

Abstract
Germline mutations in the cylindromatosis (CYLD) gene have been described in families with cylindromas, trichoepitheliomas, and/or spiradenomas. Brooke-Spiegler syndrome (BSS) is the autosomal dominant predisposition to skin appendageal neoplasms including cylindromas, trichoepitheliomas, and/or spiradenomas. We review the clinical features, molecular genetics, and the animal models of BSS. To date, a total of 51 CYLD mutations have been reported, occurring in exons 9–20, in 73 families with diverse ethnic and racial backgrounds. Of 51 mutations, 86% are expected to lead to truncated proteins. The seven missense mutations reported to date occur only within the ubiquitin-specific protease (USP) domain of the CYLD protein and most are associated exclusively with multiple familial trichoepithelioma. CYLD functions as a tumor suppressor gene. CYLD encodes a deubiquitinating (DUB) enzyme that negatively regulates the NF-kappaB and c-Jun N-terminal kinase pathways. CYLD DUB activity is highly specific for lysine 63 (K63)-linked ubiquitin (Ub) chains but has been shown to act on K48-linked Ub chains as well. In 2008 the CYLD USP domain was crystallized, revealing that the truncated Fingers subdomain confers CYLD’s unique specificity for K63-linked ubiquitin chains. Recent work using animal models revealed new roles for CYLD in immunity, lipid metabolism, spermatogenesis, osteoclastogenesis, anti-microbial defense and inflammation.
Keywords: CYLD, Brooke-Spiegler, trichoepithelioma, cylindroma, trichoepithelioma, spiroadenoma
Introduction
Brooke-Spiegler syndrome [Online Mendelian Inheritance in Man (MIM# 605041) is an autosomal dominantly inherited predisposition towards skin appendage tumors including cylindromas, trichoepitheliomas, and/or spiradenomas. Familial cylindromatosis (MIM# 132700) and multiple familial trichoepithelioma (MIM# 601606) are allelic diseases with a common genetic basis (Welch et al., 1968; Young et al., 2006; Oranje et al., 2008). Familial cylindromatosis (FC) and multiple familial trichoepithelioma (MFT) represent the two ends within the spectrum of Brooke-Spiegler syndrome but most families and patients fall somewhere in the middle. In this review, we use the term Brooke-Spiegler syndrome to include classic form as well as FC and MFT.
In 1996, the familial cylindromatosis locus was mapped to chromosome 16q12-13 by linkage analysis in two large families (Biggs et al., 1995) and later refined to a 1MB region (Takahashi et al., 2000). Bignell et al. identified the susceptibility gene for familial cylindromatosis (CYLD; MIM# 605018) by detecting germline mutations in 21 families and somatic mutations in 6 cylindromas (Bignell et al., 2000). Subsequently, Zhang et al. showed that families with multiple familial trichoepithelioma also had germline mutations in CYLD (Zhang et al., 2004). The CYLD gene is 56 kb, spanning 20 exons with the first 3 exons untranslated (GenBank NM_015247) (Figure 1). CYLD has three major splice variants. Exon 3 (in the 5’ UTR) and exon 7 exhibit alternative splicing (Bignell et al., 2000). To date, 49 unique CYLD mutations have been identified in 70 families.
Figure 1. CYLD germline mutations associated with BSS.
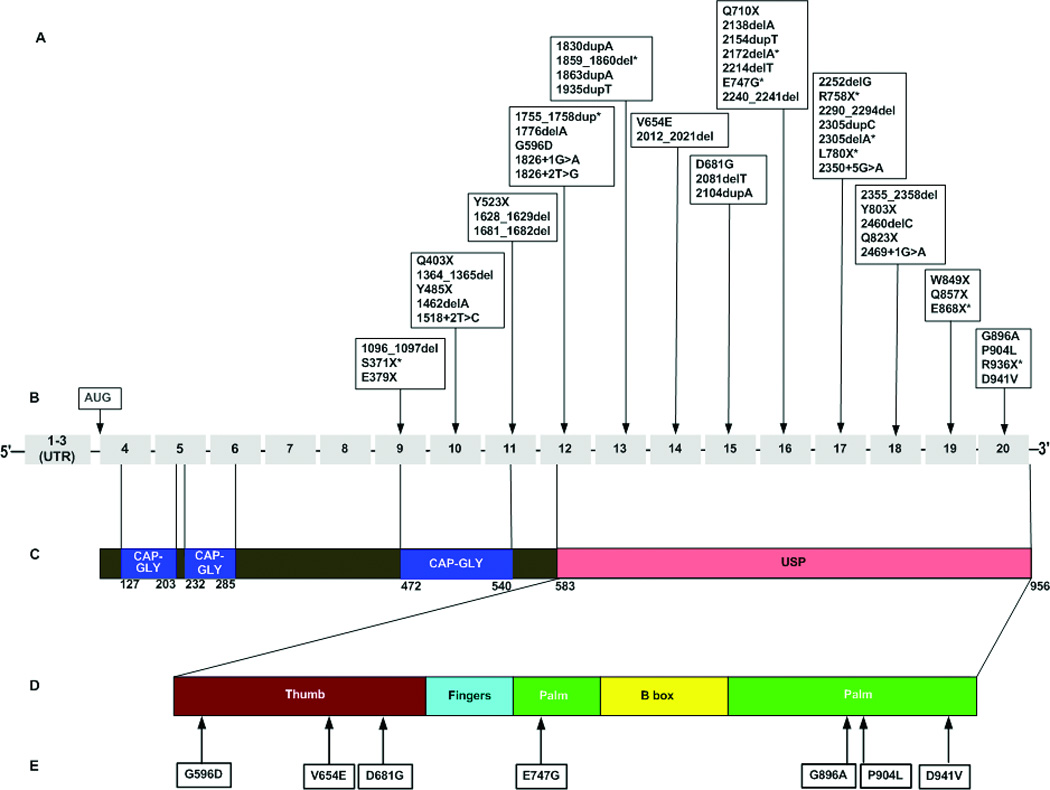
A, reported CYLD mutations, *indicates recurrent mutations; B, CYLD exons, not drawn to scale; C, CYLD protein domains, drawn to scale; D, CYLD USP subdomains, drawn to scale; E, reported CYLD missense mutations. Figure based on the published GenBank sequence NM_015247.2
The CYLD gene encodes the cylindromatosis protein (CYLD) (GenBank NP_056062), also known as ubiquitin-specific-processing protease CYLD, and ubiquitin carboxyl-terminal hydrolase CYLD. CYLD has 956 amino acids and a molecular weight of approximately 120 kDa. CYLD has moderate homology with proteins of the ubiquitin-specific protease class and is highly conserved among vertebrates. CYLD is a deubiquitinating (DUB) enzyme that removes ubiquitin chains from several proteins, notably the TNF-receptor associated factor (TRAF) 2 and TRAF6, and the NF-κB essential modulator (NEMO). Through removal of ubiquitin (Ub), CYLD regulates cell signaling including NF-κB and c-Jun N-terminal kinase (JNK) pathways. CYLD has a strong preference for activity at lysine 63 (K63)-linked Ub chains (Komander et al., 2008) but has been shown to act on K48-linked polyubiquitin chains as well (Reiley et al., 2006). K63-linked polyubiquitination affects protein interactions by acting as a specific recognition sequence for other proteins (Haglund and Dikic, 2005). CYLD is widely expressed and regulates a myriad of cell functions including inflammation and cell proliferation.
Patients with Brooke-Spiegler syndrome characteristically present in late childhood to early adulthood with cylindromas, trichoepitheliomas, and/or less commonly spiradenomas. Patients may develop any or all of these tumors at any point in their lifetime and there is variability of expression between and within families. Cylindromas and spiradenomas, trichoepietheliomas are tumors of follicular-apocrine differentiation (Uede et al., 2004). Cylindromas are slowly growing tumors that typically occur on the scalp and occasionally on the face. They are histologically characterized by dermal nodules of epithelial cells composed of large cells with abundant cytoplasm in the center and small basaloid cells in the periphery, lined by thick, basement membrane-like material, and arranged in a characteristic “jigsaw puzzle” pattern (Lian and Cockerell, 2005). Specifically, cylindromas exhibit expression patterns of highly specific hair keratins (Massoumi et al., 2006a). Trichoepitheliomas classically occur as small skin colored papules on the central face, predominantly on the nose and/or nasolabial folds. They histologically consist of uniform basaloid cells with peripheral palisades, arranged in variably sized nests or cribriform patterns surrounded by dense stroma and fibroblasts arranged in bulbs (Alsaad et al., 2007). Spiradenomas are purple to bluish nodules that are paroxysmally painful when symptomatic, usually located on the trunk and extremities. Spiradenomas are nodular neoplastic nodules composed of tumor nests of two main types of epithelial cells arranged in cords, which exhibit tubular or alveolar differentiation (Obaidat et al., 2007). Frequently spiradenomas are vascular and have glassy eosinophilic material in their interior. The tumor nests in spiradenomas are larger and have rounded outlines in contrast with the smaller, more angulated outlines seen in cylindromas (Michal et al., 1999). Some tumors, termed spiradenocylindromas, or “hybrid tumors” have features of both cylindromas and spiradenomas (Kazakov et al., 2008; Kazakov et al., 2005; Pizinger and Michal, 2000), which may suggest that both entities represent a continuous morphological and differentiation spectrum of a single disease entity. There are several reports of spiradenomas transforming into spiradenocarcinomas, and spiradenocarcinomas are likely more aggressive in the setting of BSS. (Kazakov et al., 2009; Braun-Falco et al., 2003; Ishikawa et al., 2001; Chou et al., 2004; Cooper et al., 1985; Engel et al., 1991).
CYLD Mutations
A review of the literature was performed to identify all of the known CYLD mutations. In 2000, Bignell reported CYLD mutations in 21 cylindromatosis families (Bignell et al., 2000). To date, a total of 51 distinct germline CYLD mutations have been reported in 73 families (Table 1, Figure 1). Mutations have been reported among patients with Chinese, Irish, Spanish, German, Algerian, Turkish, and Japanese backgrounds though most reported mutations are from the UK and USA. Of the 51 mutations, 41% (21) are frameshift, 35% (18) are nonsense, 14% (7) are missense, and 10% (5) are putative splice site. Eighty-six percent (44/51) of the mutations are predicted to result in truncated proteins. The seven missense mutations reported occur within the ubiquitin-specific protease (USP) domain (amino acids 583–956): p.G596D (Zuo et al., 2007), p.V654E (Kazakov et al. 2009), p.D681G (Almeida et al., 2008) p.E747G (Hu et al., 2003; Saggar et al., 2008), p.G896A (Espana et al., 2007), p.P904L (Lv et al., 2008), p.D941V (Zheng et al., 2004) (Figure 1). CYLD mutations have been solely identified in the C-terminal two-thirds of the gene (exons 9–20) even though exons 4–8 are translated (Figure 1). The most 5’ mutation (c.1096–1097delCA) occurs in exon 9 and the most 3’ mutation (c.2822A>T) occurs in exon 20. A possible explanation for the lack of reported mutations in exons 4–8 is avoidance of a dominant-negative effect that may occur with more N-terminal truncation (Bignell et al. 2000). The most common sites for CYLD mutations are exons 16 and 17 (14% each) followed by exons 18 and 10 (10% each) (Figure 1).
Table 1.
Reported CYLD Germline Mutations in BSS
| Number | Nucleotide Change (NM_015247.2)a |
Amino Acid Change (NP_056062.1)b |
Alternate (original) report |
Exon/ Intron |
Mutation Type |
References | Ethnicity/ (country of publication) |
|---|---|---|---|---|---|---|---|
| 1 | c.1096_1097del | p.Q366TfsX12 | 9 | FS | (Saggar et al. 2008) | (USA) | |
| 2 | c.1112C>A | p.S371X | 9 | NS | (Bignell et al. 2000; Bowen et al. 2005; Saggar et al. 2008) | (UK), Irish, (USA), | |
| 3 | c.1135G>T | p.E379X | c.G1526>T | 9 | NS | (Almeida et al. 2008) | (Switzerland) |
| 4 | c.1207C>T | p.Q403X | c.C1518>T, p.N403X |
10 | NS | (Almeida et al. 2008) | Irish |
| 5 | c.1364_1365del | p.Q455RfsX22 | p.454fsX476 | 10c | FS | (Liang et al. 2008) | Chinese |
| 6 | c.1455T>A | p.Y485X | 10 | NS | (Bignell et al. 2000) | (UK) | |
| 7 | c.1462delA | p.I488SfsX10 | 10d | FS | (Zheng et al. 2004) | Chinese | |
| 8 | c.1518+2T>C | ND | 10 | S | (Ly et al. 2004) | (UK) | |
| 9 | c.1569T>G | p.Y523X | 11 | NS | (Bignell et al. 2000) | (UK) | |
| 10 | c.1628_1629del | p.S543X | 11 | NS | (Saggar et al. 2008) | (USA) | |
| 11 | c.1681_1682del | p.L561SfsX8 | p.L568X | 11 | FS | (Bignell et al. 2000) | (UK) |
| 12 | c.1755_1758dup | p.M587DfsX29 | c.1758ins4, S615X | 12 | FS | (Bowen et al. 2005; Saggar et al. 2008) | Scandinavian (USA) |
| 13 | c.1776delA | p.G593AfsX11 | p.K603X | 12 | FS | (Bignell et al. 2000) | (UK) |
| 14 | c.1787G>A | p.G596D | 12e | MS | (Zuo et al. 2007) | (China) | |
| 15 | c.1826+1G>A | ND | 12 | S | (Huang et al 2009) | Taiwanese | |
| 16 | c.1826+2T>G | ND | c.1862+2T>G, | 12 | S | (Liang et al. 2005) | Chinese |
| 17 | c.1830dupA | p.F611IfsX4 | c.1830_1831insA p.V630X |
13 | FS | (Bignell et al. 2000; Saggar et al. 2008) | (UK), (USA) |
| 18 | c.1859_1860del | p.L6201TfsX2 | 13 | FS | (Bignell et al. 2000; Saggar et al. 2008) | Chinese (USA) | |
| 19 | c.1863dupA | p.L622RfsX2 | c.1863_1864insA | 13 | FS | (Saggar et al. 2008) | (USA) |
| 20 | c.1935dupT | p.N646X | 13 | NS | (Bignell et al. 2000) | (UK) | |
| 21 | c.1961T>A | p.V654E | 14 | MS | (Kazakov et al. 2009) | Czech | |
| 22 | c.2012_2021del | p.A671Dfx13 | 14 | FS | (Heinritz et al. 2006) | (Germany) | |
| 23 | c.2042A>G | p.D681G | c.A2433>G | 15 | MS | (Almeida et al. 2008) | (Switzerland) |
| 24 | c.2081delT | p.L694X | c.del2472T | 15 | NS | (Almeida et al. 2008) | (Switzerland) |
| 25 | c.2104dupA | p.I702NfsX22 | c.2104_2105insA | 15 | FS | (Salhi et al. 2004) | Algerian |
| 26 | c.2128C>T | p.Q710X | 16f | NS | (Zheng et al. 2004) | Chinese | |
| 27 | c.2138delA | p.Y713SfsX22 | 16 | FS | (Bignell et al. 2000) | (UK) | |
| 28 | c.2154dupT | p.M719EfsX5 | c.2154_2155insT, p.F738fsX723 |
16 | FS | (Saggar et al. 2008) | (USA) |
| 29 | c.2172delA | p.V725LfsX10 | 16 | FS | (Bignell et al. 2000; Scheinfeld et al. 2003; Saggar et al. 2008) | (UK), (USA), (USA), | |
| 30 | c.2214delT | p.F738LfsX6 | 16 | FS | (Saggar et al. 2008) | (USA) | |
| 31 | c.2240A>G | p.E747G | 16 | MS | (Hu et al. 2003; Saggar et al. 2008) | Turkish, (USA), | |
| 32 | c.2240_2241delA | p.E747GfsX17 | c.2241_2242delAG | 16 | FS | (Liang et al. 2005) | Chinese |
| 33 | c.2252delG | p.C751FfsX2 | c.2253delG | 17 | FS | (Poblete Gutierrez et al. 2002) | German |
| 34 | c.2272C>T | p.R758X | 17 | NS | (Bignell et al. 2000; Oiso et al. 2004; Zhang et al. 2006)g | (UK), Japanese, Chinese, | |
| 35 | c.2290_2294del | p.K764KfsX2 | 17 | FS | (Saggar et al. 2008) | (USA) | |
| 36 | c.2305_2306insC | p.I769TfsX14 | 17 | FS | (Bignell et al. 2000) | (UK) | |
| 37 | c.2305delA | p.I769FfsX7 | 17 | FS | (Bowen et al. 2005; Saggar et al. 2008)h | Spanish, (USA), | |
| 38 | c.2339T>G | p.L780X | 17 | NS | (Bowen et al. 2005; Saggar et al. 2008) | Irish, (USA), | |
| 39 | c.2350+5G>A | ND | 18 | S | (Bignell et al. 2000) | (UK) | |
| 40 | c.2355_2358del | p.R786SfsX45 | p.M785fsX45 | 18 | FS | (Zhang et al. 2004) | Chinese |
| 41 | c.2409C>G | p.Y803X | 18 | NS | (Liang et al. 2008) | Chinese | |
| 42 | c.2460delC | p.C820X | p.C831X | 18 | NS | (Bignell et al. 2000) | (UK) |
| 43 | c.2467C>T | p.Q823X | 18 | NS | (Bignell et al. 2000) | (UK) | |
| 44 | c.2469+1G>A | ND | 18 | S | (Bignell et al. 2000)i | (UK) | |
| 45 | c.2546G>A | p.W849X | c.2937G>A | 19 | NS | (Almeida et al. 2008) | (Switzerland) |
| 46 | c.2569C>T | p.Q857X | 19 | NS | (Bignell et al. 2000) | (UK) | |
| 47 | c.2602G>T | p.E868X | 19 | NS | (Bignell et al. 2000; Oranje et al. 2008) | (UK), (Netherlands) | |
| 48 | c.2687G>C | p.G896A | 20 | MS | (Espana et al. 2007) | Spanish | |
| 49 | c.2711C>T | p.P904L | 20 | MS | (Lv et al. 2008) | Chinese | |
| 50 | c.2806C>T | p.R936X | 20 | NS | (Bignell et al. 2000; Bowen et al. 2005; Young et al. 2006; Saggar et al. 2008;j Kazakov et al. 2009) | (UK), Canadian, Irish, (USA), Czech, | |
| 51 | c.2822A>T | p.D941V | 20k | MS | (Zheng et al. 2004) | Chinese |
NS: nonsense; MS: missense; FS: frame shift; S: splice-site
The nucleotide numbering is based on the published GenBank sequence NM_015247.2, and nucleotide +1 is the adenine of the ATG initiation codon.
The amino acid sequence is taken from the published GenBank sequence NP_056062.1 and begins with the translation initiation codon as 1.
Originally reported as exon 15
Originally reported as exon 9
Originally reported as exon 11
Originally reported as exon 17
Bignell reported 2 families with this mutation
Saggar reported 2 families with this mutation
Bignell reported 2 families with this mutation
Saggar reported 4 families with this mutation
Originally reported as exon 21
ND: Not determined
The most common reported mutation in CYLD is c.2806C>T (8 families) followed by c.2272C>T (4 families), c.2305delA (3 families), c.2172delA (3 families) and c.1112C>A (3 families). The ethnic and racial heritage of these families has generally not been reported, making it difficult to conjecture about potential founder effect. Haplotype analysis has been reported in four families with two different CYLD mutations. Two families of unknown background with the c.2469+1G>A mutation showed identical haplotype segregation (Bignell et al., 2000). This is the only reported founder CYLD mutation reported to date. In contrast, haplotype analysis of two families with c.2272C>T did not show a shared haplotype, excluding a possible founder (Bignell et al., 2000).
Based on the 87 BSS families in whom CYLD mutation analysis was reported (Table 1), the mutation detection rates were: 84% (73/87) for BSS overall, 88% (30/34) for familial cylindromatosis, and 72% (18/25) for multiple familial trichoepitheliomas. In contrast, previously reported CYLD mutation detection rates for familial cylindromatosis and multiple familial trichoepithelioma were 100% (3/3) and 44% (4/9), respectively (Saggar et al., 2008). The 13 families without CYLD mutations suggest possible genetic heterogeneity, as does previous mapping of a familial trichoepithelioma locus to chromosome 9p21 (Harada et al., 1996). However, the clinical and histologic differential diagnosis of multiple inherited facial papules and nodules is complex and misdiagnosis is a possibility (Crotty et al., 2003).
Genotype-Phenotype correlation
Bowen et al. described a lack of correlation between CYLD mutations and a specific phenotype in six individuals from unrelated families (3 BSS, 2 MFT, and 1 FC) (Bowen et al., 2005). Five truncating mutations were identified without any differential clustering by phenotype and one MFT family did not exhibit a CYLD mutation (Bowen et al., 2005). However, six individuals are not enough to detect a correlation. There is variability of expression between and within families with the same mutation. Oiso et al. reported a family of two patients with the c.2272C>T mutation who presented with 4 and 5 cylindromas less than 3 cm in diameter, respectively (Oiso et al., 2004). In contrast, Zhang et al. reported a patient with the same mutation but severely affected with hundreds of confluent cylindromas (Zhang et al., 2006a). Marked phenotype diversity was found in a family with the c.2252delG mutation in whom some affected family members presented only with small tumors limited to the nasolabial region, others had mild scalp involvement; and one member exhibited multiple large cylindromas on the trunk and a turban-tumor appearance (Poblete Gutierrez et al., 2002).
We investigated whether six reported CYLD missense mutations were associated with a specific phenotype since missense mutations are rare. We found that 4 of the 6 (67%) missense mutations were reported exclusively in trichoepitheliomas families. Of the remaining three mutations; one (p.D681G) occurred in a family with 3 affected members in whom 11/12 tumors analyzed were trichoepitheliomas and one individual had a solitary spiradenoma (Almeida et al., 2008). Another mutation (p.E747G) was reported in two families. One family of Turkish descent had 13 individuals with multiple trichoepitheliomas (Hu et al., 2003) including one individual with isolated papular cylindromas but no classic turban tumors. It seems that missense mutations tend to be associated with trichoepitheliomas and a milder phenotype. One additional missense was recently reported without specific histological tumor description (Kazakov et al., 2009). Furthermore, only 25% (11/44) of the truncating CYLD mutations have been reported solely in multiple familial trichoepithelioma and they are distributed exons 10, 11, 13, and 15–18. We did not observe a particular clustering or distribution of CYLD mutations associated with multiple familial trichoepithelioma. Interestingly, only 21% (8/39) of mutations reported outside of China but 92% (11/12) CYLD mutations reported in Chinese/Taiwanese families were exclusively associated with multiple familial trichoepithelioma. Ascertainment, diagnostic and/or publication bias could account for this finding. The ethnic discrepancy could also be due to inherent differences in skin type, modifier genes, and/or environmental factors.
CYLD Homologs and Structure
The Cyld protein is encoded by a single gene in the fruit fly Drosophila melanogaster (FlyBase gene ID:CG5603) and the worm Caenorhabditis elegans (F40F12.5;3K988). C. elegans Cyld is transcribed into at least two different mRNAs due to alternative splicing (Bignell et al. 2000). Using Blast (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (Altschul et al., 1997), we found CYLD conservation is striking, with 94% identity between human and mouse. The strongest homology occurs in the C-terminal half of the protein.
CYLD is a member of the ubiquitin-specific protease (USP) family of proteins. CYLD contains three cytoskeleton-associated protein glycine-rich (CAP-GLY) domains and a USP domain (Komander et al., 2008). The CAP-GLY domains are responsible for microtubule binding and the USP domain for deubiquitinating activity (Gao et al., 2008; Komander et al., 2008). CYLD is most known for its deubiquitination (DUB) function. Like all ubiquitin-specific proteases (USPs), CYLD possesses a catalytic triad (Cys601, His871, and Asp889). The cysteine and histidine amino acid residues are highly conserved amongst ubiquitin-specific proteases. The catalytic triad of CYLD removes ubiquitin chains from different substrates including BCL-3, TRAF2, TRAF6, NEMO, Lck, TRPA1 and TRAF-7 (Kovalenko et al., 2003; Trompouki et al., 2003; Jono et al., 2004; Yoshida et al., 2005; Massoumi et al., 2006b; Reiley et al., 2006).
The catalytic core of the USP family was initially defined based on the crystal structure of HAUSP/USP-7 (Hu et al. 2002). Subsequently, the crystal structures of USP2 (Renatus et al., 2006), USP8 (Avvakumov et al., 2006), and USP14 (Hu et al., 2005) were determined, further defining the catalytic core of USPs. Before the crystal structure of CYLD USP domain was known, the USP domain of HAUSP/USP-7 was used to model the CYLD mutation D681G (Almeida et al., 2008). The D681 residue is notable in that the homologous residue in USP7 (D295) (NP_003461.2) forms a hydrogen bond with leu73 of Ub and functional studies showed that the D681G mutant has diminished K63-linked DUB activity (Almeida et al., 2008). The predicted effect of other CYLD missense mutations has not been reported.
In 2008, Komander et al. determined and crystallized the CYLD USP domain (aa 583–956) (Komander et al., 2008). The CYLD USP domain is organized in four subdomains (Palm, Thumb, B box and truncated Fingers) (Figure 1). The Fingers sub domain is significantly shorter than in other USPs and likely explains the internal hydrolysis of K63-linked ubiquitin chains (Komander et al., 2008). The B box domain appears involved in cytoplasmic localization. The residues surrounding the CYLD catalytic triad are highly conserved among species but not among the family of ubiquitin-specific proteases (Komander et al., 2008). CYLD mutants lacking the extreme C-terminal 20 amino acids (937–956) are catalytically inactive (Trompouki et al., 2003).
Biological Relevance
Approximately 100 enzymes have been identified with DUB activity and CYLD is among the best studied (Nijman et al., 2005; Courtois, 2008). Ubiquitination is a reversible process by which the 76-amino-acid protein ubiquitin is covalently conjugated to a target protein. Classically viewed as a protein turnover pathway, ubiquitination is recognized to modulate protein activity with such diversity that the process has been compared to phosphorylation (Sun, 2008). Ubiquitination is important in DNA repair, nuclear translocation, and endocytosis (Haglund and Dikic, 2005). Given the important processes in which ubiquitination is involved, dynamic control is necessary. The ubiquitin-protein linkage occurs by an isopeptide bond between the target protein’s lysine residue and the C-terminal residue of ubiquitin. Polyubiquitination is achievable because ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) that serve as anchors for additional ubiquitin attachment. When polyubiquitin chains form through one of the internal lysine residues, lysine 48 (K48), the target protein is directed to degradation via the ubiquitin-proteosome pathway. CYLD DUB activity is highly specific for K63-linked chains (Komander et al., 2008) but has been shown to act on K48-linked polyubiquitin chains as well (Reiley et al., 2006). K63-linked polyubiquitination influences protein-protein interactions and has clear roles in cell signaling (Song and Rape, 2008). Yet, CYLD has been shown to modulate proteasomal degradation of some of its targets, a function classically associated with regulation of ubiquitination on K48 (Xue et al., 2007).
CYLD Expression
In Drosophila, CYLD is expressed in the ovaries, genital tract, digestive tract, fat body, and parts of the nervous system (Tsichritzis et al., 2007). CYLD is highly expressed in normal human brain, testis, and skeletal muscle (Bignell et al., 2000). Similar expression occurs in mice (Massoumi et al., 2006b). CYLD is enriched in immune cells of humans (Friedman et al., 2008) and it is also expressed in the inner root sheath of normal human scalp hair follicles (Massoumi et al., 2006a).
Regulatory roles for CYLD have been demonstrated in development, immunity, and inflammation. CYLD regulates cell proliferation (Stegmeier et al., 2007), the cell cycle (Stegmeier et al., 2007), cell survival (Brummelkamp et al., 2003; Wright et al., 2007), spermatogenesis (Wright et al., 2007), osteoclastogenesis (Jin et al., 2008), calcium channel function (Stokes et al., 2006), anti-microbial defense and inflammation (Lim et al., 2007a; Lim et al., 2007b; Lim et al., 2008a; Lim et al., 2008b), the development and activation of immune cells (Reiley et al., 2006; Jin et al., 2007; Reiley et al., 2007), and cell migration via microtubule assembly (Gao et al., 2008). Reviews of these CYLD roles and the associated molecular mechanisms have been made elsewhere (Massoumi and Paus, 2007; Courtois, 2008; Sun, 2008).
CYLD negatively regulates NF-κB activation (Figure 2). NF-κB is a transcription factor that prevents apoptosis in a variety of tissues under disparate conditions (Karin et al., 2002; Perkins, 2007). The NF-κB pathway is induced by a wide variety of stimuli including but not limited to: tumor necrosis factor-α (TNF-α), IL-1, lipopolysaccharide (LPS), dsRNA, RANK, BAFF, and CD40. Regulation of the NF-κB pathway occurs through downregulation of IKK, and deubiquitination is a key mechanism of IKK control (Hacker and Karin 2006). CYLD has several putative direct targets in the NF-κB pathway including NEMO, TRAF2, TAK1, and TRAF6 (Figure 2) (Brummelkamp et al., 2003; Kovalenko et al., 2003; Trompouki et al., 2003; Reiley et al., 2007). NEMO binds CYLD and is subsequently deubiquitinated (Brummelkamp et al., 2003). TRAF2 (Kovalenko et al., 2003) and TAK1 (Reiley et al., 2007) have also been shown to interact with CYLD. The interaction of CYLD with TRAF6 is dependent upon a signal adaptor protein p62 and this interaction drives DUB activity of CYLD (Wooten et al., 2008). The third CAP-GLY domain of CYLD interacts with a proline-rich region of IKK, a function likely conferred by the atypical resemblance of this CAP-GLY domain to an SH3 domain, which is known to bind proline-rich sequences (Saito et al., 2004). It is notable that CYLD has shown regulatory action of only the classical NF-κB pathway, not the alternative NIK pathway and that all of the CYLD regulatory targets are at or above the level of IKK. Interestingly, CYLD is a target gene of NF-κB, suggesting a negative feedback loop; when NF-κB is induced by TNF-alpha and H. flu, CYLD is induced (Jono et al., 2004). CYLD is induced by TLR2 as a component of an antiviral signaling response in which CYLD attenuates H. flu–induced TLR7 activation (Sakai et al., 2007). Furthermore, CYLD is phosphorylated by IKK, and this is required for downstream activity including TRAF2 deubiquitination (Reiley et al., 2005). TRAF-interacting protein (TRIP), a protein that regulates NF-κB induced activation by TNF, also binds CYLD (Regamey et al., 2003).
Figure 2. CYLD in cell signaling.
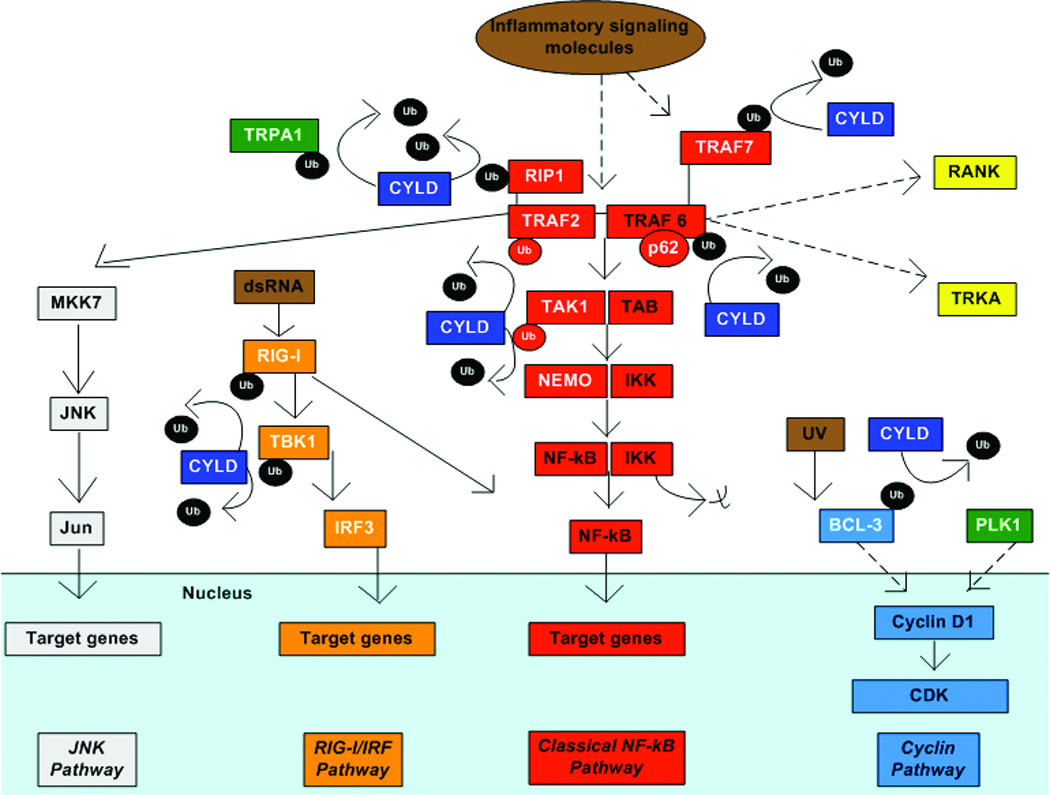
Model of how CYLD regulates different signaling pathways regulating survival, proliferation and inflammation in different cell types through deubiquitination. Dotted arrows denote multistep or incomplete pathways. CYLD and its known binding partners are in white font. Targets activated by CYLD are in green. Key targets for CYLD include removal of K63-linked Ub from TRAF2 and TRAF6, proteins that form a complex for NF-kB signaling. CYLD also deubiquitinates RIP1, a protein that associates with TRAF2. Deubiquitination of TRAF2 inhibits MKK7 activation and downstream JNK signaling. Deubiquitination of TRAF2, TRAF6, TRAF7 and TAK1 inhibit downstream dissociation of IKK from NF-kB and NF-KB signaling. In addition, deubiquitination of TRAF6 also inhibits downstream activation of RANK signaling and internalization of the TRKA receptor. CYLD interaction with TRAF6 requires the adaptor protein p62. In response to dsRNA, RIG-I is activated. CYLD DUB of RIG-I and TBK1 inhibits downstream activation of IRF3 signaling. RIG-I is a known activator of NF-kB as well. CYLD deubiquitination of BCL3 prevents its translocation from the cytoplasm to the nucleus in response to UV light. Nuclear BCL-3 activates cyclinD1 and promotes cell proliferation. CYLD also activates cyclinD1, possibly through PLK1. CYLD associates with the calcium channel, TRPA1, and maintains its activation by removing ubiquitin.
CYLD also modulates the JNK pathway, which serves important roles in apoptotic/cell survival regulation and proliferation (Figure 2). This regulation occurs in response to stimulation by several cytokines (Reiley et al., 2004). CYLD modulation of JNK likely occurs through CYLD-directed DUB of TRAF2, a requisite protein in the JNK pathway (Xue et al., 2007). CYLD reduces the activity of MKK7/JNKK2, the MAPK that phosphorylates JNK (Koga et al., 2008).
CYLD also inhibits the growth-promoting IRF3 signaling pathway by acting as a negative regulator of RIG-I (a cytoplasmic RNA sensor) (Friedman et al., 2008; Zhang et al., 2008). The mechanism is likely via CYLD deubiquitination of RIG-I and the IKK related kinases IKKε/TBK1, kinases that phosphorylate IRF3. CYLD expression is reduced in the presence of tumor necrosis factor and viral infection (Friedman et al., 2008).
In addition, CYLD localizes to microtubules in interphase, the midbody during telophase, and decreases as the cell completes mitosis, supporting a cell-cycle regulatory function (Stegmeier et al., 2007). PLK1 is a candidate target for CYLD regulation of mitotic entry. Surprisingly CYLD downregulation delays the G2/M progression, implying a tumor-promoting activity for CYLD (Stegmeier et al., 2007). CYLD interacts with microtubules, mainly through its first CAP-GLY domain (Gao et al., 2008). This interaction is critical for wound healing cell migration, occurring through enhancement of tubulin polymerization into microtubules.
Animal Models
Recently, Drosophila melanogaster has been used as an animal model to investigate the function of CYLD in development and signaling pathways. Xue and co-workers identified in vivo functions of CYLD by generating D. melanogaster CYLD (dCLYD) mutants and transgenic flies expressing wild-type or mutant dCYLD proteins (Xue et al., 2007). dCYLD mutants are normal at birth but have a shortened life span and impaired response to induced oxidative stress. The phenotype was rescued with single copy dCYLD restoration. They showed that dCYLD deubiquitinates dTRAF2, preventing degradation of dTRAF2 and that dCYLD regulates TNF-induced JNK activation and cell death. In 2007, Tsichritzis and co-workers showed that in dCYLD mutants adult flies have altered fat body morphology and increased triglycerides levels (Tsichritzis et al., 2007). In addition, flies with reduced CYLD expression were associated with susceptibility to bacterial infections. In conclusion, these investigations showed that in Drosophila CYLD is a DUB which serves functions in development, metabolism, and immunity (Xue et al., 2007; Tsichritzis et al., 2007)
Several Cyld knockout mouse models, deleting CYLD exons one (Reiley et al., 2006), 2 and 3 (Zhang et al., 2006), and 4 (Massoumi et al., 2006), have been generated to investigate the phenotypic consequences as well as the CYLD physiological functions. Cyld−/− mice are normal at birth with unremarkable prenatal history (Zhang et al., 2006b;1216 Jin et al., 2007; Massoumi et al., 2006). Cyld−/− mice exhibit increasing lymphoid hyperplasia in lymph nodes, spleen, lungs, and salivary glands with aging (Zhang et al., 2006b; Reiley et al., 2006). Cyld−/− mice develop colonic autoimmune symptoms and inflammation analogous to human inflammatory bowel disease that has been attributed to T cell dysfunction (Reiley et al., 2007). Yet, there are no reports of increased spontaneous malignancy in Cyld−/− mice. However, Cyld−/− mice subjected to azoxymethane and dextran sodium sulfate treatment, to induce colitis and colon tumors (Tanaka et al., 2003), develop more colonic tumors than wild type mice (Zhang et al., 2006). Colon tumors from Cyld−/− mice have increased expression of iNOS and COX-2, both NF-κB regulated products. Increased TRAF-2 ubiquitination and JNK activation were also observed in Cyld−/− mice (Zhang et al., 2006). Cyld−/− mice have no detectable abnormalities in naïve skin but are susceptible to chemically induced skin cancers (Massoumi,R., 2006). Cyld−/− mice treated with topical application of TPA and DMBA developed increased frequency, number, and size of skin papillomas compared to wild-type mice. Primary keratinocytes from Cyld−/− mice had increased proliferation in response to TPA. Skin tumors from Cyld−/− mice had increased activation of Bcl-3 and downstream expression of cyclin D1.
Reiley and co-workers generated Cyld−/− mice by homologous recombination in stem cells (Reiley et al., 2006). They showed that CYLD regulates TCR signaling in thymocytes. T-cells from Cyld−/− mice fail to transition from CD4+/CD8+ cells to single positive lineage and hyperproliferate following exposure to anti-CD3/CD28 antibodies. CYLD functions as a positive regulator of proximal TCR signaling by promoting recruitment of active LCK to its substrate Zap70 (Reiley et al., 2006). Subsequently, the same group showed that Cyld−/− mice have spontaneous activation of B cells as well as hyperresponsiveness to anti-IgM, LPS, or antigen exposure (Jin et al., 2007). Through the breeding process in the original study (Reiley et al., 2006) it was observed that Cyld−/− male mice were sterile, as they failed to produce offspring when mated with wild-type females. Further studies showed that loss of CYLD in testicular cells leads to aberrant germ cell apoptosis and impaired spermatogenesis (Wright et al., 2007). This study suggests that CYLD plays an essential role controlling the RIP1/NF-κB signaling axis in testis.
In 2008, Jin and co-workers showed that Cyld−/− mice have severe osteoporosis with abnormal osteoclast differentiation and activity (Jin et al., 2008). Ex-vivo Cyld−/− osteoclast precursors are hyperresponsive to RANKL-induced differentiation. Through interaction with the adaptor p62, CYLD is recruited to TRAF6 and inhibits TRAF6 ubiquitination-linked events (Jin et al., 2008). In conclusion, CYLD regulates osteoclastogenesis in mice by negative regulation of RANK signaling.
Lim and colleagues found that Cyld−/− mice inoculated with intratracheal Streptococcal pneumoniae lysate or pneumolysin (PLY) had decreased acute lung injury, mitigated microvascular leakage, hypothermia, and mortality compared to wild type mice (Lim et al., 2007a). They showed that CYLD is induced by PLY and negatively regulates MKK-p38 and its downstream products including plasminogen activator inhibitor-1. Conversely, Cyld−/− mice had a detrimental inflammatory response to nontypeable H. influenzae (Lim et al., 2007b). CYLD reduces inflammation via a deubiquitination-dependent inhibition of TRAF6/7. Lung tissue from Cyld−/− mice had increased TLR2 mRNA upregulation following S. pneumoniae inoculation (Lim et al., 2008). PLY appears to be the major virulence factor. CYLD acts as a negative regulator of S. pneumoniae-induced TLR2 up-regulation via negative-crosstalk with NF-κB signaling. A subsequent study showed that Cyld−/− mice were hypersusceptible to E. coli pneumonia and had an enhanced innate immune response to E. coli (Lim et al., 2008). Cyld−/− cells exhibited enhanced NF-κB activation upon E. coli inoculation, and the enhanced NF-κB activation by E. coli was abolished by perturbing IKK signaling. Furthermore, inhibition of IKK rescued Cyld−/− mice from lethal infection during E. coli pneumonia and reduced inflammation. Taken together, these data showed that CYLD acts as a crucial negative regulator for E. coli pneumonia through negative regulation of NF-κB.
CYLD in Health and Disease
Familial cylindromatosis and multiple familial trichoepithelioma are variants of a single disease entity: Brooke-Spiegler syndrome. There is variable phenotypic expression among and within families and patients (Bowen et al., 2005; Zhang et al., 2006a). Penetrance is quite high at increasing age. Overall, studies have reported a female predominance (van Balkom and Hennekam, 1994), which may be due to reduced penetrance in males (Anderson and Howel,l 1976). It is of interest that other benign adnexal tumors such as multiple syringomas have been reported in patients with Brooke-Spiegler syndrome (Uede et al., 2004). In these patients malignant transformation into cylindrocarcinoma has been reported and 9/15 cylindrocarcinomas that were reported in one analysis metastasized (Gerretsen et al., 1993) (Durani et al., 2001; Pizinger and Michal, 2000). Similarly, spiradenoma-like tumor degeneration into carcinoma or sarcoma has been reported in Brooke-Spiegler syndrome patients (Kazakov et al., 2009) (De Francesco et al., 2005) and in sporadic spiradenomas (Braun-Falco et al., 2003) (Kazakov et al., 2009). In addition, malignant transformation of trichoepitheliomas into BCC has been reported in Brooke-Spiegler syndrome patients (Lee et al., 2008) Pariser, 1986; Ayhan et al., 2001; Johnson and Bennett, 1993; Pincus et al., 2008). Conclusions may be presumptuous given that BCC is a common cancer (Lee et al., 2005). However, Pincus et al. reported a patient in which multiple BCCs originated exclusively within the field of his multiple trichoepitheliomas with histopathologic sections showing TE and BCC in direct continuity, supporting transformation of trichoepitheliomas to BCC (Pincus et al., 2008).
Several lines of evidence consistently support that CYLD functions as a tumor-suppressor gene. Almost 90% of the tumor-predisposing germline mutations in CYLD are truncating mutations. LOH of the wild-type allele at the CYLD locus (chromosome 16q12-13) is frequent in cylindromas and TE and some tumors without LOH have somatic mutations of CYLD (Biggs et al., 1996; Thomson et al., 1999; Bignell et al., 2000; Leonard et al., 2001). Other skin appendageal tumors such as hidrocystomas, eccrine spiradenomas, and sebaceous adenoma also have LOH at chromosome 16q12-13 (Leonard et al., 2001). Decreased CYLD expression has been shown in cylindromas by immunohistochemistry (Massoumi et al., 2006a).
Increasing evidence supports that Brooke-Spiegler syndrome is associated with salivary gland tumors, specifically basal cell monomorphic adenomas (Antonescu and Terzakis, 1997; Baican et al., 1998; Jungehulsing et al., 1999; Kakagia et al., 2004; Bowen et al., 2005; Saggar et al., 2008). Given multiple reports of the association it is suggested that patients with Brooke-Spiegler syndrome be examined for salivary gland tumors (Baican et al., 1998). Further research is needed to evaluate the risk of salivary gland tumors in these patients. Furthermore, LOH at chromosome 16q12-13 was found in 80% of salivary gland basal cell monomorphic adenomas (Choi et al., 2002). CYLD negatively regulates NF-κB signaling in human salivary gland tumor cell lines and CYLD expression is inversely correlated with NF-κB activity in salivary gland tumors, though not associated with tumor stage (Fukuda et al., 2008). These studies support that CYLD may play a role in the pathogenesis of salivary gland tumors.
Loss of CYLD has been implicated in solid tumors of the colon and liver (Hellerbrand et al., 2007), kidney (Strobel et al., 2002), cervix (Hirai et al., 2004), prostate (Kikuno et al., 2008), and lung (Zhong et al., 2007). Colon and hepatocellular cancer (HCC) cell lines show downregulation or loss of CYLD. CYLD transfection into HCC revealed that CYLD expression decreases NF-κB activity (Hellerbrand et al., 2007). Comparative genome hybridization (CGH) array revealed that over 30% of hepatitis-C associated HCC have decreased CYLD (Hashimoto et al., 2004). HCC cells treated with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) resisted apoptosis through induction of the NF-κB pathway and vector-driven expression of CYLD in HCC cells induced rapid apoptosis (Chu et al., 2006).
A genome-wide gene expression microarray found CYLD to be downregulated in inflammatory bowel disease, a finding confirmed with RT-PCR (Costello et al., 2005). The phytoestrogen genistein appears to decrease NF-κB activity in prostate cancer cells in part through demethylation and acetylation of the CYLD promoter (Kikuno et al., 2008). CYLD is epigenetically silenced in some non-small cell lung cancers (NSCLC) (Zhong et al., 2007). CYLD has been identified as inactivated by either mutation or deletion in multiple myeloma (MM) (Annunziata et al., 2007; Keats et al., 2007) and CYLD inactivation has been reported as negative predictor of survival in MM (Jenner et al., 2007). The mechanism by which CYLD appears to regulate tumorigenesis most often involves the NF-κB pathway (Massoumi et al., 2006b; Zhang et al., 2006b; Courtois, 2008), but other pathways have been implicated (Stegmeier, et al. 2007).
Genetic testing is useful in families because CYLD mutation detection rate is relatively high overall (84%), especially in familial cylindromatosis (88–100%) (Saggar et al., 2008). In contrast, the mutation detection rate for multiple familial trichoepitheliomas is lower (44–72%) (Saggar et al., 2008); this may be due to genetic heterogeneity. To date, all germline CYLD mutations reported have been identified by direct sequencing. Therefore, it is still possible that large deletions, insertions, or complex rearrangements may account for those mutations undetectable by direct sequencing. Over 75% of Brooke-Spiegler syndrome families have CYLD mutation in exons 16–20 and so it is recommended these sites be evaluated first in probands.
Prenatal diagnosis for pregnancies of Brooke-Spiegler syndrome families may be a useful endeavor in families with identified CYLD mutations in a parent or sibling. In these cases, a prenatal test showing no mutation in CYLD predicts that the offspring will not be affected due to a CYLD mutation. Options for prenatal diagnosis include chorionic villus sampling (CVS), amniocentesis, or preimplantation genetic diagnosis (PGD). CVS is performed between gestational weeks 10–13 and amniocentesis between weeks 15–18. Prenatal diagnosis may be of interest to families, especially those with family history of a severe phenotype. To our knowledge, there are no reports describing prenatal diagnosis in Brooke-Spiegler syndrome. CYLD mutation carriers should be informed that Brooke-Spiegler syndrome is highly penetrant for skin appendageal tumors though age of onset ranges from childhood to late adulthood. Early identification of mutations and management of skin appendageal tumors provides an opportunity to potentially minimize disfigurement or detect malignant lesions.
Future Directions
In less than a decade, our understanding of CYLD has evolved from its initial recognition as an uncharacterized protein encoded by a gene implicated in familial cylindromatosis to a multifunctional deubiquitinating enzyme involved in a diverse range of major cellular pathways including NF-kB, and JNK, critical in cell regulation. The important multifunctional roles of CYLD are contrasted with the relatively benign and specific phenotype characteristic of Brooke-Spiegler syndrome when the CYLD is mutated in humans. This paradox may reflect that either other clinical features have not been fully unmasked in Brooke-Spiegler syndrome or that other compensating pathways are activated when CYLD is deficient.
Germline mutations in the CYLD tumor-suppressor gene predispose individuals with Brooke-Spiegler syndrome to the development of tumors whose features recapitulate the morphogenesis of the folliculo-sebaceous-apocrine unit (Uede et al., 2004). Brooke-Spiegler syndrome-associated skin tumors derive from a single pluripotent follicular stem cell. How mutations in CYLD lead to the clinical phenotype is currently unknown. We hypothesize that CYLD may play a critical role in the stem cell folliculo-apocrine differentiation. Patients with Brooke-Spiegler syndrome have a CYLD germline mutation or “first hit” in every cell including both mesenchymal and the epithelial cells of the folliculo-sebaceous-apocrine unit. It has been shown that up to 70% of cylindromas have LOH (Bignell et al., 2000) supporting that CYLD functions as a tumor-suppressor gene. The developmental time point (embryonic vs postnatal) and the cell type (pluripotent vs. mesenchymal vs. follicular epithelial) in which the “second hit” occurs could explain in part the development of the variety of tumors. Furthermore, secondary epigenetic events downstream can lead to inappropriate activation of signaling pathways that regulate differentiation. A related and alternative explanation is that CYLD can alter normal mesenchymal-epithelial interactions. The mesenchymal-epithelial interaction entails a complex communication that includes cell surface receptors, cell adhesion molecules, extracellular matrix products, and secreted chemical messengers that can regulate cell proliferation, apoptosis, and differentiation. Furthermore, different mesenchymal-epithelial interactions may occur at different anatomic locations, explaining also in part the anatomic preference of these tumors. It is possible that modifier genes or environmental factors may also play a role. Identification of the signaling molecules and pathways in developing and postnatal hair follicles is therefore vital to our understanding of pathogenic states in the skin in patients with Brooke-Spiegler syndrome.
Moreover, given that CYLD is expressed in many tissues other than skin, it is surprising that CYLD deficiency manifests with a striking predilection for skin appendageal tumors. Future investigations of the mechanism(s) by which CYLD dysfunction leads to susceptibility for a variety of tumors may provide clues to the pathogenesis of adnexal tumors and other tumors associated with Brooke-Spiegler syndrome. Study of known CYLD targets such as TRAFs, NEMO, and BCL3 and their role in normal skin biology as well as in appendageal tumors may provide novel insights. Conversely, investigations of skin appendageal tumors may elucidate the mechanisms by which CYLD regulates tumorigenesis.
Furthermore, CYLD also serves as a potential link between two unique but related processes: inflammation and carcinogenesis. Present knowledge of the pathways that regulate CYLD is scant and identification of positive and negative regulators of CYLD could be of intense value, since they could be potentially novel anti-inflammatory or anti-neoplastic agents. Further investigations of the efficacy of salicylates (NK-kB antagonists) (Oosterkamp et al. 2006) as well as other existing anti-inflammatory therapies such as the TNF-alpha antagonists (Fisher and Geronemus, 2006;) in the treatment of adnexal tumors are warranted.
Acknowledgments
We would like to thank the Brooke-Spiegler syndrome families for their participation in research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, the Division of Cancer Epidemiology and Genetics. P.B. is a Howard Hughes Medical Institute Research Scholar.
References
- Almeida S, Maillard C, Itin P, Hohl D, Huber M. Five new CYLD mutations in skin appendage tumors and evidence that aspartic acid 681 in CYLD is essential for deubiquitinase activity. J Invest Dermatol. 2008;128:587–593. doi: 10.1038/sj.jid.5701045. [DOI] [PubMed] [Google Scholar]
- Alsaad KO, Obaidat NA, Ghazarian D. Skin adnexal neoplasms--part 1: An approach to tumours of the pilosebaceous unit. J Clin Pathol. 2007;60:129–144. doi: 10.1136/jcp.2006.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DE, Howell JB. Epithelioma adenoides cysticum: Genetic update. Br J Dermatol. 1976;95:225–232. doi: 10.1111/j.1365-2133.1976.tb07008.x. [DOI] [PubMed] [Google Scholar]
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, Zhao H, Stephens O, Santra M, Williams DR, Dang L, Barlogie B, Shaughnessy JD, Jr, Kuehl WM, Staudt LM. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu CR, Terzakis JA. Multiple malignant cylindromas of skin in association with basal cell adenocarcinoma with adenoid cystic features of minor salivary gland. J Cutan Pathol. 1997;24:449–453. doi: 10.1111/j.1600-0560.1997.tb00822.x. [DOI] [PubMed] [Google Scholar]
- Avvakumov GV, Walker JR, Xue S, Finerty PJ, Jr, Mackenzie F, Newman EM, Dhe-Paganon S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8) J Biol Chem. 2006;281:38061–38070. doi: 10.1074/jbc.M606704200. [DOI] [PubMed] [Google Scholar]
- Ayhan M, Adanali G, Senen D, Gorgu M, Erdogan B. Rarely seen cutaneous lesions in an elderly patient: Malignant transformation of multiple trichoepithelioma. Ann Plast Surg. 2001;47:98–99. doi: 10.1097/00000637-200107000-00025. [DOI] [PubMed] [Google Scholar]
- Baican A, Has C, Crisan C, Orasan R, Petrescu M, Machet L. Multiple cutaneous cylindromas associated with parotid and submandibular gland cylindromas. Ann Dermatol Venereol. 1998;125:909–911. [PubMed] [Google Scholar]
- Biggs PJ, Chapman P, Lakhani SR, Burn J, Stratton MR. The cylindromatosis gene (cyld1) on chromosome 16q may be the only tumour suppressor gene involved in the development of cylindromas. Oncogene. 1996;12:1375–1377. [PubMed] [Google Scholar]
- Biggs PJ, Wooster R, Ford D, Chapman P, Mangion J, Quirk Y, Easton DF, Burn J, Stratton MR. Familial cylindromatosis (turban tumour syndrome) gene localised to chromosome 16q12-q13: Evidence for its role as a tumour suppressor gene. Nat Genet. 1995;11:441–443. doi: 10.1038/ng1295-441. [DOI] [PubMed] [Google Scholar]
- Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, Jones C, Hansen J, Blair E, Hofmann B, Siebert R, Turner G, Evans DG, Schrander-Stumpel C, Beemer FA, van Den Ouweland A, Halley D, Delpech B, Cleveland MG, Leigh I, Leisti J, Rasmussen S. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25:160–165. doi: 10.1038/76006. [DOI] [PubMed] [Google Scholar]
- Bowen S, Gill M, Lee DA, Fisher G, Geronemus RG, Vazquez ME, Celebi JT. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: Lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124:919–920. doi: 10.1111/j.0022-202X.2005.23688.x. [DOI] [PubMed] [Google Scholar]
- Braun-Falco M, Bonel H, Ring J, Hein R. Linear spiradenoma with focal malignant transformation. J Eur Acad Dermatol Venereol. 2003;17:308–312. doi: 10.1046/j.1468-3083.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- Choi HR, Batsakis JG, Callender DL, Prieto VG, Luna MA, El-Naggar AK. Molecular analysis of chromosome 16q regions in dermal analogue tumors of salivary glands: A genetic link to dermal cylindroma? Am J Surg Pathol. 2002;26:778–783. doi: 10.1097/00000478-200206000-00012. [DOI] [PubMed] [Google Scholar]
- Chou SC, Lin SL, Tseng HH. Malignant eccrine spiradenoma: A case report with pulmonary metastasis. Pathol Int. 2004;54:208–212. doi: 10.1111/j.1440-1827.2004.01609.x. [DOI] [PubMed] [Google Scholar]
- Chu L, Gu J, He Z, Xiao T, Liu X. Adenoviral vector expressing CYLD augments antitumor activity of TRAIL by suppression of NF-kappaB survival signaling in hepatocellular carcinoma. Cancer Biol Ther. 2006;5:615–622. doi: 10.4161/cbt.5.6.2662. [DOI] [PubMed] [Google Scholar]
- Cooper PH, Frierson HF, Jr, Morrison AG. Malignant transformation of eccrine spiradenoma. Arch Dermatol. 1985;121:1445–1448. [PubMed] [Google Scholar]
- Costello CM, Mah N, Hasler R, Rosenstiel P, Waetzig GH, Hahn A, Lu T, Gurbuz Y, Nikolaus S, Albrecht M, Hampe J, Lucius R, Kloppel G, Eickhoff H, Lehrach H, Lengauer T, Schreiber S. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med. 2005;2:e199. doi: 10.1371/journal.pmed.0020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois G. Tumor suppressor CYLD: Negative regulation of NF-kappaB signaling and more. Cell Mol Life Sci. 2008;65:1123–1132. doi: 10.1007/s00018-007-7465-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty K, Dutta B, Hogan P. Multiple trichoepitheliomas in a mother and daughter. Australas J Dermatol. 2003;44:270–272. doi: 10.1046/j.1440-0960.2003.00007.x. [DOI] [PubMed] [Google Scholar]
- De Francesco V, Frattasio A, Pillon B, Stinco G, Scott CA, Trotter D, Patrone P. Carcinosarcoma arising in a patient with multiple cylindromas. Am J Dermatopathol. 2005;27:21–26. doi: 10.1097/01.dad.0000141548.69423.c7. [DOI] [PubMed] [Google Scholar]
- Durani BK, Kurzen H, Jaeckel A, Kuner N, Naeher H, Hartschuh W. Malignant transformation of multiple dermal cylindromas. Br J Dermatol. 2001;145:653–656. doi: 10.1046/j.1365-2133.2001.04460.x. [DOI] [PubMed] [Google Scholar]
- Engel CJ, Meads GE, Joseph NG, Stavraky W. Eccrine spiradenoma: A report of malignant transformation. Can J Surg. 1991;34:477–480. [PubMed] [Google Scholar]
- Ishikawa M, Nakanishi Y, Yamazaki N, Yamamoto A. Malignant eccrine spiradenoma: A case report and review of the literature. Dermatol Surg. 2001;27:67–70. [PubMed] [Google Scholar]
- Espana A, Garcia-Amigot F, Aguado L, Garcia-Foncillas J. A novel missense mutation in the CYLD gene in a Spanish family with multiple familial trichoepithelioma. Arch Dermatol. 2007;143:1209–1210. doi: 10.1001/archderm.143.9.1209. [DOI] [PubMed] [Google Scholar]
- Fisher GH, Geronemus RG. Treatment of multiple familial trichoepitheliomas with a combination of aspirin and a neutralizing antibody to tumor necrosis factor alpha: A case report and hypothesis of mechanism. Arch Dermatol. 2006;142:782–783. doi: 10.1001/archderm.142.6.782. [DOI] [PubMed] [Google Scholar]
- Friedman CS, O'Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Hiroi M, Suzuki S, Ohmori Y, Sakashita H. Loss of CYLD might be associated with development of salivary gland tumors. Oncol Rep. 2008;19:1421–1427. [PubMed] [Google Scholar]
- Gao J, Huo L, Sun X, Liu M, Li D, Dong JT, Zhou J. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J Biol Chem. 2008;283:8802–8809. doi: 10.1074/jbc.M708470200. [DOI] [PubMed] [Google Scholar]
- Gerretsen AL, van der Putte SC, Deenstra W, van Vloten WA. Cutaneous cylindroma with malignant transformation. Cancer. 1993;72:1618–1623. doi: 10.1002/1097-0142(19930901)72:5<1618::aid-cncr2820720521>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Hashimoto K, Toi Y, Yotsunoto S, Ko MS. Basal cell carcinoma occurring in multiple familial trichoepithelioma: Detection of loss of heterozygosity in chromosome 9q. Arch Dermatol. 1997;133:666–667. [PubMed] [Google Scholar]
- Hashimoto K, Mori N, Tamesa T, Okada T, Kawauchi S, Oga A, Furuya T, Tangoku A, Oka M, Sasaki K. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Mod Pathol. 2004;17:617–622. doi: 10.1038/modpathol.3800107. [DOI] [PubMed] [Google Scholar]
- Heinritz W, Grunewald S, Strenge S, Schutz A, Froster UG, Glander HJ, Paasch U, Simon JC. A case of Brooke-Spiegler syndrome with a new mutation in the CYLD gene. Br J Dermatol. 2006;154:992–994. doi: 10.1111/j.1365-2133.2006.07142.x. [DOI] [PubMed] [Google Scholar]
- Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK. Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis. 2007;28:21–27. doi: 10.1093/carcin/bgl081. [DOI] [PubMed] [Google Scholar]
- Hirai Y, Kawamata Y, Takeshima N, Furuta R, Kitagawa T, Kawaguchi T, Hasumi K, Sugai S, Noda T. Conventional and array-based comparative genomic hybridization analyses of novel cell lines harboring HPV18 from glassy cell carcinoma of the uterine cervix. Int J Oncol. 2004;24:977–986. [PubMed] [Google Scholar]
- Hu G, Onder M, Gill M, Aksakal B, Oztas M, Gurer MA, Celebi JT. A novel missense mutation in CYLD in a family with Brooke-Spiegler syndrome. J Invest Dermatol. 2003;121:732–734. doi: 10.1046/j.1523-1747.2003.12514.x. [DOI] [PubMed] [Google Scholar]
- Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/s0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- Hu M, Li P, Song L, Jeffrey PD, Chenova TA, Wilkinson KD, Cohen RE, Shi Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747–3756. doi: 10.1038/sj.emboj.7600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TM, Chao SC, Lee JY. A novel splicing mutation of the CYLD gene in a Taiwanese family with multiple familial trichoepithelioma. Clin Exp Dermatol. 2009;34:77–80. doi: 10.1111/j.1365-2230.2008.02870.x. [DOI] [PubMed] [Google Scholar]
- Jenner MW, Leone PE, Walker BA, Ross FM, Johnson DC, Gonzalez D, Chiecchio L, Dachs Cabanas E, Dagrada GP, Nightingale M, Protheroe RK, Stockley D, Else M, Dickens NJ, Cross NC, Davies FE, Morgan GJ. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood. 2007;110:3291–3300. doi: 10.1182/blood-2007-02-075069. [DOI] [PubMed] [Google Scholar]
- Jin W, Chang M, Paul EM, Babu G, Lee AJ, Reiley W, Wright A, Zhang M, You J, Sun SC. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J Clin Invest. 2008;118:1858–1866. doi: 10.1172/JCI34257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Reiley WR, Lee AJ, Wright A, Wu X, Zhang M, Sun SC. Deubiquitinating enzyme CYLD regulates the peripheral development and naive phenotype maintenance of B cells. J Biol Chem. 2007;282:15884–15893. doi: 10.1074/jbc.M609952200. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Bennett RG. Occurrence of basal cell carcinoma among multiple trichoepitheliomas. J Am Acad Dermatol. 1993;28:322–326. doi: 10.1016/0190-9622(93)70046-v. [DOI] [PubMed] [Google Scholar]
- Jono H, Lim JH, Chen LF, Xu H, Trompouki E, Pan ZK, Mosialos G, Li JD. NF-kappaB is essential for induction of CYLD, the negative regulator of NF-kappaB: Evidence for a novel inducible autoregulatory feedback pathway. J Biol Chem. 2004;279:36171–36174. doi: 10.1074/jbc.M406638200. [DOI] [PubMed] [Google Scholar]
- Jungehulsing M, Wagner M, Damm M. Turban tumour with involvement of the parotid gland. J Laryngol Otol. 1999;113:779–783. doi: 10.1017/s0022215100145190. [DOI] [PubMed] [Google Scholar]
- Kakagia D, Alexiadis G, Kiziridou A, Lambropoulou M. Brooke-Spiegler syndrome with parotid gland involvement. Eur J Dermatol. 2004;14:139–141. [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: From innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–310. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Kazakov DV, Magro G, Kutzner H, Spagnolo DV, Yang Y, Zaspa O, Mukensnabl P, Michal M. Spiradenoma and spiradenocylindroma with an adenomatous or atypical adenomatous component: A clinicopathological study of 6 cases. Am J Dermatopathol. 2008;30:436–441. doi: 10.1097/DAD.0b013e3181812729. [DOI] [PubMed] [Google Scholar]
- Kazakov DV, Soukup R, Mukensnabl P, Boudova L, Michal M. Brooke-Spiegler syndrome: Report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27–33. doi: 10.1097/01.dad.0000138049.86662.3e. [DOI] [PubMed] [Google Scholar]
- Kazakov DV, Zelger B, Rutten A, Vazmitel M, Spagnolo DV, Kacerovska D, Vanecek T, Grossmann P, Sima R, Grayson W, Calonje E, Koren J, Mukensnabl P, Danis D, Michal M. Morphologic diversity of malignant neoplasms arising in preexisting spiradenoma, cylindroma, and spiradenocylindroma based on the study of 24 cases, sporadic or occurring in the setting of Brooke-Spiegler syndrome. Am J Surg Pathol. 2009 doi: 10.1097/PAS.0b013e3181966762. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, Majid S, Igawa M, Dahiya R. Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells. Int J Cancer. 2008;123:552–560. doi: 10.1002/ijc.23590. [DOI] [PubMed] [Google Scholar]
- Koga T, Lim JH, Jono H, Ha UH, Xu H, Ishinaga H, Morino S, Xu X, Yan C, Kai H, Li JD. Tumor suppressor cylindromatosis acts as a negative regulator for streptococcus pneumoniae-induced NFAT signaling. J Biol Chem. 2008;283:12546–12554. doi: 10.1074/jbc.M710518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Lord CJ, Scheel H, Swift S, Hofmann K, Ashworth A, Barford D. The structure of the CYLD USP domain explains its specificity for Lys63-linked polyubiquitin and reveals a B box module. Mol Cell. 2008;29:451–464. doi: 10.1016/j.molcel.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–805. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- Lee DA, Grossman ME, Schneiderman P, Celebi JT. Genetics of skin appendage neoplasms and related syndromes. J Med Genet. 2005;42:811–819. doi: 10.1136/jmg.2004.025577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Kim JE, Cho BK, Kim YC, Park CJ. Malignant transformation of multiple familial trichoepithelioma: Case report and literature review. Acta Derm Venereol. 2008;88:43–46. doi: 10.2340/00015555-0322. [DOI] [PubMed] [Google Scholar]
- Leonard N, Chaggar R, Jones C, Takahashi M, Nikitopoulou A, Lakhani SR. Loss of heterozygosity at cylindromatosis gene locus, CYLD, in sporadic skin adnexal tumours. J Clin Pathol. 2001;54:689–692. doi: 10.1136/jcp.54.9.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian F, Cockerell CJ. Cutaneous appendage tumors: Familial cylindromatosis and associated tumors update. Adv Dermatol. 2005;21:217–234. doi: 10.1016/j.yadr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Liang YH, Gao M, Sun LD, Liu LJ, Cui Y, Yang S, Fan X, Wang J, Xiao FL, Zhang XJ. Two novel CYLD gene mutations in Chinese families with trichoepithelioma and a literature review of 16 families with trichoepithelioma reported in china. Br J Dermatol. 2005;153:1213–1215. doi: 10.1111/j.1365-2133.2005.06960.x. [DOI] [PubMed] [Google Scholar]
- Liang YH, Sun CS, Ye XY, Zhang W, Yang S, Zhang XJ. Novel substitution and frameshift mutations of CYLD in two Chinese families with multiple familial trichoepithelioma. Br J Dermatol. 2008;158:1156–1158. doi: 10.1111/j.1365-2133.2008.08491.x. [DOI] [PubMed] [Google Scholar]
- Lim JH, Ha UH, Woo CH, Xu H, Li JD. CYLD is a crucial negative regulator of innate immune response in Escherichia coli pneumonia. Cell Microbiol. 2008a;11:2247–2256. doi: 10.1111/j.1462-5822.2008.01204.x. [DOI] [PubMed] [Google Scholar]
- Lim JH, Ha U, Sakai A, Woo CH, Kweon SM, Xu H, Li JD. Streptococcus pneumoniae synergizes with nontypeable Haemophilus influenzae to induce inflammation via upregulating TLR2. BMC Immunol. 2008b;9:40. doi: 10.1186/1471-2172-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Jono H, Koga T, Woo CH, Ishinaga H, Bourne P, Xu H, Ha UH, Xu H, Li JD. Tumor suppressor CYLD acts as a negative regulator for non-typeable Haemophilus influenza-induced inflammation in the middle ear and lung of mice. PLoS ONE. 2007a;2:e1032. doi: 10.1371/journal.pone.0001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JH, Stirling B, Derry J, Koga T, Jono H, Woo CH, Xu H, Bourne P, Ha UH, Ishinaga H, Xu H, Andalibi A, Feng XH, Zhu H, Huang Y, Zhang W, Weng X, Yan C, Yin Z, Briles DE, Davis RJ, Flavell RA, Li JD. Tumor suppressor CYLD regulates acute lung injury in lethal Streptococcus pneumoniae infections. Immunity. 2007b;27:349–360. doi: 10.1016/j.immuni.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Lv HL, Huang YJ, Zhou D, Du YF, Zhao XY, Liang YH, Quan C, Zhang H, Zhou FS, Gao M, Zhou L, Yang S, Zhang XJ. A novel missense mutation of CYLD gene in a Chinese family with multiple familial trichoepithelioma. J Dermatol Sci. 2008;50:143–146. doi: 10.1016/j.jdermsci.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Ly H, Black MM, Robson A. Case of the Brooke-Spiegler syndrome. Australas J Dermatol. 2004;45:220–222. doi: 10.1111/j.1440-0960.2004.00101.x. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking bcl-3-dependent NF-kappaB signaling. Cell. 2006a;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Paus R. Cylindromatosis and the CYLD gene: New lessons on the molecular principles of epithelial growth control. Bioessays. 2007;29:1203–1214. doi: 10.1002/bies.20677. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Podda M, Fassler R, Paus R. Cylindroma as tumor of hair follicle origin. J Invest Dermatol. 2006b;126:1182–1184. doi: 10.1038/sj.jid.5700218. [DOI] [PubMed] [Google Scholar]
- Michal M, Lamovec J, Mukensnabl P, Pizinger K. Spiradenocylindromas of the skin: Tumors with morphological features of spiradenoma and cylindroma in the same lesion: Report of 12 cases. Pathol Int. 1999;49:419–425. doi: 10.1046/j.1440-1827.1999.00890.x. [DOI] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123:773–786. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Obaidat NA, Alsaad KO, Ghazarian D. Skin adnexal neoplasms--part 2: An approach to tumours of cutaneous sweat glands. J Clin Pathol. 2007;60:145–159. doi: 10.1136/jcp.2006.041608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oiso N, Mizuno N, Fukai K, Nakagawa K, Ishii M. Mild phenotype of familial cylindromatosis associated with an R758X nonsense mutation in the CYLD tumour suppressor gene. Br J Dermatol. 2004;151:1084–1086. doi: 10.1111/j.1365-2133.2004.06231.x. [DOI] [PubMed] [Google Scholar]
- Oosterkamp HM, Neering H, Nijman SM, Dirac AM, Mooi WJ, Bernards R, Brummelkamp TR. An evaluation of the efficacy of topical application of salicylic acid for the treatment of familial cylindromatosis. Br J Dermatol. 2006;155:182–185. doi: 10.1111/j.1365-2133.2006.07224.x. [DOI] [PubMed] [Google Scholar]
- Oranje AP, Halley D, den Hollander JC, Teepe RG, van de Graaf R, van den Ouweland A, Wagner A. Multiple familial trichoepithelioma and familial cylindroma: One cause! J Eur Acad Dermatol Venereol. 2008;22:1395–1396. doi: 10.1111/j.1468-3083.2008.02648.x. [DOI] [PubMed] [Google Scholar]
- Pariser RJ. Multiple hereditary trichoepitheliomas and basal cell carcinomas. J Cutan Pathol. 1986;13:111–117. doi: 10.1111/j.1600-0560.1986.tb01510.x. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Pincus LB, McCalmont TH, Neuhaus IM, Kasper R, Oh DH. Basal cell carcinomas arising within multiple trichoepitheliomas. J Cutan Pathol. 2008;35 Suppl 1:59–64. doi: 10.1111/j.1600-0560.2008.01002.x. [DOI] [PubMed] [Google Scholar]
- Pizinger K, Michal M. Malignant cylindroma in Brooke-Spiegler syndrome. Dermatology. 2000;201:255–257. doi: 10.1159/000018499. [DOI] [PubMed] [Google Scholar]
- Poblete Gutierrez P, Eggermann T, Holler D, Jugert FK, Beermann T, Grussendorf-Conen EI, Zerres K, Merk HF, Frank J. Phenotype diversity in familial cylindromatosis: A frameshift mutation in the tumor suppressor gene CYLD underlies different tumors of skin appendages. J Invest Dermatol. 2002;119:527–531. doi: 10.1046/j.1523-1747.2002.01839.x. [DOI] [PubMed] [Google Scholar]
- Regamey A, Hohl D, Liu JW, Roger T, Kogerman P, Toftgard R, Huber M. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor kappaB activation by tumor necrosis factor. J Exp Med. 2003;198:1959–1964. doi: 10.1084/jem.20031187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley W, Zhang M, Sun SC. Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem. 2004;279:55161–55167. doi: 10.1074/jbc.M411049200. [DOI] [PubMed] [Google Scholar]
- Reiley W, Zhang M, Wu X, Granger E, Sun SC. Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation. Mol Cell Biol. 2005;25:3886–3895. doi: 10.1128/MCB.25.10.3886-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Jin W, Lee AJ, Wright A, Wu X, Tewalt EF, Leonard TO, Norbury CC, Fitzpatrick L, Zhang M, Sun SC. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med. 2007;204:1475–1485. doi: 10.1084/jem.20062694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, Sun SC. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol. 2006;7:411–417. doi: 10.1038/ni1315. [DOI] [PubMed] [Google Scholar]
- Renatus M, Parrado SG, D'Arcy A, Eidhoff U, Gerhartz B, Hassiepen U, Pierrat B, Riedl R, Vinzenz D, Worpenberg S, Kroemer M. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Kigawa T, Koshiba S, Sato K, Matsuo Y, Sakamoto A, Takagi T, Shirouzu M, Yabuki T, Nunokawa E, Seki E, Matsuda T, Aoki M, Miyata Y, Hirakawa N, Inoue M, Terada T, Nagase T, Kikuno R, Nakayama M, Ohara O, Tanaka A, Yokoyama S. The CAP-gly domain of CYLD associates with the proline-rich sequence in NEMO/IKKgamma. Structure. 2004;12:1719–1728. doi: 10.1016/j.str.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Sakai A, Koga T, Lim JH, Jono H, Harada K, Szymanski E, Xu H, Kai H, Li JD. The bacterium, nontypeable Haemophilus influenzae, enhances host antiviral response by inducing toll-like receptor 7 expression: Evidence for negative regulation of host anti-viral response by CYLD. FEBS J. 2007;274:3655–3668. doi: 10.1111/j.1742-4658.2007.05899.x. [DOI] [PubMed] [Google Scholar]
- Salhi A, Bornholdt D, Oeffner F, Malik S, Heid E, Happle R, Grzeschik KH. Multiple familial trichoepithelioma caused by mutations in the cylindromatosis tumor suppressor gene. Cancer Res. 2004;64:5113–5117. doi: 10.1158/0008-5472.CAN-04-0307. [DOI] [PubMed] [Google Scholar]
- Scheinfeld N, Hu G, Gill M, Austin C, Celebi JT. Identification of a recurrent mutation in the CYLD gene in Brooke-Spiegler syndrome. Clin Exp Dermatol. 2003;28:539–541. doi: 10.1046/j.1365-2230.2003.01344.x. [DOI] [PubMed] [Google Scholar]
- Song L, Rape M. Reverse the curse--the role of deubiquitination in cell cycle control. Curr Opin Cell Biol. 2008;20:156–163. doi: 10.1016/j.ceb.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Sowa ME, Nalepa G, Gygi SP, Harper JW, Elledge SJ. The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci U S A. 2007;104:8869–8874. doi: 10.1073/pnas.0703268104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes A, Wakano C, Koblan-Huberson M, Adra CN, Fleig A, Turner H. TRPA1 is a substrate for de-ubiquitination by the tumor suppressor CYLD. Cell Signal. 2006;18:1584–1594. doi: 10.1016/j.cellsig.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Strobel P, Zettl A, Ren Z, Starostik P, Riedmiller H, Storkel S, Muller-Hermelink HK, Marx A. Spiradenocylindroma of the kidney: Clinical and genetic findings suggesting a role of somatic mutation of the CYLD1 gene in the oncogenesis of an unusual renal neoplasm. Am J Surg Pathol. 2002;26:119–124. doi: 10.1097/00000478-200201000-00016. [DOI] [PubMed] [Google Scholar]
- Sun SC. Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008;8:501–511. doi: 10.1038/nri2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Rapley E, Biggs PJ, Lakhani SR, Cooke D, Hansen J, Blair E, Hofmann B, Siebert R, Turner G, Evans DG, Schrander-Stumpel C, Beemer FA, van Vloten WA, Breuning MH, van den Ouweland A, Halley D, Delpech B, Cleveland M, Leigh I, Chapman P, Burn J, Hohl D, Gorog JP, Seal S, Mangion J. Linkage and LOH studies in 19 cylindromatosis families show no evidence of genetic heterogeneity and refine the CYLD locus on chromosome 16q12-q13. Hum Genet. 2000;106:58–65. doi: 10.1007/s004399900227. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003;94:965–973. doi: 10.1111/j.1349-7006.2003.tb01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SA, Rasmussen SA, Zhang J, Wallace MR. A new hereditary cylindromatosis family associated with CYLD1 on chromosome 16. Hum Genet. 1999;105:171–173. doi: 10.1007/s004399900077. [DOI] [PubMed] [Google Scholar]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–796. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- Tsichritzis T, Gaentzsch PC, Kosmidis S, Brown AE, Skoulakis EM, Ligoxygakis P, Mosialos G. A Drosophila ortholog of the human cylindromatosis tumor suppressor gene regulates triglyceride content and antibacterial defense. Development. 2007;134:2605–2614. doi: 10.1242/dev.02859. [DOI] [PubMed] [Google Scholar]
- Uede K, Yamamoto Y, Furukawa F. Brooke-Spiegler syndrome associated with cylindroma, trichoepithelioma, spiradenoma, and syringoma. J Dermatol. 2004;31:32–38. doi: 10.1111/j.1346-8138.2004.tb00501.x. [DOI] [PubMed] [Google Scholar]
- van Balkom ID, Hennekam RC. Dermal eccrine cylindromatosis. J Med Genet. 1994;31:321–324. doi: 10.1136/jmg.31.4.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JP, Wells RS, Kerr CB. Ancell-Spiegler cylindromas (turban tumours) and Brooke-Fordyce trichoepitheliomas: Evidence for a single genetic entity. J Med Genet. 1968;5:29–35. doi: 10.1136/jmg.5.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten MW, Geetha T, Babu JR, Seibenhener ML, Peng J, Cox N, Diaz-Meco MT, Moscat J. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J Biol Chem. 2008;283:6783–6789. doi: 10.1074/jbc.M709496200. [DOI] [PubMed] [Google Scholar]
- Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M, Sun SC. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13:705–716. doi: 10.1016/j.devcel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Xue L, Igaki T, Kuranaga E, Kanda H, Miura M, Xu T. Tumor suppressor CYLD regulates JNK-induced cell death in drosophila. Dev Cell. 2007;13:446–454. doi: 10.1016/j.devcel.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Jono H, Kai H, Li JD. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J Biol Chem. 2005;280:41111–41121. doi: 10.1074/jbc.M509526200. [DOI] [PubMed] [Google Scholar]
- Young AL, Kellermayer R, Szigeti R, Teszas A, Azmi S, Celebi JT. CYLD mutations underlie Brooke-Spiegler, familial cylindromatosis, and multiple familial trichoepithelioma syndromes. Clin Genet. 2006;70:246–249. doi: 10.1111/j.1399-0004.2006.00667.x. [DOI] [PubMed] [Google Scholar]
- Zhang G, Huang Y, Yan K, Li W, Fan X, Liang Y, Sun L, Li H, Zhang S, Gao M, Du W, Yang S, Liu J, Zhang X. Diverse phenotype of Brooke-Spiegler syndrome associated with a nonsense mutation in the CYLD tumor suppressor gene. Exp Dermatol. 2006a;15:966–970. doi: 10.1111/j.1600-0625.2006.00501.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Stirling B, Temmerman ST, Ma CA, Fuss IJ, Derry JM, Jain A. Impaired regulation of NF-kappaB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest. 2006b;116:3042–3049. doi: 10.1172/JCI28746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wu X, Lee AJ, Jin W, Chang M, Wright A, Imaizumi T, Sun SC. Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem. 2008;283:18621–18626. doi: 10.1074/jbc.M801451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XJ, Liang YH, He PP, Yang S, Wang HY, Chen JJ, Yuan WT, Xu SJ, Cui Y, Huang W. Identification of the cylindromatosis tumor-suppressor gene responsible for multiple familial trichoepithelioma. J Invest Dermatol. 2004;122:658–664. doi: 10.1111/j.0022-202X.2004.22321.x. [DOI] [PubMed] [Google Scholar]
- Zheng G, Hu L, Huang W, Chen K, Zhang X, Yang S, Sun J, Jiang Y, Luo G, Kong X. CYLD mutation causes multiple familial trichoepithelioma in three Chinese families. Hum Mutat. 2004;23:400. doi: 10.1002/humu.9231. [DOI] [PubMed] [Google Scholar]
- Zhong S, Fields CR, Su N, Pan YX, Robertson KD. Pharmacologic inhibition of epigenetic modifications, coupled with gene expression profiling, reveals novel targets of aberrant DNA methylation and histone deacetylation in lung cancer. Oncogene. 2007;26:2621–2634. doi: 10.1038/sj.onc.1210041. [DOI] [PubMed] [Google Scholar]
- Zuo YG, Xu Y, Wang B, Liu YH, Qu T, Fang K, Ho MG. A novel mutation of CYLD in a Chinese family with multiple familial trichoepithelioma and no CYLD protein expression in the tumour tissue. Br J Dermatol. 2007;157:818–821. doi: 10.1111/j.1365-2133.2007.08081.x. [DOI] [PubMed] [Google Scholar]