

Abstract
Toll-like receptors (TLRs) are a family of pattern-recognition receptors in innate immunity and provide a first line defense against pathogens and tissue injuries. In addition to important roles in infection, inflammation, and immune diseases, recent studies show that TLR signaling is involved in modulation of learning, memory, mood, and neurogenesis. Because MyD88 is essential for the downstream signaling of all TLRs, except TLR3, we investigated the effects of MyD88 deficiency (MyD88-/-) on behavioral functions in mice. Additionally, we recently demonstrated that a mouse model of Alzheimer’s disease (AD) deficient for MyD88 had decreases in Aβ deposits and soluble Aβ in the brain as compared with MyD88 sufficient AD mouse models. Because accumulation of Aβ in the brain is postulated to be a causal event leading to cognitive deficits in AD, we investigated the effects of MyD88 deficiency on behavioral functions in the AD mouse model at 10 months of age. MyD88 deficient mice showed more anxiety in the elevated plus-maze. In the motor coordination tests, MyD88 deficient mice remained on a beam and a bar for a longer time, but with slower initial movement on the bar. In the Morris water maze test, MyD88 deficiency appeared to improve spatial learning irrespective of the transgene. Our findings suggest that the MyD88-dependent pathway contributes to behavioral functions in an AD mouse model and its control group.
Keywords: MyD88, toll-like receptors, Alzheimer’s disease, memory, anxiety, cognitive function
1. Introduction
The innate immune system is the vital first line of defense against a wide range of pathogens and tissue injuries, which trigger inflammation through pattern-recognition receptors on the cell surface of quiescent microglia and monocytes/macrophages. Toll-like receptors (TLRs) are a family of such pattern-recognition receptors and a type I transmembrane protein consisting of an extracellular domain and a cytoplasmic domain. The extracellular domain is responsible for the recognition of TLR ligands, either exogenous (microbial components) or endogenous (host tissue components). The activation of TLRs by TLR ligands initiates innate immune responses/inflammation [1,2]. The cytoplasmic domain is referred to as Toll/interleukin-1 (IL-1) receptor (TIR) domain. The activation of TLRs by TLR ligands induces interaction of TIR domains between TLR and TIR domain-containing adaptors, including myeloid differentiation primary response protein 88 (MyD88) and Toll/interleukin-1 receptor domain-containing adaptor-inducing interferon-β (TRIF). Each TLR mediates a distinctive response through a different combination of these adaptors. Except for TLR3, all TLRs use MyD88 as an adaptor (MyD88-dependent pathway), essential for downstream signaling culminating in activation of transcription factors that induce expression of inflammatory cytokines, chemokines, and co-stimulatory molecules. TLR3 and TLR4 ligation can signal via the MyD88-independent (TRIF-dependent) pathway, leading to activation of distinct transcription factors, such as interferon regulatory factor 3 that induces expression of type I interferons.
In addition to important roles of TLRs in infection, inflammation, and immune diseases [3], recent studies show that TLR signaling is involved in modulation of learning, memory, mood, and neurogenesis [4-7]. Systemic administration in rodents of lipopolysaccharide (LPS), a TLR4 ligand, appears to cause sickness-like behavior, including decreased motor activity, social withdrawal, reduced food and water intake, and impaired cognition. LPS-induced sickness is attributed to increased levels of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) [4]. Systemic and intraventricular injections of LPS in rodents impair adult neurogenesis in the hippocampus, which is thought to be caused by activated microglia, and correlates with upregulation of TNF-α production in microglia [8-11]. TLR2 and TLR4 are also expressed in adult neural stem/progenitor cells (NPCs) [12]. TLR2 deficient (TLR2-/-) mice show decreased neuronal differentiation of NPCs in the hippocampus, while TLR4-/- and MyD88-/- mice have increased proliferation and neuronal differentiation of NPCs [12]. Thus, MyD88 signaling appears to modulate adult hippocampal neurogenesis and sickness behavior. Additionally, adult neurogenesis is considered to play important roles not only in hippocampal-dependent learning and memory but also mental health/illness [13,14]. Behavioral functions in MyD88-/- mice have not been investigated. Here, the effects of MyD88 deficiency on normal mice of the C57BL/6 strain were investigated in tests of anxiety, motor activity, and cognition.
The effects of MyD88 deficiency are also of interest in studying Alzheimer’s disease (AD), a neurodegenerative disorder characterized by progressive deteriorations in memory, learning, reasoning, judgment, and language and by deposits of aggregated amyloid β-protein (Aβ) in the brain. Fibrillar Aβ deposits are closely associated with inflammatory responses, such as activated microglia, reactive astrocytes, increased cytokines, and cytotoxic molecules [15]. Aβ aggregates can activate microglia and monocytes via interaction with multiple TLRs, including TLR2, TLR4, and TLR6, leading to upregulated Aβ phagocytosis, inflammatory cytokines and chemokines, and reactive oxygen species [16-18]. TLR signaling has been shown to modulate the amount of Aβ deposits in the brains of AD mouse models [19-27]. We recently demonstrated that an AD mouse model deficient for MyD88 had decreased diffuse and fibrillar Aβ deposits as well as buffer-soluble and insoluble Aβ in the brain compared with a MyD88 sufficient (MyD88+/+) AD model [28]. Because accumulated Aβ in the brain is postulated to be a causal event leading to AD and because clearing Aβ deposits from the brain is a viable therapeutic strategy [29], we examined whether blockade of MyD88 improves behavioral deficits in the AD model.
2. Materials and Methods
2.1. Experimental animals
A congenic C57BL/6 line of AD model, B6.Cg-Tg(APPswe, PSEN1dE9) 85Dbo/J, was purchased from Jackson Laboratory (Bar Harbor, ME) and generated by crossing transgenic males with C57BL/6 females. APP mice express chimeric mouse/human amyloid precursor protein (APP) with a double mutation (K670N and M671L) and human presenilin 1 (PS1) with a deletion of exon 9 found in familial AD patients and develop amyloid deposits composed of human Aβ in the brain, because of humanizing the Aβ sequence in the mouse APP transgene [30]. The genotyping for the APPswe/PS1dE9 transgenes was performed by the PCR-based method provided by Jackson Laboratory. MyD88-/- mice [31] were obtained from Dr. Alan Aderem, Institute for Systems Biology (Seattle, WA) and backcrossed to C57BL/6 mice for more than 10 generations. All the mice used in this study were C57BL/6 or congenic C57BL/6 mice. The genotyping for the MyD88 gene was performed by PCR of DNA extracted from mouse tails with the Extract-N-Amp tissue PCR kit (Sigma-Aldrich, St. Louis, MO). The PCR was carried out in 25 μl volume using 1 μl tail DNA extract and 200 nM each of two primers for the MyD88 deficient allele through 35 cycles, consisting of 30 s at 94°C, 30 s at 60°C, and 1.5 min at 72°C. The last elongation step was done at 72°C for 5 min. The two primer sequences, forward primer: 5’-AGC CTC TAC ACC CTT CTC TTC TCC ACA -3’ and reverse primer (neo gene): 5’-ATC GCC TTC TAT CGC CTT CTT GAC GAG-3, were used to amplify the MyD88 deficient allele. An additional reverse primer, 5’-AGA CAG GCT GAG TGC AAA CTT GTG CTG-3’, was used to amplify the wild-type allele [31]. The PCR products were subjected to electrophoresis through a 1% agarose gel. Mouse tail DNAs extracted from a MyD88-/- and C57BL/6J mouse were used as positive controls for the MyD88- and MyD88+ allele, respectively. First, we produced APP MyD88+/- (hemizygous) mice by mating APP mice with MyD88-/- mice. Next, we mated APP MyD88+/- with MyD88-/- mice to produce APP MyD88-/- mice. APP MyD88-/- mice appeared to be normal by appearance and fertile. APP MyD88-/- mice were mated with MyD88-/- mice to produce APP MyD88-/- and MyD88-/- mice. In this study, four experimental groups of 10-month-old mice differing in MyD88 genotype and the APPswe/PS1dE9 transgenes were used. The four experimental groups (N=35) were APP MyD88-/- (n = 9, 3 male and 6 female), APP (n = 10, 4 male and 6 female), MyD88-/- (n = 8, 4 male and 4 female), and control C57BL/6 (WT) (n = 8, 5 male and 3 female) mice. The mice were housed in mixed genotype groups with bedding and maintained on a 12/12 hr light-dark cycle (on at 6 AM) under a specific pathogen free condition with access to autoclaved food (NIH-31 formula) and water ad libitum. All protocols used in this study were prospectively reviewed and approved by the Institutional Animal Care and Use Committee of the University of Illinois College of Medicine at Peoria.
2.2 Behavior
2.2.1. Behavioral schedule and statistics
The behavioral alterations of experimental mice potentially associated with APPswe/PS1dE9 transgenes and/or MyD88 deficiency were tested at 10 months of age. After measuring body weight and adapting mice to handling, behavioral tests were conducted over a 17-day period. For each test day, spontaneous alternation (days 1 to 11) was evaluated first, followed by open-field (days 1 to 3), elevated plus-maze (days 4 and 5), stationary beam (day 6), coat-hanger (day 7), rotorod (days 8 to 10). The battery ended with the Morris water maze (days 12 to 17). In each test, whenever possible, the apparatus was wiped clean with a wet cloth and dried before the next mouse was introduced to minimize odor cues. Intergroup differences were assessed by a repeated measures analysis of variance (ANOVA) and two-tailed Student’s t-test for normally distributed data. In the spontaneous alternation test and the probe trial of the Morris water maze, groups were compared with the Mann-Whitney U test. Whenever possible, results are expressed as mean ± standard error of the mean (SEM). In all cases, P < 0.05 was considered to be significant.
2.2.2. Exploration and anxiety
Spontaneous alternation was measured in a T-maze, made of white acrylic and consisting of a central stem flanked on each side by 2 arms. The maze width was 9 cm, the wall height 20 cm, and each arm 30 cm in length. On the initial trial, the mice were placed in the stem with the right arm blocked by a plastic barrier (forced choice). After entering the available arm, the mice were kept in it for 1 min by closing the barrier behind them. The mice were then retrieved and after removing the barrier, placed back in the stem for a free-choice trial, either into the same arm or the opposite arm (4-paw criterion). On the following 10 days, the same 2-trial procedure was repeated, except that the blocked arm was switched from right on odd days to left on even days. The number of alternations and the latencies before responding during the choice trial were measured. In the absence of any decision within 1 min, the mice were briefly prodded from behind far from the choice point, usually not more than once, so that a response could be recorded on every trial.
Motor activity was measured in the open-field made of white acrylic with a 50 cm × 50 cm surface area. Each mouse was placed in a corner of the open-field. The activity in central (25 cm × 25 cm surface area) and peripheral zones was recorded in a 5-min session for 3 consecutive days and analyzed by video tracking software (SD Instruments, San Diego, CA). The distance travelled and the time spent resting (<2 cm/s), moving slow (2-5 cm/s), or moving fast (>5 cm/s) in each zone were measured, as well as the time spent in the periphery and center of the apparatus.
The elevated plus-maze consisted of 4 arms in a cross-shaped form 70 cm in the length with a 10 cm × 10 cm central region. Two of the arms were enclosed on 3 sides by walls (10 cm in height) facing each other, while the other two were open, except for a minimal border (0.5 cm in height) used to minimize falls. A mouse was placed in the central region and then the number of entries and the time spent inside enclosed and open arms were measured in a 5-min session on 2 consecutive days with the same video tracking system. The open/total arm entries and duration ratios were also calculated.
2.2.3. Motor coordination
The stationary beam (diameter: 2.5 cm; length: 110 cm) was made of plastic covered by white masking tape to facilitate a firm grip. The beam was divided into 11 segments along its length and placed at a 40-cm height above a cushioned floor to prevent injury. A cardboard wall was inserted at each end to prevent the mouse from escaping. A trial began by placing the mice on the middle segment. The number of segments crossed (4-paw criterion), the latencies before falling, and the number of falls were measured in a single 4-trial session, with a 1 min cut-off period and a 15-min intertrial interval.
Motor speed was measured in the coat-hanger test. The triangular-shaped coat-hanger consisted of a horizontal steel wire (diameter: 2 mm, length: 41 cm) flanked at each end by 2 side-bars (length: 19 cm; inclination: 35° from the horizontal axis). The horizontal bar was placed at a height of 40 cm above a cushioned table. The mice were placed upside-down in the middle of the horizontal wire and released only after gripping with all 4 paws. Seven types of movement time (MT) were compiled, namely latencies before reaching (snout criterion) the first 10 cm segment (MT-1) or the extremity (MT-2) of the horizontal wire, latencies before reaching either side-bar with 2, 3 or 4 paws, and latencies before reaching (snout criterion) either the midway point or the top of the side-bar. Latencies before falling and the number of falls were also compiled. A trial ended whenever the mice either fell or reached the top of the apparatus. In the latter case, a maximal score of 60 s was given for latencies before falling. This test was performed in a single 4-trial session with a 1 min cut-off period and a 15-min intertrial interval.
The accelerating rotorod (Model 7650, Stoelting, Wood Dale, IL) consisted of a beam (diameter: 3 cm) made of ribbed plastic, elevated at a height of 13.5 cm, and separated into 5 sections (width: 5.5 cm) by a plastic barrier. Facing away from the experimenter’s view, the mice were placed on top of the already revolving rod (4 rpm) in the orientation opposite to its movement, so that falls could be avoided by forward locomotion. The rotorod accelerated gradually and smoothly from 4 to 40 rpm during the 5-min trial. Latencies before falling were measured in 4-trial sessions for 3 days, with a 15-min intertrial interval. Whenever a mouse clung to the rod without moving (passive rotation) for 2 complete revolutions in succession, it was retrieved and a fall registered.
2.2.4. Spatial learning and memory
The Morris water maze consisted of a pool of blue opaque plastic, 116 cm in diameter with 75-cm high walls, filled with water (20°C) at a height of 31 cm. Powdered milk was evenly spread over the water surface to camouflage the escape platform (8 cm2) made of white plastic and covered with a wire mesh grid to ensure a firm grip. The watered milk was removed every day after a few hours of training and the pool rinsed with clean water. The pool was contained in a room with external visual cues such as light fixtures and a ladder. The mice were placed next to and facing the wall successively in north (N), east (E), south (S), and west (W) positions, with the escape platform hidden 1 cm below water level in the middle of the NW quadrant. The same video tracking equipment was used to estimate path length and escape latencies in 4-trial sessions for 5 days with a 15-min intertrial interval. The mice remained on the platform for at least 5 s. Whenever the mice failed to reach the escape platform within the 1 min cut-off period, they were retrieved from the pool and placed on the platform for 5 s. After swimming, the mice were kept dry in a plastic holding cage filled with paper towels. The morning after the acquisition phase, a probe trial was conducted by removing the platform and placing the mouse next to and facing the N side. The time spent in the previously correct quadrant was measured for a single 1-min trial. In the afternoon, the visible platform subtask was conducted, with the escape platform lifted 1 cm above water level and shifted to the SE quadrant. A 17-cm high pole was inserted on top of the escape platform as a viewing aid. The same procedure was adopted as with place learning except that the subtest was conducted on a single day.
3. Results
3.1. Exploration and anxiety
There was no intergroup difference in body weight at 10 months of age (30.9±2.4, 32.9 ± 2.4, 30.0±2.8, and 31.8 ± 1.4g for APP MyD88-/-, APP, MyD88-/-, and WT group, respectively). In the T-maze spontaneous alternation test (Table 1), all groups alternated above chance (P < 0.05, Mann-Whitney), except paradoxically the WT group, with an alternation rate slightly below the required significance (P=0.07). There was no intergroup difference in choice latencies. In the open-field (Fig. 1), day effects were observed in distances traveled in periphery, resting time, and time spent moving slow or fast in all groups (P < 0.05). Distances and time spent moving fast decreased as a function of time while time spent resting increased. There was no intergroup difference or interaction for any measure in the open-field test (P > 0.05). In the elevated plus-maze test (Table 1), mice chose to explore either safe (enclosed) or anxiogenic (open) arms. On day 1, there was an MyD88 deficiency effect on open/total arm entries (F 1,31= 5.0, P < 0.05), though not on open/total duration (F 1,31= 1.25, P > 0.05), the former measure being lower in null mutants, a sign of excessive anxiety. No intergroup difference was observed for any measure on the following day. Thus, MyD88 deficient groups were more anxious than the WT group on the initial testing day.
Table 1.
Effects of MyD88 deficiency on exploratory activity (means ± SEM)
| Test | APP MyD88 -/- | APP | MyD88 -/- | WT |
|---|---|---|---|---|
| T-maze | ||||
| Alternations (%) | 61 ± 5 | 56 ± 4 | 60 ± 3 | 59 ± 3 |
| Choice latencies/trial (s) | 15.9 ± 4 | 11.4 ± 2 | 20 ± 7 | 14.5 ± 5 |
| Elevated Plus-Maze | ||||
| Day 1 | ||||
| Open Arms | ||||
| Entries | 8. 9 ± 1.4 | 10.4 ± 1.1 | 8.6 ± 1.5 | 12.3 ± 1.0 |
| Duration (s) | 68.0 ± 17.3 | 69.8 ± 19.8 | 62.6 ± 14.9 | 100.9 ± 17.8 |
| Enclosed Arms | ||||
| Entries | 10.2 ± 1.2 | 10.1 ± 1.7 | 12.5 ± 1.1 | 9.6 ± 1.6 |
| Duration (s) | 199.4 ± 15.7 | 201.1 ± 20.8 | 202.0 ± 17.0 | 168.7 ± 18.5 |
| Open/total Ratio % | ||||
| Entries | 45.2 ± 5.1a | 50.2 ± 6.2 | 39.0 ± 3.3a | 56.0 ± 2.8 |
| Duration (s) | 22.7 ± 5.8 | 23.3 ± 6.6 | 20.9 ± 5.0 | 33.7 ± 5.9 |
| Day 2 | ||||
| Open Arms | ||||
| Entries | 2.4 ± 0.6 | 5.4 ± 2.2 | 3.1 ± 0.8 | 5.4 ± 2.0 |
| Duration (s) | 9.9 ± 3.4 | 31.1 ± 12.4 | 12.1 ± 3.6 | 26.5 ± 11.0 |
| Enclosed Arms | ||||
| Entries | 8.6 ± 1.4 | 7.8 ± 1.6 | 7.9 ± 1.6 | 8.9 ± 1.6 |
| Duration (s) | 271.5 ± 6.3 | 251.7 ± 14.8 | 268.4 ± 6.1 | 251.1 ± 12.9 |
| Open/total Ratio % | ||||
| Entries | 20.6 ± 5.2 | 32.6 ± 7.0 | 27.1 ± 6.9 | 29.2 ± 6.3 |
| Duration (s) | 3.3 ± 1.1 | 10.4 ± 4.1 | 4.0 ± 1.2 | 8.8 ± 3.7 |
P < 0.05 for either MyD88 or APP effects on ANOVA
Figure 1.
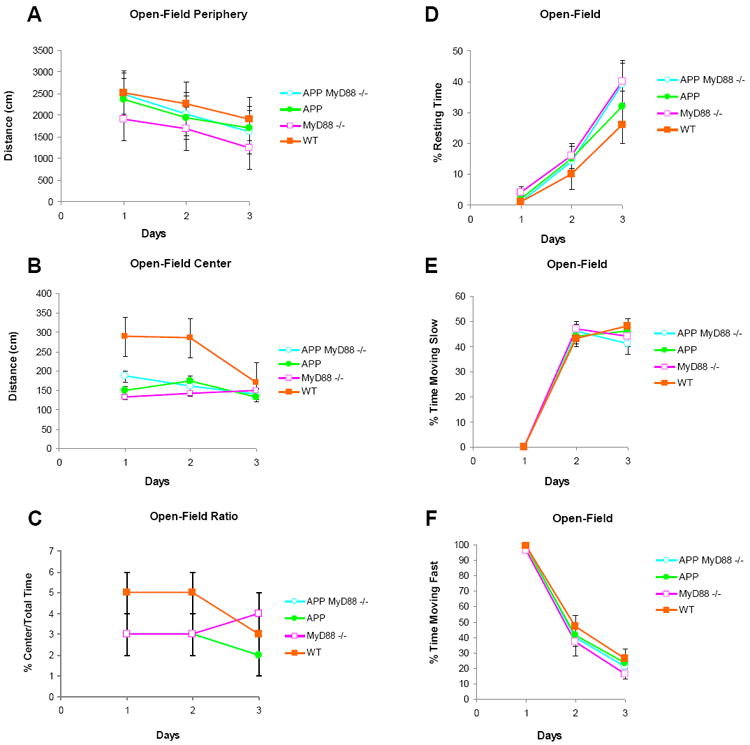
Effects of MyD88 deficiency on APP or WT mice on exploration in the open-field. A-C, Distance travelled at the periphery (A) and center (B) of the open-field as well as the center/total time ratio (C) are shown as means ± SEM in 5 min sessions for 3 days. The center/total time rations of APP MyD88-/- mice are similar to those of APP mice (C). D-F, Percent time spent resting (D), moving slow (E), and moving fast (F) in the open-field are shown as means ± SEM in 5-min sessions for 3 days.
3.2. Motor coordination
In the stationary beam test (Table 2), there was a transgene effect (F 1,31= 17.09, P < 0.001) on segments crossed, as APPswe/PS1dE9 mice walked shorter distances. There were also MyD88 deficiency effects on latencies before falling (F 1,31= 7.41, P < 0.05) and falls (F 1,31= 6.65, P < 0.05) as a result of superior performances by both knockout groups. MyD88 deficiency effects were also found on the coat-hanger (Table 2) with respect to movement times: MT-1 (F1,31=5.80, P < 0.05) and MT-2 (F1,31=8.68, P < 0.01), slower in the null mutants, as well as latencies before falling (F1,31=7.14, P < 0.05) and falls (F1,31=8.35, P < 0.01), higher in the null mutants. On the rotorod (Fig. 2), APP transgenic lines performed more poorly on trial blocks 10 (F1,31=4.80, P < 0.05) and 12 (F1,31=5.12, P < 0.05). Thus, irrespective of APPswe/PS1dE9 transgene status, MyD88 deficient (APP MyD88-/- and MyD88-/-) mice stayed longer on the stationary beam and the coat-hanger but not on the rotorod. However, they were moving slower on the coat-hanger. On the rotorod, APP transgenic lines preformed more poorly, but only on 2 of 12 trial blocks.
Table 2.
Effects of MyD88 deficiency on APP or WT mice on stationary beam and coat-hanger tests (means ± SEM)
| Measures | APP MyD88 -/- | APP | MyD88 -/- | WT |
|---|---|---|---|---|
| Stationary Beam | ||||
| Segments | 15 ± 5a | 20 ± 9a | 46 ± 11 | 68 ± 12 |
| Latencies before falling (s) | 220 ± 7a | 168 ± 16 | 223 ± 12a | 203 ± 14 |
| Falls | 0.8 ± 0.3a | 2.1 ± 0.5 | 0.4 ± 0.3a | 1.0 ± 0.4 |
| Coat-Hanger | ||||
| MT-1 (s) | 150 ± 21a | 111 ± 11 | 150 ± 20a | 99 ± 24 |
| MT-2 (s) | 200 ± 14a | 154 ± 14 | 195 ± 8a | 147 ± 24 |
| Two-paw MT (s) | 239 ± 1 | 239 ± 2 | 225 ± 6 | 223 ± 9 |
| Three-paw MT (s) | 239 ± 0.3 | 239 ± 1 | 232 ± 5 | 240 ± 0 |
| Four-paw MT (s) | 240 ± 0 | 239 ± 1 | 233 ± 5 | 240 ± 0 |
| Midway MT (s) | 240 ± 0 | 240 ± 0 | 240 ± 0 | 240 ± 0 |
| Top MT (s) | 240 ± 0 | 240 ± 0 | 240 ± 0 | 240 ± 0 |
| Fall Latencies (s) | 212 ± 9a | 174 ± 15 | 187 ± 17a | 141 ± 21 |
| Falls | 1.3 ± 0.3a | 2.7 ± 0.4 | 1.9 ± 0.5a | 2.8 ± 0.4 |
Any of these groups are significantly different from any of the other groups in the indicated measures (P < 0.05 for either MyD88 or APP effects on ANOVA).
Figure 2.
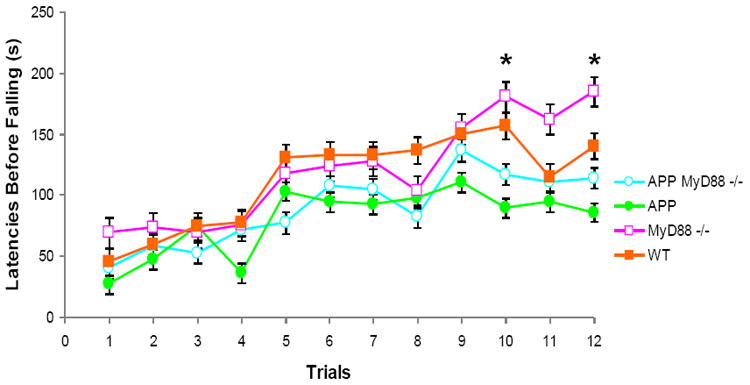
Effects of MyD88 deficiency on APP or WT mice on the rotorod. Latencies before falling (means ± SEM) from the accelerating rotorod observed in the 4 experimental mouse groups in 4-trial sessions for 3 days. * P < 0.05: APP groups vs. non-APP groups.
3.3. Spatial learning and memory
In the Morris water maze (Fig. 3), 7 mice died, but only in the two APP MyD88-/- subgroups, presumably from hypothermia, so that the new group distributions were as follows (N=28): APP MyD88-/- (n=7 instead of 9), APP (n=10), MyD88-/- (n=3 instead of 8), and WT (n=8). In the acquisition phase, there were Myd88 deficiency effects on days 1, 2, and 5 (F1,24>4.60,P < 0.05) as a result of shorter distances and escape latencies by the null mutant groups. There was also a transgene effect on day 4 (F1,24=6.44, P < 0.05) as a result of longer distances by the transgenic groups. The differences in path lengths were not observed on the final test day. Thus, MyD88 deficiency improved spatial learning irrespective of APPswe/PS1dE9 transgene status. On the contrary, there were no intergroup differences or interaction in regard to the probe trial (Fig. 3C) or the visible platform subtask (include mean values, P > 0.05).
Figure 3.
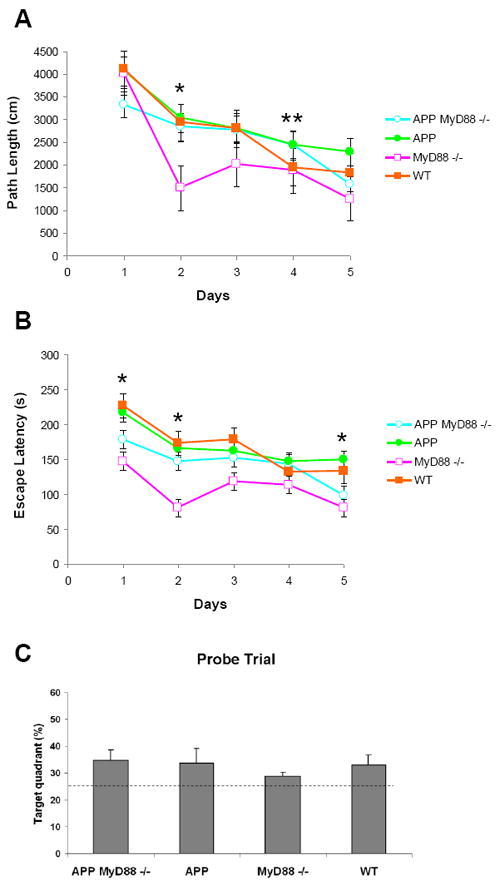
Effects of MyD88 deficiency on APP or WT mice on Morris water maze. Total cumulated scores per day for path length (cm) (A) and escape latencies (s) (B). Means ± SEM are shown during acquisition of the hidden platform task. MyD88 deficiency improved performance of APP and WT mice. (C) The probe trial of the Morris water maze. Percent of time spent in the target quadrant in which the hidden platform was previously placed. The dashed line indicates the chance level (25%). All groups spent significantly longer than chance in the target quadrant (P < 0.05) and no difference was found in any group. * P < 0.05: MyD88 deficient groups vs. MyD88 sufficient groups, ** P < 0.01: APP groups vs. non-APP groups.
4. Discussion
APPswe/PS1dE9 transgenic mice on the B6 genetic background traveled shorter distances on the stationary beam than wild-type mice, though without falling sooner, and performed more poorly on the rotorod on 2 of 12 trial blocks. These slight alterations were not observed with this model on the B6C3 background either at 7 or 12 months of age [32,33] and thus may have been amplified by the C57BL/6 background. Rotorod deficits are not a prominent feature of AD models but sometimes occur after backcrossing to the C57BL/6 line [34,35].
Irrespective of the transgene, MyD88 deficient mice stayed longer on the stationary beam and the coat-hanger but not on the rotorod. The improved performance of MyD88 deficient mice on the latter test was achieved at the expense of slower initial movement times, explainable by more cautious locomotion in view of their higher anxious levels in the elevated plus-maze. Indeed, regardless of the transgene factor, MyD88 deficiency decreased the percentage of open arm entries without increasing enclosed arm entries. The elevated plus-maze is widely used for anxiety testing in rodents [36]. Peripheral or central administration of pro-inflammatory cytokines such as IL-1β and TNF-α in rodents decreased percentages of open arm entries without affecting enclosed arm entries [37-39]. Likewise, intraperitoneal injection of LPS, a TLR4 ligand inducing TNF-α and IL-1β release from immune cells [40], decreased open arm entries and the percentage of duration on the open arms in mice [41,42]. In an opposite manner, mice deficient for IL-1β showed increased open arm duration, indicating that a decrease in a proinflammatory cytokine signaling reduces anxiety [43]. Central administration of IL-10, an anti-inflammatory cytokine, counteracts LPS-induced behavioral alterations by inhibiting proinflammatory cytokine production and signaling [4,44]. The balance between pro-and anti-inflammatory cytokines is postulated to modulate animal behavior [4]. Thus, increases in anxiety in MyD88 deficient mice may be caused by an imbalance between pro- and anti-inflammatory cytokines. MyD88 is essential for downstream signaling of all TLRs except TLR3. Depending on TLRs, their ligands, and cell types, distinct genes are upregulated, resulting in differences in cytokine profiles. Additionally, microorganisms and tissue injuries activate multiple TLRs and other pattern recognition receptors, further diversifying expression profiles [2]. Constitutive loss of MyD88 signaling may shift the balance toward proinflammatory cytokines. It is noteworthy that TLR3 deficiency in mice reduces anxiety in the plus-maze and open-field tests [6]. Therefore, it is possible that TRIF-dependent signaling increases anxiety in MyD88 deficient mice so that it would be interesting to determine whether TRIF deficiency reduces anxiety.
MyD88 deficient mice had shorter path lengths and escape latencies than wild-type mice during acquisition of the Morris water maze task regardless of the transgene, indicating improved spatial learning in 10-month-old mice on a C57BL/6 congenic line. A transgene effect was found only on a single day during acquisition, whereas the deficit was more evident in 12-month-old though not in 7-month-old APPswe/PS1dE9 mice on the B6C3 background [32,33]. However, MyD88 deficiency in the current study did not improve the probe test of long-term memory. A point of concern is the mortality found in the MyD88 deficient groups, probably from hypothermia caused by swimming in cool water (20°C), which was lower than our previous study (22°C). It is possible that MyD88 deficiency causes hypothermia, and so this study must be repeated at higher temperatures. Should this result be confirmed, faster learning may be due to enhanced adult hippocampal neurogenesis reported in MyD88-/- mice [12]. The acquisition phase of the Morris water maze correlated with genetically determined baseline levels of adult hippocampal neurogenesis, but the probe trial did not [45]. Contrariwise, the PS1dE9 transgene impaired adult neurogenesis in mice and the impairment in neurogenesis appeared to be caused by soluble signaling factors (cytokines, chemokines, and growth factors) secreted from activated microglia with the transgene [46,47]. Furthermore, transplantation of MyD88-/- bone marrow cells into an AD mouse model decreased cerebral Aβ load and improved cognitive deficits more efficiently than MyD88+/+ cells. Such beneficial effects were associated with increased synaptophysin levels [48]. Consistent with this view, there was a trend of increased synaptophysin in the hippocampus of APP MyD88-/- mice compared with APP mice (data not shown). Therefore, MyD88 deficiency may alter soluble signaling factors secreted from microglia bearing the PS1dE9 transgene, resulting in improved neurogenesis and, ultimately, cognitive functions. Our results support the notion that anti-inflammatory drugs with pleiotropic effects of promoting neurogenesis and neurite outgrowth ameliorate cognitive deficits in AD patients [49].
Highlights.
MyD88 deficiency in mice increases anxiety in the elevated plus-maze.
MyD88 deficiency in mice slows movement in the stationary beam and coat hanger tests.
MyD88 deficiency in mice enhances spatial learning performance in Morris water maze.
MyD88 deficiency ameliorates spatial cognitive deficits in an Alzheimer mouse model.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (AG030399, AG031979, AG029818 and EY018478) and the Alzheimer’s Association (IIRG-07-59494). We thank Dr. Alan Aderem, Institute for Systems Biology, for providing a MyD88-/- mouse line and Linda Walter for assistance in preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 2.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Keogh B, Parker AE. Toll-like receptors as targets for immune disorders. Trends Pharmacol Sci. 2011 doi: 10.1016/j.tips.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 6.Okun E, Griffioen K, Barak B, Roberts NJ, Castro K, Pita MA, et al. Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2010;107:15625–30. doi: 10.1073/pnas.1005807107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;121:367–87. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–7. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonde S, Ekdahl CT, Lindvall O. Long-term neuronal replacement in adult rat hippocampus after status epilepticus despite chronic inflammation. Eur J Neurosci. 2006;23:965–74. doi: 10.1111/j.1460-9568.2006.04635.x. [DOI] [PubMed] [Google Scholar]
- 10.Wu CW, Chen YC, Yu L, Chen HI, Jen CJ, Huang AM, et al. Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory. J Neurochem. 2007;103:2471–81. doi: 10.1111/j.1471-4159.2007.04987.x. [DOI] [PubMed] [Google Scholar]
- 11.Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31:149–60. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–8. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 13.Marin-Burgin A, Schinder AF. Requirement of adult-born neurons for hippocampus-dependent learning. Behav Brain Res. 2011 doi: 10.1016/j.bbr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Eisch AJ, Cameron HA, Encinas JM, Meltzer LA, Ming GL, Overstreet-Wadiche LS. Adult neurogenesis, mental health, and mental illness: hope or hype? J Neurosci. 2008;28:11785–91. doi: 10.1523/JNEUROSCI.3798-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128:1778–89. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- 17.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–61. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci. 2009;29:11982–92. doi: 10.1523/JNEUROSCI.3158-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–12. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 20.Herber DL, Roth LM, Wilson D, Wilson N, Mason JE, Morgan D, et al. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–53. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–42. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Quinn J, Montine T, Morrow J, Woodward WR, Kulhanek D, Eckenstein F. Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer’s disease. J Neuroimmunol. 2003;137:32–41. doi: 10.1016/s0165-5728(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 23.Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi KI. Role of toll-like receptor signalling in A{beta} uptake and clearance. Brain. 2006;129:3006–19. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richard KL, Filali M, Prefontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J Neurosci. 2008;28:5784–93. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29:1846–54. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doi Y, Mizuno T, Maki Y, Jin S, Mizoguchi H, Ikeyama M, et al. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid {beta} neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. Am J Pathol. 2009;175:2121–32. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim J, Kou J, Song M, Pattanyak A, Jin J, Lalonde R, et al. MyD88 deficiency ameliorates beta-amyloidosis in an animal model of Alzheimer’s disease. Am J Pathol. 2011 doi: 10.1016/j.ajpath.2011.05.045. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 30.Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–70. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 31.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 32.Lalonde R, Kim HD, Fukuchi K. Exploratory activity, anxiety, and motor coordination in bigenic APPswe + PS1/DeltaE9 mice. Neurosci Lett. 2004;369:156–61. doi: 10.1016/j.neulet.2004.07.069. [DOI] [PubMed] [Google Scholar]
- 33.Lalonde R, Kim HD, Maxwell JA, Fukuchi K. Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci Lett. 2005;390:87–92. doi: 10.1016/j.neulet.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 34.Van Dam D, D’Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–96. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- 35.Lee KW, Lee SH, Kim H, Song JS, Yang SD, Paik SG, et al. Progressive cognitive impairment and anxiety induction in the absence of plaque deposition in C57BL/6 inbred mice expressing transgenic amyloid precursor protein. J Neurosci Res. 2004;76:572–80. doi: 10.1002/jnr.20127. [DOI] [PubMed] [Google Scholar]
- 36.File SE. Factors controlling measures of anxiety and responses to novelty in the mouse. Behav Brain Res. 2001;125:151–7. doi: 10.1016/s0166-4328(01)00292-3. [DOI] [PubMed] [Google Scholar]
- 37.Anisman H, Merali Z. Anhedonic and anxiogenic effects of cytokine exposure. Adv Exp Med Biol. 1999;461:199–233. doi: 10.1007/978-0-585-37970-8_12. [DOI] [PubMed] [Google Scholar]
- 38.Connor TJ, Song C, Leonard BE, Merali Z, Anisman H. An assessment of the effects of central interleukin-1beta, -2, -6, and tumor necrosis factor-alpha administration on some behavioural, neurochemical, endocrine and immune parameters in the rat. Neuroscience. 1998;84:923–33. doi: 10.1016/s0306-4522(97)00533-2. [DOI] [PubMed] [Google Scholar]
- 39.Cragnolini AB, Schioth HB, Scimonelli TN. Anxiety-like behavior induced by IL-1beta is modulated by alpha-MSH through central melanocortin-4 receptors. Peptides. 2006;27:1451–6. doi: 10.1016/j.peptides.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 40.Balaram P, Kien PK, Ismail A. Toll-like receptors and cytokines in immune responses to persistent mycobacterial and Salmonella infections. Int J Med Microbiol. 2009;299:177–85. doi: 10.1016/j.ijmm.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Lacosta S, Merali Z, Anisman H. Behavioral and neurochemical consequences of lipopolysaccharide in mice: anxiogenic-like effects. Brain Res. 1999;818:291–303. doi: 10.1016/s0006-8993(98)01288-8. [DOI] [PubMed] [Google Scholar]
- 42.Swiergiel AH, Dunn AJ. Effects of interleukin-1beta and lipopolysaccharide on behavior of mice in the elevated plus-maze and open field tests. Pharmacol Biochem Behav. 2007;86:651–9. doi: 10.1016/j.pbb.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koo JW, Duman RS. Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci Lett. 2009;456:39–43. doi: 10.1016/j.neulet.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bluthe RM, Castanon N, Pousset F, Bristow A, Ball C, Lestage J, et al. Central injection of IL-10 antagonizes the behavioural effects of lipopolysaccharide in rats. Psychoneuroendocrinology. 1999;24:301–11. doi: 10.1016/s0306-4530(98)00077-8. [DOI] [PubMed] [Google Scholar]
- 45.Kempermann G, Gage FH. Genetic determinants of adult hippocampal neurogenesis correlate with acquisition, but not probe trial performance, in the water maze task. Eur J Neurosci. 2002;16:129–36. doi: 10.1046/j.1460-9568.2002.02042.x. [DOI] [PubMed] [Google Scholar]
- 46.Choi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM, Ramirez JM, et al. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron. 2008;59:568–80. doi: 10.1016/j.neuron.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veeraraghavalu K, Choi SH, Zhang X, Sisodia SS. Presenilin 1 mutants impair the self-renewal and differentiation of adult murine subventricular zone-neuronal progenitors via cell-autonomous mechanisms involving notch signaling. J Neurosci. 2010;30:6903–15. doi: 10.1523/JNEUROSCI.0527-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hao W, Liu Y, Liu S, Walter S, Grimm MO, Kiliaan AJ, et al. Myeloid differentiation factor 88-deficient bone marrow cells improve Alzheimer’s disease-related symptoms and pathology. Brain. 2011;134:278–92. doi: 10.1093/brain/awq325. [DOI] [PubMed] [Google Scholar]
- 49.Frautschy SA, Cole GM. Why pleiotropic interventions are needed for Alzheimer’s disease. Mol Neurobiol. 2010;41:392–409. doi: 10.1007/s12035-010-8137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]