303. doi: 10.1016/j.pneurobio.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 66.Caviness JN. Pathophysiology and treatment of myoclonus. Neurol Clin. 2009;27:757–77. vii. doi: 10.1016/j.ncl.2009.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.