

Abstract
While L-3,4-dihydroxyphenylalanine (L-DOPA) remains the standard treatment for Parkinson’s disease (PD), long-term efficacy is often compromised by L-DOPA-induced dyskinesia (LID). Recent research suggests that targeting the noradrenergic (NE) system may provide relief from both PD and LID, however, most PD patients exhibit NE loss which may modify response to such strategies. Therefore this investigation aimed to characterize the development and expression of LID and the anti-dyskinetic potential of the α2- and β-adrenergic receptor antagonists idazoxan and propranolol, respectively, in rats receiving 6-OHDA lesions with (DA lesion) or without desipramaine protection (DA + NE lesion). Male Sprague–Dawley rats (N = 110) received unilateral 6-hydroxydopamine lesions. Fifty-three rats received desipramine to protect NE neurons (DA lesion) and 57 received no desipramine reducing striatal and hippocampal NE content 64% and 86% respectively. In experiment 1, the development and expression of L-DOPA-induced abnormal involuntary movements (AIMs) and rotations were examined. L-DOPA efficacy using the forepaw adjusting steps (FAS) test was also assessed in DA- and DA + NE-lesioned rats. In experiment 2, DA- and DA + NE-lesioned rats received pre-treatments of idazoxan or propranolol followed by L-DOPA after which the effects of these adrenergic compounds were observed. Results demonstrated that moderate NE loss reduced the development and expression of AIMs and rotations but not L-DOPA efficacy while anti-dyskinetic efficacy of α2- and β-adrenergic receptor blockade was maintained. These findings suggest that the NE system modulates LID and support the continued investigation of adrenergic compounds for the improved treatment of PD.
Keywords: Parkinson’s disease, L-DOPA, Dyskinesia, Norepinephrine, Alpha-adrenergic, Beta-adrenergic
1. Introduction
Parkinson’s disease (PD) is widely recognized by the preferential loss of dopamine (DA) neurons located within the substantia nigra pars compacta (SNc) leading to motor deficits typified by rigidity, resting tremor, and akinesia (Dauer and Przedborski, 2003). DA replacement therapy with L-3,4-dihydroxyphenylalanine (L-DOPA) alleviates many of these motor impairments and has become the gold standard treatment for PD. Unfortunately, chronic pharmacotherapy with L-DOPA often results in side effects such as motor fluctuations and L-DOPA-induced dyskinesia (LID; Obeso et al., 2000).
In addition to SNc DA cell loss, norepinephrine (NE) neurons of the locus coeruleus (LC) degenerate, even preceding the death of DA neurons (Fornai et al., 2007; Hornykiewicz and Kish, 1987; McMillan et al., 2011; Zarow et al., 2003). Post-mortem estimates for NE loss in PD brains range from 60 to 90% (Zarow et al., 2003) and a number of preclinical studies suggest that NE cell death increases the vulnerability of DA neurons (Rommelfanger and Weinshenker, 2007; Srinivasan and Schmidt, 2003). Within the past few decades, there have been attempts to reduce motor symptoms of PD by increasing NE activity. For instance, Narabayashi and colleagues decreased akinesia with DL-threo-3,4-dihydroxyphenylserine (L-DOPS; Droxidopa), a precursor to NE that increases its activity (Narabayashi et al., 1984, 1991). Still others have used the α2-adrenergic receptor antagonists such as idazoxan (IDZ), atipamezole and fipamezole to modulate NE function and improve PD treatment (Domino et al., 2003; Haapalinna et al., 2003; Johnston et al., 2010; Rascol et al., 2001; Yavich et al., 2003).
NE compounds have also been shown to modulate the expression of LID. For example, the non-selective β-adrenergic receptor antagonist (±)propranolol (PRO) significantly attenuated established LID in PD patients (Carpentier et al., 1996), an effect supported in both rodents (Buck and Ferger, 2010; Dekundy et al., 2007; Lindenbach et al., 2011) and primates (Gomez-Mancilla and Bedard, 1993). Anti-dyskinetic properties have also been shown with α1- and α2-adrenergic receptor antagonists (Buck and Ferger, 2010; Buck et al., 2010; Fox et al., 2001; Grondin et al., 2000; Rascol et al., 2001; Savola et al., 2003) and fipamezole, an α2-adrenergic receptor antagonist, has demonstrated therapeutic efficacy in persons suffering from LID and is currently in the process of being brought to market. Other drugs acting on NE transmission such as duloxetine, atomoxetine and droxidopa, are in various stages of development for the treatment of symptoms of PD (clinicaltrials.gov).
Still, there is much to be learned regarding the NE system and its role in LID. Indeed, studies investigating LID rarely consider NE cell loss as a modulatory factor in LID development, expression, or treatment or are contradictory. For example, in the abnormal involuntary movements (AIMs) rat model of LID, the NE reuptake inhibitor desipramine is often given to protect NE neurons during 6-hydroxydopamine (6-OHDA) lesion surgery (Barnum et al., 2008; Putterman et al., 2007). Other studies suggest that NE lesions either exacerbate L-DOPA and DA agonist-induced dyskinesia (Fulceri et al., 2007; Wang et al., 2010) or have no effect (Marin et al., 2008; Perez et al., 2009). Currently no studies have systematically addressed the effects of NE loss on adrenergic anti-dyskinetic efficacy. Therefore, we characterized the development and expression of LID and the anti-dyskinetic potential of the α2- and β-adrenergic receptor antagonists IDZ and PRO, respectively, in rats receiving 6-OHDA lesions with (DA lesion) or without desipramaine protection (DA + NE lesion).
2. Materials and methods
2.1. Animals
Adult male Sprague–Dawley rats were used (225–250 g upon arrival; Taconic Farms, NY, USA). Animals were housed in plastic cages (22 cm high, 45 cm deep and 23 cm wide) and had free access to standard lab chow (Rodent Diet 5001; Lab Diet, Brentwood, MO, USA) and water. The colony room was maintained on a 12/12 h light/-dark cycle (lights on at 0700 h) at a temperature of 22–23 °C. Animals were maintained in accordance with the guidelines of the Institutional Animal Care and Use Committee of Binghamton University and the “Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Academic Press; NIH publication number 85–23, revised 1996).
2.2. 6-Hydroxydopamine lesion surgeries
One week after arrival, most rats (N = 110) received unilateral 6-hydroxydopamine (6-OHDA; Sigma, St. Louis, MO, USA) lesions of the left medial forebrain bundle to destroy DA neurons or DA + NE neurons (Barnum et al., 2008; Bishop et al., 2009; Lindenbach et al., 2011). Fifty-three rats received DA lesions only. In these rats, desipramine HCl (25 mg/kg, ip; Sigma) was given 30 min prior to 6-OHDA injection to protect the loss of NE neurons. The remaining subjects (n = 57) did not receive desipramine prior to 6-OHDA surgery in order to destroy both DA and NE neurons. To do this, rats were anesthetized with inhalant isoflurane (2–3%; Sigma) in oxygen (2.5 L/min), and placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). The coordinates for 6-OHDA injections were AP: −1.8 mm, ML: +2.0 mm, DV: −8.6 mm relative to bregma with the incisor bar positioned 3.3 mm below the interaural line (Paxinos and Watson, 1998). Using a 10 μl Hamilton syringe attached to a 26 gage needle, 6-OHDA (12 μg) dissolved in 0.9% NaCl + 0.1% ascorbic acid was infused through a small burr hole in the skull at a rate of 2 μl/min for a total volume of 4 μl. The needle was withdrawn 5 min later. Following surgery, all rats were placed in clean cages on a warming pad for recovery, after which they were returned to group-housing (2 rats/cage). Soft chow was provided as needed to facilitate recovery during the first week after surgery. To minimize pain, rats were injected with buprenorphine (0.03 mg/kg) prior to surgery, 6 h after surgery, and the following morning. All rats were allowed to recover for 3 weeks before testing commenced.
2.3. Pharmacological treatments and design
2.3.1. L-DOPA priming
Beginning 3 weeks after 6-OHDA lesion surgery, 6-OHDA-lesioned rats were administered L-DOPA methyl ester (L-DOPA; 12 mg/kg, s.c.; Sigma) + DL-serine 2-(2,3,4-trihydroxybenzyl) hydrazide hydrochloride (benserazide; 15 mg/kg, s.c.; Sigma) once daily for 7 days to induce stable and reliable LID (Barnum et al., 2008; Bishop et al., 2009). L-DOPA and benserazide were dissolved in Vehicle (0.9% NaCl containing 0.1% ascorbic acid) and administered at a volume of 1 mL/kg. All lesioned rats displaying cumulative axial, limb, and orolingual AIMs (ALO AIMs) score of >30 on the 7th day of priming were used in the current study (106 of 110). Experiments outlined below continued on day 8 and testing occurred every 3rd or 4th day until studies were completed.
2.4. Experiment 1
2.4.1. Effects of NE loss on dyskinesia development and expression LID development in DA and DA + NE rats
The goal of the first experiment was to determine whether NE lesions modified the development of ALO AIMs and rotations during the 7 day priming period. To do this, DA- (n = 7) and DA + NE- (n=12) lesioned rats were injected (s.c.) with 12 mg/kg L-DOPA + 15 mg/kg Benserazide each day for 7 days and ALO AIMs and rotations were scored (every 10 min for 3 h) on days 1, 4, and 7.
2.4.2. L-DOPA dose–response in L-DOPA-primed DA and DA + NE rats
To determine whether L-DOPA-induced ALO AIMs and rotations varied between DA- and DA + NE-lesioned rats as a function of L-DOPA dose, L-DOPA primed DA (n=13) and DA + NE (n=9) rats were injected with Vehicle, 2, 3, 4, 6, and 12 mg/kg L-DOPA + 15 mg/kg benserazide using a counterbalanced within subjects design and scored every 20 min for 2 h for ALO AIMs and rotations.
2.4.3. L-DOPA efficacy in L-DOPA-primed DA and DA + NE rats
To determine whether the antiparkinsonian efficacy of L-DOPA was compromised by NE loss, L-DOPA-primed DA- (n=10) and DA + NE- (n=9) lesioned rats were injected with Vehicle or 4 mg/kg L-DOPA + 15 mg/kg benserazide using a counterbalanced within subjects design. L-DOPA efficacy was examined 60 min post-injection by using the forepaw adjusting steps test (Bishop et al., 2009; Chang et al., 1999).
2.5. Experiment 2
2.5.1. Effects of NE loss on the anti-dyskinetic efficacy of β- and α-adrenergic antagonists PRO and IDZ
To examine the effects of NE integrity on purported anti-dyskinetic adrenergic compounds, the β-adrenergic receptor antagonist PRO and the α2-adrenergic antagonist IDZ were employed at doses employed in previous rodent work (Buck and Ferger, 2010; Lindenbach et al., 2011). A 2 (dose of L-DOPA) × 3 (dose of PRO/IDZ) design was utilized for both DA and DA + NE-lesioned rats. The expression of ALO AIMs and rotations were examined in both DA (PRO, n=10; IDZ, n=5) and DA + NE (PRO, n=9; IDZ, n=6) rats pretreated with PRO (0, 5 and 20 mg/kg; i.p.; Sigma; dissolved in 10% DMSO and 90% sterile saline) 10 min prior to 4 and 12 mg/kg L-DOPA injection or IDZ (0, 2.5, and 10 mg/kg; i.p.; Sigma; dissolved in sterile saline) 5 min prior to 4 and 12 mg/kg L-DOPA injection. Following L-DOPA treatment, rats were scored for ALO AIMs and rotations every 10 min for 3 h.
2.6. Behavioral analyses
2.6.1. AIMs and rotations
Rats were monitored for AIMs using a procedure described previously (Barnum et al., 2008) and similar to that initially depicted by Lundblad et al. (2002) with slight modifications. On test days (0900–1400 h), rats were individually placed in plexiglass cylinders (22.2 cm diameter, 25.4 cm height; Thermo Fisher Scientific, Rochester, NY, USA) 5 min prior to pretreatments. Following L-DOPA injection, a trained observer blind to treatment condition assessed each rat for exhibition of axial, limb, and orolingual AIMs. Each new rater was trained for a minimum of 3 sessions and then correlated with a well-trained instructor. A correlation of ≥90% with the instructor is required before new raters can score AIMs. In addition, contralateral rotations, defined as complete 360 degree turns away from the lesioned side of the brain, were tallied. Dystonic posturing of the neck and torso, involving positioning of the neck and torso in a twisted manner directed toward the side of the body contralateral to the lesion, were referred to as “axial” AIMs. “Forelimb” AIMs were defined as rapid, purposeless movements of the forelimb located on the side of the body contralateral to the lesion. “Orolingual” AIMs were composed of repetitive openings and closings of the jaw and tongue protrusions. The movements are considered abnormal since they occur at times when the rats are not chewing or gnawing on food or other objects. For the L-DOPA dose response experiment, ALO AIMs and rotations were recorded for 1 min, every 20th min for 2 h. For all other experiments, AIMs and rotations were recorded for 1 min, every 10th min for 3 h. During the AIMs observation period a severity score of 0–4 was assigned for each AIMs category: 0 = not present, 1 = present for less than 50% of the observation period (i.e., 1–29 s), 2 = present for more than 50% or more of the observation period (i.e., 30–59 s), 3 = present for the entire observation period (i.e., 60 s) and interrupted by a loud stimulus (a tap on the cylinder), or 4 = present for the entire observation period but not interrupted by a loud stimulus. For each AIMs category, the scores were summed for the entire 2 or 3 h period. Thus, the theoretical maximum score for each type of AIM was 24 (4 × 6 period) for the 2 h test and 72 (4 × 18 period) for the 3 h test although observed scores were never this severe.
2.6.2. Forepaw adjusting steps
The FAS test is a measure of forelimb akinesia and rats with >80% unilateral DA depletion performed poorly on the test with the lesioned side of the body (Chang et al., 1999). L-DOPA reduces this deficit so the test can be used to determine if an L-DOPA adjunct is interfering with the relief of PD symptoms provided by L-DOPA (Eskow et al., 2007). To do so, an experimenter held the rat’s hindlimbs and one forelimb such that the free forelimb was forced to bear the rat’s body weight. Rats were then moved laterally for 90 cm over 10 s across a marked surface while another experimenter counted the number of steps taken in the forehand direction (defined as movement toward the rat’s midline) and backhand direction (movement distal to the rat’s midline). Each FAS test consisted of 3 backhand and 3 forehand trials with each limb, for a total of 12 trials per rat. The score for percent intact stepping was derived by summing the total steps with the lesioned forepaw, dividing by the number of steps with the unlesioned forepaw, and multiplying this number by 100. Lower percent intact scores indicate greater forelimb akinesia. Prior to FAS testing with the compounds of interest, all rats received 3 separate acclimation periods to reduce potential practice effects.
2.7. High-performance liquid chromatography
To determine monoamine and metabolite levels following DA (n = 8) and DA + NE (n = 12) lesions, MFB-lesioned were killed by rapid decapitation at least 48 h after their last L-DOPA treatment. In order to confirm the protective effects of desipramine against 6-OHDA-induced NE loss, a group of age and gender matched untreated, naïve rats (n = 9) was also killed by rapid decapitation. The striata and/or hippocampi were dissected, immediately frozen on dry ice, and stored at −80 °C. These samples were then homogenized in a solution of ice-cold perchloric acid (0.1 M), 1% (v:v) ethanol, and 0.02% (v:v) EDTA. The homogenates were spun for 30 min at 14,400 g with the temperature maintained at 4 °C. Aliquots of supernatant were then analyzed for abundance of NE, DA, 3,4-dihydroxyphenylacetic acid (DOPAC), 5-HT, and 5-hydroxyindole-3-acetic acid (5-HIAA) using reverse-phase high performance liquid chromatography coupled to electrochemical detection according to the protocol of Kilpatrick et al. (1986). The system employed consisted of an ESA autoinjector (Model 542), an ESA solvent delivery system (Model 582), an ESA Guard-Pak column, a C-18 (100 × 4.6 mm, 5 μm packing) column (ESA), a Coulochem III electrochemical detector (ESA) connected to an analytical cell (ESA model 5011A) located immediately after the column, an ESA Model 5020 guard cell positioned prior to the autoinjector, and an external pulse dampener (ESA). Samples were chromatographically separated using a mobile phase composed of 90 mM sodium dihydrogen phosphate (monobasic, anhydrous), 0.05 mM EDTA, 1.7 mM octane sulfonic acid, 10% (v:v) acetonitrile, and adjusted to pH 3.0 with o-phosphoric acid. The first electrode of the analytical cell was set to a potential of −100 mV and the second electrode to +250 mV. The guard cell had a potential of 350 mV. As monoamines and metabolites eluted from the column and passed through the analytical cell, they were oxidized at the second electrode and a spike in current was generated. This signal was recorded as a trace of electrode current versus time by EZChrom Elite software via a Scientific Software, Inc. (SS420χ) module. Each compound was detected as a peak in this trace, and the EZChrom Elite software was used to calculate peak areas. Peak areas were converted to picograms (pg) of compound using a standard curve made from samples of known monoamine and metabolite concentrations (1e–6 M to 1e–9 M), normalized to striatal and hippocampal tissue weights, and expressed as picograms (pg) of compound per milligrams (mg) of tissue (mean ± standard error of the mean; S.E.M.).
2.8. Statistical analyses
All parametric data (HPLC, rotations and FAS) are expressed as mean ± standard error of the mean (S.E.M.). Non-parametric ALO AIMs data are expressed as medians ± median absolute deviation (M.A.D.). HPLC-derived striatal monoamine and metabolite levels were analyzed by a 2-factor (Side × Lesion) analyses of variance (ANOVA) or independent t-tests. In experiment 1, ALO AIMs were analyzed by nonparametric Mann–Whitney tests while rotations and FAS data were analyzed by parametric 2-factor ANOVAs. In experiment 2, ALO AIMs were analyzed with nonparametric Friedman ANOVAs at each time point or over the 3 h period. Where appropriate, nonparametric Wilcoxon post hoc tests or parametric LSD post hoc comparisons were employed. All statistical analyses were performed with the use of Statistica software version 9 (Statsoft Inc., Tulsa, OK). Alpha was set at 0.05.
3. Results
3.1. Monoamine and metabolite levels
At least 48 h following the final L-DOPA treatment, rat striata and hippocampi were examined for NE, DA, DOPAC, 5-HT and 5-HIAA content via HPLC. A 2 (Side; Intact vs. Lesion) × 2 (Lesion; DA vs. DA + NE) ANOVA was used to analyze potential lesion-induced differences (Table 1). In striatum, main effects of Side (Intact vs. Lesion) were observed for DA, DOPAC (F1,31 = 652.2, p<0.0001, F1,31 = 43.9, p<0.0001) and 5-HIAA (F1,31 = 21.1, p<0.0001) with post hocs indicating toxin-induced depletion ipsilateral to the 6-OHDA lesion (both p<0.05). There were also main effects of Lesion (DA vs. DA + NE) on striatal DA, DOPAC and NE (F1,31 = 10.29, p<0.003, F1,31 = 7.35, p<0.011, F1,31 = 4.36, p<0.045, respectively). Post-hoc tests revealed more overall DA and DOPAC loss in DA-lesioned rats while conversely, DA + NE lesions produced a 64% reduction in striatal NE content (all p<0.05). Finally, a significant Side × Lesion interaction was found upon analysis of DA (F1,31 = 7.40, p<0.01) which indicated DA depletion was more pronounced on the side ipsilateral to DA + NE lesion (p<0.05). In the hippocampus, a region that receives direct innervation from the LC via the MFB (Arvin et al., 1992), main effects of Side (Intact vs. Lesion) were observed for DOPAC only (F1,31 = 6.94, p<0.02) and post hoc analysis revealed a significant reduction in DOPAC ipsilateral to the lesion (p<0.05). Importantly, a main effect of lesion (DA vs. DA + NE) was also observed for hippocampal NE (F1,31 = 12.08, p<0.002). Post hoc analysis revealed that hippocampal NE was reduced bilaterally (86%) in DA + NE lesioned rats compared to DA lesioned rats alone (p<0.05). Collectively, bilateral forebrain NE loss suggested that ascending LC efferents affected by unilateral MFB 6-OHDA decussate. As a final control measure to determine whether desipramine protected against 6-OHDA-induced NE loss, monoamine levels from naïve control rats were compared to levels in the contralateral striata of DA-lesioned rats. T-tests revealed no differences in NE, DOPAC or 5-HT (ns) however, striatal DA content in the contralateral striata of DA-lesioned rats was ~30% lower, perhaps reflecting lesion procedure-induced DA compromise (Table 1; both p<0.05).
Table 1.
Concentrations striatal (A) and hippocampal (B) Norepinephrine (NE), Dopamine (DA), 3,4-Dihydroxyphenylacetic acid (DOPAC), Serotonin (5-HT) and 5-Hydroxyindoleacetic acid (5-HIAA) in naïve control rats or those receiving unilateral medial forebrain bundle (MFB) 6-hydroxydopamine (n = 6–12/group) treated with Vehicle or the NE reuptake inhibitor desipramine (25 mg/kg, ip) 30 min prior to lesion. Values (as means ± S.E.M.) are expressed as picograms monoamine or metabolite per milligram wet weight of tissue. Differences between Side (Contralateral vs. Ipsilateral to 6-OHDA) and Lesion (Desipramine + 6-OHDA vs. 6-OHDA) were determined by 2-way ANOVA and planned comparison post-hocs. Differences between Desipramine-treated striatum contralateral to 6-OHDA vs. naïve control striata were examined with independent t-tests.
| A. ASTRIATUM
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Contralateral to 6-OHDA | NE | DA | DOPAC | 5-HT | 5-HIAA | |||||
| Desipramine-treated | 33±7 | 10185±639 | 2663±463 | 590±141 | 338±45 | |||||
| Vehicle-treated | 13±4* | 12605±551 | 4879±914 | 890±177 | 430±53 | |||||
| Ipsilateral to 6-OHDA | ||||||||||
| Desipramine-treated | 30±10 | 18±10+ | 31±11+ | 763 ±217 | 204±11 | |||||
| Vehicle-treated | 10±3* | 14±10+ | 138±47+ | 852±231 | 174±28+ | |||||
| Naïve Control | 36±3 | 14715±632# | 3160±307 | 508±59 | 557±55# | |||||
| B. HIPPOCAMPUS
|
||||||||||
| Contralateral to 6-OHDA | NE | DA | DOPAC | 5-HT | 5-HIAA | |||||
|
| ||||||||||
| Desipramine-treated | 733±307 | N/D | 182±67 | 812±91 | 300±85 | |||||
| Vehicle-treated | 88±23* | N/D | 120±36 | 860±319 | 163±68 | |||||
| Ipsilateral to 6-OHDA | ||||||||||
| Desipramine-treated | 703±212 | N/D | 50±19+ | 583±43 | 311±90 | |||||
| Vehicle-treated | 125±35* | N/D | 56±11+ | 330±184 | 248±92 | |||||
p<0.05 vs. Desipramine-treated.
p<0.05 vs. Contralateral side.
p<0.05 vs. Desipramine-treated Contralateral side.
3.2. Experiment 1
3.2.1. NE lesions delay the development of ALO AIMS and rotations
DA- (n = 7) and DA + NE- (n = 12) lesioned rats were primed for 7 days with 12 mg/kg L-DOPA + 15 mg/kg Benserazide and ALO AIMs and rotations were recorded on days 1, 4, and 7. As demonstrated in Fig. 1A, DA + NE-lesioned rats were less dyskinetic than DA-lesioned rats on day 1 of L-DOPA-priming (U = 67.5, p<0.05). However, this difference in ALO AIMs was gone on days 4 and 7 (U = 130 and 142.5, ns) and all rats met the inclusion criterion of ALO AIMs >30 by day 7 and were therefore subject to further study. Analyses of rotations indicated main effects of Lesion (F1,35 = 32.14, p<0.001) and Day (F2,70 = 16.64, p<0.001) as well as a significant Lesion × Day interaction (F2,70 = 10.1, p<0.001; Fig. 1B) with post hoc analyses revealing that DA + NE-lesioned rats rotated significantly less than DA-lesioned rats on each successive day of L-DOPA priming (p<0.05).
Fig. 1.
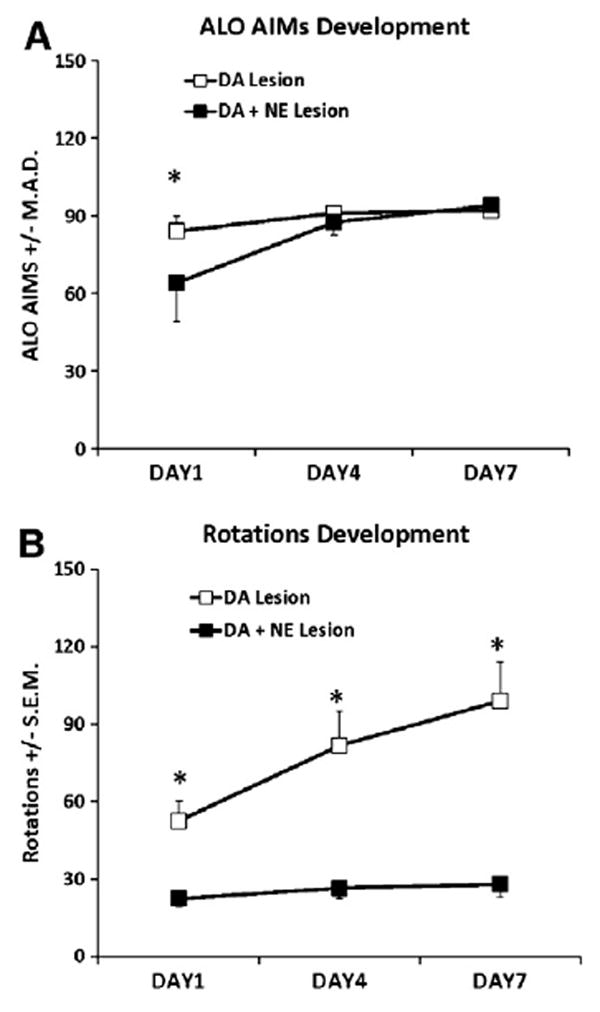
Development of ALO AIMs and rotations during L-DOPA priming in DA (n = 7) and DA + NE (n = 12) lesioned rats. Rats were primed with 12 mg/kg L-DOPA + 15 mg/kg benserazide for 7 consecutive days and ALO AIMs and rotations were scored for 1 min every 10 min for 3 h on days 1, 4, and 7. Differences in ALO AIMs between DA and DA + NE rats were analyzed on each day using nonparametric Mann Whitney U-test whereas a 2 (Lesion) × 3 (Day) ANOVA was used to determine potential differences in rotations. DA + NE-lesioned rats showed fewer ALO AIMs on day 1 (A) and less rotations on days 1, 4, and 7 (B). *P<0.05 vs. DA + NE Lesion on same day of testing. Data presented as median ALO AIMs ± M.A.D or Rotations ± S.E.M.
3.2.2. NE lesions blunt the expression of L-DOPA-induced ALO AIMs and rotations
L-DOPA-primed DA- (n = 13) and DA + NE- (n = 9) lesioned rats were tested for ALO AIMs and rotations following 2, 3, 4, 6 or 12 mg/kg of L-DOPA in a counterbalanced within subjects design. Mann Whitney tests revealed reduced ALO AIMs at 2, 3, 4 and 6 mg/kg L-DOPA in DA + NE-lesioned rats (U<77, p<0.05 for all; Fig. 2A). L-DOPA-induced rotations as shown in Fig. 2B were also significantly modified by Lesion (F1,31 = 31.61, p<0.001) and Dose (F4,124 = 48.97, p<0.001). In addition, a significant Lesion × Dose interaction (F4,124 = 8.16, p<0.001) and subsequent post hoc analyses revealed that DA + NE-lesioned rats rotated less at each dose of L-DOPA (all p<0.05).
Fig. 2.
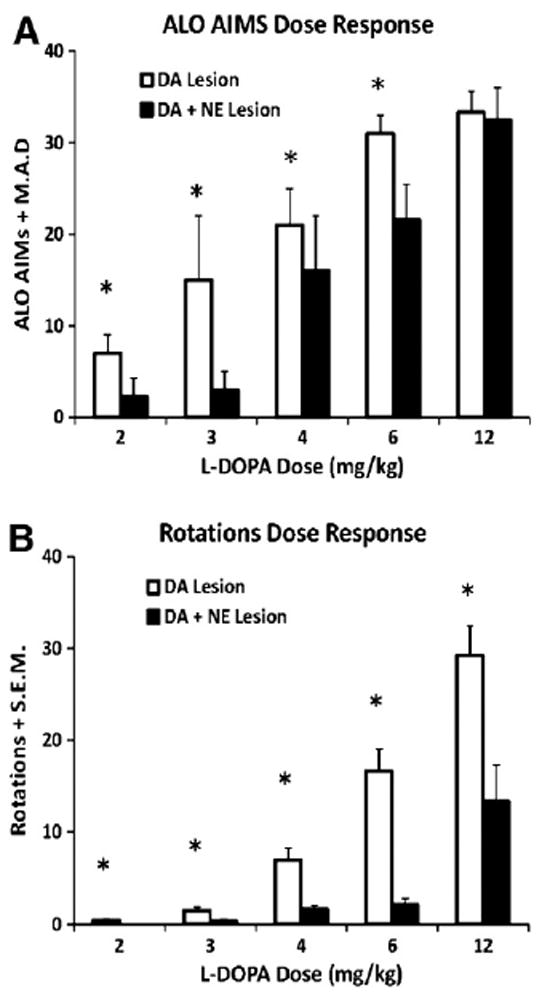
Expression of ALO AIMs and rotations following 2, 3, 4, 6, and 12mg/kg + 15 mg/kg benserazide of L-DOPA in L-DOPA-primed DA (n = 13) and DA + NE (n = 9) lesioned rats. ALO AIMs and rotations were measured for 1 min every 20 min for 2 h. Nonparametric Mann Whitney U-tests were used to analyze potential differences in ALO AIMs between DA and DA + NE-lesioned rats for each dose of L-DOPA. Differences in rotations were analyzed using a 2 (Lesion) × 5 (L-DOPA dose) ANOVA. DA + NE-lesioned rats showed attenuated ALO AIMs following 2, 3, 4 and 6 mg/kg L-DOPA (A) and fewer rotations with each dose of L-DOPA (B). *P<0.05 vs. DA + NE Lesion at same dose of L-DOPA. Data presented as median ALO AIMs ± M.A.D or Rotations ± S.E.M.
3.2.3. L-DOPA efficacy is not altered by NE lesions
L-DOPA-primed DA- (n = 10) and DA + NE- (n = 9) lesioned rats were tested for L-DOPA efficacy 60 min after 4 mg/kg of L-DOPA. A 2-factor ANOVA revealed only a main effect of Treatment (F1,31 = 13.64, p<0.001). As shown in Fig. 3, subsequent post hoc analyses revealed that L-DOPA treatment improved stepping in both DA or DA + NE lesioned rats (both p<0.05).
Fig. 3.
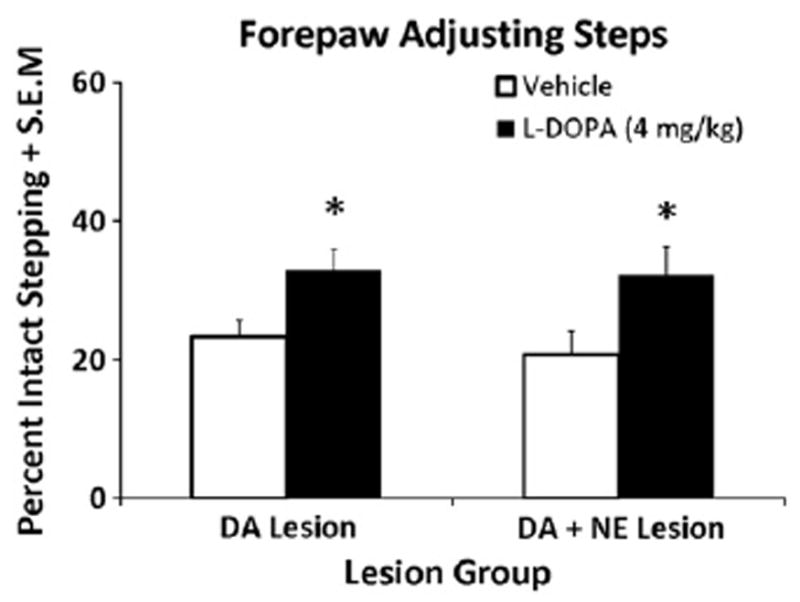
Forepaw adjusting steps test (FAS) in L-DOPA-primed DA (n = 10) and DA + NE (n = 9) lesioned rats. Both lesion groups received either Vehicle or L-DOPA (4 mg/kg + Bensarazide 15 mg/kg) 60 min prior to the FAS test in a counterbalanced design. Bars show the effects of treatments on FAS performance expressed as mean percentages of intact forepaw adjusting steps ± S.E.M. Effects were analyzed by a 2 (Lesion) × 2 (Treatment) ANOVA. In both Lesion groups, L-DOPA improved stepping vs. Vehicle. *P<0.05 vs Vehicle.
3.3. Experiment 2
3.3.1. The β-adrenergic receptor antagonist PRO reduces AIMs in DA- and DA + NE-lesioned rats
3.3.1.1. DA-lesioned rats
L-DOPA-primed DA-lesioned rats were pretreated with PRO (0, 5 or 20 mg/kg) 10 min before L-DOPA (4 and 12 mg/kg) to determine the effects of β-adrenergic receptor blockade on ALO AIMs and rotations. Significant treatment effects were observed for ALO AIMs over the entire 3 h period (χ2 = 10.9, P<0.05; Fig. 4A, Inset). Further time analysis found significant effects at 20–40, and 70 min after 12 mg/kg of L-DOPA (χ2 = 6.0, 11.0, 6.8, and 6.4, respectively, all p<0.05; Fig. 4A). Post hoc analyses revealed that 20 mg/kg PRO (at 20, 30, 40 and 70 min) reduced ALO AIMs compared to VEH pretreatment while over the testing period, 20 mg/kg PRO reduced ALO AIMs compared to both Vehicle and 5 mg/kg PRO (both P<0.05). As seen in Fig. 4B (Inset), significant treatment effects were also demonstrated for ALO AIMs when rats received 4 mg/kg L-DOPA (χ2 = 10.9, P<0.05). Further analyses over time demonstrated widespread effects (20–150 min, not 130 min; all χ2>6.0; all P<0.05). Post hoc analyses revealed dose-dependent effects of PRO with higher doses providing greater anti-dyskinetic efficacy.
Fig. 4.
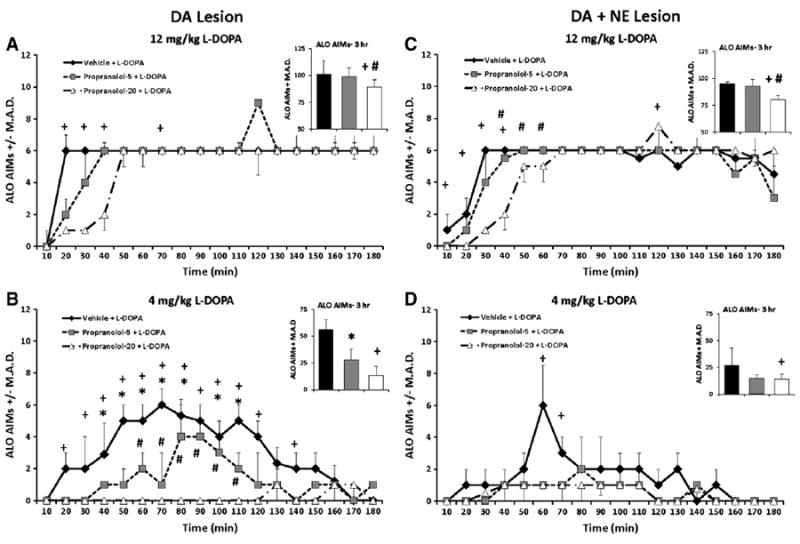
ALO AIMs in rats pretreated with 0, 5, and 20 mg/kg propranolol (PRO) in L-DOPA-primed DA (n = 10) and DA + NE (n = 9) lesioned rats prior to 4 and 12 mg/kg L-DOPA. Nonparametric Friedman ANOVAs were used to analyze potential treatment differences in ALO AIMs at each time point (10–180 min). In the DA lesion group, pretreatment with 20 mg/kg PRO reduced ALO AIMs at 20–40 and 70 min following 12 mg/kg L-DOPA (A). Following 4 mg/kg L-DOPA, 20 mg/kg of PRO attenuated ALO AIMs from 20 to 120 min and 140 to 150 min, while 10 mg/kg conveyed anti-dyskinetic effected from 40 to 110 min and at 150. PRO 5 and 20 mg/kg differed from 60 to 110 min (B). In the DA + NE lesion group, 20 mg/kg PRO dose-dependently reduced ALO AIMs 10–40 and at 120 min following 12 mg/kg L-DOPA (C), while reduced ALO AIMs were observed following 20 mg/kg PRO and 4 mg/kg L-DOPA only at 60 and 70 min time points (D). *P<0.05 for Vehicle vs. PRO-5, +P<0.05 for Vehicle vs. PRO-20 and #P<0.05 for PRO-5 vs. PRO-20. All significant differences were between groups at the same time point (line graphs) or total time (insets). Data presented as median ALO AIMs ± M.A.D.
3.3.1.2. DA + NE-lesioned rats
Using the same doses and procedure as described above, ALO AIMs were also examined in DA + NE-lesioned rats. Significant treatment-related effects of PRO were found upon analysis of ALO AIMs in rats administered 12 mg/kg L-DOPA overall (χ2 = 7.8, P<0.05; Fig. 4C, Inset) and at the 10–60 and 120 min time points (χ2>6.0; all p<0.05; Fig. 4C). Overall, post hocs demonstrated that 20 mg/kg PRO reduced AIMs vs. both Vehicle and 5 mg/kg PRO (both P<0.05). According to post hoc analyses of time effects (all P<0.05), 20 mg/kg PRO reduced ALO AIMs compared to Vehicle (10–40 min) and 5mg/kg PRO(40–60 min). At 120 min, the high dose of PRO exacerbated dyskinesia. In DA + NE-lesioned rats receiving 4 mg/kg L-DOPA, overall main effects of treatment (χ2 = 7.8, P<0.05 Fig. 4D, Inset) were also shown as well as specific time effects at the 60 and 70 min time points (χ2 = 7.3, 6.6, respectively, both P<0.05; Fig. 4D). Post hocs revealed anti-dyskinetic effects of high doses of PRO (all P<0.05).
3.3.2. The α2-adrenergic receptor antagonist IDZ reduces AIMs in both DA- and DA + NE-lesioned rats
3.3.2.1. DA-lesioned rats
L-DOPA-primed DA-lesioned rats were pretreated with IDZ (0, 2.5, and 10 mg/kg) 10 min before receiving L-DOPA (4 and 12 mg/kg) to determine the effects of α2-adrenergic receptor blockade on ALO AIMs. As seen in Fig. 5A (Inset), significant overall treatment effects were observed on ALO AIMs in rats receiving 12 mg/kg L-DOPA (χ2 > 10.0; all P<0.05; Fig. 5A). Analyses of distinct time points revealed significant effects at 30–50, 80, 100, 110 and 140 min (All χ2 > 6.0; all P<0.05). Of note, post hoc analysis indicated that ALO AIMs were reduced in rats receiving 10 mg/kg IDZ (at 30, 40, 50, 80, 100, 110, and 140 min) compared to rats treated with Vehicle. While no significant IDZ treatment-induced changes where observed when ALO AIMs were summed over 3 h, significant effects were seen at individual time points (50–100 and 130–180 min) after 4 mg/kg of L-DOPA (χ2 > 6.1; all P<0.05; Fig. 5B) with post hoc analyses demonstrating that ALO AIMs were reduced in rats receiving 10 mg/kg IDZ compared to Vehicle at 50–100 min time points (P<0.05). Interestingly from 130 to 180 min, ALO AIMs were higher in rats receiving IDZ treatment (all P<0.05).
Fig. 5.
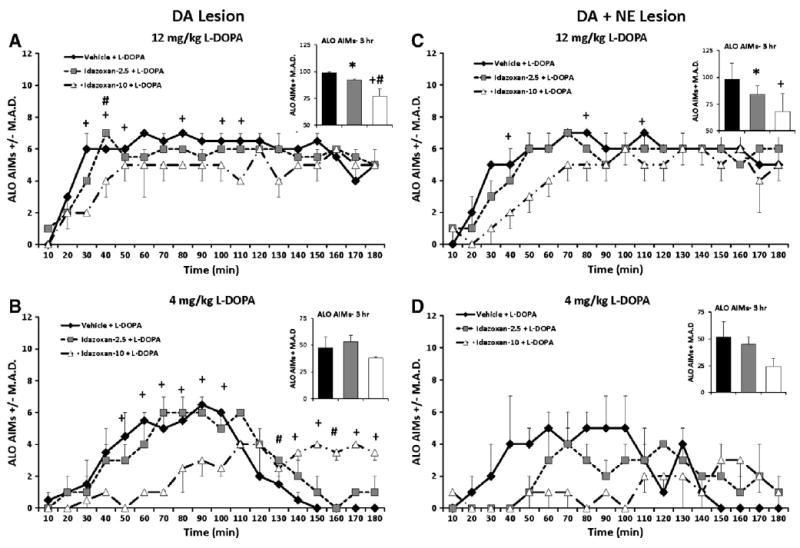
ALO AIMs in L-DOPA-primed DA- (n = 5) and DA + NE- (n = 6) lesioned rats pretreated with 0, 2.5 and 10 mg/kg Idazoxan (IDZ) prior to 4 and 12 mg/kg L-DOPA. Nonparametric Friedman ANOVA was used to analyze treatment differences in ALO AIMs at each time point (10–180 min). A dose-dependent reduction in ALO AIMs was observed 30, 50, 80, 100, 110 and 140 min following 12 mg/kg L-DOPA in DA-lesioned rats (A). After 4 mg/kg L-DOPA, the DA lesion group showed an early IDZ-10-induced reduction in ALO AIM from 50 to 100 min followed by an increase in AIMs that was observed from 140 180 min. In addition IDZ-2.5 pretreatment worsened late AIMs at 130 and 160 min time points (B). In DA + NE lesioned rats, the high dose of IDZ dose-dependently reduced ALO AIMs at 40, 80 and 110 min following 12 mg/kg L-DOPA (C). No IDZ-induced treatment changes in ALO AIMs were observed following 4 mg/kg L-DOPA (D). *P<0.05 for Vehicle vs. PRO-5, +P<0.05 for Vehicle vs. PRO-20 and #P<0.05 for PRO-5 vs. PRO-20. All significant differences were between groups at the same time point (line graphs) or total time (insets). Data presented as median ALO AIMs ± M.A.D.
3.3.2.2. DA + NE-lesioned rats
The impact of IDZ on ALO AIMs was also examined in DA + NE-lesioned rats. A significant reduction in ALO AIMs were observed in rats receiving 12 mg/kg L-DOPA overall (χ2 > 6.1; all P<0.05; Fig. 5C, Inset), and at the 40, 80, and 110 min time points (χ2 = 6.2, 10.9, and 7.6 respectively; all P<0.05; Fig. 5C). Post hoc analyses revealed that overall both 2.5 and 10 mg/kg IDZ reduced rat ALO AIMs compared to Vehicle, but that at individual time points only 10 mg/kg IDZ effectively reduced LID (40, 80, and 110 min; all p<0.05). No significant changes in ALO AIMs were observed in DA + NE rats receiving 4 mg/kg L-DOPA (Fig. 5D).
4. Discussion
In the current series of studies, we sought to clarify the contribution of the NE system to the development and expression of LID and determine how moderate NE loss affects the therapeutic benefits of NE antagonists in an animal model of LID. In order to modify NE, rats received unilateral MFB 6-OHDA lesions with or without the NE reuptake inhibitor desipramine. HPLC analysis demonstrated that striatal NE content in rats receiving desipramine did not differ from naïve control NE levels. However, subjects not protected with desipramine displayed 66% and 82% reductions in striatal and hippocampal NE ipsilateral to the lesion, a range of loss comparable to what has been reported in the human literature (Zarow et al., 2003). Interestingly, these rats also displayed reduced NE within the contralateral striatum and hippocampus, supporting evidence that LC efferents to these regions decussate at ascending commissural points (Jones and Moore, 1977; Kitt and Brauth, 1986; Lindvall and Bjorklund, 1974; McBride and Sutin, 1976).
With this model, we first examined whether an impaired NE system would modulate the development and/or expression of AIMs and rotations since current work on this topic has been contradictory (Fulceri et al., 2007; Perez et al., 2009). In the current study, all rats developed maximal AIMs by day 7 of L-DOPA priming; however, peak AIMs were slightly delayed in rats with a combined DA + NE lesion. In addition, fully primed DA + NE-lesioned rats displayed fewer AIMs when treated with low to moderate doses of L-DOPA (2–6 mg/kg). These findings corroborate the attenuated locomotor stimulating effects of L-DOPA in MPTP-treated mice with NE lesions (Nishi et al., 1991), but oppose previous work. For example, Fulceri et al. (2007) demonstrated accelerated AIMs onset and severity in DA + NE-lesioned Wistar rats administered 10 mg/kg L-DOPA daily for 22 days. The reasons for such opposing results are not clear, however, strain and methodological differences likely contribute. For example, Wistar rats display NE hyperplasticity when subjected to stressful stimuli (Tejani-Butt et al., 1994) that may make them more susceptible to NE lesions than Sprague Dawley rats. Furthermore, differences in striatal NE depletion between the present study (64%) compared to Fulceri et al. (2007; 85%) may have further contributed to the observed differences. In addition, the most pronounced effects in the present study were seen in the L-DOPA dose response study, where a dynamic range of L-DOPA was employed for tests of AIMs expression. Such an approach may be necessary to observe discrete lesioninduced differences. Finally, HPLC data suggest that like NE loss, DA depletion between the present study and Fulceri et al. (2007) were dissimilar (>95% vs. ~80%, respectively). It is also noteworthy Perez et al. (2009), who found no effects of NE lesion on AIMs, employed the NE neurotoxin (N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4). DSP-4 is a potent but transient NE neurotoxin (Hughes and Stanford, 1998; Szot et al., 2010) and while some evidence of LC cell loss was demonstrated it was not clear what level of NE depletion was achieved. Collectively these findings suggest that the extent of DA and NE loss likely contributes to LID trajectory.
Interestingly, rotations, a conventional proxy of LID (Cenci et al., 2002), were markedly reduced in DA + NE-lesioned rats (compared to DA lesion rats) and did not appear to change during the development of AIMs. Furthermore, only a high dose of L-DOPA (12 mg/kg) elicited increased rotations. Thus, in an animal model more akin to PD (i.e., depleted DA + NE), rotations appear to dissociate from LID. The fact that PD patients also display NE cell loss but animal models of dyskinesia often do not speaks more to the validity, or lack thereof, of the widely used animal models of dyskinesia when NE cells are protected. In fact, based on our data, we would suggest that L-DOPA-induced rotations better align with the status of the NE system in the DA-depleted brain.
In order to determine whether moderate NE loss also altered L-DOPA efficacy, L-DOPA-primed DA and DA + NE-lesioned rats were subject to the FAS test which is sensitive to both lesion induced deficits and their reversal by L-DOPA (Chang et al., 1999; Eskow et al., 2007). Interestingly, both lesion groups demonstrated stepping deficits that were similarly improved by an acute low dose of L-DOPA (4 mg/kg). These findings indicate that moderate NE loss may not significantly impact the anti-parkinsonian effects of L-DOPA, but when combined with other work (Nishi et al., 1991; Srinivasan and Schmidt, 2003; Rommelfanger et al., 2007), may suggest a critical threshold where NE loss attenuates L-DOPA efficacy.
Our next goal was to determine whether the anti-dyskinetic potential of the pan β-adrenergic receptor antagonist PRO and the α2-adrenergic receptor antagonist IDZ were differentially affected by NE loss. Both PRO and IDZ were effective in alleviating the expression of AIMs in DA-lesioned rats, consistent with previous experimental studies (Gomez-Mancilla and Bedard, 1993; Henry et al., 1999; Savola et al., 2003; Dekundy et al., 2007; Buck et al., 2010; Lindenbach et al., 2011). Generally, both adrenergic receptor antagonists also conveyed anti-dyskinetic effects in DA + NE-lesioned rats. There were however, some subtle differences in the magnitude of attenuation. For example, at the lower dose of L-DOPA, PRO reduced AIMs to a greater extent in DA-lesioned rats summing ALO AIMs over the course of 3 h (Fig. 4 C–D; inset) although, a floor effect in AIMs induction in DA + NE-lesioned rats was likely. In regards to IDZ, AIMs summed over 3 h looked virtually identical in both DA- and DA + NE-lesioned rats at the higher L-DOPA (Fig. 5A, C; inset), while IDZ co-administered with a 4 mg/kg L-DOPA exacerbated AIMs in the DA-lesioned group. Whether or not these subtle differences would be clinically relevant is unknown; but these data suggest that NE and/or DA loss may alter the expression/activity of adrenergic receptors and their ability to modulate subsequent behavior. For example, β1 and β2 adrenergic receptors are found in high concentrations in basal ganglia (Nicholas et al., 1993; Rainbow et al., 1984; Waeber et al., 1991) and may upregulate following SNc and/or LC lesions (Johnson et al., 1989; Lorton et al., 1988). The putative pro-dyskinetic effects of IDZ in DA-lesioned rats may relate to preferential antagonism at presynaptic LC autoreceptors (NE) or SNc heteroreceptors (DA), thereby increasing NE, DA or both (Gobert et al., 2004; Yavich et al., 1997). These empirical questions may have important consequences regarding the most appropriate adrenergic LID treatment.
The mechanism(s) by which NE lesion, PRO and IDZ reduce LID are unknown, although the NE transporter and/or receptor(s) may be instrumental. For example, it has recently been reported that L-DOPA can be taken up by the NE transporter (Arai et al., 2008) where it may then be converted to DA, released, and ultimately contributes to pulsatile extracellular levels of DA believed to underlie LID (Cenci and Lindgren, 2007). Therefore LC loss may remove the influence of NE terminal release of L-DOPA-derived DA leading to reduced LID (Lategan et al., 1990, 1992). However, this does not explain the effects of IDZ on AIMs. Because most α2-adrenergic receptors are thought to be inhibitory autoreceptors or heteroreceptors modulating NE or DA respectively (Yavich et al., 1997; Gobert et al., 2004), antagonists would be more likely to increase DA release and exacerbate LID, an effect only seen with lower L-DOPA doses. Moreover, Buck and Ferger (2009) recently demonstrated that IDZ (at 9 mg/kg) reduced L-DOPA-induced striatal DA and associated AIMs. Given that IDZ effects may differ across lesion groups and doses, it seems as though multiple mechanisms may be contributing. More recently it has been suggested that post-synaptic striatal β1- and α2-adrenergic receptors may actually mediate striatal plasticity associated with PD and LID (Hara et al., 2010; Holmberg et al., 1999; Meitzen et al., 2011). A second possibility lies in the affinity of NE and adrenergic antagonists for DA receptors. There is ample evidence that NE has affinity for DA receptors (Lanau et al., 1997; Luscombe et al., 1979; Newman-Tancredi et al., 1997). However, how these affinities change following DA denervation and subsequent LID remain unexplored. Some indirect evidence might be gleaned from Buck and Ferger (2009) who show that NE alone, when infused via microdialysis probe into the striatum, potently induced AIMs. These surprising data indicate that striatal DA receptors may become more sensitive to NE or like compounds following DA depletion. Future studies will be necessary to confirm these hypotheses.
The repeated success of these NE compounds in alleviating LID makes them strong candidates for human clinical trials. This is particularly true since the drugs employed in the current work and prior preclinical and clinical investigations either do not hinder, or may even improve L-DOPA efficacy (Buck and Ferger, 2010; Carpentier et al., 1996; Henry et al., 1999; Lindenbach et al., 2011) Currently, however, only the α2-adrenergic receptor antagonist fipamezole is in clinical trials for treatment of LID (“Santhera licenses novel dyskinesia therapy to Biovail”, 2009). Interestingly, there is no indication that β-adrenergic receptor antagonists are being considered for LID, though favorable outcomes with PRO have been noted in both experimental and clinical studies. This may, in part, be due to potential cardiovascular complications (Prichard et al., 2001), since PD patients are particularly susceptible to orthostatic hypotension (Oka et al., 2007). However, in a small study lasting approximately 6weeks, Carpentier et al. (1996) found no adverse cardiovascular side effects, warranting a more long-term investigation. Furthermore, identifying the contribution of specific β-adrenergic receptors might ultimately yield a bettermechanistic target with fewer, if any, adverse cardiovascular effects.
In summary, it is important to consider the importance of protecting, or not protecting, NE neurons in animal models of PD and LID. Accumulating experimental and clinical evidence indicates that NE neuron integrity modulates SNc DA degeneration (Bing et al., 1994; Fornai et al., 1995; Marien et al., 1993; Mavridis et al., 1991). Likewise, our current findings demonstrate that the status of the NE system also plays in integral role in the development, expression and treatment of LID, supporting the continued investigation of adrenergic compounds for the improved treatment of PD.
Acknowledgments
This work was supported by NIH NS059600 (C. Bishop) and the Center for Development and Behavioral Neuroscience at Binghamton University. The authors would especially like to thank Thomas Button for his technical assistance and Karen L. Eskow-Jaunarajs, Kristin B. Dupre, and Corinne Y. Ostock, for their help with behavioral scoring and surgery.
References
- Arai A, Tomiyama M, Kannari K, Kimura T, Suzuki C, Watanabe M, et al. Reuptake of L-DOPA-derived extracellular DA in the striatum of a rodent model of Parkinson’s disease via norepinephrine transporter. Synapse. 2008;62(8):632–5. doi: 10.1002/syn.20535. [DOI] [PubMed] [Google Scholar]
- Arvin B, Le Peillet E, Dürmüller N, Chapman AG, Meldrum BS. Electrolytic lesions of the locus coeruleus or 6-hydroxydopamine lesions of the medial forebrain bundle protect against excitotoxic damage in rat hippocampus. Brain Res. 1992;579(2):279–84. doi: 10.1016/0006-8993(92)90061-d. [DOI] [PubMed] [Google Scholar]
- Barnum CJ, Eskow KL, Dupre K, Blandino P, Deak T, Bishop C. Exogenous corticosterone reduces L-DOPA-induced dyskinesia in the hemi-parkinsonian rat: role for interleukin-1beta. Neuroscience. 2008;156(1):30–41. doi: 10.1016/j.neuroscience.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing G, Zhang Y, Watanabe Y, McEwen BS, Stone EA. Locus coeruleus lesions potentiate neurotoxic effects of MPTP in dopaminergic neurons of the substantia nigra. Brain Res. 1994;668(1-2):261–5. doi: 10.1016/0006-8993(94)90534-7. [DOI] [PubMed] [Google Scholar]
- Bishop C, Krolewski DM, Eskow KL, Barnum CJ, Dupre KB, Deak T, et al. Contribution of the striatum to the effects of 5-HT1A receptor stimulation in L-DOPA-treated hemiparkinsonian rats. J Neurosci Res. 2009;87(7):1645–58. doi: 10.1002/jnr.21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck K, Ferger B. Comparison of intrastriatal administration of noradrenaline and l-DOPA on dyskinetic movements: a bilateral reverse in vivo microdialysis study in 6-hydroxydopamine-lesioned rats. Neuroscience. 2009;159(1):16–20. doi: 10.1016/j.neuroscience.2008.12.026. [DOI] [PubMed] [Google Scholar]
- Buck K, Ferger B. The selective alpha1 adrenoceptor antagonist HEAT reduces L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Synapse. 2010;64(2):117–26. doi: 10.1002/syn.20709. [DOI] [PubMed] [Google Scholar]
- Buck K, Voehringer P, Ferger B. The alpha(2) adrenoceptor antagonist idazoxan alleviates L-DOPA-induced dyskinesia by reduction of striatal dopamine levels: an in vivo microdialysis study in 6-hydroxydopamine-lesioned rats. J Neurochem. 2010;112(2):444–52. doi: 10.1111/j.1471-4159.2009.06482.x. [DOI] [PubMed] [Google Scholar]
- Carpentier AF, Bonnet AM, Vidailhet M, Agid Y. Improvement of levodopa-induced dyskinesia by propranolol in Parkinson’s disease. Neurology. 1996;46(6):1548–51. doi: 10.1212/wnl.46.6.1548. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lindgren H. Advances in understanding L-DOPA-induced dyskinesia. Curr Opin Neurobiol. 2007;17(6):655–71. doi: 10.1016/j.conb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Whishaw IQ, Schallert T. Animal models of neurological deficits: how relevant is the rat. Nat Rev Neurosci. 2002;3(7):574–9. doi: 10.1038/nrn877. [DOI] [PubMed] [Google Scholar]
- Chang J, Wachtel S, Kang U. Biochemical and anatomical characterization of forepaw adjusting steps in rats models of Parkinson’s disease: studies on medial forebrain bundle and striatal lesions. Neuroscience. 1999;88(2):617–28. doi: 10.1016/s0306-4522(98)00217-6. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dekundy A, Lundblad M, Danysz W, Cenci M. Modulation of L-DOPA-induced abnormal involuntary movements by clinically tested compounds: further validation of the rat dyskinesia model. Behav Brain Res. 2007;179:76–89. doi: 10.1016/j.bbr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Domino EF, Ni L, Colpaert F, Marien M. Effects of (+/−)-idazoxan alone and in combination with L-DOPA methyl ester in MPTP-induced hemiparkinsonian monkeys. Receptors Channels. 2003;9(5):335–8. doi: 10.3109/713745180. [DOI] [PubMed] [Google Scholar]
- Eskow K, Gupta V, Alam S, Park J, Bishop C. The partial 5-HT1A agonist buspirone reduces the expression and development of l-DOPA-induced dyskinesia in rats and improves l-DOPA efficacy. Pharmacol Biochem Behav. 2007;87:306–14. doi: 10.1016/j.pbb.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Fornai F, Bassi L, Torracca MT, Scalori V, Corsini GU. Norepinephrine loss exacerbates methamphetamine-induced striatal dopamine depletion in mice. Eur J Pharmacol. 1995;283(1-3):99–102. doi: 10.1016/0014-2999(95)00313-a. [DOI] [PubMed] [Google Scholar]
- Fornai F, di Poggio AB, Pellegrini A, Ruggieri S, Paparelli A. Noradrenaline in Parkinson’s disease: from disease progression to current therapeutics. Curr Med Chem. 2007;14(22):2330–4. doi: 10.2174/092986707781745550. [DOI] [PubMed] [Google Scholar]
- Fox SH, Henry B, Hill MP, Peggs D, Crossman AR, Brotchie JM. Neural mechanisms underlying peak-dose dyskinesia induced by levodopa and apomorphine are distinct: evidence from the effects of the alpha(2) adrenoceptor antagonist idazoxan. Mov Disord. 2001;16(4):642–50. doi: 10.1002/mds.1148. [DOI] [PubMed] [Google Scholar]
- Fulceri F, Biagioni F, Ferrucci M, Lazzeri G, Bartalucci A, Galli V, et al. Abnormal involuntary movements (AIMs) following pulsatile dopaminergic stimulation: severe deterioration and morphological correlates following the loss of locus coeruleus neurons. Brain Res. 2007;1135(1):219–29. doi: 10.1016/j.brainres.2006.12.030. [DOI] [PubMed] [Google Scholar]
- Gobert A, Billiras R, Cistarelli L, Millan MJ. Quantification and pharmacological characterization of dialysate levels of noradrenaline in the striatum of freely-moving rats: release from adrenergic terminals and modulation by alpha2-autoreceptors. J Neurosci Method. 2004;140(1-2):141–52. doi: 10.1016/j.jneumeth.2004.04.040. [DOI] [PubMed] [Google Scholar]
- Gomez-Mancilla B, Bedard PJ. Effect of nondopaminergic drugs on L-dopa-induced dyskinesias in MPTP-treated monkeys. Clin Neuropharmacol. 1993;16(5):418–27. doi: 10.1097/00002826-199310000-00004. [DOI] [PubMed] [Google Scholar]
- Grondin R, Hadj Tahar A, Doan VD, Ladure P, Bedard PJ. Noradrenoceptor antagonism with idazoxan improves L-dopa-induced dyskinesias in MPTP monkeys. Naunyn Schmiedebergs Arch Pharmacol. 2000;361(2):181–6. doi: 10.1007/s002109900167. [DOI] [PubMed] [Google Scholar]
- Haapalinna A, Leino T, Heinonen E. The alpha 2-adrenoceptor antagonist atipamezole potentiates anti-Parkinsonian effects and can reduce the adverse cardiovascular effects of dopaminergic drugs in rats. Naunyn Schmiedebergs Arch Pharmacol. 2003;368(5):342–51. doi: 10.1007/s00210-003-0827-z. [DOI] [PubMed] [Google Scholar]
- Hara M, Fukui R, Hieda E, Kuroiwa M, Bateup HS, Kano T, et al. Role of adrenoceptors in the regulation of dopamine/DARPP-32 signaling in neostriatal neurons. J Neurochem. 2010;113(4):1046–59. doi: 10.1111/j.1471-4159.2010.06668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry B, Fox SH, Peggs D, Crossman AR, Brotchie JM. The alpha2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of L-dopa in the MPTP-lesioned primate model of Parkinson’s disease. Mov Disord. 1999;14(5):744–53. doi: 10.1002/1531-8257(199909)14:5<744::aid-mds1006>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Holmberg M, Scheinin M, Kurose H, Miettinen R. Adrenergic alpha2C-receptors reside in rat striatal GABAergic projection neurons: comparison of radioligand binding and immunohistochemistry. Neuroscience. 1999;93(4):1323–33. doi: 10.1016/s0306-4522(99)00260-2. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O, Kish SJ. Biochemical pathophysiology of Parkinson’s disease. Adv Neurol. 1987;45:19–34. [PubMed] [Google Scholar]
- Hughes ZA, Stanford SC. A partial noradrenergic lesion induced by DSP-4 increases extracellular noradrenaline concentration in rat frontal cortex: a microdialysis study in vivo. Psychopharmacology (Berl) 1998;136(3):299–303. doi: 10.1007/s002130050569. [DOI] [PubMed] [Google Scholar]
- Johnson EW, Wolfe BB, Molinoff PB. Regulation of subtypes of beta-adrenergic receptors in rat brain following treatment with 6-hydroxydopamine. J Neurosci. 1989;9(7):2297–305. doi: 10.1523/JNEUROSCI.09-07-02297.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston TH, Fox SH, Piggott MJ, Savola JM, Brotchie JM. The alpha adrenergic antagonist fipamezole improves quality of levodopa action in Parkinsonian primates. Mov Disord. 2010;25(13):2084–93. doi: 10.1002/mds.23172. [DOI] [PubMed] [Google Scholar]
- Jones BE, Moore RY. Ascending projections of the locus coeruleus in the rat. II. Autoradiographic study. Brain Res. 1977;127(1):25–53. [PubMed] [Google Scholar]
- Kilpatrick IC, Jones MW, Phillipson OT. A semiautomated analysis method for catecholamines, indoleamines, and some prominent metabolites in microdissected regions of the nervous system: an isocratic HPLC technique employing coulometric detection and minimal sample preparation. J Neurochem. 1986;46(6):1865–76. doi: 10.1111/j.1471-4159.1986.tb08506.x. [DOI] [PubMed] [Google Scholar]
- Kitt CA, Brauth SE. Telencephalic projections from midbrain and isthmal cell groups in the pigeon. I. Locus coeruleus and subcoeruleus. J Comp Neurol. 1986;247(1):69–91. doi: 10.1002/cne.902470105. [DOI] [PubMed] [Google Scholar]
- Lanau F, Zenner MT, Civelli O, Hartman DS. Epinephrine and norepinephrine act as potent agonists at the recombinant human dopamine D4 receptor. J Neurochem. 1997;68(2):804–12. doi: 10.1046/j.1471-4159.1997.68020804.x. [DOI] [PubMed] [Google Scholar]
- Lategan AJ, Marien MR, Colpaert FC. Effects of locus coeruleus lesions on the release of endogenous dopamine in the rat nucleus accumbens and caudate nucleus as determined by intracerebral microdialysis. Brain Res. 1990;523(1):134–8. doi: 10.1016/0006-8993(90)91646-x. [DOI] [PubMed] [Google Scholar]
- Lategan AJ, Marien MR, Colpaert FC. Suppression of nigrostriatal and mesolimbic dopamine release in vivo following noradrenaline depletion by DSP-4: a microdialysis study. Life Sci. 1992;50(14):995–9. doi: 10.1016/0024-3205(92)90093-5. [DOI] [PubMed] [Google Scholar]
- Lindenbach D, Ostock CY, Eskow Jaunarajs KL, Dupre KB, Barnum CJ, Bhide N, et al. Behavioral and cellular modulation of L-DOPA-induced dyskinesia by β-adrenoceptor receptor blockade in the 6-OHDA lesioned rat. J Pharmacol Exp Ther. 2011;337(3):755–65. doi: 10.1124/jpet.111.179416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindvall O, Bjorklund A. The organization of the ascending catecholamine neuron systems in the rat brain as revealed by the glyoxylic acid fluorescence method. Acta Physiol Scand. 1974;(Suppl 412):1–48. [PubMed] [Google Scholar]
- Lorton D, Bartolome J, Slotkin TA, Davis JN. Development of brain beta-adrenergic receptors after neonatal 6-hydroxydopamine treatment. Brain Res Bull. 1988;21(4):591–600. doi: 10.1016/0361-9230(88)90198-0. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci M. Pharmacological validation of behavioral measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci. 2002;15(1):120–32. doi: 10.1046/j.0953-816x.2001.01843.x. [DOI] [PubMed] [Google Scholar]
- Luscombe G, Clow A, Jenner P, Marsden CD. Antagonism by propranolol of central dopamine receptor stimulation is not related to beta-adrenergic blockade. J Pharm Pharmacol. 1979;31(5):355–6. doi: 10.1111/j.2042-7158.1979.tb13522.x. [DOI] [PubMed] [Google Scholar]
- Marien M, Briley M, Colpaert F. Noradrenaline depletion exacerbates MPTP-induced striatal dopamine loss in mice. Eur J Pharmacol. 1993;236(3):487–9. doi: 10.1016/0014-2999(93)90489-5. [DOI] [PubMed] [Google Scholar]
- Marin C, Aguilar E, Bonastre M. Effect of locus coeruleus denervation on levodopa-induced motor fluctuations in hemiparkinsonian rats. J Neural Transm. 2008;115(8):1133–9. doi: 10.1007/s00702-008-0060-5. [DOI] [PubMed] [Google Scholar]
- Mavridis M, Degryse AD, Lategan AJ, Marien MR, Colpaert FC. Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in monkeys: a possible role for the locus coeruleus in the progression of Parkinson’s disease. Neuroscience. 1991;41(2-3):507–23. doi: 10.1016/0306-4522(91)90345-o. [DOI] [PubMed] [Google Scholar]
- McBride RL, Sutin J. Projections of the locus coeruleus and adjacent pontine tegmentum in the cat. J Comp Neurol. 1976;165(3):265–84. doi: 10.1002/cne.901650302. [DOI] [PubMed] [Google Scholar]
- McMillan PJ, White SS, Franklin A, Greenup JL, Leverenz JB, Raskind MA, et al. Differential response of the central noradrenergic nervous system to the loss of locus coeruleus neurons in Parkinson’s disease and Alzheimer’s disease. Brain Res. 2011;1373:240–52. doi: 10.1016/j.brainres.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitzen J, Luoma JI, Stern CM, Mermelstein PG. beta1-Adrenergic receptors activate two distinct signaling pathways in striatal neurons. J Neurochem. 2011;116(6):984–95. doi: 10.1111/j.1471-4159.2010.07137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narabayashi H, Kondo T, Nagatsu T, Hayashi A, Suzuki T. DL-threo-3,4-dihydroxyphenylserine for freezing symptom in parkinsonism. Adv Neurol. 1984;40:497–502. [PubMed] [Google Scholar]
- Narabayashi H, Yokochi F, Ogawa T, Igakura T. Analysis of L-threo-3, 4-dihydroxyphenylserine effect on motor and psychological symptoms in Parkinson’s disease. No To Shinkei. 1991;43(3):263–8. [PubMed] [Google Scholar]
- Newman-Tancredi A, Audinot-Bouchez V, Gobert A, Millan MJ. Noradrenaline and adrenaline are high affinity agonists at dopamine D4 receptors. Eur J Pharmacol. 1997;319(2-3):379–83. doi: 10.1016/s0014-2999(96)00985-5. [DOI] [PubMed] [Google Scholar]
- Nicholas AP, Pieribone VA, Hokfelt T. Cellular localization of messenger RNA for beta-1 and beta-2 adrenergic receptors in rat brain: an in situ hybridization study. Neuroscience. 1993;56(4):1023–39. doi: 10.1016/0306-4522(93)90148-9. [DOI] [PubMed] [Google Scholar]
- Nishi K, Kondo T, Narabayashi H. Destruction of norepinephrine terminals in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice reduces locomotor activity induced by L-dopa. Neurosci Lett. 1991;123(2):244–7. doi: 10.1016/0304-3940(91)90941-l. [DOI] [PubMed] [Google Scholar]
- Obeso J, Rodriguez-Oroz M, Chana P, Lera G, Rodriguez M, Olanow C. The evolution and origin of motor complications in Parkinson’s disease. Neurology. 2000;55(11 Suppl 4):S13–23. [PubMed] [Google Scholar]
- Oka H, Yoshioka M, Onouchi K, Morita M, Mochio S, Suzuki M, et al. Characteristics of orthostatic hypotension in Parkinson’s disease. Brain. 2007;130(Pt 9):2425–32. doi: 10.1093/brain/awm174. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson W. The rat brain in stereotaxic coordinates. 4. San Diego: Academic Press; 1998. pp. 1–456. [Google Scholar]
- Perez V, Sosti V, Rubio A, Barbanoj M, Gich I, Rodriguez-Alvarez J, et al. Noradrenergic modulation of the motor response induced by long-term levodopa administration in Parkinsonian rats. J Neural Transm. 2009;116(7):867–74. doi: 10.1007/s00702-009-0242-9. [DOI] [PubMed] [Google Scholar]
- Prichard BN, Cruickshank JM, Graham BR. Beta-adrenergic blocking drugs in the treatment of hypertension. Blood Press. 2001;10(5-6):366–86. doi: 10.1080/080370501753400665. [DOI] [PubMed] [Google Scholar]
- Putterman D, Munhall A, Kozell L, Belknap J, Johnson S. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2007;323(1):277–84. doi: 10.1124/jpet.107.126219. [DOI] [PubMed] [Google Scholar]
- Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of beta 1- and beta 2-adrenergic receptors in rat brain. Proc Natl Acad Sci USA. 1984;81(5):1585–9. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascol O, Arnulf I, Peyro-Saint Paul H, Brefel-Courbon C, Vidailhet M, Thalamas C, et al. Idazoxan, an alpha-2 antagonist, and L-DOPA-induced dyskinesias in patients with Parkinson’s disease. Mov Disord. 2001;16(4):708–13. doi: 10.1002/mds.1143. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Weinshenker D. Norepinephrine: the redheaded stepchild of Parkinson’s disease. Biochem Pharmacol. 2007;74(2):177–90. doi: 10.1016/j.bcp.2007.01.036. [DOI] [PubMed] [Google Scholar]
- Rommelfanger KS, Edwards GL, Freeman KG, Liles LC, Miller GW, Weinshenker D. Norepinephrine loss produces more profound motor deficits than MPTP treatment in mice. Proc Natl Acad Sci USA. 2007;104(34):13804–9. doi: 10.1073/pnas.0702753104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhera licenses novel dyskinesia therapy to Biovail. Nat Rev Drug Discov. 2009;8(10):762. doi: 10.1038/nrd3009. [DOI] [PubMed] [Google Scholar]
- Savola JM, Hill M, Engstrom M, Merivuori H, Wurster S, McGuire SG, et al. Fipamezole (JP-1730) is a potent alpha2 adrenergic receptor antagonist that reduces levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Mov Disord. 2003;18(8):872–83. doi: 10.1002/mds.10464. [DOI] [PubMed] [Google Scholar]
- Srinivasan J, Schmidt W. Functional recovery of locus coeruleus noradrenergic neurons after DSP-4 lesion: effects on dopamine levels and neuroleptic induced-parkinsonian symptoms in rats. J Neural Transm. 2003;111(1):13–26. doi: 10.1007/s00702-003-0062-2. [DOI] [PubMed] [Google Scholar]
- Szot P, Miguelez C, White SS, Franklin A, Sikkema C, Wilkinson CW, et al. A comprehensive analysis of the effect of DSP4 on the locus coeruleus noradrenergic system in the rat. Neuroscience. 2010;166(1):279–91. doi: 10.1016/j.neuroscience.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejani-Butt SM, Pare WP, Yang J. Effect of repeated novel stressors on depressive behavior and brain norepinephrine receptor system in Sprague–Dawley and Wistar Kyoto (WKY) rats. Brain Res. 1994;649(1-2):27–35. doi: 10.1016/0006-8993(94)91045-6. [DOI] [PubMed] [Google Scholar]
- Waeber C, Rigo M, Chinaglia G, Probst A, Palacios JM. Beta-adrenergic receptor subtypes in the basal ganglia of patients with Huntington’s chorea and Parkinson’s disease. Synapse. 1991;8(4):270–80. doi: 10.1002/syn.890080405. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Yao YS, Wang HX. Comparing effects of U50488H, prazosin and/or propranolol on cardiac hypertrophy induced by NE in rat. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2010;26(1):82–5. [PubMed] [Google Scholar]
- Yavich L, Lappalainen R, Sirvio J, Haapalinna A, MacDonald E. Alpha2-adrenergic control of dopamine overflow and metabolism in mouse striatum. Eur J Pharmacol. 1997;339(2-3):113–9. doi: 10.1016/s0014-2999(97)01375-7. [DOI] [PubMed] [Google Scholar]
- Yavich L, Sirvio J, Haapalinna A, Ylinen A, Mannisto PT. Atipamezole, an alpha2-adrenoceptor antagonist, augments the effects of L-DOPA on evoked dopamine release in rat striatum. Eur J Pharmacol. 2003;462(1-3):83–9. doi: 10.1016/s0014-2999(03)01324-4. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60(3):337–41. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]