

Abstract
The role of autophagy, traditionally considered a cellular homeostatic and recycling mechanism, has expanded dramatically to include an involvement in discrete stages of tumor initiation and development. Gliomas are the most aggressive and also the most common brain malignancies. Current treatment modalities have only a modest effect on patient outcomes. Resistance to apoptosis, a hallmark of most cancers, has driven the search for novel targets in cancer therapy. The autophagy lysosomal pathway is one such target that is being explored in multiple cancers including gliomas and is a promising avenue for further therapeutic development. This review summarizes our current understanding of the autophagic process and its potential utility as a target for glioma therapy.
Keywords: BCL‐2, BECLIN1, Combinatorial therapy, Glioblastoma, LC3
INTRODUCTION
Malignant neoplasms constitute the second most common cause of death in the United States (41) and malignant brain tumors contribute to 2.4% of cancer‐related deaths (6). According to the American Cancer Society, an estimated 20 340 new cases of primary central nervous system tumors will be diagnosed in 2011 in the United States alone and will result in 13 110 deaths. Gliomas are the most common brain malignancies. Glioblastoma (GBM), classified as grade IV astrocytoma by the World Health Organization, is the most aggressive and accounts for 54% of all gliomas (6). Despite considerable advances in multimodality treatment of tumors in the last five decades, there has been only a minimal improvement in the median survival time of GBM patients (from approximately 12 to 14 months) or the 5‐year survival rate (less than five percent) (88). Causative factors for the poor survival rate include the highly invasive nature of GBMs making them intractable to complete surgical resection, and resistance to standard chemotherapy and radiotherapy, which primarily depend on triggering tumor cell apoptosis. Evading apoptosis is one of the major hallmarks of cancer (32) and it has been shown that defective apoptosis contributes to both tumorigenesis and chemoresistance 42, 73. This underscores the need to target non‐apoptotic death pathways and prosurvival signaling mechanisms that contribute to resistance to conventional therapies. For these reasons, autophagy, which can be either survival promoting or death inducing depending on cellular context, has received increasing scientific attention.
Autophagy is a highly conserved cellular homeostatic process. The term “autophagy” was coined by Christian de Duve and means self (auto)–eating (phagy) (95). There are three main types of autophagy—macroautophagy, microautophagy and chaperone‐mediated autophagy. Macroautophagy (hereby referred to as autophagy) is a catabolic pathway in which cytoplasmic contents including organelles are sequestered within double‐membrane vesicles (autophagosomes) and targeted to the lysosomes for degradation and recycling. Though originally suggested to be a mechanism for nonspecific bulk segregation and degradation of cytoplasmic contents in the lysosome (14), the current perception of autophagy has greatly evolved to include roles in development, maintenance of genomic stability and protein and organelle quality control. In the context of tumors, reduced autophagic activity in carcinogen treated rat hepatocytes was observed over 25 years ago (77). The report in 1999 that the autophagy‐related gene BECLIN1 functions as a tumor suppressor (54) stimulated significant interest from cancer biologists on this previously unexplored process. Subsequent reports demonstrated an accelerated rate of spontaneous tumor development in murine models with defects in autophagy‐related genes thereby implicating a tumor suppressive role for autophagy in general 58, 71, 98. Of particular interest are recent findings that high‐grade gliomas have lower expression of autophagy‐related proteins when compared with low‐grade gliomas (69) and that progression of astrocytic tumors is associated with a decrease in autophagic capacity (36). Several studies have also shown that modulation of autophagy sensitizes brain tumor cells to standard chemotherapy and radiotherapy induced death. The increasingly recognized relevance of autophagy to tumorigenesis, tumor progression, tumor suppression and ultimately, tumor therapy, make it an extremely important investigative focus.
MECHANISM OF AUTOPHAGY
Autophagy is a tightly regulated process controlled by a number of highly conserved autophagy‐related genes known as ATGs (for AuTophaGy gene) (87). Genetic screens in yeast have led to the identification of over 30 ATGs and many of their homologs have been identified and characterized in mammalian cells. The ATG proteins function at several discrete but continuous steps in autophagy, which include induction or selection/packaging of cargo, vesicle nucleation, vesicle elongation, vesicle docking and fusion with lysosomes and degradation of vesicular contents.
Autophagy is induced by various cellular stress mediated signaling pathways involved in nutrient signaling, growth factor status, energy sensing, hypoxia, oxidative and ER stress and pathogen infection (33). The input from these multiple upstream signal transduction pathways is integrated by the serine/threonine protein kinase target of rapamycin (TOR). TOR acts upstream of the ATG proteins as a central inhibitor of autophagy under nutrient rich conditions, negatively regulating ATG1 and preventing the ATG1‐ATG13‐ATG17 scaffold formation (43) (phosphorylation of the ULKs‐ATG13‐FIP200 complex in mammalian cells). Activity of TOR in response to diverse stimuli is positively regulated by the GTPase Ras homolog enriched in brain (Rheb) and mediated through the class I phosphoinositide 3‐kinase (PI3‐K)–protein kinase B (AKT) pathway in response to growth factor signaling (51). Negative regulation of TOR, resulting in the induction of autophagy, is mediated through energy‐sensing kinase—adenosine monophosphate‐activated protein kinase (AMPK) and the eukaryotic initiation factor 2α (EIF2α) 51, 83. Tumor suppressor phosphatase and tensin homolog (PTEN), a negative regulator of PI3‐K/AKT signaling pathway, negatively regulates TOR and can induce autophagy when over‐expressed (3). The tumor suppressor protein TP53 has also been shown to be involved in the regulation of autophagy indirectly via the regulation of the TOR pathway by promoting the transcription of various negative regulators, such as AMPKβ and PTEN 21, 22.
Vesicle nucleation requires the formation of a class III phosphatidylinositol 3‐kinase (PI3‐K) complex, which is composed of PI3‐K/Vps34 (vacuolar protein sorting 34), BECLIN1 (mammalian ortholog of ATG6) and p150 (myristoylated serine/threonine kinase Vps15 in yeast) (79). Various binding partners of BECLIN1—ultraviolet (UV) radiation resistance‐associates gene (UVRAG) 37, 53, ATG14L/Barkor 62, 99 and AMBRA1 (activating molecule in beclin‐regulated autophagy) (23)—which positively regulate BECLIN1 activity at different steps of autophagy have been recently identified. Autophagy is negatively regulated at this step by the anti‐apoptotic BCL‐2 family members, BCL‐2 and BCL‐xL, which physically bind to BECLIN1 and prevent the formation of the PI3K core complex (57). However, the BCL‐2/BECLIN1 interaction is disrupted by a pro‐apoptotic BCL‐2 family member—BCL‐2/adenovirus E1B 19 kd‐interacting protein3 (BNIP3), which binds to BCL‐2 thus releasing BECLIN1 (72). Recently, another molecule Rubicon (RUN domain and cysteine‐rich domain containing, BECLIN1‐interacting protein) has been found to negatively regulate BECLIN1 (99), whereas the mitogen‐activated protein kinase/extracellular signal‐regulated kinase pathway has been shown to positively affect BECLIN1 levels (92).
Vesicle elongation involves two ubiquitin‐like conjugation systems 66, 67. The first system involves the formation of the ATG5‐ATG12 complex via the E1‐ and E2‐like actions of ATG7 and ATG10, respectively. The ATG5‐ATG12 complex then binds ATG16, resulting in a large multimeric ATG16L complex. The second system involves the cleavage of LC3/ATG8 by the protease ATG4, which is followed by the conjugation of the cleaved ATG8 to the lipid phosphotidylethanolamine (PE) via ATG7 and E2‐like enzyme, ATG3. This results in the conversion of the soluble LC3/ATG8 (LC3‐1) to LC3‐II, which is recruited to both the outer and inner surfaces of the autophagosomal membrane. It has been reported that the ATG16L complex acts as an E3 that promotes the lipidation of LC3/ATG8 25, 31. Ultimately, the autophagosome docks and fuses with the lysosome, resulting in the formation of a single membrane‐bound acidic vesicle called the autolysosome. The contents of the autolysosome are degraded by the proteolytic activity of the lysosomal hydrolases and then recycled. Complete details of the various steps involved in the molecular machinery of autophagy have yet to be defined.
AUTOPHAGY IN TUMORIGENESIS
The current opinion regarding the role of autophagy in tumorigenesis is that autophagy can be either a tumor suppressor or promoter depending on the tumor type and stage. The observation that BECLIN1 is monoallelically deleted in a high percentage of human breast, ovarian and prostate cancers provided the first genetic link between autophagy and cancer (54). Subsequently, heterozygous disruption of the Beclin1 gene in mice was shown to result in increased cell proliferation and tumorigenesis, suggesting that Beclin1 is a haploinsufficient tumor suppressor gene 71, 98. In the context of brain tumors, cytoplasmic levels of BECLIN1 protein and mRNA were found to be lower in GBMs relative to lower grade astrocytomas as well as normal brain tissue (64). In addition, high cytoplasmic levels of BECLIN1 protein have been found to bear a positive correlation with patient survival and performance status (Karnofski classification), whereas low expression of BECLIN1 protein correlates with an increase in cell proliferation and a decrease in apoptosis (69). Interestingly, high LC3 expression was associated with improved survival in GBM patients with poor performance scores, whereas in patients with normal performance scores, low LC3 expression correlates with better survival (2). As low levels of BECLIN1 and LC3B‐II protein are found in higher grade astrocytomas (36), it has been suggested that a decrease in autophagic activity may drive progression of astrocytic tumors. Existing knowledge about the involvement of autophagy‐related proteins in brain tumorigenesis is largely limited to BECLIN1 and LC3. Proteins interacting with BECLIN1, such as UVRAG and BIF1 (bax interacting factor 1), have been implicated in gastric carcinoma tumorigenesis 50, 52, and could have a similar role in brain tumors, but no direct studies have addressed this possibility. Additionally, frameshift mutations of other ATG genes—ATG2B, ATG5, ATG9B and ATG12—reported in gastric and colorectal cancers (45) need to be assessed in brain tumors.
Indirect evidence suggests that autophagy may be highly relevant to gliomas. Genes such as epidermal growth factor receptor (EGFR), neurofibromin 1 (NF1), PTEN, AKT and TP53 whose alterations/mutations are frequently associated with brain tumors, are known to be involved in regulation of autophagy (Figure 1). The amplification of the receptor tyrosine kinase (RTK) EGFR, often found in gliomas, is known to suppress autophagy. EGFR suppresses autophagy by maintaining the basal intracellular glucose level through physically interacting with the sodium/glucose cotransporter (SGLT) (94). PTEN, a known tumor suppressor, has long been known to positively regulate autophagy by inhibiting the PI3‐K/AKT pathway (3). However, comutations of PTEN and NF1 are frequently seen in gliomas and result in constitutive activation of the PI3‐K/AKT/TOR signaling, which could suppress autophagy. TP53, another tumor suppressor that is frequently mutated in brain tumors, has been suggested to have a dual role in the regulation of autophagy. Nuclear TP53 has been shown to promote autophagy through transcriptional regulation of damage‐regulated autophagy modulator (DRAM) (11) whereas cytoplasmic TP53 can negatively regulate autophagy (84). In the specific context of brain tumors, the frequent alterations/mutations of tumor suppressors known to positively regulate autophagy suggest a tumor suppressor role of autophagy. Furthermore, the role of other ATG genes, apart from BECLIN1 and LC3, in brain tumors needs to be investigated as they could represent potentially useful prognostic markers and/or therapeutic targets.
Figure 1.
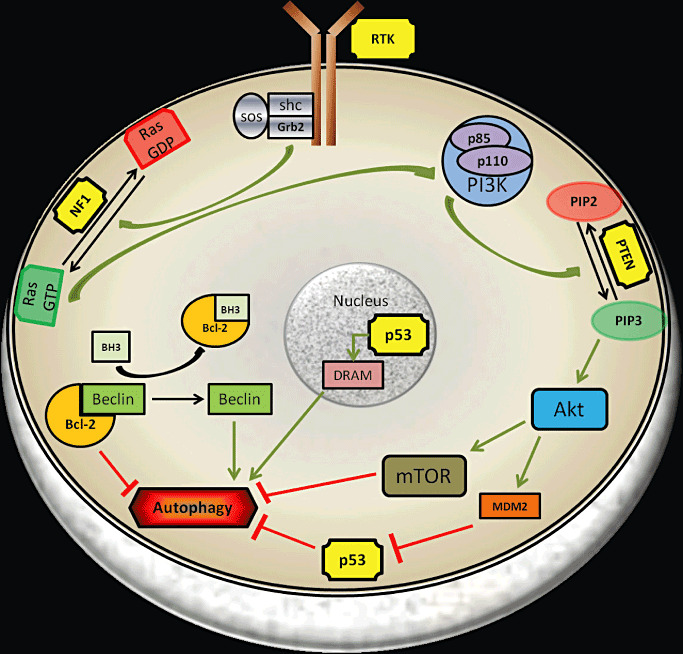
Schematic illustration of common mutations seen in gliomas and their role in autophagy regulation. Mutations in receptor tyrosine kinases (EGFR, PDGFR), phosphatase and tensin homolog (PTEN), NF1 and p53 are frequently observed in gliomas (yellow boxes). Aberrant signals from the RTK and/or Ras pathways feed into the PI3K–AKT–mTOR axis altering the autophagic machinery of the tumor cell. Nuclear p53 induces autophagy whereas cytoplasmic localization of p53 negatively regulates autophagy. BCL‐2 is bound to BECLIN, which is required for induction of autophagy and BH3‐only molecules like BNIP3 release BECLIN from BCL‐2 resulting in autophagy induction. Abbreviations: DRAM, damage‐regulated autophagy modulator; GDP, guanosine diphosphate; GTP, guanosine triphosphate; mTOR, mammalian target of rapamycin; PIP3, phosphatidylinositol (3,4,5)‐triphosphate; PIP2, phosphatidylinositol (4,5)‐bisphosphate; RTK, receptor tyrosine kinase; SOS, son of sevenless.
Several mechanisms have been suggested to explain the role of autophagy in suppression of tumorigenesis. Maintenance of genomic stability by autophagy is considered to be one of the major mechanisms of tumor suppression (60). The role of the tumor suppressor TP53 in maintaining genomic integrity is well established and inactivation or loss of TP53 is a common finding in multiple tumors. Therefore tumor cells with an additional defect in autophagy may further accumulate significantly higher DNA double strand breaks and gene amplifications, along with damaged mitochondria. Reduced clearance of damaged mitochondria and protein aggregates can potentially lead to increased DNA damage presumably because of elevated levels of reactive oxygen species (ROS) (61). However, these observations were made in cells with defects in multiple genes including TP53, therefore the role of autophagy as a primary contributor in preventing tumor initiation in normal cells by maintaining genome stability is yet to be definitively established (75). Interestingly, limiting necrosis in tumor cells has been suggested as an additional mechanism by which autophagy suppresses tumorigenesis. Tumor cells that are defective in apoptosis can die by necrosis when exposed to excessive metabolic stress. Active autophagy in these cells prevents necrosis and hence suppresses the resultant inflammation, which is known to increase tumor growth (15). Another mechanism by which autophagy could negatively regulate tumor initiation is through oncogene induced senescence. Oncogene‐induced senescence is a state of cell cycle arrest by which malignant transformation of cells is blocked in response to activation of oncogenes. The role of autophagy in oncogene‐induced senescence was established by knockdown studies of ATG5 and ATG7, which resulted in significant bypass of senescence (97).
Although reduced autophagic capacity has been shown to be associated with tumor progression and poor prognosis in various tumors, it is widely appreciated that cancer cells can use autophagy as a survival mechanism to cope with diverse stresses in the tumor microenvironment. Following extracellular matrix detachment, autophagy has been shown to protect cells from anoikis (26). This ability to evade anoikis provides the tumor cells the opportunity to invade neighboring tissue and form diffuse tumors, a characteristic feature of GBMs. Recent studies have proposed a new model for autophagy in tumor promotion known as the “autophagic tumor stroma model of cancer”(59). These studies propose that tumor cells induce oxidative stress and autophagy in the adjacent stromal fibroblasts leading to the stromal overproduction of recycled nutrients. Tumor cells use these recycled nutrients to promote their growth (59). Whether malignant glial cells can similarly induce such a response from non‐malignant glial cells is unknown.
Facilitating glycolysis has been suggested as another novel tumor‐promoting function for autophagy. Autophagy inhibition resulted in decreased proliferation of Ras‐transformed cells and autophagy deficient cells were reported to have reduced glycolytic capacity (55). The central regions of solid tumors are frequently hypoxic as a result of the tumor outgrowing existing vasculature. In the hypoxic core of tumors, autophagy is induced as a survival mechanism by the tumor cells (72). Overexpression of hypoxia inducible factor‐1α (HIF1α) in the hypoxic regions of the tumor, results in the upregulation of BNIP3 and BNIP3L. Autophagy is induced by BNIP3 by disrupting the BECLIN1‐BCL‐2 complex and has been shown to be an adaptive survival response during prolonged hypoxia (4).
Sufficient evidence is available to support the dual nature of autophagy in cancers as well as the significance of tumor cell type and context. The complex relationship between autophagy and tumorigenesis and the potential avenues it presents for cancer therapy are currently the focus of intense investigation.
ROLE OF AUTOPHAGY IN BRAIN TUMOR THERAPY
The poor response of brain tumors to current treatment modalities that primarily promote apoptosis makes it important to target autophagy as an alternative death pathway. Frequently observed mutations/alterations in brain tumors such as EGFR, NF1, AKT, PTEN and TP53 are also known to be involved in autophagy regulation. Recently the cancer genome atlas consortium has classified GBMs into four different molecular subtypes—proneural, classical, mesenchymal and neural—based on the frequency of mutations in platelet derived growth factor receptor (PDGFR), TP53, NF1, PTEN and EGFR (90). Interestingly, we observed differences in the basal level expression of LC3 protein in xenografts derived from these different molecular subtypes (Figure 2A). These differences could be attributed to the role of specific characteristic mutations, on which the classification was based, in the regulation of autophagy. These observations suggest that a combinatorial approach targeting these pathways along with the autophagy lysosomal pathway (ALP) may lead to development of effective subtype specific therapies. Moreover, autophagy has been shown to be induced by a number of current and experimental glioma therapies. While autophagy mediates tumor cell resistance and survival in some therapies, in other therapies autophagy contributes to the cytotoxic and/or cytostatic response. The specific role of autophagy in promoting survival or death in various therapeutic settings is yet to be clearly elucidated and would play a crucial role in designing effective therapeutic combinations. In this section, we review the current literature pertaining to treatment outcomes following autophagy modulation in various combinatorial regimes in brain tumors (gliomas) and discuss the role of autophagy in current treatment modalities.
Figure 2.
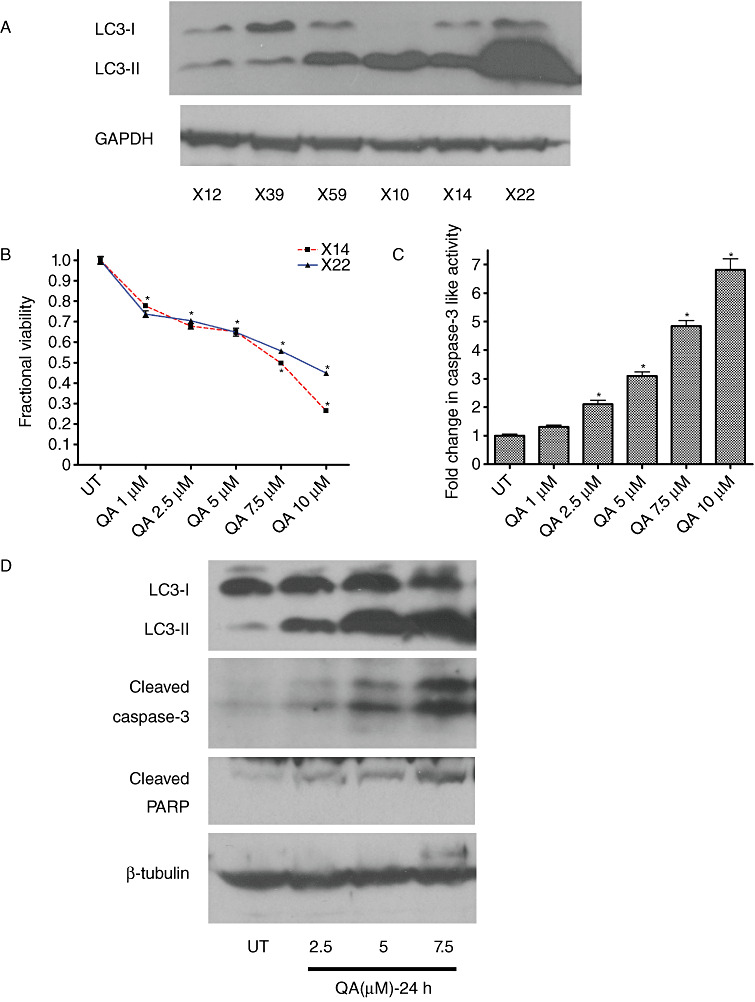
Assessment of baseline level and quinacrine (QA)‐induced autophagy and tumoricidal activity in xenograft derived glioma cells in vitro. Baseline levels of LC3 I and II vary in xenografts derived from different molecular subtypes of gliomas (A). X12 is a xenograft classified as proneural, X39 and X59 are classified as classical and X10, X14 and X22 are classified as mesenchymal subtype. QA induces a concentration‐dependent (B) decrease in viability and an increase in levels of (C) caspase‐3 like activity (D) LC3‐II, cleaved caspase 3 and cleaved poly (ADP‐ribose) polymerase (PARP). Data shown in (C) and (D) are from xenografts X14 and X12, respectively. Error bars represent standard deviation (SD) measurements that are less than 5% of the mean value (*P < 0.05).
PI3K‐AKT‐mTOR PATHWAY: ROLE OF EGFR, NF‐1 AND PTEN MUTATIONS AND AUTOPHAGY MODULATION
The most common genetic mutations/alterations seen in gliomas are the amplification of EGFR, expression of EGFR vIII mutant and homozygous/hemizygous deletion of PTEN and NFI (90). The aberrant signals that result from these mutations interact with the PI3K‐AKT‐mTOR pathway and are responsible for promoting survival and chemo‐resistance in gliomas (9). Therefore targeting the RTKs with either monoclonal antibodies or small molecule inhibitors emerged as a promising therapeutic strategy (Figure 3). However, initial clinical studies with small molecule inhibitors of EGFR, such as erlotinib and gefitinib, have been disappointing in gliomas 24, 70, 74. Similarly, monoclonal antibodies against EGFR, cetuximab and panitumumab, have only a cytostatic effect in glioma cell lines 5, 68. The abundance of multiple types of RTKs along with the frequent deletion of PTEN in gliomas may account for the lack of effectiveness of single agent tyrosine kinase inhibitors (TKIs) 19, 63, 80, 93. In addition, preclinical studies with individual inhibitors of PI3K and mTOR have shown only modest efficacy in gliomas. Phase II studies with temsirolimus (CCI‐779), an analog of allosteric mTOR inhibitor rapamycin, did not provide any survival benefit in recurrent GBM patients 7, 27. However, a dual inhibitor of PI3K and mTOR, PI‐103 had potent antiproliferative activity in gliomas in preclinical models by eliminating activation of AKT often observed with mTOR inhibitors (18). Therapies targeting components of the RTK‐PI3K‐AKT‐mTOR axis typically promote autophagy induction, which has been suggested to play a cytoprotective role. Therefore, combining late‐stage autophagy inhibitors with agents promoting autophagy induction could enhance cytotoxicity in gliomas by inducing autophagic stress. In fact, this concept was tested when combining PI3K‐mTOR/AKT inhibitors (PI‐103 and AKT‐1/2), with the lysosomotropic agent chloroquine (CQ), which inhibits lysosomal protease activity, accelerated cell death in gliomas (16). Additionally, a dual PI3K and mTOR inhibitor, NVP‐BEZ235, which is currently in clinical trials for use in solid tumors (34) induces autophagy and works synergistically in combination with CQ by inducing apoptosis in glioma cells (20). Combining bafilomycin A1 or monensin (late‐stage inhibitors of autophagy) with PI‐103 or Ku‐0063794, an mTOR kinase inhibitor, also enhanced glioma cell death by inducing apoptosis (20). Similarly, autophagy inhibition has been reported to synergize with erlotinib to induce GBM cell death (17). In the case of imatinib mesylate, another TKI, the stage at which autophagy is inhibited resulted in different outcomes in gliomas. Suppression of autophagy at an early stage with 3‐methyladenine (3‐MA) or small interfering RNA against ATG5 attenuated cytotoxicity of imatinib, whereas late‐stage inhibition with bafilomycin A1 enhanced the cytotoxicity through induction of apoptosis (78). The differing outcomes of autophagy inhibition in various therapies might depend on the nature of the autophagy initiator, the combination therapy used and other factors that are not yet clearly understood.
Figure 3.
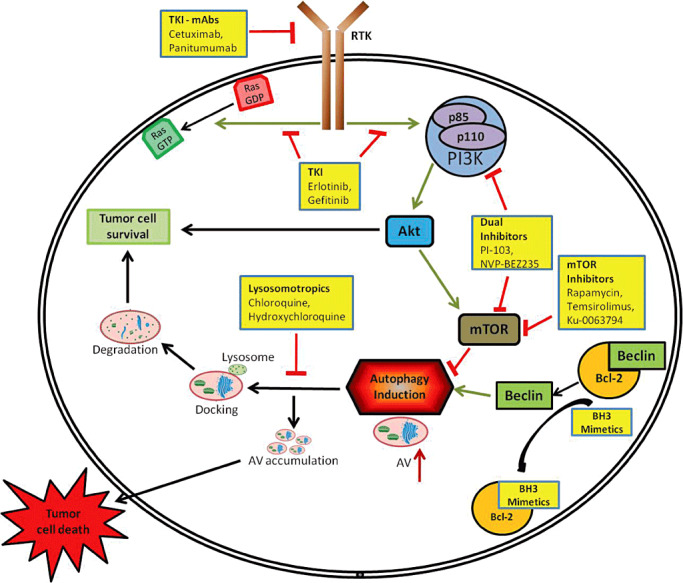
Therapies modulating the autophagy lysosomal pathway. Tyrosine kinase inhibitors (monoclonal antibodies and small molecules) block RTK/Ras signaling upstream of the PI3‐K–AKT–mTOR axis. Inhibitors for PI3‐K and/or mTOR act on their specific targets resulting in the inhibition of mTOR, which induces autophagy. BH3 mimetics induce autophagy by disrupting the BECLIN–BCL‐2 interaction. These agents when combined with lysosomotropic drugs result in the disruption of the autophagy‐lysosomal pathway, leading to accumulation of autophagic vacuoles ultimately resulting in tumor cell death. Abbreviations: DRAM; damage‐regulated autophagy modulator; GDP, guanosine diphosphate; GTP, guanosine triphosphate; mTOR, mammalian target of rapamycin; PI3‐K, phosphoinositide 3‐kinase; RTK, receptor tyrosine kinase.
MANIPULATING THE AUTOPHAGY LYSOSOMAL PATHWAY TO INDUCE APOPTOTIC CELL DEATH IN GLIOMAS
Several studies have explored the potential of combining therapies known to modulate autophagy to produce a synergistic effect on tumor cell death by inducing apoptosis. The ubiquitin‐proteasome system is a major intracellular protein degradation pathway that is known to functionally complement autophagy. Inhibition of either pathway induces a feedback up‐regulation of the other (65). Therefore, proteasome inhibitors have been used to indirectly modulate the autophagy lysosomal pathway and hence serve as adjuvants in therapy. The proteasome inhibitor (MG‐132) induces autophagy and when combined with 3‐MA increases cell death in human glioma cells (28). Another combinatorial therapy explored the effect of autophagy inhibition on N‐(4‐hydroxyphenyl)retinamide (4‐HPR)‐induced cytotoxicity. 4‐HPR, a known inducer of apoptosis, also induces autophagy at lower concentrations in gliomas. 4‐HPR induced cytotoxicity was enhanced by inhibiting autophagy using both 3‐MA and bafilomycin A1 (85). While combining different classes of drugs has been shown to be effective in gliomas, quinacrine (QA), an acridine derivative and lysosomotropic agent, alone has been reported to decrease tumor growth in vivo (89). Moreover, we observed that QA induces cell death in glioma xenografts accompanied by autophagic vacuole (AV) accumulation and concomitant caspase‐3 activation (Figure 2B–D). These studies suggest potential new avenues for the development of novel therapeutic strategies for treating gliomas.
THERAPIES TARGETING INDUCTION OF AUTOPHAGY‐DEPENDENT CELL DEATH IN GLIOMAS
Even though combining drugs inducing autophagy with agents blocking the completion of autophagy has shown promise, and some drug combinations are being clinically evaluated, there are other therapies where induction of autophagy promotes autophagy‐dependent cell death in gliomas. In such instances, addition of an autophagy inducer to the regime should enhance cytotoxicity. This phenomenon is usually observed in cells that are resistant to apoptotic cell death.
Various agents that have been implicated in eliciting autophagy‐dependent cell death in different cancers include actinomycin D, arsenic trioxide, tamoxifen, vitamin D analogs, resveratrol and IFN‐γ. In malignant glioma cell lines, arsenic trioxide [As(2)O(3)] has been reported to induce autophagic cell death through an upregulation of the BCL‐2 family member, BNIP3 and its homolog BNIP3L. As previously mentioned, BNIP3 promotes autophagy by displacing BECLIN1 from its complex with BCL‐2. Moreover, exogenous expression of BNIP3 by itself also induced autophagic cell death in these cells (47). Similarly, BNIP3 plays a pivotal role in ceramide‐induced autophagic cell death in malignant glioma cells (12). Another therapy that induces autophagic cell death in malignant gliomas is the inorganic compound, sodium selenite, through superoxide mediated mitochondrial damage (49). While arsenic trioxide, ceramide and sodium selenite induce autophagic cell death through mitochondrial damage, Δ9‐tetrahydrocannabinol (THC) leads to autophagy‐dependent cell death in glioma cell lines by inducing ER stress (76).
GBMs are characterized by regions of hypoxia and often large, necrotic areas within the tumors. Enhanced expression of anti‐apoptotic BCL‐2 family members in gliomas results in tumor resistance to anoxia (35) and may contribute to resistance to therapy. BH3 mimetics are a class of small molecules that can selectively bind to the BH3 binding groove of the anti‐apoptotic BCL‐2 proteins. Thus by disrupting the BCL‐2/BAX and BCL‐2/BECLIN1 interactions, BH3 mimetics can induce either apoptosis or autophagy in various cancers. BH3 mimetics have been found to induce autophagic cell death in anoxia resistant malignant glioma cells (35). Additionally, gossypol, a BH3 mimetic, has been reported to exclusively induce caspase‐independent autophagic cell death in malignant gliomas (91).
TP53 is mutated in approximately a third of gliomas (82) and often renders tumor cells resistant to apoptotic stimuli. Previously, we reported that CQ induces autophagic cell death in gliomas independent of TP53 status (29). However, p53 has been shown to play a role in regulation of autophagy in some therapies. Induction of DNA damage and subsequent autophagy has been reported with selective cyclooxygenase (COX)‐2 inhibitors like celecoxib in glioma cells, mediated via a functional TP53 (44).
Autophagy plays a significant role in viral‐mediated gene therapy approaches. Pathogen infection is one of the known causes of autophagy induction in eukaryotic cells. Depending on the type of virus, autophagy can either facilitate or impede replication. In the case of oncolytic adenovirus, excessive autophagy observed at the end of adenoviral infection results in cell death 39, 40. Additionally, adenovirus mediated therapies when combined with autophagy induction have a synergistic effect on glioma cell death. Therapies with adenovirus—Delta‐24‐RGD and OBP‐405—when combined with RAD001 (everolimus), have shown to enhance the cytotoxic effect through induction of autophagy‐dependent cell death in gliomas 1, 96.
ROLE OF AUTOPHAGY IN CURRENT TREATMENT REGIME FOR GLIOMAS
Over the last decade, the standard treatment regime for GBMs has been surgical resection of the tumor followed by concurrent radiation therapy and chemotherapy with temozolomide (TMZ). The combination of radiation therapy and temozolomide significantly improves survival of patients compared with radiation therapy alone (81). TMZ, an alkylating agent, acts by methylating the guanine residues resulting in DNA damage and ultimately, cell death. Similarly, ionizing radiation (IR) causes DNA damage in cells. Both TMZ and IR also induce autophagy in glioma cells 38, 46, 48, 56, but the importance of autophagy induction in these therapies is currently unresolved. Inhibition of autophagy induction by 3‐MA suppressed the anti‐tumor effect of TMZ, whereas blocking autophagy completion with bafilomycin A1 enhanced the cytotoxicity of TMZ by inducing apoptosis (46). However, TMZ‐induced autophagy also resulted in a cytoprotective adenosine triphosphate (ATP) surge in glioma cells and inhibition of autophagy with 3‐MA abolished the ATP surge (48). These contradictory findings may be attributed to the timing of 3‐MA delivery to inhibit autophagy. When used concurrently with TMZ, 3‐MA rescued the tumor cells from death, whereas use of 3‐MA after TMZ enhanced tumor cell death. A recent study also showed that shRNA mediated knock down of BECLIN1 or ATG5 protected glioma cells from TMZ induced death (91). This study also reported that combining TMZ with gossypol potentiated the cell death induced by TMZ via autophagic cell death. Another combinatorial regime of TMZ with cannabinoids was found to have a strong anti‐tumoral action in glioma xenografts through the induction of autophagy (86). Antimalarial drugs CQ and hydroxycholoroquine (HCQ), which block the completion of autophagy by interfering with lysosomal acidification, are currently in clinical trials in combination with temozolomide and radiation therapy in GBMs (8). While awaiting the results from these trials, which would prove the effectiveness of autophagy inhibition in glioma therapy, we should remember that CQ and HCQ, while indirectly inhibiting autophagy, also have several other mechanisms of action including DNA intercalation and production of ROS.
Similarly, the role of radiation‐induced autophagy in glioma cells is not clearly understood. Conflicting observations portray both a protective as well as a death inducing role for radiation‐induced autophagy. While malignant glioma cells lacking DNA‐dependent protein kinase undergo autophagic cell death upon radiation (13), inhibition of autophagy at any stage sensitizes radiation resistant glioma cells to such therapy (38). There are multiple studies attributing contrasting roles for autophagy in radiation therapy. This contrast could be a function of the specific context in which autophagy was modulated. In the case of glioma stem cells (CD133‐positive cells), induction of autophagy contributes to radioresistence and inhibition of autophagy by bafilomycin A1 or silencing of BECLIN1 and ATG5 sensitized the cells to gamma‐radiation (56). However, combining arsenic trioxide with IR enhances autophagy and leads to cell death (10). Moreover, a recent study reported that autophagy induced by rapamycin in glioma stem cells could trigger differentiation and enhance their radiosensitization in vitro and in an immunocompromised mouse model (100). Further studies are needed to address the specific role of radiation‐induced autophagy in gliomas to facilitate the development of combination therapy of IR with drugs that would either induce or inhibit autophagy to achieve greater efficiency.
CONCLUSION AND FUTURE DIRECTIONS
There is a growing body of evidence to indicate that in non‐transformed cells, the autophagy lysosomaI pathway integrates signals from multiple upstream signaling cascades thereby resulting in an intricate regulatory mechanism. An additional layer of complexity is added when studying autophagy regulation in cancer cells against a background of functional mutations that alter major signaling events. Activation of specific oncogenes has been shown to alter the autophagy dependence of cancer cells (30). Therefore, the genetic makeup of a particular cancer cell determines how the cell responds to autophagy modulation and this knowledge is critical while formulating therapeutic strategies targeting the autophagy lysosomal pathway. This becomes increasingly important in the context of glioma cells, which display immense genetic heterogeneity. The recent subtyping of glioma cells based on representative mutations affords an opportunity to study specific alterations in the ALP in subtypes against a background of common mutations found in gliomas.
Funding: This work was supported by NIH grants NS41962, CA134773 and P50CA097247 (Kevin A Roth).
Conflict of interest: No conflict of interest exists.
REFERENCES
- 1. Alonso MM, Jiang H, Yokoyama T, Xu J, Bekele NB, Lang FF et al (2008) Delta‐24‐RGD in combination with RAD001 induces enhanced anti‐glioma effect via autophagic cell death. Mol Ther 16:487–493. [DOI] [PubMed] [Google Scholar]
- 2. Aoki H, Kondo Y, Aldape K, Yamamoto A, Iwado E, Yokoyama T et al (2008) Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule‐associated protein 1 light chain 3. Autophagy 4:467–475. [DOI] [PubMed] [Google Scholar]
- 3. Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier‐Denis E (2001) The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3‐kinase/protein kinase B pathway. J Biol Chem 276:35243–35246. [DOI] [PubMed] [Google Scholar]
- 4. Bellot G, Garcia‐Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM (2009) Hypoxia‐induced autophagy is mediated through hypoxia‐inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29:2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carrasco‐Garcia E, Saceda M, Grasso S, Rocamora‐Reverte L, Conde M, Gomez‐Martinez A et al (2011) Small tyrosine kinase inhibitors interrupt EGFR signaling by interacting with erbB3 and erbB4 in glioblastoma cell lines. Exp Cell Res 317:1476–1489. [DOI] [PubMed] [Google Scholar]
- 6. CBTRUS (2011) CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2007. Hinsdale, IL. [Google Scholar]
- 7. Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C et al (2005) Phase II study of CCI‐779 in patients with recurrent glioblastoma multiforme. Invest New Drugs 23:357–361. [DOI] [PubMed] [Google Scholar]
- 8. Chen N, Karantza V (2011) Autophagy as a therapeutic target in cancer. Cancer Biol Ther 11:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng CK, Fan QW, Weiss WA (2009) PI3K signaling in glioma—animal models and therapeutic challenges. Brain Pathol 19:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chiu HW, Ho SY, Guo HR, Wang YJ (2009) Combination treatment with arsenic trioxide and irradiation enhances autophagic effects in U118‐MG cells through increased mitotic arrest and regulation of PI3K/Akt and ERK1/2 signaling pathways. Autophagy 5:472–483. [DOI] [PubMed] [Google Scholar]
- 11. Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR et al (2006) DRAM, a p53‐induced modulator of autophagy, is critical for apoptosis. Cell 126:121–134. [DOI] [PubMed] [Google Scholar]
- 12. Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S (2004) Pivotal role of the cell death factor BNIP3 in ceramide‐induced autophagic cell death in malignant glioma cells. Cancer Res 64:4286–4293. [DOI] [PubMed] [Google Scholar]
- 13. Daido S, Yamamoto A, Fujiwara K, Sawaya R, Kondo S, Kondo Y (2005) Inhibition of the DNA‐dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res 65:4368–4375. [DOI] [PubMed] [Google Scholar]
- 14. De Duve C, Wattiaux R (1966) Functions of lysosomes. Annu Rev Physiol 28:435–492. [DOI] [PubMed] [Google Scholar]
- 15. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G et al (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY et al (2008) Akt inhibition promotes autophagy and sensitizes PTEN‐null tumors to lysosomotropic agents. J Cell Biol 183:101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eimer S, Belaud‐Rotureau MA, Airiau K, Jeanneteau M, Laharanne E, Veron N et al (2011) Autophagy inhibition cooperates with erlotinib to induce glioblastoma cell death. Cancer Biol Ther 11:1017–1027. [DOI] [PubMed] [Google Scholar]
- 18. Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D et al (2006) A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9:341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, Weiss WA (2007) A dual phosphoinositide‐3‐kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN‐mutant glioma. Cancer Res 67:7960–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T et al (2010) Akt and autophagy cooperate to promote survival of drug‐resistant glioma. Sci Signal 3:ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feng Z, Zhang H, Levine AJ, Jin S (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 102:8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ (2007) The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF‐1‐AKT‐mTOR pathways. Cancer Res 67:3043–3053. [DOI] [PubMed] [Google Scholar]
- 23. Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R et al (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447:1121–1125. [DOI] [PubMed] [Google Scholar]
- 24. Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D et al (2007) Gefitinib in patients with progressive high‐grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro‐Oncologia (GICNO). Br J Cancer 96:1047–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19:2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fung C, Lock R, Gao S, Salas E, Debnath J (2008) Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 19:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J et al (2005) Phase II trial of temsirolimus (CCI‐779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol 23:5294–5304. [DOI] [PubMed] [Google Scholar]
- 28. Ge PF, Zhang JZ, Wang XF, Meng FK, Li WC, Luan YX et al (2009) Inhibition of autophagy induced by proteasome inhibition increases cell death in human SHG‐44 glioma cells. Acta Pharmacol Sin 30:1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Geng Y, Kohli L, Klocke BJ, Roth KA (2010) Chloroquine‐induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro-oncol 12:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli‐Uzunbas G et al (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T et al (2007) The Atg12‐Atg5 conjugate has a novel E3‐like activity for protein lipidation in autophagy. J Biol Chem 282:37298–37302. [DOI] [PubMed] [Google Scholar]
- 32. Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70. [DOI] [PubMed] [Google Scholar]
- 33. He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herrera VA, Zeindl‐Eberhart E, Jung A, Huber RM, Bergner A (2011) The dual PI3K/mTOR inhibitor BEZ235 is effective in lung cancer cell lines. Anticancer Res 31:849–854. [PubMed] [Google Scholar]
- 35. Hetschko H, Voss V, Senft C, Seifert V, Prehn JH, Kogel D (2008) BH3 mimetics reactivate autophagic cell death in anoxia‐resistant malignant glioma cells. Neoplasia 10:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang X, Bai HM, Chen L, Li B, Lu YC (2010) Reduced expression of LC3B‐II and Beclin 1 in glioblastoma multiforme indicates a down‐regulated autophagic capacity that relates to the progression of astrocytic tumors. J Clin Neurosci 17:1515–1519. [DOI] [PubMed] [Google Scholar]
- 37. Itakura E, Kishi C, Inoue K, Mizushima N (2008) Beclin 1 forms two distinct phosphatidylinositol 3‐kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19:5360–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y (2005) Radiation‐induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol 26:1401–1410. [PubMed] [Google Scholar]
- 39. Ito H, Aoki H, Kuhnel F, Kondo Y, Kubicka S, Wirth T et al (2006) Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J Natl Cancer Inst 98:625–636. [DOI] [PubMed] [Google Scholar]
- 40. Jiang H, Gomez‐Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F et al (2007) Examination of the therapeutic potential of Delta‐24‐RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst 99:1410–1414. [DOI] [PubMed] [Google Scholar]
- 41. Xu J, Kochanek KD, Murphy SL, Tejada‐Vera B (2010) Final Data for 2007. National Vital Statistic Reports. [PubMed]
- 42. Johnstone RW, Ruefli AA, Lowe SW (2002) Apoptosis: a link between cancer genetics and chemotherapy. Cell 108:153–164. [DOI] [PubMed] [Google Scholar]
- 43. Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000) Tor‐mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150:1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kang KB, Zhu C, Yong SK, Gao Q, Wong MC (2009) Enhanced sensitivity of celecoxib in human glioblastoma cells: Induction of DNA damage leading to p53‐dependent G1 cell cycle arrest and autophagy. Mol Cancer 8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS et al (2009) Frameshift mutations of autophagy‐related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 217:702–706. [DOI] [PubMed] [Google Scholar]
- 46. Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S (2004) Role of autophagy in temozolomide‐induced cytotoxicity for malignant glioma cells. Cell Death Differ 11:448–457. [DOI] [PubMed] [Google Scholar]
- 47. Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S (2005) Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 14:980–991. [DOI] [PubMed] [Google Scholar]
- 48. Katayama M, Kawaguchi T, Berger MS, Pieper RO (2007) DNA damaging agent‐induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 14:548–558. [DOI] [PubMed] [Google Scholar]
- 49. Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS (2007) Sodium selenite induces superoxide‐mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res 67:6314–6324. [DOI] [PubMed] [Google Scholar]
- 50. Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ (2008) Frameshift mutation of UVRAG, an autophagy‐related gene, in gastric carcinomas with microsatellite instability. Hum Pathol 39:1059–1063. [DOI] [PubMed] [Google Scholar]
- 51. Klionsky DJ (2005) The molecular machinery of autophagy: unanswered questions. J Cell Sci 118(Pt 1):7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee JW, Jeong EG, Soung YH, Nam SW, Lee JY, Yoo NJ, Lee SH (2006) Decreased expression of tumour suppressor Bax‐interacting factor‐1 (Bif‐1), a Bax activator, in gastric carcinomas. Pathology 38:312–315. [DOI] [PubMed] [Google Scholar]
- 53. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU (2006) Autophagic and tumour suppressor activity of a novel Beclin1‐binding protein UVRAG. Nat Cell Biol 8:688–699. [DOI] [PubMed] [Google Scholar]
- 54. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676. [DOI] [PubMed] [Google Scholar]
- 55. Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J (2011) Autophagy facilitates glycolysis during Ras‐mediated oncogenic transformation. Mol Biol Cell 22:165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN et al (2009) The induction of autophagy by gamma‐radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 125:717–722. [DOI] [PubMed] [Google Scholar]
- 57. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P et al (2007) Functional and physical interaction between Bcl‐X(L) and a BH3‐like domain in Beclin‐1. EMBO J 26:2527–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marino G, Salvador‐Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez‐Otin C (2007) Tissue‐specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin‐3. J Biol Chem 282:18573–18583. [DOI] [PubMed] [Google Scholar]
- 59. Martinez‐Outschoorn UE, Whitaker‐Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T et al (2010) The autophagic tumor stroma model of cancer or “battery‐operated tumor growth”: a simple solution to the autophagy paradox. Cell Cycle 09:4297–4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K et al (2007) Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21:1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY et al (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N et al (2009) Two Beclin 1‐binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396. [DOI] [PubMed] [Google Scholar]
- 63. Mellinghoff IK, Cloughesy TF, Mischel PS (2007) PTEN‐mediated resistance to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res 13:378–381. [DOI] [PubMed] [Google Scholar]
- 64. Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I et al (2007) Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol 30:429–436. [PubMed] [Google Scholar]
- 65. Nedelsky NB, Todd PK, Taylor JP (2008) Autophagy and the ubiquitin‐proteasome system: collaborators in neuroprotection. Biochim Biophys Acta 1782:691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ohsumi Y (2001) Molecular dissection of autophagy: two ubiquitin‐like systems. Nat Rev Mol Cell Biol 2:211–216. [DOI] [PubMed] [Google Scholar]
- 67. Ohsumi Y, Mizushima N (2004) Two ubiquitin‐like conjugation systems essential for autophagy. Semin Cell Dev Biol 15:231–236. [DOI] [PubMed] [Google Scholar]
- 68. Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA et al (2009) The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia 11:448–458, 2 pp. following 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pirtoli L, Cevenini G, Tini P, Vannini M, Oliveri G, Marsili S et al (2009) The prognostic role of Beclin 1 protein expression in high‐grade gliomas. Autophagy 5:930–936. [DOI] [PubMed] [Google Scholar]
- 70. Prados MD, Lamborn KR, Chang S, Burton E, Butowski N, Malec M et al (2006) Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro-oncol 8:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A et al (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112:1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC et al (2000) BNIP3 heterodimerizes with Bcl‐2/Bcl‐X(L) and induces cell death independent of a Bcl‐2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem 275:1439–1448. [DOI] [PubMed] [Google Scholar]
- 73. Reed JC (2002) Apoptosis‐based therapies. Nat Rev Drug Discov 1:111–121. [DOI] [PubMed] [Google Scholar]
- 74. Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL et al (2004) Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol 22:133–142. [DOI] [PubMed] [Google Scholar]
- 75. Roy S, Debnath J (2010) Autophagy and tumorigenesis. Semin Immunopathol 32:383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Salazar M, Carracedo A, Salanueva IJ, Hernandez‐Tiedra S, Lorente M, Egia A et al (2009) Cannabinoid action induces autophagy‐mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 119:1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schwarze PE, Seglen PO (1985) Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogen‐treated rats. Exp Cell Res 157:15–28. [DOI] [PubMed] [Google Scholar]
- 78. Shingu T, Fujiwara K, Bogler O, Akiyama Y, Moritake K, Shinojima N et al (2009) Inhibition of autophagy at a late stage enhances imatinib‐induced cytotoxicity in human malignant glioma cells. Int J Cancer 124:1060–1071. [DOI] [PubMed] [Google Scholar]
- 79. Simonsen A, Tooze SA (2009) Coordination of membrane events during autophagy by multiple class III PI3‐kinase complexes. J Cell Biol 186:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R et al (2007) Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318:287–290. [DOI] [PubMed] [Google Scholar]
- 81. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 82. Szybka M, Zawlik I, Kulczycka D, Golanska E, Jesien E, Kupnicka D et al (2008) Elimination of wild‐type P53 mRNA in glioblastomas showing heterozygous mutations of P53. Br J Cancer 98:1431–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tallóczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ et al (2002) Regulation of starvation‐ and virus‐induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99:190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri‐Mergny M, D'Amelio M et al (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10:676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tiwari M, Bajpai VK, Sahasrabuddhe AA, Kumar A, Sinha RA, Behari S, Godbole MM (2008) Inhibition of N‐(4‐hydroxyphenyl)retinamide‐induced autophagy at a lower dose enhances cell death in malignant glioma cells. Carcinogenesis 29:600–609. [DOI] [PubMed] [Google Scholar]
- 86. Torres S, Lorente M, Rodriguez‐Fornes F, Hernandez‐Tiedra S, Salazar M, Garcia‐Taboada E et al (2011) A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther 10:90–103. [DOI] [PubMed] [Google Scholar]
- 87. Tsukada M, Ohsumi Y (1993) Isolation and characterization of autophagy‐defective mutants of Saccharomyces cerevisiae. FEBS Lett 333:169–174. [DOI] [PubMed] [Google Scholar]
- 88. Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ (2010) Exciting new advances in neuro‐oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 60:166–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Vassey JW, Edmonds J, Irvin JL, Green JA, Irvin EM (1955) Studies on the administration of quinacrine to tumor‐bearing mice. Cancer Res 15:573–578. [PubMed] [Google Scholar]
- 90. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Voss V, Senft C, Lang V, Ronellenfitsch MW, Steinbach JP, Seifert V, Kogel D (2010) The pan‐Bcl‐2 inhibitor (‐)‐gossypol triggers autophagic cell death in malignant glioma. Mol Cancer Res 8:1002–1016. [DOI] [PubMed] [Google Scholar]
- 92. Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T (2009) A non‐canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem 284:21412–21424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM et al (2006) Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN‐deficient and PTEN‐intact glioblastoma cells. Cancer Res 66:7864–7869. [DOI] [PubMed] [Google Scholar]
- 94. Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC (2008) Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 13:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12:814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yokoyama T, Iwado E, Kondo Y, Aoki H, Hayashi Y, Georgescu MM et al (2008) Autophagy‐inducing agents augment the antitumor effect of telerase‐selve oncolytic adenovirus OBP‐405 on glioblastoma cells. Gene Ther 15:1233–1239. [DOI] [PubMed] [Google Scholar]
- 97. Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF et al (2009) Autophagy mediates the mitotic senescence transition. Genes Dev 23:798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100:15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT et al (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1‐phosphatidylinositol‐3‐kinase complex. Nat Cell Biol 11:468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhuang W, Li B, Long L, Chen L, Huang Q, Liang Z (2011) Induction of autophagy promotes differentiation of glioma‐initiating cells and their radiosensitivity. Int J Cancer 129:2720–2731. [DOI] [PubMed] [Google Scholar]