

Abstract
Improved treatments for prostate cancer are in great need to overcome lethal recurrence and metastasis. Targeting the tumorigenic cancer stem cells (CSCs) with self-renewal and differentiation capacity appears to be a promising strategy. Blockade of the hedgehog (Hh) signaling pathway, an important pathway involved in stem cell self-renewal, by cyclopamine leads to long-term prostate cancer regression without recurrence, strongly suggesting the connection between Hh pathway and prostate CSCs. Here we designed a HPMA (N-(2-hydroxypropyl)methacrylamide)-based cyclopamine delivery system as a CSC-selective macromolecular therapeutics with improved drug solubility and decreased systemic toxicity. To this end, HPMA and N-methacryloylglycylphenylalanylleucylglycyl thiazolidine-2-thione were copolymerized using the RAFT (reversible addition-fragmentation chain transfer) process, followed by polymer-analogous attachment of cyclopamine. The selectivity of the conjugate toward CSCs was evaluated on RC-92a/hTERT cells, the human prostate cancer epithelial cells with human telomerase reverse transcriptase transduction. The use of RC-92a/hTERT cells as an in vitro CSC model was validated by stem cell marker expression and prostasphere culture. The bioactivity of cyclopamine was retained after conjugation to the polymer. Furthermore, HPMA polymer-conjugated cyclopamine showed anti-CSC efficacy on RC-92a/hTERT cells as evaluated by decreased stem cell marker expression and CSC viability.
1. Introduction
Prostate cancer is the most commonly diagnosed and second lethal malignancy in men in the United States. Progressive prostate cancer is mostly treated with androgen deprivation therapy, however, nearly all patients eventually relapse and develop to an androgen-independent state of cancer, resulting in death due to widespread metastases [1]. Similarly, docetaxel, considered as the traditional first-line chemotherapy for advanced prostate cancer, does not provide satisfactory progression free survival for patients with advanced prostate cancer [2]. Contemporary anticancer therapies mostly fail to eliminate prostate cancer, but result in lethal recurrence and metastasis. Improved approaches are urgently needed that will result in long-term survival. In the field of nanomedicine, efforts have been made to improve the pharmacokinetics, lower the systemic toxicity and overcome drug resistance of traditional chemotherapeutics, thereby improving their anticancer efficacy. However, the biological/functional heterogeneity of tumors challenges the traditional anticancer therapeutics, suggesting the necessity to develop therapeutics that target different subpopulations in cancer [3,4]. Recently, the Cancer Stem Cell (CSC) hypothesis was revived. Based on this hypothesis, CSCs, a subset of cells within the tumor that can drive tumorigenesis with the ability to self-renew and give rise to phenotypically diverse tumor cells, contribute to lethal cancer recurrence and metastasis [3-7]. It favors the possibility that the failure of traditional anticancer therapies is due to the failure to kill CSCs. Therefore, targeting CSCs or in combination with traditional anticancer therapeutics represents a promising strategy to improve cancer patient survival.
Tumorigenic CSCs in hematopoietic system malignancies and solid tumors could be identified based on marker expression profiles that are distinct from non-tumorigenic cancer cells [3,5]. In prostate cancer, recent evidence showed that the androgen-independent CSCs, identified from both primary prostate tumors and the majority of metastases, have a basal profile of marker expressions similar to normal prostatic stem cells [8,9]. Individual or combinations of CD133, CD44, integrin α2β1, ABCG2, CK5/14, bmi-1, and CD49f have been used depending on different cancer models [10-12]. These putative prostate CSCs, isolated from clinical samples, xenograft tumors, or cancer cell lines, showed evidence of CSC properties, including in vitro clonogenicity, sphere formation and differentiation capacity as well as in vivo tumorigenicity by recapitulating a cellular hierarchy of the original parental tumor [9,10,13].
A critical hurdle to eliminate CSCs in the clinics lies in its inherent resistance to various conventional chemo- or radiotherapies, which mainly target differentiated cancer cells [14-16]. Thus, therapeutics to which CSCs are sensitive should be employed to eliminate CSCs. Noticeably, the role of Hedgehog (Hh) pathway, crucial in regulating stem cells during embryonic development, has been implicated in the proliferation, progression and metastasis of prostate cancer in clinical samples and xenografts [17,18]. More importantly, studies have shown connections between Hh signaling and CSCs [17-20]. Blockade of Hh pathway led to down-regulation of stem cell self-renewal gene expressions, along with complete and long-term prostate cancer regression without recurrence [17]. Thus, Hh pathway can be a useful target for CSC elimination.
Among several Hh inhibitors identified, the inhibitory effect of cyclopamine has been proved in vivo in prostate cancer models [17,21]. Cyclopamine, a natural steroidal alkaloid, inhibits the Hh pathway by directly binding to a membrane receptor Smoothened (SMO), suppressing SMO and its downstream activities, eventually leading to apoptotic cell deaths [21,22]. Despite the promising in vivo efficacy of cyclopamine, its application for cancer treatment is limited by the high hydrophobicity, systemic toxicity and poor pharmacokinetics [23].
N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer conjugates carrying anticancer drugs have shown significant antiproliferative effects in cancer cells and tumor growth suppression in animal models [24,25]. Aiming to improve the outcome of prostate cancer treatments by targeting CSCs, we designed and synthesized a water soluble macromolecular drug carrier based on HPMA copolymer, by RAFT (reversible addition-fragmentation chain transfer) copolymerization. Cyclopamine was attached to the end of GFLG (glycylphenylalanylleucylglycyl) biodegradable tetrapeptide side chains via reaction of secondary amine in cyclopamine with thiazolidine-2-thione (TT) reactive groups. We evaluated the CSC inhibitory effects of the HPMA copolymer-cyclopamine conjugate in an in vitro prostate cancer epithelial cell model, RC-92a/hTERT cells, with stem cell properties [26,27]. RC-92a/hTERT cells were chosen since the CD133+/integrin α2β1hi/CD44+ putative prostate CSCs within the whole cell line could be enriched to 5%, higher than that reported on primary prostate cancer cells or other established prostate cancer cell lines [26]. Cell surface marker expression analysis and cytotoxicity studies following drug and conjugate treatments on RC-92a/hTERT cells supported the anti-CSC efficacy of the designed macromolecular therapeutics.
2 Experimental
2.1. Materials
Cyclopamine (free base) was purchased from LC Laboratories (Woburn, MA). 2,2’-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) and 2,2’-azobis(2,4-dimethyl valeronitrile) (V-65) were from Wako Chemicals (Richmond, VA). Dichloromethane (DCM) was obtained from Mallinckrodt Baker (Phillipsburg, NJ). Papain (from papaya latex) and cathepsin B (from bovine spleen) were purchased from Sigma-Aldrich (St. Louis, MO). 4-cyanopentanoic acid dithiobenzoate [28], N-(2-hydroxypropyl)methacrylamide (HPMA) [29] and N-methacryloylglycylphenylalanylleucylglycyl thiazolidine-2-thione (MA-GFLG-TT) [30] were synthesized as previously described. All other solvents were obtained from Sigma-Aldrich as the highest purity available.
2.2. Synthesis of HPMA copolymer containing thiazolidine-2-thione (TT) reactive groups (P-GFLG-TT)
HPMA copolymer containing TT side-chain reactive groups was prepared in methanol at 40°C by RAFT copolymerization of HPMA and MA-GFLG-TT using VA 044 as initiator and 4-cyanopentanoic acid dithiobenzoate as chain transfer agent [31]. HPMA (134.6 mg, 0.94 mmol) and MA-GFLG-TT (33.7 mg, 0.06 mmol) were added into an ampoule attached to the Schlenk-line. After three vaccum-nitrogen cycles to remove oxygen, 0.3 mL degassed methanol, VA 044 solution (0.14 mg in 60 μL) and 4-cyanopentanoic acid dithiobenzoate solution (0.42 mg in 60 μL) in methanol were added via syringe. The mixture was bubbled with nitrogen for 15 min before sealing the ampoule and the copolymerization was performed at 40°C for 20 h. The copolymer was obtained by precipitation into acetone and purified by dissolution-precipitation in methanol-acetone twice and dried under vacuum. The molecular weight and molecular weight distribution was determined by size-exclusion chromatography (SEC) on an AKTA FPLC system (Pharmacia) equipped with miniDAWN and OptilabEX detectors with acetate/30% acetonitrile as mobile phase. Superose 6 HR10/30 column (Pharmacia) was used. Yield of P-GFLG-TT was 75 mg (45%) (Mw 50.8 kDa, PDI 1.07).
To remove the dithiobenzoate groups from HPMA copolymer chain ends, radical-induced end-group modification was conducted by utilizing high excess of free radical species. Briefly, HPMA copolymer (70 mg, Mn 47.7 kDa, 1.47 μmol) and V65 (20x excess, 7.3 mg, 29.4 μmol) were added into an ampoule. After three vacuum-nitrogen cycles to remove oxygen, 0.6 mL methanol was added. The solution was bubbled with nitrogen for 15 min, sealed and reacted at 50°C for 3 h. The end-modified copolymer was purified by precipitation into acetone twice and then dried under vacuum (yield 60 mg). The content of TT in the copolymer was determined by UV spectrophotometry in methanol (ε305= 10,800 L mol-1 cm-1).
2.3. Synthesis of HPMA copolymer-cyclopamine conjugate (P-GFLG-Cyclopamine)
HPMA copolymer-cyclopamine conjugate was obtained by the reaction of end-modified P-GFLG-TT with cyclopamine in pyridine. P-GFLG-TT (30 mg, TT content 12 μmol) and cyclopamine (Mw 411.6, 3x excess of TT mol content, 15 mg, 36 μmol) were added into an ampoule, followed by addition of 0.3 mL pyridine to dissolve the mixture. The ampoule was bubbled with nitrogen for 30 min, sealed and then reacted at 50°C for 28 h. Then excess of 1-amino-2-propanol was added to remove the remaining TT groups. The conjugate was precipitated into ether, and then re-dissolved and precipitated in methanol-ether/acetone (1:1) twice. The dried product was then dissolved in deionized water (DI H2O), purified through PD-10 column and freeze-dried. Yield: 21 mg.
2.4. Enzyme-catalyzed cleavage of cyclopamine from HPMA copolymer-cyclopamine conjugate
To assure the conjugation of cyclopamine to the HPMA copolymer, model enzymes papain and cathepsin B were used to cleave the GFLG tetrapeptide between HPMA copolymer main chain and the conjugated drug. The cleavage was performed in McIlvaine’s buffer (50 mM citrate/0.1 M phosphate) at pH 6.0 (for papain) and 5.0 (for cathepsin B) at 37°C [32]. The exact concentrations of papain and cathepsin B solutions were determined by UV spectrophotometer at 278 nm using A1%=25 for papain and 20 for cathepsin B. Papain (8.6 mg/mL, 50 μL) and cathepsin B (0.6 mg/mL, 50 μL) was preincubated with McIlvaine’s buffer containing 5 mM glutathione (50 μL) for 5 min at 37°C. Then 1.5 mg of HPMA copolymer-cyclopamine conjugate in 50 μL buffer was added into the mixture and incubated at 37°C. At different time points 60 μL mixture was withdrawn, and mixed with 60 μL methanol as solvent for cyclopamine before analyzing on HPLC. The release of cleaved cyclopamine was analyzed by HPLC (Agilent Technologies 1100 series, Zorbax C8 column 4.6 × 150 mm) using flow rate 1.0 mL/min and gradient elution from 10% to 50% of buffer B within 30 min (Buffer A: DI H2O with 0.1% TFA, Buffer B: acetonitrile with 0.1% TFA).
2.5. Cell line
RC-92a/hTERT cells, the human prostate cancer epithelial cells derived from patient with human telomerase reverse transcriptase (hTERT) transduction, was a gift of Dr. Rhim (Center for Prostate Disease Research, Uniformed Services University of the Health Sciences, Bethesda, MD) [26,27]. Cells were grown and maintained in a keratinocyte serum-free medium (K-SFM) supplemented with bovine pituitary extract and recombinant epidermal growth factor (EGF; KGM; Life Technologies, Gaithersburg, MD) at 37°C, 5% CO2. The cells were routinely trypsinized and subcultured when reaching 90% confluency.
2.6. Flow cytometry and Fluorescence-activated cell sorting (FACS)
Flow cytometry and Fluorescence-activated cell sorting (FACS) were used for determination of cell surface marker expressions and CD133+ cell sorting. To determine the expression levels of CD133, CD44 and integrin α2β1, monoclonal antibodies mouse anti-human CD133/1 (allophycocyanin (APC) labeled; Miltenyi Biotec, Inc., Auburn, CA), mouse anti-human CD44 (clone 515; phycoerythrin (PE) labeled; Becton Dickinson PharMingen), and mouse anti-human integrin α2β1 (Chemicon, Millipore) were used. A typical antibody staining procedure was as follows: Cells at different passages growing in log phase were trypsinized using 0.25% trypsin EDTA (Gibco, Invitrogen) and harvested. Cells were resuspended at the density of up to 1×107/mL in cold staining buffer (phosphate buffered saline pH 7.2, 0.5% bovine serum albumin, 2 mM EDTA), preblocked with 3% bovine serum albumin, and then stained with any of the monoclonal antibodies (the concentrations used according to manufacturer’s instructions) described above in the dark at 4°C for 15 min. Cells were agitated intermittently during staining. For integrin α2β1 staining, cells were stained for another 30 min using Alexa Fluor® 488 labeled goat anti-mouse IgG secondary antibody (Molecular Probes®, Invitrogen) at the concentration of 10 μg/mL. After staining, cells were thoroughly washed twice using staining buffer before flow cytometry analysis. The corresponding isotype controls (mouse IgG1, κ) were included in each staining condition. Samples were analyzed with FACScan flow cytometer (Becton Dickinson) and data were analyzed by FCS Express (De Novo Software, Los Angeles, CA). CD133+/- Cells were sorted by FACSAria (Becton Dickinson) following the same staining procedures as described above. Cells were gated on the forward and side-scatter plot and sort gates were defined on the dot plot of CD133 (APC).
For the analysis of CD133 expressions following regrowth of sorted CD133+ and CD133-cells, sorted cells were seeded in 25 cm2 tissue culture flasks at the density of 5×105/flask. After two passages, cells were harvested, stained and subjected to flow cytometry analysis.
2.7. Prostasphere culture
RC-92a/hTERT cells or sorted CD133+ and CD133- cells were prepared into single cell suspensions. The whole RC-92a/hTERT cells were plated at the density of 1,000 cells/well in 6-well ultra low attachment plates (Corning, NY), growing in K-SFM supplemented with 10 ng/mL EGF, and 10 ng/mL basic fibroblast growth factor and B27 (all from Invitrogen, Carlsbad, CA). For sorted CD133+ and CD133- cells, cells were plated at the initial densities of 50, 100, 500/well respectively in 24-well plates. Each group was plated in triplicate. After 10 days of growth under the above culture condition, the numbers of nonadherent spheres formed were counted under the microscope [33]. To access CD133 expression, prostaspheres were collected and dissociated with TrypLE Express (Gibco, Invitrogen). The dissociated cells were stained and filtered before analyzed by flow cytometry.
2.8. Determination of CD133 expressions in RC-92a/hTERT cells following drug or conjugate treatments
RC-92a/hTERT cells growing in log phase were trypsinized and seeded in 6-well plates at the initial density of 5×104 cells/well. After growing for 24 h, growth medium were changed to drug/conjugate-containing medium with different concentrations of free cyclopamine (2 μM, 5 μM, 10 μM), HPMA copolymer-cyclopamine conjugate (free cyclopamine equivalent 5 μM, 10 μM, 20 μM, 40 μM, 80 μM) and free docetaxel (0.5 nM, 1 nM, 2 nM). Free cyclopamine and docetaxel stock solutions were dissolved in dimethylsulfoxide (DMSO) prior to dilution. HPMA copolymer-cyclopamine conjugate was dissolved directly in growth medium. After 72 h incubation, drug or conjugate solutions were discarded and cells were washed and allowed to recover for 48 h in drug/conjugate-free medium. Both DMSO treated and untreated cells were used as controls. For flow cytometry, cells were harvested and stained with anti-CD133 APC as described above. Unstained cells in each group were used as negative controls for instrumental settings. To access cell viabilities, 2 μg/mL propidium iodide (Sigma-Aldrich, St Louis, MO) was incubated with cells 15 min prior to flow cytometry analysis.
2.9. Confocal laser scanning microscopy
Cells were plated onto sterile 35 mm glass bottom dishes with 14 mm microwells (MatTek Corporation, Ashland, MA) at a density of 1 × 105 cells per well for 24 h before experiment. Then growth medium containing HPMA copolymer-cyclopamine conjugate labeled with FITC (P-Cyclopamine-FITC, 0.5 mg/mL, Mw 45 kDa, FITC content 3.6×10-5 mmol/mg) were incubated with cells at 37 °C, 5% CO2 for 2 h. HPMA copolymer-FITC (P-FITC, 0.5 mg/mL, Mw 45 kDa, FITC content 4.76×10-5 mmol/mg) was used as control. After incubation, conjugate containing media were discarded and cells were mounted with Hoechst 33342 (Molecular Probes®, Invitrogen) containing fresh media for 10 min and then washed thoroughly with PBS twice. Live-cell fluorescence imaging was performed immediately using Olympus laser scanning confocal microscopy (FV 1000).
2.10. Cytotoxicity assay
To access cytotoxicities following the treatments of free cyclopamine, docetaxel and HPMA copolymer-cyclopamine conjugate, cell viability assay was performed using cell counting Kit-8 (CCK-8, Dojindo, Japan). Cells were seeded in a 96-well plate in triplicate at the density of 5,000/well and grew for 24 h. Then different concentrations of free cyclopamine, docetaxel and HPMA copolymer-cyclopamine conjugate were incubated with cells for 72 h. After incubation, drug/conjugate-containing medium was discarded and replaced with 100 μL fresh growth medium each well, followed by the addition of 50 μL 5x diluted CCK-8 solution. Dehydrogenase activities in live cells converted the water-soluble tetrazolium salt WST-8 into a soluble yellow-color formazan dye. After the incubation of cells at 37°C, 5% CO2 for 2 h, the absorbance was measured via a microplate reader at 450 nm (630 nm as reference). Untreated or DMSO treated control cells were set as 100% viable.
2.11. Statistical analyses
Statistical analyses were done using the Student’s t test of paired samples to compare the results between two groups or one-way analysis of variance to compare three or more groups, with P values of <0.05 indicating statistically significant differences.
3. Results and discussion
3.1 Design, synthesis, and characterization of HPMA copolymer-cyclopamine conjugate
The synthetic route of HPMA copolymer-cyclopamine conjugate is shown in Figure 1. In order to attach the Hh pathway inhibitor cyclopamine to the side chains, HPMA copolymer precursor containing thiazolidine-2-thione (TT) functional groups was firstly synthesized by RAFT copolymerization. 4-Cyanopentanoic acid dithiobenzoate used here was a commonly used RAFT chain transfer agent for HPMA polymerization with excellent control of molecular weight and molecular weight distribution [31]. The resulting copolymer has a narrow molecular weight distribution, with an actual molecular weight close to the theoretical molecular weight as shown in Table 1. The molar ratio of TT to HPMA in P-GFLG-TT was similar to the feed ratio. Cyclopamine was conjugated to P-GFLG-TT under basic condition via the substitution of TT groups with the secondary amine group in cyclopamine. The successful conjugation of cyclopamine to P-GFLG-TT was confirmed by papain and cathepsin B cleavage of the conjugate. The GFLG tetrapeptide side chain, as the substrate of papain and cathepsin B, can be cleaved, leading to the release of cyclopamine from the conjugate. Interestingly, enzyme cleavage led to two components-free cyclopamine ([CYP+H]+, 412.1) and glycine-cyclopamine ([Gly-CYP+H]+, 469.4) as confirmed by HPLC and LC-MS. Due to the different preferential cleavage sites of cathepsin B and papain, the majority of the cleaved product was glycine-cyclopamine following papain cleavage, while cathepsin B cleavage led to more free cyclopamine fraction as observed by HPLC. Papain has a preference for phenylalanine in position P2 (nomenclature of Schechter and Berger [34]) of the substrate. Consequently, two modes of arrangement of the GFLG side-chains in the active site of papain occurred (F in P2 and F in P3) in spite of the increased steric hindrance of the HPMA copolymer backbone on the formation of papain-substrate complex for the former arrangement (F in P2). These results are in agreement with those obtained in papain-catalyzed cleavage of branched HPMA copolymers with oligopeptide crosslinks [35]. The amount of cyclopamine within the conjugate was estimated from the peak area under the curve of cyclopamine according to the standard curve constructed from different concentrations of free cyclopamine determined by HPLC. The conversion of TT groups to cyclopamine was 50%, that is, 7.5 wt% in the final HPMA copolymer-cyclopamine conjugate.
Figure 1.
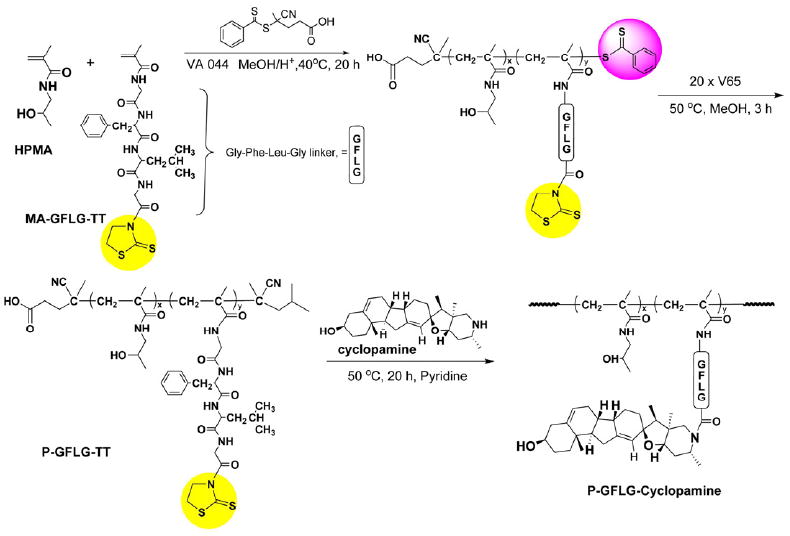
Synthesis of HPMA copolymer-cyclopamine conjugate by RAFT copolymerization of HPMA and MA-GFLG-TT, followed by conjugation of cyclopamine to GFLG side chains.
Table 1.
Characterization of HPMA copolymer containing thiazolidine-2-thione reactive group
| [CTA]0/[I]0 | Mth (kDa) | Mn (kDa) | Mw (kDa) | Mw/Mn | Conversion | TT mol% |
|---|---|---|---|---|---|---|
| 4 | 45 | 47.7 | 50.8 | 1.066 | 45% | 6.1 |
CTA .. chain transfer agent; Mth .. theoretical molecular weight.
3.2 Characterization of CSC properties in RC-92a/hTERT cells
3.2.1. CSC surface marker expression
Based on previous characterizations in Rhim’s group, prostate CSC-enriched population in RC-92a/hTERT cells has a CD133+/integrin α2β1hi/CD44+ surface marker profile, with enhanced colony and sphere forming ability in vitro and increased tumorigenicity in vivo [26,27]. Here we further characterized the cell line to facilitate the conjugate evaluation. To validate the use of surface markers, we determined the CD133, CD44 and integrin α2β1 expression levels in the RC-92a/hTERT cells. As shown in Figure 2a,b, only a small proportion of cells (around 5%) expressed the CD133 surface marker, while the majority (>90%) of cells within RC-92a/hTERT cells expressed CD44 and integrin α2β1. These results are in agreement with previous studies [26]. Multicolor flow cytometry analysis showed that all CD133+ cells express CD44 and integrin α2β1; however, CD44 and integrin α2β1 were not exclusively expressed in CD133+ cells (Figure 2c). Therefore, among the three cell surface markers, CD133+ was the most specific to define cancer stem-like cells in this cell line, although CSCs has a marker profile of CD133+/integrin α2β1hi/CD44+. In accordance with the previous results, CD133 was stably expressed throughout different passages of RC-92a/hTERT cells (Figure 3). This permits the use of these cells at different passages for conjugate evaluations. In addition, we separated the CD133+ and CD133- cells by FACS and seeded cells separately in monolayer for regrowth. CD133 expression in CD133+ cells still remained enriched up to 30% within first two passages (Figure 4), although gradually decreased with further passages. The purity of sorted CD133+ and CD133- cells was confirmed by reanalyzing marker expression by flow cytometry immediately after sorting. Therefore, the gradual loss of CD133 marker expression was due to the long-term monolayer culture condition of CD133+ cells, instead of the impurity of sorted CD133+ cells immediately after FACS. On the other hand, CD133 expression in cells subcultured from CD133- cells remained close to the background level of unstained CD133- cells within two passages (Figure 4).
Figure 2.
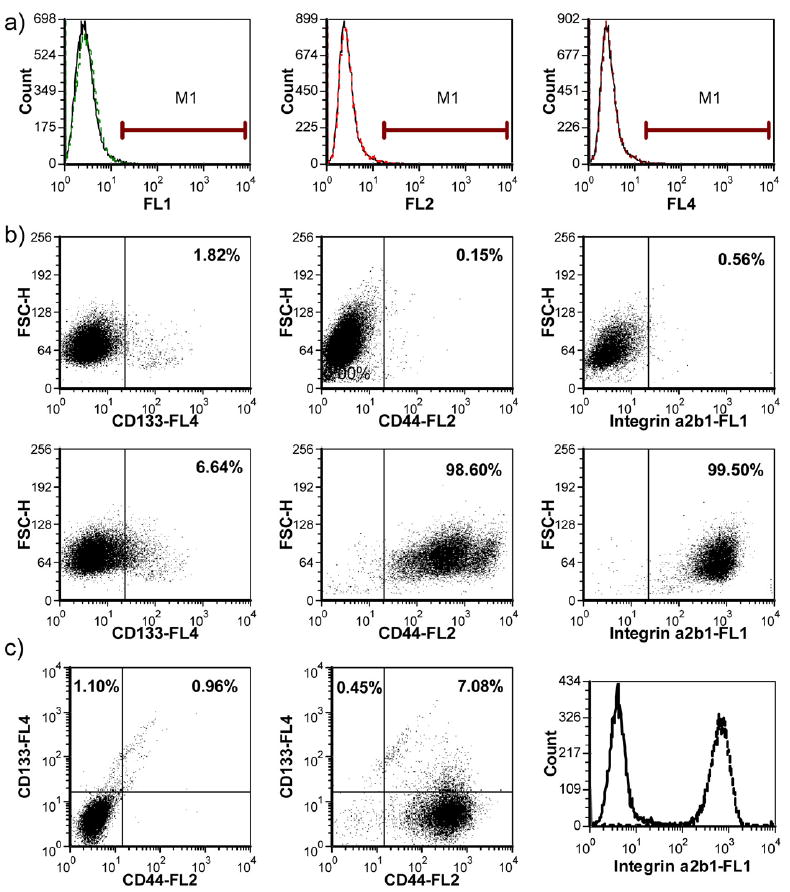
Characterization of cell surface marker expressions on RC-92a/hTERT cells. a) Isotype control profiles on all channels (dashed line: isotype; solid line: negative control without staining); b) single color staining of CD133, CD44 and integrin α2β1 (the upper panels show the negative control profiles; the lower panels show the stained samples); c) multicolor staining; the left profile shows the negative control; the middle profile shows the CD133 and CD44 double color trace; the right profile shows the integrin expression in CD133+/CD44+ cells (solid line: negative control; dotted line: integrin staining).
Figure 3.
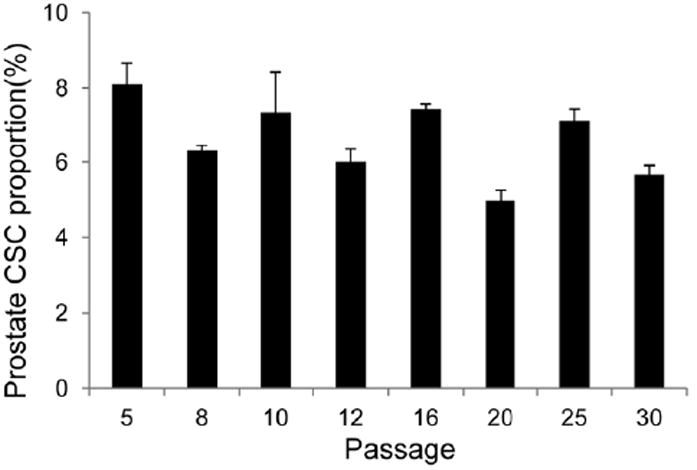
Expression pattern of CD133 in RC-92a/hTERT cells at different passages.
Figure 4.
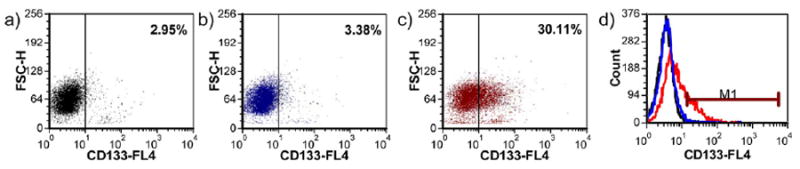
CD133 expression in sorted cells after regrowth following FACS. a) Sorted CD133-cells stained with anti CD133-APC; b) negative control of sorted CD133+ cells; c) sorted CD133+ cells stained with anti CD133-APC; d) histogram overlay.
3.2.2. Prostasphere formation
To correlate the CD133 expression with CSC functional properties, we performed the prostasphere-forming assay on both RC-92a/hTERT cells and sorted cells. Sphere forming assay has been used as a standard in vitro method to characterize stem cell-like behavior in both tumor and non-tumor tissues [36]. Prostaspheres refer to spherical clusters clonally generated from single prostate cancer cells grown under nonadherent culture condition. Theoretically, under this condition, the cells with self-renewal and differentiation abilities can grow and expand their population but cells in a more differentiated state lose the self-renewal capacity and hardly grow. Therefore, sphere-forming capacity can be used as a measure of stem cell properties. As shown in Figure 5a, single cell suspension of RC-92a/hTERT cells could grow into spheres within 10 days of growth. The cells within the spheres showed an enriched CD133 expression (30% of the whole population) compared to cells grown in monolayer.
Figure 5.
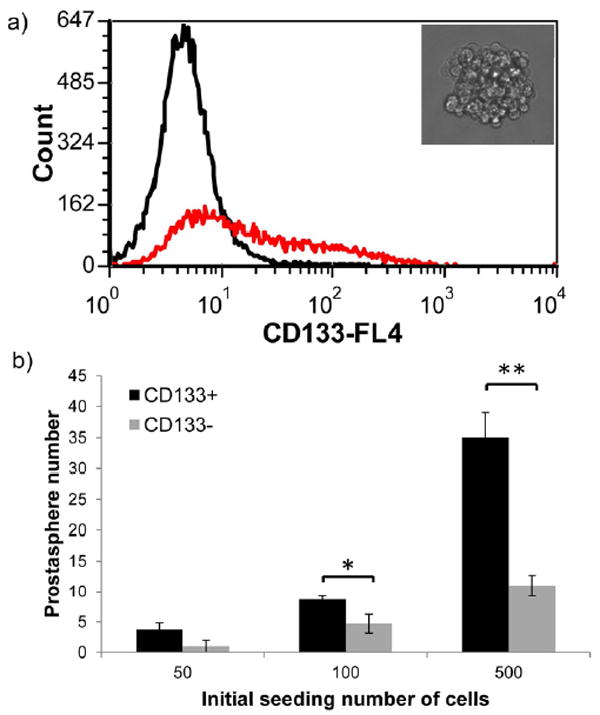
Prostasphere formation of RC-92a/hTERT cells and sorted CD133+/- cells. a) The morphology of prostasphere and CD133 expression in the cells dissociated from prostaspheres (black line: negative control; red line: cells stained with anti CD133-APC); b) prostasphere forming efficiency of CD133+ and CD133- cells at different initial seeding densities. Experiments were done in triplicate. *, p<0.05; **, p<0.01.
To further characterize which subpopulation contributed to the sphere growth, sphere-forming abilities of sorted CD133+ and CD133- cells were compared by seeding the same number of viable CD133+ and CD133- cells. An intrinsic limitation of the sphere forming assay is the possible occurrence of sphere fusion (or sphere aggregation) [36,37]. The lower the cell seeding density, the lesser the chance to observe sphere aggregation. To minimize the influence of cell aggregations on the evaluation of stem cell properties, we compared the sphere formations of cells at three different low initial seeding densities. We observed that the numbers of spheres formed from CD133+ cells were roughly proportional to the initial cell seeding densities (Figure 5b). At all three seeding densities, CD133+ cells showed enhanced prostasphere forming ability than CD133- cells. These results further confirmed that CD133 could be used to mark a subpopulation with stem-like functions that differ from the majority of prostate cancer cells within this cell line.
Studies of CSCs have found that CSCs share similar biomarker profiles as well as functional properties with normal stem cells from the same organs. This is the main reason that the initial identifications of CSCs used similar biomarker profiles to normal stem cells. In the case of prostate, it has long been observed that a subset of cells, enriched in the basal compartment, play important roles in the organ homeostasis maintenance and tissue regeneration [38]. These putative human adult prostatic stem cells express several basal and stem cell markers including CD133, CD44, integrin α2β1 and CK5/14 [9]. Therefore, the identification of prostate CSC subpopulation was attempted using these markers [8-10]. The most important step to validate the use of markers to define prostate CSCs is to perform a variety of in vitro and in vivo functional assays to confirm the stem cell properties. Once the significantly enhanced clonogenicity in vitro and tumorigenicity in vivo were proved, researchers would use the corresponding cell markers to enrich the CSC population in a certain cancer model.
We are aware that the so called “prostate CSC subpopulation” defined by certain marker expressions is not a pure CSC population, but a cell population with highly enriched CSC properties, as in many CSCs identified in other cancer models of other organs [37]. Moreover, certain markers do not necessarily reveal the origin of CSCs, or do not clearly link to CSC properties [7,37]. It was suggested that CD133, which is located in the apical protrusions of epithelial cell membranes, might be important in regulating cell polarity and migration [39]. However, a clear role of CD133 in the maintenance of self-renewal, differentiation and tumorigenic potential is yet to be defined.
Despite the current limitations in using cell markers to define CSCs, the identification of CSCs based on cell surface marker expressions is still the most common and validated method to date [40]. Cells isolated with putative CSC markers defined and characterized previously do represent a cell population in the expected undifferentiated cell state, with different global gene expression profiles from bulk cancer cell population [40].
3.3. CSC inhibitory effect upon HPMA copolymer-cyclopamine conjugate treatment
3.3.1. HPMA copolymer-cyclopamine conjugate binding to cells
The Hh inhibitor-cyclopamine inhibits the hedgehog pathway by directly binding to and suppressing the SMO membrane receptor [21]. Normally, Hh pathway is regulated by Hh ligands, Sonic hedgehog (SHh) being the most widely expressed including in the prostate. In the absence of ligands, a 12-transmembrane protein receptor Patched (PTCH) silences the pathway by repression of the 7-transmembrane protein SMO. Upon binding of Hh ligands to PTCH, the inhibitory effect of PTCH on SMO is relieved. The activated SMO eventually induces the activation of GLI transcription factors and transcription of multiple genes, including cyclin D1, N-Myc, Bcl-2, Bmi-1 and Snail. Based on the mechanism of cyclopamine action, a binding assay using BODIPY-cyclopamine, a fluorescence labeled derivative of cyclopamine, has been used to evaluate the biological activity of free cyclopamine [41]. The bioactivity of cyclopamine remained in the BODIPY-cyclopamine since conjugation of the fluorescent dye did not interfere with the 3’-hydroxyl bioactive site of cyclopamine [41]. The bioactivity of cyclopamine is revealed by the intensity and distribution of BODIPY fluorescent signals on SMO-expressing cell surfaces. Similar to this assay, we evaluated the binding ability of FITC labeled HPMA copolymer-conjugated cyclopamine to cell surfaces. The incubation time was selected based on BODIPY-cyclopamine binding assay. Shorter time frame was used to minimize the extent of intracellular uptake of FITC labeled control HPMA copolymer. Our data showed positive FITC fluorescent signals on the cell membranes following incubation with cyclopamine conjugate for 2 h, which demonstrated the retained activity of cyclopamine in HPMA copolymer drug conjugate. On the other hand, minimal binding signal on cell surfaces was shown for the cells incubated with FITC labeled HPMA copolymers without cyclopamine (Figure 6). To be noted, it is reasonable not to observe the HPMA copolymer-cyclopamine conjugate binding to the majority or all of the cultured cells, since the Hh elements including SMO transmembrane receptors were heterogeneously expressed within the whole cell line.
Figure 6.
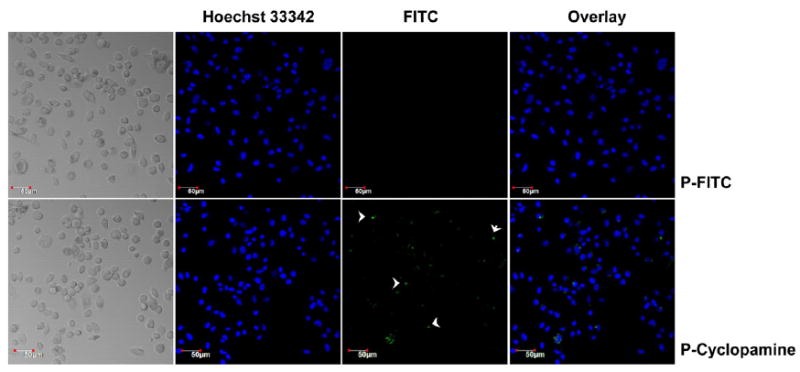
Binding of HPMA copolymer-cylcopamine conjugate to cell surfaces. P-FITC: FITC-labeled HPMA copolymer; P-Cyclopamine: HPMA copolymer-cyclopamine conjugate.
3.3.2. CSC marker expression following HPMA copolymer-cyclopamine conjugate treatment
We examined whether the HPMA copolymer-cyclopamine conjugate possesses inhibitory effect on CD133+ putative prostate CSCs by reducing the amount of prostate CSCs within the whole cancer cell population. Based on Rhim’s group and our characterization of RC-92a/hTERT prostate cancer epithelial cells, CD133 marks the subpopulation enriched with tumorigenic prostate CSCs. So, if prostate CSC population changes following drug or conjugate treatment of the whole cell line, changes of CD133 expression level within the viable cells before and after therapeutic interventions should be observed. As shown in Figure 7, HPMA copolymer-cyclopamine conjugate treatment resulted in decreased fraction of CD133+ cells within the whole surviving cancer cells, in a dose-dependent manner. As revealed in previous studies [22], the decrease of CD133+ cell content should not be due to the induction of cell differentiation. The inhibition of Hh pathway by cyclopamine down-regulates the proliferation and arrest the growth of prostate CSCs by inducing apoptotic cell death [21,22].
Figure 7.
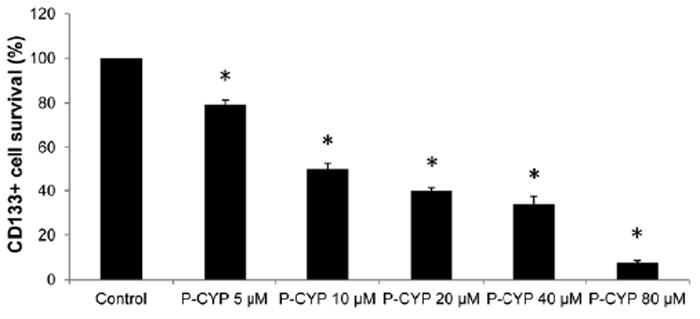
CD133 expression levels following HPMA copolymer-cyclopamine conjugate treatments. Data represent mean±SD of three independent experiments. *, p<0.01 compared to control.
We chose to indirectly evaluate the inhibitory effects of drug/conjugate on CD133+ prostate CSC fractions by treating the whole cell line, instead of directly treating isolated CD133+/- cells. A similar strategy has been used to access the sensitivity of different cell subpopulations to chemotherapeutic drugs [40]. This approach circumvents the potential problem observed in many cancer cell models, that the isolated CSC population growing in monolayer may quickly change their phenotypic and/or functional properties. Indeed, our characterization of sorted CD133+ cells showed gradual loss of CD133 marker expression during in vitro culture. In contrast, CD133 expression level in the whole RC-92a/hTERT cell line remains stable when cultured in vitro; we used the same batch of cells in each set of experiment so that the initial cell differentiation status were the same among different treatment groups. Treated cells were allowed to recover in drug/conjugate free condition for additional two days before analysis, to ensure any possible further cytotoxicity of drug/conjugate did not confound the results during analysis [42].
3.3.3. Comparison of the therapeutic effects of HPMA copolymer-cyclopamine conjugate and docetaxel on prostate CSCs
To evaluate whether the prostate cancer cells respond differently to various therapeutics, we compared the sensitivity of RC-92a/hTERT cells to HPMA copolymer-cyclopamine conjugate and to a traditional first line chemotherapeutic for prostate cancer-docetaxel, with free cyclopamine serving as positive control. Since the potency (or IC50) of cyclopamine and docetaxel are different, we chose the drug or conjugate concentrations, which resulted in similar cell cytotoxicity levels to help normalize the comparison of CD133 level changes [42]. In addition, the concentration range chosen for free cyclopamine or cyclopamine equivalent in the conjugate was similar to the effective doses used previously in other prostate cancer models [20,22]. The results showed a more evidently decreased percentage of viable CD133+ prostate CSC-enriched population within the whole cancer cell population in cyclopamine and cyclopamine conjugate treated groups than in the docetaxel treated group (Figure 8). We observed 20% and 50% decreases of CD133 proportion within the residual cancer cells at the cyclopamine equivalent concentrations of 5 μM and 10 μM in cyclopamine conjugate treatment group, while the whole cell viability still remained to be very high, around 98% and 91% as normalized to untreated group. Similar decreases in the fraction of CD133+ cells were observed in free cyclopamine treated cells in contrast to slight cytotoxicity for bulk cancer cells. On the contrary, 0.5 nM, 1 nM and 2 nM docetaxel treatment caused around 10%, 20% and 45% RC-92a/hTERT cell deaths, however, no evident decrease of the fraction of CD133+ cells in the residual cancer cells was observed (Fig. 8). These data suggested that prostate CSCs, as characterized in this RC-92a/hTERT prostate cancer in vitro model, are more sensitive to cyclopamine and HPMA copolymer-cyclopamine conjugate than to traditional first line chemotherapeutics docetaxel. The growth of prostate CSCs was suppressed while bulk cancer cells appeared to survive following cyclopamine or cyclopamine conjugate treatment at their effective doses. Following docetaxel treatment, bulk prostate cancer cells could be eradicated, but prostate CSCs survived. HPMA copolymer-cyclopamine conjugate has the potential to shrink the CSC population, which contributes to refractory and metastatic prostate cancers, thus, improve the outcomes of prostate cancer treatment. Moreover, the selective cytotoxic effects of conjugated cyclopamine and docetaxel on different subpopulations of prostate cancer cells suggest a potential combination therapeutic strategy to eliminate both bulk and cancer stem cells. The recent finding that cyclopamine can down-regulate multidrug resistance transporters in androgen-independent prostate cancer also opens the possibility to sensitize multidrug resistant cancer cells and enhance traditional anticancer therapeutic effects [43].
Figure 8.
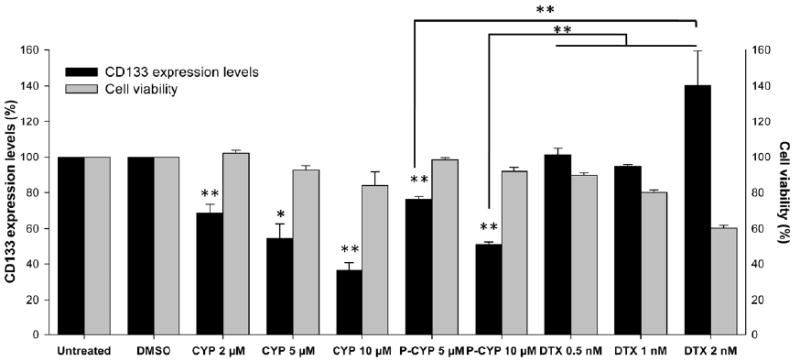
Summary of CD133 changes and cell viability. Black columns: CD133 expression level (%); gray columns: cell viability (%). The data are presented as mean±SD of the experiments done in triplicate. *, p<0.05; **, p<0.01. Free cyclopamine and docetaxel treatments were compared to DMSO treatment; HPMA copolymer-cyclopamine conjugate was compared to untreated group.
To develop effective anti-cancer treatments able to overcome recurrence and metastasis, understanding the question-which cancer cells contribute to cancer progression is critical. An explanation of CSC theory is that tumor heterogeneity, similar to the hierarchical organization of normal tissues, is maintained by CSCs, which are intrinsically different from bulk cancer cells [4]. It is supported by several lines of evidence that a subset of cells within hematopoietic malignancies and solid tumors showed self-renewal, differentiation and tumorigenic capacity, whereas the other cancer cells were depleted of this property [7]. However, limitations of current assays (including in vivo transplantation) evaluating CSC properties cast doubt on the strict definition of CSCs [3,7]. On the other hand, stochastic model states that cancer cells are randomly chosen to gain the tumorigenic capacity, presenting functional heterogeneity within the whole tumor [3,4]. Despite of the hot debate around the two proposed tumor models, it is for sure that subsets of functional CSCs were observed in numerous studies. The most important implication from CSC hypothesis is the presence of cancer cells, which are not only tumorigenic but also inherently resistant to various anticancer therapeutics [3,4,7,40,42]. It is beneficial to search for therapeutics targeting CSCs, which are not the object of current anticancer treatments. Indeed, clinical practice has shown relative expansions of CSCs in the residual tumor tissue after standard chemotherapy than prior to the treatment [44]. Our results suggested that putative prostate CSCs were not sensitive to docetaxel treatment; while cyclopamine and its polymer conjugate administration did shrink the prostate CSC compartment. In future work, we would like to evaluate the long-term effects of prostate CSC inhibition by cyclopamine on prostate cancer regression. As suggested by some studies, CSCs may be generated de novo from non-CSCs [40]; thus it is probable that targeting CSCs alone within a tumor will not lead to complete cancer regression in long-term. We propose that it would be worthy to study the effect of cyclopamine conjugate in combination with another traditional chemotherapeutics, such as docetaxel.
4. Conclusions
The stem cell property of an enriched subset of CD133+ cells within a prostate cancer epithelial cell line RC-92a/hTERT was validated by confirming the correlation between the marker expression and stem cell functions, using FACS and prostasphere forming assay. HPMA copolymer-cyclopamine conjugate, designed to eliminate putative prostate CSCs, was synthesized and characterized. The HPMA copolymer-cyclopamine conjugate, like free cyclopamine, showed selective inhibitory effect on prostate CSCs than on bulk cancer cells in the in vitro prostate cancer model. In contrast, docetaxel, a traditional chemotherapeutic agent for prostate cancer, showed preferential cytotoxicity to bulk cancer cells. These results suggest the treatment potential of a combination macromolecular therapeutics targeting both bulk tumor cells and CSCs.
Acknowledgments
The research was supported in part by NIH grant RO1 CA132831 from the National Cancer Institute. We thank Dr. J.S. Rhim for the gift of RC92a/hTERT cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64(24):9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 2.Pienta KJ, Smith DC. Advances in prostate cancer chemotherapy: a new era begins. CA Cancer J Clin. 2005;55(5):300–318. doi: 10.3322/canjclin.55.5.300. [DOI] [PubMed] [Google Scholar]
- 3.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138(5):822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 4.Dick JE. Looking ahead in cancer stem cell research. Nat Biotechnol. 2009;27(1):44–46. doi: 10.1038/nbt0109-44. [DOI] [PubMed] [Google Scholar]
- 5.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 6.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;234(26):1670–1677. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 8.Kelly K, Yin J. Prostate cancer and metastasis initiating stem cells. Cell Res. 2008;18(5):528–537. doi: 10.1038/cr.2008.50. [DOI] [PubMed] [Google Scholar]
- 9.Collins AT, Berry PA, Hyde C. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65(23):10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 10.Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT. CD133, a novel marker for human prostatic epithelial stem cells. J Cell Sci. 2004;117(16):3539–3545. doi: 10.1242/jcs.01222. [DOI] [PubMed] [Google Scholar]
- 11.Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem. 2008;283(25):17635–17651. doi: 10.1074/jbc.M800109200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding XW, Wu JH, Jiang CP. ABCG2: a potential marker of stem cells and novel target in stem cell and cancer therapy. Life Sci. 2010;86(17-18):631–637. doi: 10.1016/j.lfs.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 13.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25(12):1696–1708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 14.Dean M, Fojo T, Bates S. Tumor stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 15.Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. A distinct ‘side population’ of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101(39):14228–14233. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 17.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, et al. Hedgehog signaling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci USA. 2004;101(34):12561–12566. doi: 10.1073/pnas.0404956101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. 2007;104(10):4048–4053. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25(10):2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altaba AR, Sánchez P, Dahmane N. Hedgehog signaling in cancer formation and maintenance. Nat Rev Cancer. 2003;3(12):903–911. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- 22.Mimeault M, Johansson SL, Henichart JP, Depreux P, Batra SK. Cytotoxic effects induced by Docetaxel, Gefitinib, and Cyclopamine on side population and nonside population cell fractions from human invasive prostate cancer cells. Mol Cancer Ther. 2010;9(3):617–630. doi: 10.1158/1535-7163.MCT-09-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scales SJ, De Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30(6):303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Říhová B, Strohalm J, Hoste K, Jelínková M, Hovorka O, Kovář M, et al. Immunoprotective therapy with targeted anticancer drugs. Macromolecular Symposia. 2001;172(1):21–28. [Google Scholar]
- 25.Nishiyama N, Nori A, Malugin A, Kasuya Y, Kopečková P, Kopeček J. Free and N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer-bound geldanamycin derivative induce different stress responses in A2780 human ovarian carcinoma cells. Cancer Res. 2003;63(22):7876–7882. [PubMed] [Google Scholar]
- 26.Miki J, Furusato B, Li H, Gu Y, Takahashi H, Egawa S, et al. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res. 2007;67(7):3153–3161. doi: 10.1158/0008-5472.CAN-06-4429. [DOI] [PubMed] [Google Scholar]
- 27.Gu Y, Li H, Miki J, Kim KH, Furusato B, Sesterhenn IA, et al. Phenotypic characterization of telomerase-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines. Exp Cell Res. 2006;312(6):831–843. doi: 10.1016/j.yexcr.2005.11.029. [DOI] [PubMed] [Google Scholar]
- 28.Mitsukami Y, Donovan MS, Lowe AB, McCormick CL. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules. 2001;34(7):2248–2256. [Google Scholar]
- 29.Kopeček J, Bažilová H. Poly[N-(2-hydroxypropyl)methacrylamide]. 1. Radical polymerization and copolymerization. Eur Polym J. 1973;9(1):7–14. [Google Scholar]
- 30.Šubr V, Ulbrich K. Synthesis and properties of new N-(2-hydroxypropyl)methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React Funct Polym. 2006;66(12):1525–1538. [Google Scholar]
- 31.Yang J, Luo K, Pan H, Kopečková P, Kopeček J. Synthesis of biodegradable multiblock copolymers by click coupling of RAFT-generated heterotelechelic polyHPMA conjugates. React Funct Polym. 2011;71(3):294–302. doi: 10.1016/j.reactfunctpolym.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dvořák M, Kopečková P, Kopeček J. High-molecular weight HPMA copolymer-adriamycin conjugates. J Controlled Release. 1999;60(2-3):321–332. doi: 10.1016/s0168-3659(99)00087-5. [DOI] [PubMed] [Google Scholar]
- 33.Wang S. Anchorage-independent growth of prostate cancer stem cells. Methods Mol Biol. 2009;568:151–160. doi: 10.1007/978-1-59745-280-9_9. [DOI] [PubMed] [Google Scholar]
- 34.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27(2):157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 35.Ulbrich K, Zacharieva EI, Obereigner B, Kopeček J. Polymers containing enzymatically degradable bonds. 5. Hydrophilic polymers degradable by papain. Biomaterials. 1980;1(4):199–204. doi: 10.1016/0142-9612(80)90017-4. [DOI] [PubMed] [Google Scholar]
- 36.Singec I, Knoth R, Meyer RP, Maciaczyk J, Volk B, Nikkhah G, et al. Defining the actual sensitivity and specificity of the neurosphere assay in stem cell biology. Nat Methods. 2006;10(3):801–806. doi: 10.1038/nmeth926. [DOI] [PubMed] [Google Scholar]
- 37.Wan F, Zhang S, Xie R, Gao B, Campos B, Herold-Mende C. The utility and limitations of neurosphere assay, CD133 immunophenotyping and side population assay in glioma stem cell research. Brain Pathol. 2010;20(5):877–889. doi: 10.1111/j.1750-3639.2010.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leong KG, Wang BE, Johnson L, Gao WQ. Generation of a prostate from a single adult stem cell. Nature. 2008;456(7223):804–808. doi: 10.1038/nature07427. [DOI] [PubMed] [Google Scholar]
- 39.Kosodo Y, Roper K, Haubensak W, Marzesco AM, Corbeil D, Huttner WB. Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 2004;23(11):2314–2324. doi: 10.1038/sj.emboj.7600223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146(4):633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 41.Sinha S, Chen JK. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat Chem Biol. 2006;2(1):29–30. doi: 10.1038/nchembio753. [DOI] [PubMed] [Google Scholar]
- 42.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–659. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic hegdehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007;26(38):5674–5679. doi: 10.1038/sj.onc.1210356. [DOI] [PubMed] [Google Scholar]
- 44.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. Let-7 regulates self-renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]