

Abstract
Legius syndrome, caused by SPRED1 mutations, has phenotypic overlap with neurofibromatosis type 1 (NF1) without tumorigenic manifestations. Patients fulfilling the NIH diagnostic criteria for NF1 were enrolled from the University of Utah NF Clinic and SPRED1 mutation analysis performed in order to identify the frequency of Legius syndrome within an NF1 clinic population.
SPRED1 sequencing was performed on 151 individuals with the clinical diagnosis of NF1 and 2 individuals (1.3%) were found to have novel SPRED1 mutations, p.R18X and p.Q194X. The phenotypes for the two individuals with SPRED1 mutations included altered pigmentation without tumorigenesis. A specific SPRED1 haplotype allele was identified in 27 individuals.
The frequency of SPRED1 mutations in patients meeting diagnostic criteria for NF1 in a hospital-based clinic is 1–2%. The likelihood an individual is harboring a SPRED1 mutation increases with age if multiple, non-pigmentary NF1 findings are absent. Legius syndrome patients may benefit from altered medical surveillance.
Keywords: neurofibromatosis type 1 (NF1), Legius syndrome, Ras pathway, SPRED1
Introduction
Neurofibromatosis type 1 (NF1) is a relatively common autosomal dominant disorder occurring in 1 out of every 3000 births. NF1 shows complete penetrance by adulthood yet there is a high degree of variable clinical expressivity, both inter- and intra-familial variability. Cardinal clinical manifestations are café-au-lait spots and neurofibromas, and other features include Lisch nodules, optic pathway gliomas, malignant peripheral nerve sheath tumors (MPNSTs), learning disorders, and bone abnormalities. While the clinical diagnosis for NF1 is based on NIH clinical diagnostic criteria, other disorders have overlapping phenotypes with NF1. The overlapping phenotype can potentially complicate diagnostic evaluations and lead to medically inappropriate screening protocols. Examples of these disorders include Noonan syndrome, LEOPARD syndrome, cardio-facio-cutaneous (CFC) syndrome, Costello syndrome (Stevenson et al., 2008), and Legius syndrome (Brems et al., 2007; Pasmant et al., 2009; Spurlock et al., 2009). The causative genes for all of these disorders encode proteins within the Ras-MAPK signaling pathway.
Legius syndrome, originally termed “neurofibromatosis type 1-like syndrome”, is caused by SPRED1 mutations on chromosome 15q13.2 (Brems et al., 2007). Individuals with SPRED1 mutations frequently fulfill the NIH diagnostic criteria for NF1 based on pigmentary manifestations of café-au-lait spots and distinctive freckling patterns (Brems et al., 2007; Pasmant et al., 2009; Spurlock et al. 2009). This overlapping phenotype is likely due to increased Ras signal propagation caused by inactivating SPRED1 mutations. Normally, SPRED1 protein binds to Ras and blocks phosphorylation of Raf, which diminishes downstream ERK activation (Brems et al., 2007).
The phenotype due to SPRED1 mutations as described by Brems et al. (2007) includes café-au-lait spots (98%), freckling (30%), and macrocephaly ≥97th centile (42%). Of the 44 reported cases (age ranges 1 to 66 years) in these 5 families, Brems et al. (2007) reported 14 patients with lipomas, 6 with learning problems, 5 with Noonan-like facial features, 3 with depigmented spots, 2 with attention deficit and hyperactivity disorder, and 3 with pectus excavatum. Neurofibromas, Lisch nodules, optic pathway tumors, or malignant tumors were not identified. Two additional studies by Pasmant and Spurlock (Pasmant et al., 2009; Spurlock et al., 2009) reported similar phenotypes in patients with SPRED1 mutations. Spurlock et al. (2009) tested 85 unrelated cases without detectable NF1 mutations or neurofibromas, and identified 6 individuals with SPRED1 mutations. They then screened family members of these 6 cases and identified a total of 12 individuals with SPRED1 mutations. All had café-au-lait macules, approximately two-thirds had intertriginous freckling, and there was no report of learning problems, Lisch nodules, neurofibromas, or optic gliomas. Pasmant et al. (2009) tested 61 index cases with the clinical diagnosis of NF1 without an identifiable NF1 mutation and found 5 individuals with SPRED1 mutations. They also screened additional family members from these 5 cases and identified a total of 18 individuals from 5 families with SPRED1 mutations. From the report (Pasmant et al, 2009), 9 of the 18 individuals with SPRED1 mutations fulfilled the NF1 diagnostic criteria assuming that individuals in their age categories of 0–4, 5–9, and 10–14 years are not post-pubertal. This assumption is important given that the NIH diagnostic criteria for café-au-lait macules states “six or more café-au-lait macules over 5mm in diameter in prepubertal individuals and over 15mm in greatest diameter in postpubertal individuals” (NIH, 1988; Gutmann et al., 1997). All 18 individuals had at least 2 café-au-lait macules, 72% had intertriginous freckling, 2 had lipomas, 4 had learning disabilities, 1 had epilepsy, 1 had monoblastic acute leukemia, and none had Lisch nodules, neurofibromas or optic gliomas. A comparison of some of the clinical features of SPRED1 and NF1 are summarized in Table 1. It appears from the few reports to date that patients with Legius syndrome do not develop some of the more morbid complications seen with NF1, particularly tumor formation, and an altered health care management plan may be appropriate.
Table 1.
Clinical Features of NF1 vs SPRED1
| NF1 | SPRED1 | |
|---|---|---|
| Café-au-lait spots | + | + |
| Intertriginous freckling | + | + |
| Lisch nodules | + | |
| Neurofibromas | + | |
| Tibial bowing | + | |
| Pseudarthrosis | + | |
| Macrocephaly | + | + |
| Learning disabilities | + | + |
| MPNST | + | |
| Optic glioma | + |
❖ Comparison of some features reported to be associated with the diagnosis of neurofibromatosis type 1 (NF1) and Legius syndrome. MPNST: malignant peripheral nerve sheath tumor
These previous studies (Brems et al., 2007; Pasmant et al., 2009; Spurlock et al. 2009) evaluated cohorts of individuals with clinical features of NF1 who did not have identifiable NF1 mutations. Approximately 5% of patients who meet the NIH clinical diagnostic criteria for NF1 do not have an identifiable NF1 mutation by exhaustive mutation analysis (Messiaen et al., 2000). Our aim was to identify the frequency of SPRED1 mutations in individuals with a clinical diagnosis of NF1 within an unselected Neurofibromatosis Clinic population.
Patients and Methods
NF1 patient phenotyping
Individuals meeting NIH clinical diagnostic criteria for neurofibromatosis type 1 (NF1) evaluated at the University of Utah Neurofibromatosis Clinic were offered enrollment. Prior to SPRED1 sequencing, a detailed physical examination and review of medical records were performed on all NF1 individuals by one physician investigator between 2002 and 2009 (DS). A standardized NF1 history and exam form created for use for the University of Utah NF1 Registry and modeled after information requested by the Children’s Tumor Foundation (formerly National Neurofibromatosis Foundation) International Database (Friedman and Birch, 1997), was modified and used to document the phenotype. For purposes of this study probands were defined as the individual within a family who was first enrolled. Approval by the Institutional Review Board at the University of Utah was obtained.
SPRED1 gene Sequencing
Primer sets were designed for the coding regions and intron/exon boundaries of SPRED1. These were designed using LightScanner Primer DesignR software by Idaho Technology Inc. (Salt Lake City, Utah). Selected primer sets were supplied by Integrated DNA Technologies (Coralville, Iowa). Primer sequences are available upon request. Genomic DNAs were isolated from peripheral blood using Puregene DNA extraction kit (Gentra System Inc, Minnesota). Each patient sample was amplified by polymerase chain reaction (PCR). All post PCR products were checked for the appropriate band size using capillary electrophoresis by the Qiagen eGene. PCR primers were then degraded using exoSAP-ITR (USB Corporation, Cleveland, OH), after which bidirectional Sanger sequencing was performed followed by sephadex cleanup. Products were then assessed by an ABI 3730 analyzer. All sequence analysis was performed using Mutation Surveyor® software (SoftGenetics, State College, PA) and compared with the GenBank reference sequence NC_000015. All amplicons were sequenced in both the forward and reverse directions.
Control Population Sample
Normal control population allele studies were performed using DNA from 32 anonymous healthy individuals from the ARUP clinical laboratory (Salt Lake City, UT). These samples were only tested for the specific exons required to identify the SPRED1 haplotype of interest.
Results
The SPRED1 gene was sequenced for 151 individuals with the clinical diagnosis of NF1. Demographics are described in Table 2. Within this cohort, 2 prepubertal unrelated individuals with novel SPRED1 mutations, p.R18X and p.Q194X, were identified.
Table 2.
Clinic Cohort Demographics
| Individuals Enrolled | 151 |
|
| |
| males | 79 |
| females | 72 |
|
| |
| Age Range | 1 to 34 years |
|
| |
| Average Age | 10.9 years |
|
| |
| Ethnicity
|
|
| white | 122 |
| black | 1 |
| Hispanic | 6 |
| Asian | 1 |
| >1 ethnicity indicated | 18 |
| unknown | 3 |
|
| |
| Inheritance Pattern
|
|
| Sporadic cases | 72 |
| Familial cases | 73 |
| Unknown pattern | 6 |
❖ All individuals included meet NIH clinical criteria for the diagnosis of NF1.
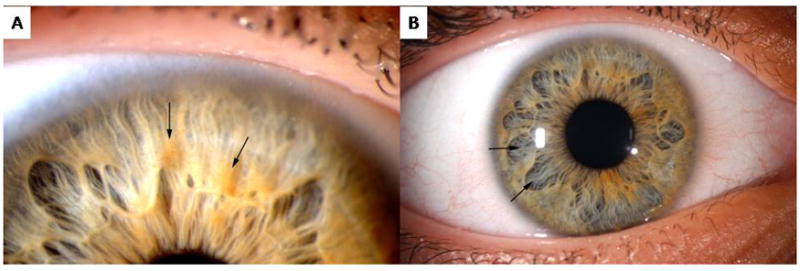
The first individual had a mutation in SPRED1 with a C>T nucleotide substitution at position c.52, generating a premature stop codon at p.18 (p.R18X). This patient’s mutation was confirmed on a clinical basis by the University of Alabama Medical Genomics Laboratory (Birmingham, AL). The observed phenotype for this individual, age 12 years, included 10–20 café-au-lait spots, subtle inguinal and axillary freckling, learning disabilities requiring intervention, and no neurofibromas (Figure 1A,B). His occipital frontal circumference (OFC) was at the 95th centile, weight was at the 75th centile, and height was at the 25th centile. He was reported to have a single Lisch nodule by slit lamp examination, which was clinically reported by an ophthalmologist at an outside institution over multiple evaluations. Given that no other individual with a SPRED1 mutation has been reported to have Lisch nodules, the patient was evaluated at the University of Utah by one of the authors (DD), a pediatric ophthalmologist who follows a large number of individuals with NF1, for further investigation of the ophthalmologic findings. The patient did not have classic Lisch nodules; however, he had an unusual pattern of iris pigmentation with multiple iris nevi of varying size (Figure 2A) and abnormally large and wide iris crypts (Figure 2B).
Fig. 1.
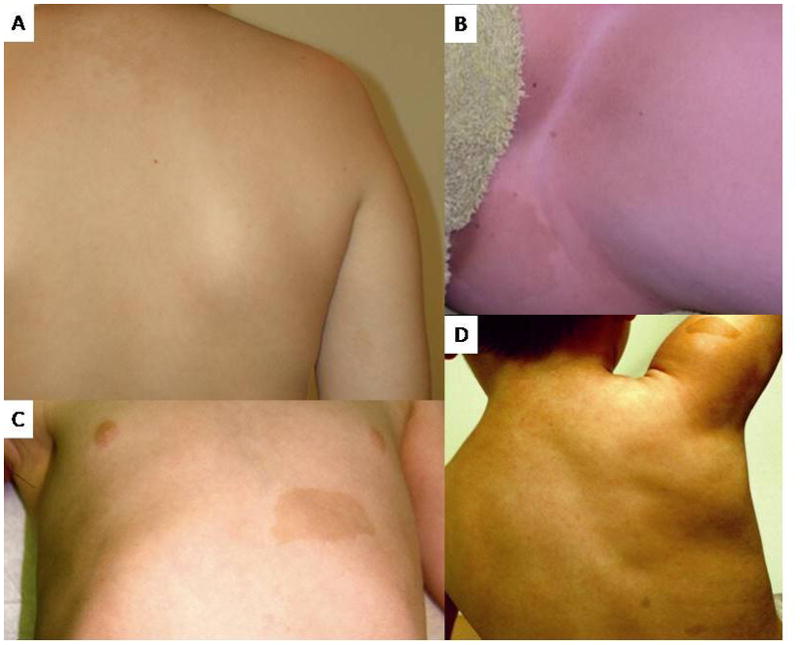
Fig. 2.
The second individual had a mutation in SPRED1 with a C>T nucleotide substitution at position c.580, resulting in a premature stop codon at p.194 (p.Q194X). The phenotype for this individual, age 10 years, included more than 10 café-au-lait spots, axillary and groin freckling, learning disabilities requiring intervention, and no neurofibromas (Figure 1C,D). His OFC was >98th centile, height was at the 95th centile, and weight was >98th centile. Ophthalmologic examination at 7 years of age showed no Lisch nodules; however, this individual was reported by a pediatric ophthalmologist at the University of Utah to have “the sort of very convoluted brown iris making detection of Lisch nodules very difficult.”
Demographic and phenotypic information for the clinic cohort was used for comparison between individuals meeting the NIH diagnostic criteria for NF1 with and without a SPRED1 mutation. Based on the presence or absence of a specific finding or combination of specific findings, the likelihood of having a SPRED1 mutation is outlined in Figure 3. Within our clinic cohort of individuals fulfilling NIH diagnostic criteria for NF1, we identified 1.32% with SPRED1 mutations, and 2.60% if selected for only sporadic cases. Selecting for those individuals who lacked Lisch nodules, optic gliomas, neurofibromas, long bone dysplasia and sphenoid wing dysplasia or a family history, this percentage increases to 20%. This percentage increases further with age to 50% for those 10 years or older.
Fig. 3.

In establishing an assay to identify SPRED1 mutations, we identified a common haplotype allele matching GenBank sequence NG_008980. This haplotype allele (SPRED12) is defined by 4 single nucleotide polymorphisms (SNPs), differing from the more common SPRED11 reference sequence NC_000015. The SPRED12 allele was identified in 27 of the 151 individuals sequenced (17.9%). Twenty-five NF1 individuals were heterozygous for SPRED12 while two patients were homozygous. The overall SPRED12 allele frequency was 9.6%. When only probands are used to calculate the haplotype allele frequency (excluding the affected related individuals), 25 of 127 individuals carry the SPRED12 haplotype allele [19.7%, allele frequency 10.6%]. Two of the single nucleotide polymorphisms were found within exons and caused synonymous changes. The first exonic SNP is located in exon 4 (c.291A>G; p.K97K) and the second exonic SNP is located in exon 8 (c.1044C>T; p.V348V). Both intronic SNPs are located within the intronic region between exons 5 and 6 (c.424-8A>C and c.424-98C>T). Neither of these two intronic SNPs is predicted to cause splice site alterations (http://www.fruitfly.org/seq_tools/splice.html). None of these 4 SNPs were seen as an isolated event in any of the 151 patients sequenced; indicating that this haplotype block is in high disequilibrium. The allele frequency in our unselected normal population was slightly decreased from the NF1 cohort [12.5% (4/32, p=0.049)]. No SPRED12 allele homozygotes were identified in the unselected population. The overall allele frequency was 6.3%, (4/64 alleles). The difference in allele frequency between the NF1 probands only versus the unselected control population is statistically significant (p=0.02). Demographic data was not available for the normal population.
Discussion
The overlap of disease phenotype within the disorders of the Ras-MAPK pathway is well established (Brems et al., 2007; Pasmant et al., 2009; Spurlock et al. 2009; Stevenson et al., 2008; Stevenson et al., 2006; Nyström et al., 2003; Johnson et al., 1998; Lopez-Rangel, 2007; Diglio et al., 2002; Tassabehji et al., 1993; Allanson et al., 1991). One of the most common Ras-MAPK pathway disorders is NF1. In individuals fulfilling the NIH diagnostic criteria for NF1, 95% have identifiable mutations of the NF1 gene (Messiaen et al. Hum Mut: 2000). Thus NF1 mutations are not identified in ~5% of patients who meet NIH clinical diagnostic criteria for NF1. If diagnostic criteria are met solely by pigmentary findings (with or without a first-degree relative), a proportion of these individuals could have Legius syndrome with mutations in SPRED1. It is also possible that the remaining individuals have an NF1 mutation that is undetectable by current methods, a different overlapping Ras pathway disorder, or mutations in other unknown genes. There are several reports of families with isolated multiple café-au-lait macules inherited in an autosomal dominant pattern (Charrow et al., 1993; Arnsmeier et al., 1994; Nyström et al., 2009; Abeliovich et al., 1995; Brunner et al., 1993). Several of these families showed no linkage to the NF1 locus (Charrow et al., 1993; Nyström et al., 2009; Brunner et al., 1993), and one family’s phenotype did not segregate with SPRED1 (Nyström et al., 2009). This suggests that other unknown genes can result in a similar pigmentary phenotype similar to Legius syndrome and NF1.
Our data show that 1.3% of individuals who meet the clinical diagnostic criteria for NF1 and seeking medical care at an NF Clinic have SPRED1 mutations. This percentage increases if the cohort is stratified to eliminate those with an affected parent, an optic pathway tumor, bona fide Lisch nodules, neurofibromata, long bone dysplasia or sphenoid wing dysplasia. In this circumstance, the percentage of patients with SPRED1 mutations is 20%. Because of the age-related penetrance of non-pigmentary manifestations of NF1, as an individual’s age increases the absence of certain characteristic features of NF1 becomes more predictive of Legius syndrome. For individuals 10-years of age or older who do not have other NF1 features, yet fulfill diagnostic criteria for NF1 by pigmentary features, 50% harbor a SPRED1 mutation (Figure 3). While, the chance of having both an NF1 mutation and a SPRED1 mutation is possible, co-occurrences of 2 rare disorders are extremely infrequent (Wilken et al., 2009).
The phenotypes of the 2 individuals with SPRED1 mutations were consistent with the previous reports to date (Table 1) (Brems et al., 2007; Pasmat et al., 2009; Spurlock et al., 2009), including café-au-lait spots, intertriginous freckling, learning disabilities, and no neurofibromas. Both children were older than 10 years of age and had not yet developed neurofibromas. One patient with a SPRED1 mutation was initially reported to have a single identified Lisch nodule, although a subsequent evaluation did not confirm the findings. To date, there are no reported cases of individuals with SPRED1 mutations who have Lisch nodules, including those adults (>18-years-old) identified by previous authors (Brems et al., 2007; Pasmat et al., 2009; Spurlock et al., 2009).
As shown in Figure 2, it is possible that individuals with Legius syndrome have increased pigment deposits in the iris which may lead some physicians to confuse increased pigment with Lisch nodules, especially if one does not frequently evaluate patients with NF1. It is important to accurately diagnose Lisch nodules in individuals with isolated café-au-lait macules and/or intertriginous freckling, as the presence of Lisch nodules would impact the delineation of Legius syndrome versus NF1, and the diagnosis could subsequently change medical management and counseling. Since fewer than 90 Legius syndrome patients have been reported, it may be that a small percentage of patients can develop increased pigment deposits or other iris findings. Further investigation is necessary to determine the degree of association and types of ocular abnormalities for individuals with SPRED1 mutations. The absence of a Lisch nodule in the above reported patient is consistent with Legius syndrome.
The phenotype of Legius syndrome is currently based on a cross-sectional phenotype of a small number of patients (Brems et al., 2007; Pasmat et al., 2009; Spurlock et al., 2009). The clinical manifestations of Legius syndrome will continue to become better defined with the identification of more individuals. Long-term studies will be required to confirm the current suggested phenotype and establish the lifetime risk of tumorigenesis and whether it is increased compared to the general population. Within our cohort, the phenotype primarily consists of altered pigmentary patterns and learning disorders as demonstrated in previously reported cases (Brems et al., 2007; Pasmant et al., 2009; and Spurlock et al., 2009). This is also consistent with the relative lack of manifestations of tumorigenesis in Legius syndrome. Pasmant et al. identified one SPRED1 mutation positive patient with acute myelogenous leukemia. While no second hit in SPRED1 could be identified in this patient, the authors could not definitively discern causality in relation to Legius syndrome. Our data support the lack of tumorigenesis in Legius syndrome.
An altered management plan may be appropriate for young individuals fulfilling the pigmentary diagnostic criteria for NF1 who have SPRED1 mutations rather than NF1 mutations. Since the absence of characteristic NF1 findings appears consistent across multiple studies, this absence may help identify individuals with SPRED1 mutations. For those patients meeting NIH diagnostic criteria for NF1 based on pigmentary findings alone, the likelihood of an individual having a SPRED1 mutation increases as multiple additional characteristic NF1 findings are absent (Figure 3), with increasing age. However, the possibility of an individual mildly affected with NF1 or NF1 mosaicism must always be considered.
A common SPRED1 haplotype allele was identified in 17.9% of our NF1 cohort (allele frequency of 9.6%), which is significantly more frequent than the 12.5% of an unselected population (allele frequency of 6.3%) (p=0.049). However, because demographic data for the unselected population are not available, ethnicity may be a confounding factor. In addition, it is possible that this is a founder effect within the Utah population. SPRED1 sequence variants could potentially modify the NF1 phenotype and future studies will be important in examining the role of the SPRED1 haplotypes on the clinical manifestations in individuals with NF1.
Previous reports have shown that many individuals with SPRED1 mutations do not meet the NIH diagnostic criteria for NF1 (Brems et al., 2007; Pasmat et al., 2009; Spurlock et al., 2009). Our cohort only included individuals who fulfilled the NIH diagnostic criteria. Hence, the incidence of SPRED1 mutations in individuals without the clinical diagnosis of NF1 cannot be determined from our results. There is also a potential bias in this study as the NF1 Clinic is staffed primarily by pediatricians at a tertiary children’s hospital, thus the cohort is primarily a pediatric population.
Conclusion
In summary, 1–2% of patients meeting NIH clinical diagnostic criteria for NF1 in a hospital-based clinic may harbor SPRED1 mutations. Patients who fulfill only the pigmentary component of the NF1 diagnostic criteria are more likely to have a SPRED1 mutation, especially at an older age. Patients with SPRED1 mutations may require altered medical surveillance, based on the current Legius syndrome phenotype showing altered pigmentation with a lack of tumorigenesis.
Acknowledgments
Funding: This research was completed thanks to a generous Young Investigator Award from the University of Utah Department of Pathology and NIH NINDS K23 NS052500. This investigation was supported in part by the Center for Clinical and Translational Sciences at the University of Utah through the Public Health Services research grant numbers UL1-RR025764 and C06-RR11234 from the National Center for Research Resources. Dr. Stevenson is a recipient of a Doris Duke Clinical Scientist Development Award and this work was supported in part by the Doris Duke Charitable Foundation.
We acknowledge Melinda Procter, Lan-Szu Chou, and Brian Shirts for their support within the research laboratory. We acknowledge John Carey, Alan Rope, Susan Lewin, Steve Bleyl, Heather Hanson, Stephanie Bauer, Lisa Smith, and the Clinical Genetics Research Program Phenotyping Core for support in enrolling participants and obtaining samples.
References
- Stevenson DA, Swensen J, Viskochil DH. Neurofibromatosis type 1 and other syndromes of the Ras pathway. In: Kaufmann D, editor. Neurofibromatoses. Monographs in Human Genetics. Vol. 16. Basel; Karger: 2008. pp. 32–45. [Google Scholar]
- Brems H, Chmara M, Sahbatou M, et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007;39(9):1120–1126. doi: 10.1038/ng2113. [DOI] [PubMed] [Google Scholar]
- Pasmant E, Sabbagh A, Hanna N, et al. SPRED1 germline mutations caused a neurofibromatosis type 1 overlapping phenotype. J Med Genet. 2009;46(7):425–430. doi: 10.1136/jmg.2008.065243. [DOI] [PubMed] [Google Scholar]
- Spurlock G, Bennett E, Chuzhanova N, et al. SPRED1 mtuations (Legius syndrome): another clinically useful genotype for dissecting the NF1 phenotype. J Med Genet. 2009;46(7):431–437. doi: 10.1136/jmg.2008.065474. [DOI] [PubMed] [Google Scholar]
- Neurofibromatosis Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45:575–578. [PubMed] [Google Scholar]
- Gutmann DH, Aylsworth A, Carey JC, Korf B, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. [PubMed] [Google Scholar]
- Friedman JM, Birch PH. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet. 1997;70:138–43. doi: 10.1002/(sici)1096-8628(19970516)70:2<138::aid-ajmg7>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- http://www.fruitfly.org/seq_tools/splice.html
- Stevenson DA, Viskochil DH, Rope AF, Carey JC. Clinical and molecular aspects of an informative family with neurofibromatosis type 1 and Noonan phenotype. Clin Genet. 2006;69:246–253. doi: 10.1111/j.1399-0004.2006.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyström AM, Annerén G, Bondeson ML. Noonan syndrome with café-au-lait spots and LEOPARD syndrome: evidence for genetic heterogeneity. Am J Hum Genet. 2003;73(supplement):725. [Google Scholar]
- Johnson JP, Golabi M, Norton ME, Rosenblatt RM, Feldman GM, Yang SP, Hall BD, Fries MH, Carey JC. Costello syndrome: phenotype, natural history, differential diagnosis, and possible cause. J Pediatr. 1998;133:441–8. doi: 10.1016/s0022-3476(98)70284-7. [DOI] [PubMed] [Google Scholar]
- Lopez-Rangel E. Overlapping clinical phenotypes: the road to identifying dysmorphology signaling pathways and their associated risks. Clin Genet. 2007;71:43–44. [Google Scholar]
- Diglio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–394. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassabehji M, Strachan T, Sharland M, Colley A, Donnai D, Harris R, Thakker N. Tandem duplication within a neurofibromatosis type I (NF1) gene exon in a family with features of Watson syndrome and Noonan syndrome. Am J Hum Genet. 1993;53:90–95. [PMC free article] [PubMed] [Google Scholar]
- Allanson JE, Upadhyaya M, Watson GH, Partington M, MacKenzie A, Lahey D, MacLeod H, Sarfarazi M, Broadhead W, Harper PS, Huson SM. Watson syndrome: is it a subtype of type 1 neurofibromatosis? J Med Genet. 1991;28:752–756. doi: 10.1136/jmg.28.11.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messiaen LM, Callens T, Mortier G, et al. Exhaustive Mutation Analysis of the NF1 Gene Allows Identification of 95% of Mutations and Reveals a High Frequency of Unusual Splicing Defects. Hum Mutat. 2000;15:541–555. doi: 10.1002/1098-1004(200006)15:6<541::AID-HUMU6>3.0.CO;2-N. 7 (DS) intake forms. [DOI] [PubMed] [Google Scholar]
- Charrow J, Listernick R, Ward K. Autosomal dominant multiple café-au-lait spots and neurofibromatosis-1: evidence of non-linkage. Am J Med Genet. 1993;45 :606–8. doi: 10.1002/ajmg.1320450518. [DOI] [PubMed] [Google Scholar]
- Arnsmeier SL, Riccardi VM, Paller AS. Familial multiple cafe au lait spots. Arch Dermatol. 1994;130:1425–6. [PubMed] [Google Scholar]
- Nyström AM, Ekvall S, Strömberg B, Holmström G, Theuresson AC, Annerén G, Bondeson ML. A severe form of Noonan syndrome and autosomal dominant café-au-lait spots – evidence for different genetic origins. Acta Paediatr. 2009;98:693–8. doi: 10.1111/j.1651-2227.2008.01170.x. [DOI] [PubMed] [Google Scholar]
- Abeliovich D, Gelman-Kohan Z, Silverstein S, Lerer I, Chemke J, Merin S, Zlotogora J. Familial cafe au lait spots: a variant of neurofibromatosis type 1. J Med Genet. 1995;32:985–6. doi: 10.1136/jmg.32.12.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner HG, Hulsebos T, Steijlen PM, der Kinderen DJ, vd Steen A, Hamel BC. Exclusion of the neurofibromatosis 1 locus in a family with inherited café-au-lait spots. Am J Med Genet. 1993;46:472–4. doi: 10.1002/ajmg.1320460428. [DOI] [PubMed] [Google Scholar]
- Wilken TC, Zenker M, Sticht H, et al. Independent NF1 and PTPN11 mutations in a family with neurofibromatosis-Noonan syndrome. Am J Med Genet A. 2009;149A(6):1263–1267. doi: 10.1002/ajmg.a.32837. [DOI] [PubMed] [Google Scholar]