

Abstract
Regulatory gene circuits enable stem and progenitor cells to detect and process developmental signals and make irreversible fate commitment decisions. To gain insight into the gene circuits underlying the T-cell specification decision in progenitor cells, we generated an updated T lymphocyte developmental gene regulatory network from genes and connections found in the literature. This reconstruction allowed us to identify candidate regulatory gene circuit elements underlying T-cell fate decision making. Here, we examine the roles of these circuits in facilitating different aspects of the decision making process, and discuss experiments to further probe their structure and function.
Keywords: Transcription factor, Notch, IL-7 receptor, Feed-forward circuit, Lineage commitment
Haematopoietic stem and progenitor cells utilize networks of interacting genes and proteins to make cell fate decisions. Regulatory gene circuits within these networks enable progenitors to sense and process external developmental signals, choose between alternate competing fates, and irreversibly commit to a single fate once a decision has been made. The connectivity between genes constituting a circuit determines its dynamic behavior and function, and specific connectivity patterns, or motifs, are thought to enable cells to perform specific tasks and operations [1]. Circuits with positive feedback loops can generate multiple stable cellular states, and mediate irreversible commitment in a variety of developmental contexts, including haematopoietic fate decision making [2-5]. On the other hand, negative feedback loops and feed-forward loops can perform different processing operations on input signals [6, 7], and has been proposed to operate in a variety of signaling pathways [8, 9]. We here review the underlying gene network structure that contributes to a particularly intricate cell fate decision, the decision of haematopoietic progenitors to abandon other developmental options and undertake the T-lymphocyte fate.
Outline of T-cell development
T cell lineage identity is forged by a gene regulatory network that is initially triggered by environmental signals (Figure 1). Haematopoietic progenitors begin T-cell development as they enter the thymus and activate the Notch-Delta signaling pathway in response to a Delta family molecule in the thymic stroma (Notch ligand, probably Delta-like 4) [10, 11]. Upon sustained exposure to Notch-Delta signaling, progenitors enter a developmental stage where they up-regulate certain T-cell identity genes, but maintain expression of stem-cell genes and plasticity to some alternative fates (DN2A, [12]). We will refer to these two pre-commitment stages of T-cell development collectively as the Phase 1 stage (Figure 1, [13]). Upon further Notch-Delta signaling, progenitors undergo commitment to the T-cell fate, undergo major changes in their gene regulatory state (DN2B), and eventually re-arrange T-cell receptor genes (DN3). We will refer to these post-commitment stages collectively as the Phase 2 stage. Sustained Notch-Delta signaling across Phase 1 is required for fate commitment: progenitors transiently exposed to Delta-ligand can turn on T-cell genes, but fail to undergo commitment and may instead differentiate into other lineages, including dendritic, myeloid and NK cells and initially B cells as well [12, 14-22]. Note that Notch signaling first inhibits and then excludes all these non-T fate alternatives at different developmental stages, through distinct regulatory mechanisms, the details of which are reviewed elsewhere [23]. After commitment, Notch signals remain important for viability support and then become dispensable.
Figure 1.

Stages of T-cell specification and commitment. Double negative (CD4-CD8-DN) developmental stages before T-cell fate commitment (Phase 1), and immediately after commitment (Phase 2) are shown. Straight arrows represent signal-dependent developmental transitions between successive stages, and the curved arrow represents cytokine-dependent proliferation in the Phase 1 stage. Gray dashed lines represent the commitment checkpoint (left), and two TCR-dependent developmental checkpoints (right). [all figures are original]
The ability to commit only upon sustained signaling may reflect the action of coherent feed-forward regulatory circuits, where a signal regulates a target gene both through a direct input and also indirectly through an intermediate regulator (Figure 2A). Coherent feed-forward loops act as persistence detectors, allowing cells to filter out transient signals and respond only to sustained stimulation [24, 25]. In T-cell progenitors, different Notch target genes can show very different induction kinetics after the onset of Notch-Delta signaling [15], consistent with the existence of coherent feed-forward loops regulating their expression.
Figure 2.
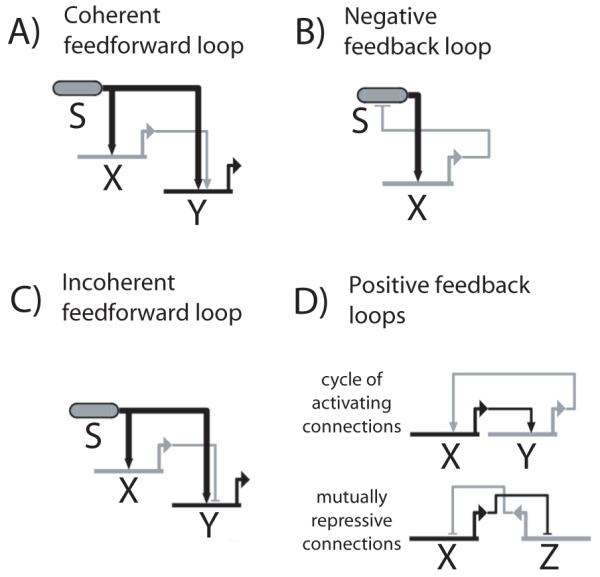
Regulatory circuit motifs. A) Coherent feedforward loop. B) Negative feedback loop (top). C) Incoherent feedforward loop. D) Positive feedback loop, consisting of a cycle of positive connections (top), or a cycle of repressive connections (bottom). In the former circuit, X and Y correspond to regulators of the same fate; in the latter circuit, X and Z are regulators of alternate fates that are expressed in a mutually exclusive manner (bottom).
Besides Delta ligand, cytokines from the thymic microenvironment (i.e. Kit ligand, interleukin (IL)-7, and possibly also thymic stromal lymphopoietin) play critical roles in early T-cell development [26, 27]. Flt3 ligand is also very important for prethymic precursors, although its role within the thymus proper is less clear. While these cytokines are primarily thought to support viability and proliferation of early progenitors (Figure 1), recent work suggests that dynamic IL-7 signals may also play a major role in regulating the commitment transition itself [28]. The IL-7 signaling pathway may be inactive in the earliest T-cell progenitors (ETP) [29-31], but is strongly activated in uncommitted DN2 progenitors, in which it supports survival and proliferation [32]. It is then inactivated after commitment [33-35], possibly also affected by cell non-autonomous mechanisms [36]. Recently, Ikawa and colleagues found in a feeder-free in vitro system that a reduction of IL-7 signal levels promoted the commitment of uncommitted DN2 progenitors [28]. Progenitors experiencing uniform IL-7 levels remained arrested at the DN2 stage and retained the potential to undergo myeloid differentiation. These results suggest that T-cell progenitors can sense changes in IL-7 signaling activity and make commitment decisions accordingly.
How do cells perceive changes in external signal levels? Both the negative feedback loop and the incoherent feedforward loop can, under certain parameter regimes, show sensitivity not to absolute levels of upstream signal, but to changes in the level of signal [6, 7]. In a negative feedback loop, a signal activates a downstream target, which in turn feeds back to negatively modulate the signaling pathway’s sensitivity to signal (Figure 2B) In an incoherent feedforward loop, the signal regulates a downstream target through two different connections that have opposing signs (Figure 2C). Both these circuits can adjust their regulatory state to maintain sensitivity to signal level changes over a range of signal strengths, a phenomenon known as adaptation. Such regulatory circuit motifs may play a role in sensing IL-7 level changes during T-cell fate decision making.
By committing to the T-cell fate, progenitors suppress alternate fates and stably express T-cell identity genes, features that are subsequently maintained even after withdrawal of environmental signals. Stable maintenance of fate identity in committed progenitors is thought to arise through the action of positive feedback loops. These positive feedback loops are engaged or triggered in response to upstream signaling inputs (i.e. Notch and IL-7 signaling in the case of T-cell development), and stably maintain a cell regulatory state even if these signaling inputs are withdrawn. In a developmental context, positive feedback loops may consist of a cycle of activating connections, either from a single fate identity gene onto itself, or between multiple fate identity genes (Figure 2D, top). Such loops enable self-sustaining expression of fate-identity genes in the absence of signaling inputs, and occur frequently in embryonic gene regulatory networks [5, 37]. They have also been found in the context of early B-cell development [38, 39]. Alternatively, positive feedback loops may be built from mutually repressive connections between genes associated with alternate fates (Figure 2D, bottom). Several well-studied hematopoietic cell fate decisions can also be explained by minimal gene regulatory networks in which mutual repression between two transcriptional regulators forms the core. These apparently govern binary choices between erythroid and myeloid fates [3, 40, 41], between granulocyte and macrophage fates [4], and between myeloid and B-cell fates [42].
The decision of a precursor to become a T cell appears to be more complex in regulatory terms. In particular, more than one alternative fate is excluded by the T lineage commitment process, arguing against a simple binary switch decision mechanism. Even so, the early T-cell transcription factor repertoire includes regulatory genes that either activate other T-cell genes or repress non-T genes, and could thus participate in these positive feedback loops to maintain T-cell identity. Recent studies have identified the T-cell specific transcription factors TCF-1 and Bcl11b as key regulators of T-cell specification and commitment [28, 43-45]. Interestingly, these two transcription factors appear to perform complementary functions during T-cell development - TCF-1 acts to turn on T-cell specific genes, whereas Bcl11b may primarily function to repress alternate fate genes. However, it is still unclear how TCF-1, Bcl11b and other T-cell fate regulators interact with each other and work together on a circuit level to maintain fate identity in committed T-cell progenitors.
To gain insight into the regulatory gene circuits mediating the T-cell fate decision, we present here a new construction of the gene regulatory network that guides T-cell development. This reconstruction builds upon an earlier model of regulatory network interactions during T-cell commitment [46], and incorporates recently-identified genes and connections. This network reconstruction enables us to identify candidate regulatory circuits that may play roles in processing developmental signals and maintaining stable T-cell identity. We speculate on the functional significance of these regulatory circuits, and discuss further experiments that would enable us to examine their structure and function.
Data for a T-cell specification network model
T cell developmental progression depends not only on environmental signals, but also on temporally and quantitatively precise control of transcription factor activities. The same factors have different roles at different stages, and their effects in any given stage can depend sensitively upon level of expression. For example, several crucial regulatory genes are haploinsufficient, while the same factors that are required at a given stage can inhibit that same stage if they are overexpressed. This makes it challenging to design the experimental perturbations that are needed to evaluate regulatory network connections. Ideally, the immediate gene-expression consequences of changing the activity of a regulatory factor should be monitored within a well-defined developmental stage when the only difference from the control is the one targeted molecule. Ideally also, gain of function tests, which induce their effects quickly, should stay within a dosage range that is physiologically relevant, to avoid off-target effects.
The strategies used to generate samples for analysis of the T-cell gene network are summarized in Figure 3, where the caveats of different perturbation strategies within developmental stages of interest (Figure 3A) are noted. Germline or conditional knockout phenotypes in vivo are often regarded as a gold standard for assessing loss of function effects in otherwise normal conditions; however, the in vivo steady state is often complicated by compensatory mechanisms or more profound developmental deviation (Figure 3B, yellow circle), especially if deletion occurs long before the stage being assayed, and under these circumstances inference of regulatory gene connections is not reliable. In vitro differentiation systems for T-cell development allow for a wide variety of perturbations to be applied in a more stage-specific manner [47]. These systems allow for stage-specific addition or removal of environmental signals and gene over-expression, as well as deletion or neutralization of endogenous genes by transgenic expression of recombinase, retroviral introduction of recombinase, or introduction of shRNA (Figure 3C). Even in these more accessible systems, in order to identify direct regulatory connections reliably, the effects of such perturbations on downstream gene expression must be measured before uncontrolled changes in cell-type identity occur (Figure 3C, yellow circle). Measurements of gene expression changes in response to regulatory gene deletion or neutralization can be particularly difficult to time, as additional factors such as protein and mRNA stability may cause a time-delay in the loss of transcriptional factor activity.
Figure 3.

Strategies used for inferring regulatory gene connections. A) Natural developmental progression, showing developmental stages of interest. B) In vivo conditional or germline deletion experiments. Upper: ideal in vivo stage-specific gene deletion experiment, where development before the stage of interest is not compromised and compensatory mechanisms do not come into play. Lower: Complication of in vivo germline or conditional deletion experiments, if normal developmental sequence is deranged long before the stages of interest. Gene expression pattern in cell state generated as a result of altered developmental progression (yellow circle) is not strictly comparable with control. C) In vitro T-cell differentiation experiment. Short-term effects of stage-specific environmental signal or gene perturbations (gain or loss of function) remain comparable with controls. Altered cell state (yellow circle), in some cases transformation to a different lineage, can be generated due to prolonged effects of the applied perturbations. Gene expression in such cases may not simply reflect gain or loss of function of the targeted regulator. Dosage effects (not shown) can also yield developmentally inappropriate responses.
One recent tool that will have a large impact on network modeling is genome-wide analysis of transcription factor binding, as measured by chromatin immune precipitation and microarray hybridization (ChIP-chip) or deep sequencing (ChIP-seq) (e.g. [48-51]). The data are extremely useful to reveal where the binding sites may be through which a particular transcription factor acts on a particular target gene. However, binding alone does not tell whether a factor is acting as activator or repressor. Furthermore, it is emerging from these studies that there are often thousands of genomic sites bound by particular factors where, despite exquisite sequence-specificity, the factors exert no detectable regulatory influence at all. ChIP-chip and ChIP-seq evidence is still far from superseding the need to use actual experimental perturbations to determine a factor’s functional effects. It is the combination of direct, stage-specific measurements of protein/DNA binding together with functional data that will ultimately enable solution of the T-cell network in a definitive form.
The data sources used for this T-cell network reconstruction are listed in Supplementary Table 1, together with the approaches used to evaluate regulatory network linkages in each study [28, 35, 42, 44, 52-96]. In fact, all the available data sources on which T-cell network construction must currently be based include compromises with ideal conditions. As a result, the network relationships described here should still be regarded as a model and a guide to further experimental work.
Reconstruction of a T-Cell Developmental Network Model
The network model focuses on regulatory genes and connections that are active in the Phase 1 and 2 pro-T cell stages (Figure 1), i.e., from when cells begin to respond to Notch signals to after they commit to becoming T-cells. To build this network, we first searched the literature for recently-identified genes and connections, as listed in Table 1. We obtained most of the genes and connections for our regulatory network from experiments done on mouse models of fetal or adult haematopoiesis; we do note, however, that there may be differences in the regulatory circuit architecture that have arisen during the course of evolution or even between adult development and ontogeny. These nodes and edges were then combined with relationships from an earlier model of the network [46]. Network building was performed using Biotapestry [97, 98], a software for the representation and visualization of developmental gene regulatory networks.
Genes in the T-cell developmental network can be divided into the following groups broadly based on their function and expression patterns [13] (Figure 4): (1) Effectors or regulators of Notch, IL-7 and pre-TCR signaling, three signaling pathways that are important for early T-cell development (top). (2) Regulatory genes associated with T-cell developmental stage progression and fate commitment (middle). These genes are either up-regulated during the course of development (e.g. Bcl11b and Lef1) or kept at a constant expression level during the developmental stage progression (e.g. Ikaros and Runx1). (3) Regulatory genes that are down-regulated during T-cell development, and are associated with the either stem cells or non T-cell fates (bottom). Genes in this group generally antagonize T-cell developmental stage progression and commitment; however, some of them are also necessary for T-cell production in vivo as they play important roles at the stem or early progenitor stages (e.g. PU.1). Thus, the down-regulated group may actually comprise two subgroups: one consisting of the drivers of competing fates, but the other consisting of genes that sustain proliferation or other functions of early T cells while delaying commitment [13].
Figure 4.

Reconstruction of the T-Cell Developmental Gene Regulatory Network. Regulatory network encompasses Notch signaling (blue), IL-7 signaling (dark green), pre-TCR signaling (gray), T-cell regulatory genes (light green), and stem and alternate fate regulatory genes (red). T-cell regulatory genes sharply up-regulated prior to commitment are enclosed in a yellow box. Genes and connections active during the earlier T-cell developmental stages are placed to the left, whereas genes and connections active during later stages are placed to the right. The prevailing connections between the genes in different groups and their signs are summarized in the inset. An online version of this network is available at http://www.its.caltech.edu/~tcellgrn/.
Genes within each of these functional groups are further arranged from left to right based on the temporal sequence of expression and/or activity level changes during T-cell development (Figure 4). At the top left are Notch and IL-7 signaling pathway components (e.g. IL-7Rα and Notch1), which are active prior to T-cell commitment (Phase 1, Figure 4); components required for the pre-TCR signaling pathway (e.g. pTα, Tcr-β and Lck), which are T-cell specific genes turned on only after commitment (Phase 2, Figure 4), are placed to the right. Within the T-cell regulatory gene group (middle), the left hand cluster contains genes already expressed prior to commitment (e.g. Gata3, Ikaros and Runx1), while genes up-regulated after commitment are placed towards the right (e.g. Id3 and Ets1). A number of regulatory genes are up-regulated sharply immediately prior to commitment (e.g. Bcl11b and HEB-Alt); these genes, poised to promote T-cell fate commitment, are placed in the center and boxed. Stem and alternate fate regulatory genes are placed at the bottom, with those already being silenced prior to commitment to the left (e.g. Gata2, C/EBPα and Ebf1), and those not down-regulated until after commitment in the center and right (e.g. PU.1, Lyl1 and Kit).
We note that, besides Notch and IL-7, a number of other cytokine signaling pathways – such as the Kit or the Flt3 pathway – may also be important for the viability and proliferation of uncommitted Phase I progenitors (Figure 1). All stages of T-cell development require viability functions, however, and in this network reconstruction, we have not included cell cycle or apoptosis genes regulated by these pathways (or by the Notch or IL-7 pathways themselves). This network model instead focuses on the regulatory genes that have been implicated in the commitment transition itself. Nevertheless, Kit signaling could also contribute to the Stat activation function in parallel with IL7Rα in Phase 1 cells. Similarly, although the crucial T-lineage transcription factors TCF-1 (encoded by Tcf7) and Lef1 have important roles as mediators of Wnt/β-catenin signaling, their roles in early T-cell development may not depend on this pathway [99], and so Wnt pathway inputs have also been omitted.
Genes in the T-cell developmental network interact extensively with one another, and specific patterns of connections within or between functional groups can be observed (see Figure 4 inset for a summary). The Notch-Delta signaling pathway provides direct or indirect positive regulatory inputs into many T-cell regulatory genes, including those activated at the earliest stages, consistent with its role in promoting T-cell fate. It also positively regulates genes encoding IL-7, Notch and pre-TCR signaling pathway components. Nevertheless, some pre-TCR signaling pathway genes, although directly targeted by Notch, turn on only after commitment. These reflect a requirement for persistent, sustained Notch signals to complete proper T-cell fate commitment, most likely enforced by network subcircuit architecture as described below. In contrast, IL-7 signaling may negatively regulate T-cell regulatory genes (Bcl11b, TCF-1 and Lef1) in uncommitted DN2 cells, either directly or indirectly, as well as positively regulate stem and alternate fate genes (Kit, PU.1 and C/EBPα), consistent with its role in antagonizing or at least delaying commitment [28].
T-cell regulatory genes generally provide positive inputs into Notch and pre-TCR signaling genes (e.g. Gata3–Notch3; E protein activity–pTα; Runx1-Tcr–β), positive inputs into other T-cell regulatory genes (e.g. TCF-1–Bcl11b; HEBAlt–HEBAlt), and/or negative inputs into stem and alternate fate genes (e.g. Bcl11b–Id2; Gata3–PU.1, Runx1–PU.1), consistent with their roles in promoting the T-cell fate and repressing alternate cell fates. However, we found that several T-cell regulatory genes, most notably the crucial transcription factor Gata3, can also form negative connections with other T-cell regulatory genes (e.g. Gata3–Lef1, Gata3–TCF-1, E protein activity–Gata3), while forming positive connections with stem and alternate fate genes (e.g. Gata3–Scl/Tal1, Gata3–Kit, Gata3–Cpa3), especially when overexpressed. Such ‘incoherent’ connections may constitute part of the regulatory circuitry involved in sensing IL-7 signal changes during development (see below). Stem and alternate fate regulatory genes generally form negative connections with T-cell regulatory genes and pre-TCR signaling genes (e.g. Gfi1b–Gata3; PU.1–Bcl11b; Scl/Tal1–pTα) and positive connections amongst themselves (PU.1–Bcl11a; Lmo2–Lyl1), consistent with their role in delaying or antagonizing the T-cell fate and promoting stem and alternate fates.
Regulatory Circuits Underlying the T-Cell Fate Decision
The reconstructed T-cell gene regulatory network presented above enables us to identify regulatory circuits that are important for T-cell fate decision making. In this section, we use the reconstructed network to identify candidate regulatory circuits that may facilitate three crucial aspects of the fate decision making process: 1) Detection of persistence in Notch-Delta signaling in early multipotent progenitors, 2) Detection of dynamic changes in IL-7 signaling in uncommitted T-cell progenitors, and 3) Maintenance of T-cell identity and silencing of alternate cell fates in committed progenitors.
Detection of Persistence in Notch Signaling
Proper commitment to the T-cell fate requires sustained Notch-Delta signaling throughout the Phase 1 stage. Transient Notch-Delta signaling causes up-regulation of early T-cell genes, but is insufficient for promoting commitment [14, 15]. Detection of such signal persistence can be performed by coherent feed-forward loops, which can generate time delays using multiple inputs from a signal onto a target gene. In the coherent feedforward loop example shown (Figure 2A), a signal S provides a positive input into a target gene Y, and also into another gene X that provides another input into Y. In order for Y to be completely unresponsive to transient signals, it must be respond to X and S with strict AND logic (that is, both S and X must be on for Y to be on). We note, however, that persistence detection can still take place even if X and S do not operate in such a boolean manner, but synergize to up-regulate Y; or, alternatively, if heightened threshold levels of Y, attained only when X and S are both present, are required for promoting downstream developmental transitions.
Our regulatory network reconstruction reveals several coherent feed-forward loops that may mediate the response of T-cell regulatory genes to Notch signaling. Bcl11b, an important regulator of T-cell commitment, is thought to be a direct Notch target [44]; however, it remains off even when other Notch target genes such as CD25 and Hes1 have already turned on in response to Notch-Delta signaling [15, 28, 100], and turns on only immediately prior to commitment. In our network re-construction, the Notch signaling-induced gene TCF-1 provides a second arm of positive inputs from Notch to Bcl11b (Figure 5A, left), which may provide the time-delayed inputs that enable Bcl11b to respond only after sustained and persistent Notch signaling. There is also a similar coherent feed-forward loop regulating GATA3 expression (Figure 5A) 1, which may possibly mediate the up-regulation of GATA3 that occurs post-commitment [101]. We also found that the T-cell identity genes pTα and Rag-1 may also be subject to coherent feedforward regulation through E protein activity (Figure 5A, right). In a third possible example (not shown), Notch-induced TCF-1 can provide direct positive input into a major promoter of the Notch-dependent gene Lef1 [102]. This regulatory circuit architecture may explain why these genes, though being Notch targets, do not turn on until after T-cell commitment. Interestingly, TCRβ, another T-cell identity gene that is up-regulated only after commitment, does not appear to receive inputs directly from Notch signaling, but only indirectly through Gata3 (Figure 5A, right), suggesting that not all T-cell identity genes may be subject to such coherent feedforward regulation.
Figure 5.

Regulatory Gene Circuits Underlying T-Cell Fate Decision Making. A) Coherent feedforward loops for detection of persistent Notch signaling. Shown here are coherent feedforward loops mediating inputs from Notch signaling to Bcl11b (left), as well as to CD25, pTa and Rag1 (right). B) Incoherent feedforward loops and negative feedback loops for detection of IL-7 signal level changes. Shown here is a negative feedback loop regulating expression of IL-7Ra (top), and an incoherent feedforward loop regulating expression of Tcf7 and Lef1 (bottom). C) Positive feedback loops for T-cell fate commitment and exclusion of alternate fates. Shown here are a series of positive feedback loops involving mutual repression between PU.1 and the T-cell regulators TCF-1, Bcl11b, E2A/HEB and Gfi1. Note that TCF-1, Bcl11b, PU.1, HEBAlt and E2A also receive inputs from Notch and/or IL-7 signaling and may hence mediate engagement of the positive feedback loops in response to upstream signals.
To establish whether a given gene is indeed subject to such coherent feed-forward regulation, we need to demonstrate that it receives two independent inputs, and that it turns on only when both are present. As evidence for direct regulation is currently lacking in many of the feedforward inputs discussed above (Figure 5, dashed and dotted lines), it is not known whether the two inputs into the target genes are indeed independent --- an apparent feedforward input into a target gene could turn out to be an indirect input mediated completely through the second coincident input. We will discuss experiments that could distinguish between these two possibilities below.
Detection of Dynamic IL-7 Signaling
Besides persistent Notch signaling, recent work suggests that a drop in IL-7 signal levels can promote fate commitment in uncommitted DN2 progenitors [28]. Detecting input signal changes requires circuits that can adapt to different baseline levels and thus maintain sensitivity to signal level changes over a range of signal strengths. Two types of regulatory circuits are capable of undergoing such adaptation – negative feedback loops and incoherent feedforward loops [7]. Negative feedback loops perform adaptation by altering a signaling pathway’s sensitivity to signal inputs in response to the input signal itself. Negative feedback connections can occur directly through biochemical interactions within a signaling pathway, as is frequently observed [103], or indirectly through transcriptional regulation of signaling pathway components. In an example of the latter (Figure 2B), signal S provides a positive input into X, which is an inhibitory component of the S signaling pathway. Incoherent feedforward loops perform adaptation through independent opposing inputs from a signal onto a target gene. In the example shown (Figure 2C), gene Y receives a positive input from a signal S, but also receives a negative input from gene X, which also receives a positive input from S. These two opposing regulatory inputs into Y can cancel each other out at steady state, allowing the circuit to maintain a constant level of Y amidst different baseline values of S, and hence maintain sensitivity to signal level changes over a range of signal changes.
Negative feedback loops in IL-7 signaling may occur either within the signaling pathway itself or on a transcriptional level. Negative feedback within the IL-7 signaling pathway could be mediated by SOCS1, a general suppressor of cytokine signaling that is induced in T-cell precursors in response to IL-7 [104]. An alternative mode of negative feedback may involve internalization and degradation of the IL-7 receptor, which has been shown to be triggered by IL-7 ligand binding [105, 106]. Examination of the regulatory network also revealed a potential transcriptional circuit that involves negative feedback regulation of IL-7Rα, the receptor chain that receives IL-7 signals (Figure 5B, top). IL-7 signaling may provide a positive input into GATA-3. GATA-3 in turn appears to provide negative regulatory inputs into IL-7Rα [69, 81], thereby providing a negative modulatory feedback into the IL-7 pathway. Negative feedback regulation of IL-7Rα expression at the transcriptional level has also been observed in mature T-cells [106, 107], as a prominent response triggered either by contact with IL-7 or during T-cell receptor activation by antigen. Such negative feedback connections may help the IL-7 signaling pathway adapt to different IL-7 baseline levels and maintain sensitivity to changes in IL-7 levels2.
From our network reconstruction, we also found examples of the kinds of incoherent feedforward circuits that may potentially facilitate IL-7 signal adaptation on the level of T-cell regulatory gene expression (Figure 5B, bottom). IL-7 signaling can provide negative inputs into TCF-1, either indirectly or through Gata3 itself. However, our network reconstruction suggests the existence of other net positive inputs from IL-7 signaling to TCF-1, for example through Gata3-mediated antagonism of PU.1. Such opposing inputs would form incoherent feedforward loops that may allow the expression of TCF-1 to adapt in response to changing IL-7 levels.
The regulatory circuits discussed here respond to signal level changes in only a transient manner – in the absence of other regulatory mechanisms, these circuits could eventually undergo adaptation and reset their regulatory state around the new baseline signal levels. To carry out a commitment decision, progenitors need to convert these transient responses into stable and irreversible changes in their regulatory state, as is the case when progenitors transition across developmental stages and make fate commitment decisions. In the next section, we examine and discuss regulatory circuits that may enable progenitors to undergo such stable and irreversible commitment to the T-cell fate.
Commitment and Maintenance of T-Cell Identity
In response to appropriate Notch and IL-7 signals, progenitors switch irreversibly into a committed T-cell regulatory state, where all other non T-cell fates are silenced. This regulatory state is upheld throughout all subsequent stages of T-cell development and effector function, and is stably maintained even when initial Notch and IL-7 signals are no longer present. As noted above, stable maintenance of fate identity is likely to involve action of positive feedback loops, which are initially triggered by upstream signals and then maintain a committed cellular state upon signal withdrawal. Positive feedback loops can consist of a cycle of positive connections; as an example, gene X provides a positive input to gene Y, which in turn provides a positive input back to X (Figure 2D, top). When X turns on in response to a signal S, it up-regulates Y, which in turn feeds back to up-regulate X, allowing it to remain on even when the signal S has been withdrawn. Alternatively, a feedback loop that is net positive in sign can arise from mutually repressive connections between pairs of genes; in this case, X provides a negative input to Z, which in turn provides a negative input back to X (Figure 2D, bottom). Activation of X by S causes down-regulation of Z, which in turn relieves X of repression by Z. This positive feedback into X allows the circuit to stably express X while keeping Z silent at the same time. In context of cell fate decision making, X and Z usually represent opposite fate regulatory genes that are expressed in a mutually exclusive manner.
The T-cell gene regulatory network revealed a number of candidate positive feedback circuits that may play a role in enforcing stable T-cell fate commitment. However, while positive connections exist between different T-cell regulators (e.g. TCF-1–GATA3, TCF-1–Bcl11b) [45], we could not find complete positive feedback loops consisting of cycles of activating connections between the T-cell regulatory genes themselves that have been reported to date. The only apparent exceptions are the positive auto-regulatory loops involving HEBAlt or TCF-1. The dearth of known positive loops between T-cell regulatory genes, which is apparent from the network reconstruction (Figure 4), stands in contrast to other developmental systems, notably the B-cell developmental network, where key fate regulators positively regulate each other [38, 108]. Instead, most positive feedback loops here involve mutual repression between T-cell regulatory genes and stem cell or alternate fate genes (Figure 5C). Gfi1, TCF-1 and Bcl11b all form mutually antagonistic loops with PU.1 (Figure 5C). E proteins may also participate in some of these positive feedback loops – HEB/E2A dimers positively regulate Gfi1, and are also negatively regulated by PU.1, either through transcriptional repression or through up-regulation of Id2, an E protein inhibitor and PU.1 target. Mutual antagonism between these T-cell regulatory genes and PU.1 may enable progenitors to maintain a stable regulatory state where T-cell identity genes are expressed, and stem and alternate fate genes such as PU.1 are silenced. As many of the T-cell regulatory genes discussed here also participate in IL-7 and Notch signal detection (see above), they are good candidates for mediating commitment in response to upstream signals.
Conclusion and Future Directions
Here, we have presented a new model of the gene regulatory network underlying T-cell development in haematopoietic progenitor cells. Using this network reconstruction, we have identified candidate regulatory gene circuits for facilitating the following aspects of the T-cell fate decision making process: 1) Detection of persistence in Notch-Delta signaling, 2) Detection of IL-7 signal level changes, and 3) Maintenance of T-cell identity and silencing of alternate cell fates upon commitment. These regulatory circuits provide a basis for understanding how regulatory genes work together to mediate a developmental cell fate decision.
To further understand these regulatory gene circuits, experiments will need to be performed that probe their structure and function. The gene circuits we describe here contain many connections not yet tested on a cis-regulatory level (Figure 4, dashed and dotted lines). To validate regulatory gene circuit structure, it will be necessary to determine whether these connections indeed represent direct regulatory inputs; if not, the identities of the regulatory genes that do mediate these indirect connections will need to be resolved. This knowledge is particularly important for validating the structure of coherent feedforward loops (Figure 5A), where an apparent feedforward input into a gene could in fact be mediated completely through the second coincident input. One way to test whether a transcription factor targets a gene directly is to determine whether it binds to sites on the target gene locus. Transcription factor binding sites can now be identified on a genome-wide level using chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-Seq), and this approach has been used to elucidate the structure of the regulatory networks underlying B- and myeloid cell development [50, 51]. We are currently using ChIP-Seq to determine the binding sites and putative target genes of different transcription factors at several finely-divided sub-stages in T-cell development (J. Zhang, A. Mortazavi, B. Williams, B. J. Wold, and E. V. Rothenberg, in preparation). This approach is yielding insights not only into the structure of the T-cell regulatory network, but also its dynamic behavior during the process of fate commitment.
Validating a direct regulatory connection between a transcription factor and its putative target requires not only evidence for transcription factor binding on the gene locus, but also evidence that the binding site is active during development. To show that a binding site is active, it is important to show that its mutation or removal affects proper regulation of the target gene. Binding site mutagenesis can be performed on endogenous gene loci using gene targeting [109], or, alternatively, on reporter transgenes [110]. The transcription factor itself also needs to be shown to act on the binding site to control target gene expression, and to exert its gain and loss of function effects in a target element-dependent way. These types of experiments are needed to validate the structure of the proposed regulatory circuits, and also to identify new circuits possibly involved in fate decision making.
To examine the function of these regulatory gene circuits, gene circuit activity will eventually need to be followed closely over time, under conditions where input signal levels and/or regulatory gene expression levels are varied in a quantitative manner. Such close monitoring of gene circuit activity has been greatly facilitated by the development of fluorescent gene expression reporters, which allow expression levels of individual genes to be measured in single cells in a quantitative and non-invasive manner [111, 112]. For regulatory circuits involved in processing Notch-Delta or IL-7 signals, measurements of their input/output relationships will yield insights into the signal processing operations that they perform. Furthermore, input/output measurements performed in conjunction with gene perturbations that alter connectivity of these circuits will enable direct tests of the roles of circuit structure in signal processing. Similar measurements will also yield insights into the behavior of positive feedback circuits mediating cell fate commitment. For example, signal levels can be experimentally varied and the effects on the levels of gene expression within the positive feedback loop measured, to assess the stability of a positive feedback loop to fluctuating signals. Furthermore, by perturbing genes and connections within the positive feedback loop and measuring resultant changes in circuit activity, it will be possible to determine their roles in maintaining regulatory state stability. Such experiments will help to develop an understanding of how regulatory gene circuits mediate different aspects of the cell fate decision making process, and more generally, how they function during mammalian development.
Supplementary Material
Acknowledgements
We particularly thank Avinash Bhandoola and Brittany Weber (University of Pennsylvania), for sharing of data before publication and valuable criticism of our models. We also thank Rothenberg lab members for useful advice and discussions, as well as Sagar Damle and Bill Longabaugh for advice on Biotapestry. We also thank Mary Yui for a critical reading of this manuscript. This work was supported by a CRI/Irvington Postdoctoral Fellowship to H.Y.K., and NIH grants to E.V.R (RC2 CA148278, R33 HL089123, and R01 CA90233), the Albert Billings Ruddock Professorship, the Al Sherman Foundation, and the Louis A. Garfinkle Memorial Laboratory Fund.
Footnotes
Regulation of GATA3 by Notch signaling may be indirect and/or specific to a mouse context, as studies using human thymocytes did not identify GATA3 as a Notch target [111-112].
We note that IL-7Rα expression levels eventually drop after commitment as the cells pass the first T-cell receptor-dependent checkpoint to exit from the late DN3 stage. The negative feedback circuits presented here do not explain this post-commitment drop IL-7Rα receptor levels, which more likely arises as a result of cellular responses to T-cell receptor triggering.
Contributor Information
Hao Yuan Kueh, Division of Biology 156-29, California Institute of Technology, Pasadena, CA 91125 USA; kueh@caltech.edu.
Ellen V. Rothenberg, Division of Biology 156-29, California Institute of Technology, Pasadena, CA 91125 USA; evroth@its.caltech.edu
References
- 1.Shoval O, Alon U. SnapShot: network motifs. Cell. 2010;143(2):326–e1. doi: 10.1016/j.cell.2010.09.050. [DOI] [PubMed] [Google Scholar]
- 2.Xiong W, Ferrell JE., Jr. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature. 2003;426(6965):460–5. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- 3.Cantor AB, Orkin SH. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene. 2002;21(21):3368–76. doi: 10.1038/sj.onc.1205326. [DOI] [PubMed] [Google Scholar]
- 4.Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126(4):755–66. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 5.Levine M, Davidson EH. Gene regulatory networks for development. Proc Natl Acad Sci U S A. 2005;102(14):4936–42. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goentoro L, Shoval O, Kirschner MW, Alon U. The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol Cell. 2009;36(5):894–9. doi: 10.1016/j.molcel.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138(4):760–73. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krejci A, Bernard F, Housden BE, Collins S, Bray SJ. Direct response to Notch activation: signaling crosstalk and incoherent logic. Sci Signal. 2009;2(55):ra1. doi: 10.1126/scisignal.2000140. [DOI] [PubMed] [Google Scholar]
- 9.Cohen-Saidon C, Cohen AA, Sigal A, Liron Y, Alon U. Dynamics and variability of ERK2 response to EGF in individual living cells. Mol Cell. 2009;36(5):885–93. doi: 10.1016/j.molcel.2009.11.025. [DOI] [PubMed] [Google Scholar]
- 10.Sambandam A, Maillard I, Zediak VP, Xu L, Gerstein RM, Aster JC, Pear WS, Bhandoola A. Notch signaling controls the generation and differentiation of early T lineage progenitors. Nat Immunol. 2005;6(7):663–70. doi: 10.1038/ni1216. [DOI] [PubMed] [Google Scholar]
- 11.Koch U, Fiorini E, Benedito R, Besseyrias V, Schuster-Gossler K, Pierres M, Manley NR, Duarte A, Macdonald HR, Radtke F. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J Exp Med. 2008;205(11):2515–23. doi: 10.1084/jem.20080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yui MA, Feng N, Rothenberg EV. Fine-scale staging of T cell lineage commitment in adult mouse thymus. J Immunol. 2010;185(1):284–93. doi: 10.4049/jimmunol.1000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothenberg EV, Zhang J, Li L. Multilayered specification of the T-cell lineage fate. Immunol Rev. 2010;238(1):150–68. doi: 10.1111/j.1600-065X.2010.00964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt TM, Ciofani M, Petrie HT, Zuniga-Pflucker JC. Maintenance of T cell specification and differentiation requires recurrent notch receptor-ligand interactions. J Exp Med. 2004;200(4):469–79. doi: 10.1084/jem.20040394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taghon TN, David ES, Zuniga-Pflucker JC, Rothenberg EV. Delayed, asynchronous, and reversible T-lineage specification induced by Notch/Delta signaling. Genes Dev. 2005;19(8):965–78. doi: 10.1101/gad.1298305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinzel K, Benz C, Martins VC, Haidl ID, Bleul CC. Bone marrow-derived hemopoietic precursors commit to the T cell lineage only after arrival in the thymic microenvironment. J Immunol. 2007;178(2):858–68. doi: 10.4049/jimmunol.178.2.858. [DOI] [PubMed] [Google Scholar]
- 17.Wada H, Masuda K, Satoh R, Kakugawa K, Ikawa T, Katsura Y, Kawamoto H. Adult T-cell progenitors retain myeloid potential. Nature. 2008;452(7188):768–72. doi: 10.1038/nature06839. [DOI] [PubMed] [Google Scholar]
- 18.Bell JJ, Bhandoola A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature. 2008;452(7188):764–7. doi: 10.1038/nature06840. [DOI] [PubMed] [Google Scholar]
- 19.Porritt HE, Rumfelt LL, Tabrizifard S, Schmitt TM, Zuniga-Pflucker JC, Petrie HT. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity. 2004;20(6):735–45. doi: 10.1016/j.immuni.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Carlyle JR, Michie AM, Furlonger C, Nakano T, Lenardo MJ, Paige CJ, Zuniga-Pflucker JC. Identification of a novel developmental stage marking lineage commitment of progenitor thymocytes. J Exp Med. 1997;186(2):173–82. doi: 10.1084/jem.186.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikawa T, Kawamoto H, Fujimoto S, Katsura Y. Commitment of common T/Natural killer (NK) progenitors to unipotent T and NK progenitors in the murine fetal thymus revealed by a single progenitor assay. J Exp Med. 1999;190(11):1617–26. doi: 10.1084/jem.190.11.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodewald HR, Moingeon P, Lucich JL, Dosiou C, Lopez P, Reinherz EL. A population of early fetal thymocytes expressing Fc gamma RII/III contains precursors of T lymphocytes and natural killer cells. Cell. 1992;69(1):139–50. doi: 10.1016/0092-8674(92)90125-v. [DOI] [PubMed] [Google Scholar]
- 23.Rothenberg EV. T cell lineage commitment: identity and renunciation. J Immunol. 2011;186(12):6649–55. doi: 10.4049/jimmunol.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangan S, Zaslaver A, Alon U. The coherent feedforward loop serves as a sign-sensitive delay element in transcription networks. J Mol Biol. 2003;334(2):197–204. doi: 10.1016/j.jmb.2003.09.049. [DOI] [PubMed] [Google Scholar]
- 25.Swiers G, Patient R, Loose M. Genetic regulatory networks programming hematopoietic stem cells and erythroid lineage specification. Dev Biol. 2006;294(2):525–40. doi: 10.1016/j.ydbio.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 26.Massa S, Balciunaite G, Ceredig R, Rolink AG. Critical role for c-kit (CD117) in T cell lineage commitment and early thymocyte development in vitro. Eur J Immunol. 2006;36(3):526–32. doi: 10.1002/eji.200535760. [DOI] [PubMed] [Google Scholar]
- 27.Jensen CT, Boiers C, Kharazi S, Lubking A, Ryden T, Sigvardsson M, Sitnicka E, Jacobsen SE. Permissive roles of hematopoietin and cytokine tyrosine kinase receptors in early T-cell development. Blood. 2008;111(4):2083–90. doi: 10.1182/blood-2007-08-108563. [DOI] [PubMed] [Google Scholar]
- 28.Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, Kominami R, Katsura Y, Kawamoto H. An essential developmental checkpoint for production of the T cell lineage. Science. 2010;329(5987):93–6. doi: 10.1126/science.1188995. [DOI] [PubMed] [Google Scholar]
- 29.Schlenner SM, Madan V, Busch K, Tietz A, Laufle C, Costa C, Blum C, Fehling HJ, Rodewald HR. Fate mapping reveals separate origins of T cells and myeloid lineages in the thymus. Immunity. 2010;32(3):426–36. doi: 10.1016/j.immuni.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Allman D, Sambandam A, Kim S, Miller JP, Pagan A, Well D, Meraz A, Bhandoola A. Thymopoiesis independent of common lymphoid progenitors. Nat Immunol. 2003;4(2):168–74. doi: 10.1038/ni878. [DOI] [PubMed] [Google Scholar]
- 31.Ceredig R, Rolink T. A positive look at double-negative thymocytes. Nat Rev Immunol. 2002;2(11):888–97. doi: 10.1038/nri937. [DOI] [PubMed] [Google Scholar]
- 32.von Freeden-Jeffry U, Solvason N, Howard M, Murray R. The earliest T lineage-committed cells depend on IL-7 for Bcl-2 expression and normal cell cycle progression. Immunity. 1997;7(1):147–54. doi: 10.1016/s1074-7613(00)80517-8. [DOI] [PubMed] [Google Scholar]
- 33.Balciunaite G, Ceredig R, Fehling HJ, Zuniga-Pflucker JC, Rolink AG. The role of Notch and IL-7 signaling in early thymocyte proliferation and differentiation. Eur J Immunol. 2005;35(4):1292–300. doi: 10.1002/eji.200425822. [DOI] [PubMed] [Google Scholar]
- 34.Wojciechowski J, Lai A, Kondo M, Zhuang Y. E2A and HEB are required to block thymocyte proliferation prior to pre-TCR expression. J Immunol. 2007;178(9):5717–26. doi: 10.4049/jimmunol.178.9.5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braunstein M, Anderson MK. HEB-deficient T-cell precursors lose T-cell potential and adopt an alternative pathway of differentiation. Mol Cell Biol. 2011;31(5):971–82. doi: 10.1128/MCB.01034-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7(2):144–54. doi: 10.1038/nri2023. [DOI] [PubMed] [Google Scholar]
- 37.Peter IS, Davidson EH. Modularity and design principles in the sea urchin embryo gene regulatory network. FEBS Lett. 2009;583(24):3948–58. doi: 10.1016/j.febslet.2009.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh H, Medina KL, Pongubala JM. Contingent gene regulatory networks and B cell fate specification. Proc Natl Acad Sci U S A. 2005;102(14):4949–53. doi: 10.1073/pnas.0500480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandel EM, Grosschedl R. Transcription control of early B cell differentiation. Curr Opin Immunol. 2010;22(2):161–7. doi: 10.1016/j.coi.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 40.Chickarmane V, Enver T, Peterson C. Computational modeling of the hematopoietic erythroid-myeloid switch reveals insights into cooperativity, priming, and irreversibility. PLoS Comput Biol. 2009;5(1):e1000268. doi: 10.1371/journal.pcbi.1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laiosa CV, Stadtfeld M, Graf T. Determinants of lymphoid-myeloid lineage diversification. Annu Rev Immunol. 2006;24:705–38. doi: 10.1146/annurev.immunol.24.021605.090742. [DOI] [PubMed] [Google Scholar]
- 42.Spooner CJ, Cheng JX, Pujadas E, Laslo P, Singh H. A recurrent network involving the transcription factors PU.1 and Gfi1 orchestrates innate and adaptive immune cell fates. Immunity. 2009;31(4):576–86. doi: 10.1016/j.immuni.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science. 2010;329(5987):89–93. doi: 10.1126/science.1188989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, Lu D, Campos L, Goulding D, Ng BL, Dougan G, Huntly B, Gottgens B, Jenkins NA, Copeland NG, Colucci F, Liu P. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science. 2010;329(5987):85–9. doi: 10.1126/science.1188063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber BN, Chi AW, Chavez A, Y Y-O, Yang Q, Shestova O, Bhandoola A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476(7358):63–68. doi: 10.1038/nature10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Georgescu C, Longabaugh WJ, Scripture-Adams DD, David-Fung ES, Yui MA, Zarnegar MA, Bolouri H, Rothenberg EV. A gene regulatory network armature for T lymphocyte specification. Proc Natl Acad Sci U S A. 2008;105(51):20100–5. doi: 10.1073/pnas.0806501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmitt TM, Zuniga-Pflucker JC. T-cell development, doing it in a dish. Immunol Rev. 2006;209:95–102. doi: 10.1111/j.0105-2896.2006.00353.x. [DOI] [PubMed] [Google Scholar]
- 48.Young RA. Control of the embryonic stem cell state. Cell. 2011;144(6):940–54. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oram SH, Thoms JA, Pridans C, Janes ME, Kinston SJ, Anand S, Landry JR, Lock RB, Jayaraman PS, Huntly BJ, Pimanda JE, Gottgens B. A previously unrecognized promoter of LMO2 forms part of a transcriptional regulatory circuit mediating LMO2 expression in a subset of T-acute lymphoblastic leukaemia patients. Oncogene. 2010;29(43):5796–808. doi: 10.1038/onc.2010.320. [DOI] [PubMed] [Google Scholar]
- 50.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, Ideker T, Glass CK, Murre C. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11(7):635–43. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kastner P, Chan S, Vogel WK, Zhang LJ, Topark-Ngarm A, Golonzhka O, Jost B, Le Gras S, Gross MK, Leid M. Bcl11b represses a mature T-cell gene expression program in immature CD4(+)CD8(+) thymocytes. Eur J Immunol. 2010;40(8):2143–54. doi: 10.1002/eji.200940258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Izon DJ, Aster JC, He Y, Weng A, Karnell FG, Patriub V, Xu L, Bakkour S, Rodriguez C, Allman D, Pear WS. Deltex1 redirects lymphoid progenitors to the B cell lineage by antagonizing Notch1. Immunity. 2002;16(2):231–43. doi: 10.1016/s1074-7613(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 54.Jia J, Dai M, Zhuang Y. E proteins are required to activate germline transcription of the TCR Vbeta8.2 gene. Eur J Immunol. 2008;38(10):2806–20. doi: 10.1002/eji.200838144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gregoire JM, Romeo PH. T-cell expression of the human GATA-3 gene is regulated by a non-lineage-specific silencer. J Biol Chem. 1999;274(10):6567–78. doi: 10.1074/jbc.274.10.6567. [DOI] [PubMed] [Google Scholar]
- 56.Xu W, Kee BL. Growth factor independent 1B (Gfi1b) is an E2A target gene that modulates Gata3 in T-cell lymphomas. Blood. 2007;109(10):4406–14. doi: 10.1182/blood-2006-08-043331. [DOI] [PubMed] [Google Scholar]
- 57.Ikawa T, Kawamoto H, Goldrath AW, Murre C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. J Exp Med. 2006;203(5):1329–42. doi: 10.1084/jem.20060268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwartz R, Engel I, Fallahi-Sichani M, Petrie HT, Murre C. Gene expression patterns define novel roles for E47 in cell cycle progression, cytokine-mediated signaling, and T lineage development. Proc Natl Acad Sci U S A. 2006;103(26):9976–81. doi: 10.1073/pnas.0603728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Claus CL, Vaccarelli G, Braunstein M, Schmitt TM, Zuniga-Pflucker JC, Rothenberg EV, Anderson MK. The basic helix-loop-helix transcription factor HEBAlt is expressed in pro-T cells and enhances the generation of T cell precursors. J Immunol. 2006;177(1):109–19. doi: 10.4049/jimmunol.177.1.109. [DOI] [PubMed] [Google Scholar]
- 60.Dias S, Mansson R, Gurbuxani S, Sigvardsson M, Kee BL. E2A proteins promote development of lymphoid-primed multipotent progenitors. Immunity. 2008;29(2):217–27. doi: 10.1016/j.immuni.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yashiro-Ohtani Y, He Y, Ohtani T, Jones ME, Shestova O, Xu L, Fang TC, Chiang MY, Intlekofer AM, Blacklow SC, Zhuang Y, Pear WS. Pre-TCR signaling inactivates Notch1 transcription by antagonizing E2A. Genes Dev. 2009;23(14):1665–76. doi: 10.1101/gad.1793709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.San-Marina S, Han Y, Suarez Saiz F, Trus MR, Minden MD. Lyl1 interacts with CREB1 and alters expression of CREB1 target genes. Biochim Biophys Acta. 2008;1783(3):503–17. doi: 10.1016/j.bbamcr.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 63.Donaldson IJ, Chapman M, Kinston S, Landry JR, Knezevic K, Piltz S, Buckley N, Green AR, Gottgens B. Genome-wide identification of cis-regulatory sequences controlling blood and endothelial development. Hum Mol Genet. 2005;14(5):595–601. doi: 10.1093/hmg/ddi056. [DOI] [PubMed] [Google Scholar]
- 64.Wilson NK, Miranda-Saavedra D, Kinston S, Bonadies N, Foster SD, Calero-Nieto F, Dawson MA, Donaldson IJ, Dumon S, Frampton J, Janky R, Sun XH, Teichmann SA, Bannister AJ, Gottgens B. The transcriptional program controlled by the stem cell leukemia gene Scl/Tal1 during early embryonic hematopoietic development. Blood. 2009;113(22):5456–65. doi: 10.1182/blood-2009-01-200048. [DOI] [PubMed] [Google Scholar]
- 65.Lecuyer E, Herblot S, Saint-Denis M, Martin R, Begley CG, Porcher C, Orkin SH, Hoang T. The SCL complex regulates c-kit expression in hematopoietic cells through functional interaction with Sp1. Blood. 2002;100(7):2430–40. doi: 10.1182/blood-2002-02-0568. [DOI] [PubMed] [Google Scholar]
- 66.Herblot S, Steff AM, Hugo P, Aplan PD, Hoang T. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain expression. Nat Immunol. 2000;1(2):138–44. doi: 10.1038/77819. [DOI] [PubMed] [Google Scholar]
- 67.Landry JR, Kinston S, Knezevic K, de Bruijn MF, Wilson N, Nottingham WT, Peitz M, Edenhofer F, Pimanda JE, Ottersbach K, Gottgens B. Runx genes are direct targets of Scl/Tal1 in the yolk sac and fetal liver. Blood. 2008;111(6):3005–14. doi: 10.1182/blood-2007-07-098830. [DOI] [PubMed] [Google Scholar]
- 68.Kim WY, Sieweke M, Ogawa E, Wee HJ, Englmeier U, Graf T, Ito Y. Mutual activation of Ets-1 and AML1 DNA binding by direct interaction of their autoinhibitory domains. EMBO J. 1999;18(6):1609–20. doi: 10.1093/emboj/18.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taghon T, Yui MA, Rothenberg EV. Mast cell lineage diversion of T lineage precursors by the essential T cell transcription factor GATA-3. Nat Immunol. 2007;8(8):845–55. doi: 10.1038/ni1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang HC, Perry SS, Sun XH. Id1 attenuates Notch signaling and impairs T-cell commitment by elevating Deltex1 expression. Mol Cell Biol. 2009;29(17):4640–52. doi: 10.1128/MCB.00119-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang XO, Doty RT, Hicks JS, Willerford DM. Regulation of T-cell receptor D beta 1 promoter by KLF5 through reiterated GC-rich motifs. Blood. 2003;101(11):4492–9. doi: 10.1182/blood-2002-08-2579. [DOI] [PubMed] [Google Scholar]
- 72.Wei W, Wen L, Huang P, Zhang Z, Chen Y, Xiao A, Huang H, Zhu Z, Zhang B, Lin S. Gfi1.1 regulates hematopoietic lineage differentiation during zebrafish embryogenesis. Cell Res. 2008;18(7358):677–85. doi: 10.1038/cr.2008.60. [DOI] [PubMed] [Google Scholar]
- 73.Koldehoff M, Zakrzewski JL, Klein-Hitpass L, Beelen DW, Elmaagacli AH. Gene profiling of growth factor independence 1B gene (Gfi-1B) in leukemic cells. Int J Hematol. 2008;87(1):39–47. doi: 10.1007/s12185-007-0013-z. [DOI] [PubMed] [Google Scholar]
- 74.Doan LL, Porter SD, Duan Z, Flubacher MM, Montoya D, Tsichlis PN, Horwitz M, Gilks CB, Grimes HL. Targeted transcriptional repression of Gfi1 by GFI1 and GFI1B in lymphoid cells. Nucleic Acids Res. 2004;32(8):2508–19. doi: 10.1093/nar/gkh570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chari S, Winandy S. Ikaros regulates Notch target gene expression in developing thymocytes. J Immunol. 2008;181(9):6265–74. doi: 10.4049/jimmunol.181.9.6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kikuchi K, Lai AY, Hsu CL, Kondo M. IL-7 receptor signaling is necessary for stage transition in adult B cell development through up-regulation of EBF. J Exp Med. 2005;201(8):1197–203. doi: 10.1084/jem.20050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu Q, Erman B, Park JH, Feigenbaum L, Singer A. IL-7 receptor signals inhibit expression of transcription factors TCF-1, LEF-1, and RORgammat: impact on thymocyte development. J Exp Med. 2004;200(6):797–803. doi: 10.1084/jem.20032183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grutz GG, Bucher K, Lavenir I, Larson T, Larson R, Rabbitts TH. The oncogenic T cell LIM-protein Lmo2 forms part of a DNA-binding complex specifically in immature T cells. EMBO J. 1998;17(16):4594–605. doi: 10.1093/emboj/17.16.4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCormack MP, Young LF, Vasudevan S, de Graaf CA, Codrington R, Rabbitts TH, Jane SM, Curtis DJ. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science. 2010;327(5967):879–83. doi: 10.1126/science.1182378. [DOI] [PubMed] [Google Scholar]
- 80.Maurice D, Hooper J, Lang G, Weston K. c-Myb regulates lineage choice in developing thymocytes via its target gene Gata3. EMBO J. 2007;26(15):3629–40. doi: 10.1038/sj.emboj.7601801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Franco CB, Scripture-Adams DD, Proekt I, Taghon T, Weiss AH, Yui MA, Adams SL, Diamond RA, Rothenberg EV. Notch/Delta signaling constrains reengineering of pro-T cells by PU.1. Proc Natl Acad Sci U S A. 2006;103(32):11993–8. doi: 10.1073/pnas.0601188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maillard I, Tu L, Sambandam A, Yashiro-Ohtani Y, Millholland J, Keeshan K, Shestova O, Xu L, Bhandoola A, Pear WS. The requirement for Notch signaling at the beta-selection checkpoint in vivo is absolute and independent of the pre-T cell receptor. J Exp Med. 2006;203(10):2239–45. doi: 10.1084/jem.20061020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gonzalez-Garcia S, Garcia-Peydro M, Martin-Gayo E, Ballestar E, Esteller M, Bornstein R, de la Pompa JL, Ferrando AA, Toribio ML. CSL-MAML-dependent Notch1 signaling controls T lineage-specific IL-7R{alpha} gene expression in early human thymopoiesis and leukemia. J Exp Med. 2009;206(4):779–91. doi: 10.1084/jem.20081922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krebs LT, Deftos ML, Bevan MJ, Gridley T. The Nrarp gene encodes an ankyrin-repeat protein that is transcriptionally regulated by the notch signaling pathway. Dev Biol. 2001;238(1):110–9. doi: 10.1006/dbio.2001.0408. [DOI] [PubMed] [Google Scholar]
- 85.Pirot P, van Grunsven LA, Marine JC, Huylebroeck D, Bellefroid EJ. Direct regulation of the Nrarp gene promoter by the Notch signaling pathway. Biochem Biophys Res Commun. 2004;322(2):526–34. doi: 10.1016/j.bbrc.2004.07.157. [DOI] [PubMed] [Google Scholar]
- 86.Reizis B, Leder P. Direct induction of T lymphocyte-specific gene expression by the mammalian Notch signaling pathway. Genes Dev. 2002;16(3):295–300. doi: 10.1101/gad.960702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakagawa M, Ichikawa M, Kumano K, Goyama S, Kawazu M, Asai T, Ogawa S, Kurokawa M, Chiba S. AML1/Runx1 rescues Notch1-null mutation-induced deficiency of para-aortic splanchnopleural hematopoiesis. Blood. 2006;108(10):3329–34. doi: 10.1182/blood-2006-04-019570. [DOI] [PubMed] [Google Scholar]
- 88.Yun TJ, Bevan MJ. Notch-regulated ankyrin-repeat protein inhibits Notch1 signaling: multiple Notch1 signaling pathways involved in T cell development. J Immunol. 2003;170(12):5834–41. doi: 10.4049/jimmunol.170.12.5834. [DOI] [PubMed] [Google Scholar]
- 89.Chang HC, Han L, Jabeen R, Carotta S, Nutt SL, Kaplan MH. PU.1 regulates TCR expression by modulating GATA-3 activity. J Immunol. 2009;183(8):4887–94. doi: 10.4049/jimmunol.0900363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DeKoter RP, Lee HJ, Singh H. PU.1 regulates expression of the interleukin-7 receptor in lymphoid progenitors. Immunity. 2002;16(2):297–309. doi: 10.1016/s1074-7613(02)00269-8. [DOI] [PubMed] [Google Scholar]
- 91.Landry JR, Bonadies N, Kinston S, Knezevic K, Wilson NK, Oram SH, Janes M, Piltz S, Hammett M, Carter J, Hamilton T, Donaldson IJ, Lacaud G, Frampton J, Follows G, Kouskoff V, Gottgens B. Expression of the leukemia oncogene Lmo2 is controlled by an array of tissue-specific elements dispersed over 100 kb and bound by Tal1/Lmo2, Ets, and Gata factors. Blood. 2009;113(23):5783–92. doi: 10.1182/blood-2008-11-187757. [DOI] [PubMed] [Google Scholar]
- 92.Chan WY, Follows GA, Lacaud G, Pimanda JE, Landry JR, Kinston S, Knezevic K, Piltz S, Donaldson IJ, Gambardella L, Sablitzky F, Green AR, Kouskoff V, Gottgens B. The paralogous hematopoietic regulators Lyl1 and Scl are coregulated by Ets and GATA factors, but Lyl1 cannot rescue the early Scl−/− phenotype. Blood. 2007;109(5):1908–16. doi: 10.1182/blood-2006-05-023226. [DOI] [PubMed] [Google Scholar]
- 93.Okuno Y, Huang G, Rosenbauer F, Evans EK, Radomska HS, Iwasaki H, Akashi K, Moreau-Gachelin F, Li Y, Zhang P, Gottgens B, Tenen DG. Potential autoregulation of transcription factor PU.1 by an upstream regulatory element. Mol Cell Biol. 2005;25(7):2832–45. doi: 10.1128/MCB.25.7.2832-2845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zarnegar MA, Chen J, Rothenberg EV. Cell-type-specific activation and repression of PU.1 by a complex of discrete, functionally specialized cis-regulatory elements. Mol Cell Biol. 2010;30(20):4922–39. doi: 10.1128/MCB.00354-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yannoutsos N, Barreto V, Misulovin Z, Gazumyan A, Yu W, Rajewsky N, Peixoto BR, Eisenreich T, Nussenzweig MC. A cis element in the recombination activating gene locus regulates gene expression by counteracting a distant silencer. Nat Immunol. 2004;5(4):443–50. doi: 10.1038/ni1053. [DOI] [PubMed] [Google Scholar]
- 96.Rosenbauer F, Owens BM, Yu L, Tumang JR, Steidl U, Kutok JL, Clayton LK, Wagner K, Scheller M, Iwasaki H, Liu C, Hackanson B, Akashi K, Leutz A, Rothstein TL, Plass C, Tenen DG. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat Genet. 2006;38(1):27–37. doi: 10.1038/ng1679. [DOI] [PubMed] [Google Scholar]
- 97.Longabaugh WJ, Davidson EH, Bolouri H. Computational representation of developmental genetic regulatory networks. Dev Biol. 2005;283(1):1–16. doi: 10.1016/j.ydbio.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 98.Longabaugh WJ, Davidson EH, Bolouri H. Visualization, documentation, analysis, and communication of large-scale gene regulatory networks. Biochim Biophys Acta. 2009;1789(4):363–74. doi: 10.1016/j.bbagrm.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jeannet G, Scheller M, Scarpellino L, Duboux S, Gardiol N, Back J, Kuttler F, Malanchi I, Birchmeier W, Leutz A, Huelsken J, Held W. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood. 2008;111(1):142–9. doi: 10.1182/blood-2007-07-102558. [DOI] [PubMed] [Google Scholar]
- 100.Tydell CC, David-Fung ES, Moore JE, Rowen L, Taghon T, Rothenberg EV. Molecular dissection of prethymic progenitor entry into the T lymphocyte developmental pathway. J Immunol. 2007;179(1):421–38. doi: 10.4049/jimmunol.179.1.421. [DOI] [PubMed] [Google Scholar]
- 101.Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol. 2008;8(1):9–21. doi: 10.1038/nri2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li TW, Ting JH, Yokoyama NN, Bernstein A, van de Wetering M, Waterman ML. Wnt activation and alternative promoter repression of LEF1 in colon cancer. Mol Cell Biol. 2006;26(14):5284–99. doi: 10.1128/MCB.00105-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alon U. Chapman & Hall/CRC mathematical and computational biology series. xvi. Chapman & Hall/CRC; Boca Raton, FL: 2007. An introduction to systems biology: design principles of biological circuits; p. 301.p. 4. of plates. [Google Scholar]
- 104.Saba I, Kosan C, Vassen L, Moroy T. IL-7R-dependent survival and differentiation of early T-lineage progenitors is regulated by the BTB/POZ domain transcription factor Miz-1. Blood. 2011;117(12):3370–81. doi: 10.1182/blood-2010-09-310680. [DOI] [PubMed] [Google Scholar]
- 105.Henriques CM, Rino J, Nibbs RJ, Graham GJ, Barata JT. IL-7 induces rapid clathrin-mediated internalization and JAK3-dependent degradation of IL-7Ralpha in T cells. Blood. 2010;115(16):3269–77. doi: 10.1182/blood-2009-10-246876. [DOI] [PubMed] [Google Scholar]
- 106.Alves NL, van Leeuwen EM, Derks IA, van Lier RA. Differential regulation of human IL-7 receptor alpha expression by IL-7 and TCR signaling. J Immunol. 2008;180(8):5201–10. doi: 10.4049/jimmunol.180.8.5201. [DOI] [PubMed] [Google Scholar]
- 107.Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL, Singer A. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 2004;21(2):289–302. doi: 10.1016/j.immuni.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 108.Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol. 2004;22:55–79. doi: 10.1146/annurev.immunol.22.012703.104807. [DOI] [PubMed] [Google Scholar]
- 109.Huang G, Zhang P, Hirai H, Elf S, Yan X, Chen Z, Koschmieder S, Okuno Y, Dayaram T, Growney JD, Shivdasani RA, Gilliland DG, Speck NA, Nimer SD, Tenen DG. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat Genet. 2008;40(1):51–60. doi: 10.1038/ng.2007.7. [DOI] [PubMed] [Google Scholar]
- 110.Decker T, Pasca di Magliano M, McManus S, Sun Q, Bonifer C, Tagoh H, Busslinger M. Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity. 2009;30(4):508–20. doi: 10.1016/j.immuni.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 111.Nutt SL, Metcalf D, D’Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med. 2005;201(2):221–31. doi: 10.1084/jem.20041535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Back J, Allman D, Chan S, Kastner P. Visualizing PU.1 activity during hematopoiesis. Exp Hematol. 2005;33(4):395–402. doi: 10.1016/j.exphem.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 113.Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, Paul W. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A. 2009;106(32):13463–8. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Van de Walle I, De Smet G, De Smedt M, Vandekerckhove B, Leclercq G, Plum J, Taghon T. An early decrease in Notch activation is required for human TCR-alphabeta lineage differentiation at the expense of TCR-gammadelta T cells. Blood. 2009;113(13):2988–98. doi: 10.1182/blood-2008-06-164871. [DOI] [PubMed] [Google Scholar]
- 115.Weerkamp F, Luis TC, Naber BA, Koster EE, Jeannotte L, van Dongen JJ, Staal FJ. Identification of Notch target genes in uncommitted T-cell progenitors: No direct induction of a T-cell specific gene program. Leukemia. 2006;20(11):1967–77. doi: 10.1038/sj.leu.2404396. [DOI] [PubMed] [Google Scholar]
- 116.Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, Leeds KE, Du H, Wu B, Waterman ML, Zhu Z, Sen JM. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009;10(9):992–9. doi: 10.1038/ni.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Further Reading
- 117.Davidson EH. The regulatory genome: gene regulatory networks in development and evolution. Academic Press; San Diego, CA: 2006. [Google Scholar]
- 118.Rothenberg EV, Anderson MK. Elements of transcription factor network design for T-lineage specification. Devel Biol. 2002;246(1):29–44. doi: 10.1006/dbio.2002.0667. [DOI] [PubMed] [Google Scholar]
- 119.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8(6):450–61. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 120.Enver T, Pera M, Peterson C, Andrews PW. Stem cell states, fates, and the rules of attraction. Cell Stem Cell. 2009;4(5):387–397. doi: 10.1016/j.stem.2009.04.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.