

Abstract
Autosomal recessive congenital ichthyosis (ARCI) is a heterogeneous group of rare cornification diseases. Germline mutations in TGM1 are the most common cause of ARCI in the US. TGM1 encodes for the TGase-1 enzyme that functions in the formation of the cornified cell envelope. We review the clinical manifestations as well as the molecular genetics of ARCI. In addition, we characterized 115 TGM1 mutations reported in 234 patients from diverse racial and ethnic backgrounds (Caucasion Americans, Norwegians, Swedish, Finnish, German, Swiss, French, Italian, Dutch, Portuguese, Hispanics, Iranian, Tunisian, Moroccan, Egyptian, Afghani, Hungarian, African-Americans, Korean, Japanese and South African). We report 23 novel mutations: 71 (62%) missense; twenty (17%) nonsense; nine (8%) deletion; eight (7%) splice-site and seven (6%) insertion. The c.877-2A>G was the most commonly reported TGM1 mutation accounting for 34 % (147/435) of all TGM1 mutant alleles reported to date. It had been shown that this mutation is common among North American and Norwegian patients due to a founder effect. Thirty-one percent (36/115) of all mutations and 41% (29/71) of missense mutations occurred in arginine residues in TGase-1. Forty-nine percent (35/71) of missense mutations were within CpG dinucleotides, and 74% (26/35) of these mutations were C> T or G>A transitions. We constructed a model of human TGase-1 and showed that all mutated arginines that reside in the two beta-barrel domains and two (R142 and R143) in the beta-sandwich are located at domain interfaces. In conclusion, this study expands the TGM1 mutation spectrum, summarizes the current knowledge of TGM1 mutations. The high frequency of mutated arginine codons in TGM1 may be due to the deamination of 5′ methylated CpG dinucleotides. Structurally defective or attenuated cornified cell envelop have been shown in epidermal scales and appendages of ARCI patients with TGM1 mutations.
Keywords: Autosomal recessive congenital ichthyosis, ARCI, TGM1 gene, TGAse-1, molecular modeling, mutation update
INTRODUCTION
Autosomal recessive congenital ichthyosis (ARCI; MIM#s 190195, 242100, 242300) is a clinically heterogenous group of cornification diseases. ARCI is rare, with estimated incidence rates of 1:200,000–1:300,000 in the USA and as high as 1:91,000 in Norway due to a founder effect [Bale and Doyle, 1994; Pigg et al., 1998].
While the clinical hallmark of ARCI is epidermal scaling, patients may also have collodion membrane at birth, ectropion, eclabium, alopecia, palmar-plantar hyperkeratosis, hypohidrosis, and/or variable erythema. ARCI is classically divided into lamellar ichthyosis (LI; MIM# 242300) and non-bullous congenital ichthyosiform erythroderma (NBCIE; MIM# 242100) [Williams and Elias, 1985]. Patients with LI have large, dark, plate-like cutaneous scales with minimal erythema, while patients with NBCIE have erythroderma with overlying fine white scales [Williams and Elias 1985]. LI and NBCIE both have their own clinical spectrum within ARCI.
In 1983, absence of a cornified cell envelope (CCE) was shown in the epidermins of patients with LI suggesting that LI was a clinical consequence of missing the CCE [Kanerva et al., 1983]. In 1993, altered expression of loricrin and involucrin, CCE precursor proteins, and transglutaminase were shown in patients with LI suggesting that in LI disturbed membrane anchorage of transglutaminase could alter loricrin and involucrin cross-linkage and the formation of the CCE [Hohl et al., 1993]. Subsequently, an ARCI locus was linked to the transglutaminase-1 (TGM1) gene located on chromosome 14q11.2, and germline mutations in TGM1 were identified as a genetic cause of ARCI [Huber et al., 1995; Russell et al., 1995]. TGM1 has 14,095 bp, including 2454bp of coding sequence, and spans 15 exons (GenBank NM_000359.2). TGM1 encodes for the transglutaminase-1 (TGase-1) enzyme, which has 817 amino acid residues and a molecular weight of ~89 kD (GenBank NM_000359.2) [Kim et al., 1991; Kim et al., 1992]. Ninety-two unique TGM1 germline mutations have been reported to date [Farasat et al., 2008]. We recently showed that in the United States 55% of patients affected with ARCI have germline mutations in TGM1 [Farasat et al., 2008]. Thus, germline mutations in TGM1 are the most common cause of ARCI in the United States. TGase-1 functions in the formation of the cornified cell envelope (CCE), a structure important for the skin’s barrier function [Eckert et al., 1997; Boeshans et al., 2007]. The CCE, which measures approximately 15-nm, has a lipid extracellular layer and an inner layer containing proteins cross-linked by Nε(γ-glutamyl) lysine bonds that are deposited on the inner surface of the keratinocytes’ plasma membrane [Rothnagel and Rogers, 1984; Kalinin et al., 2002]. TGase-1 is responsible for catalyzing the Nε-(γ-glutamyl) lysine crosslinking of precursor proteins, such as involucrin, loricrin, and small protein rich proteins, during the formation of the cytoplasmic layer of the CCE. [Steinert and Marekov, 1995; Robinson et al., 1997]. It has been proposed that in the formation of the CCE, ω-hydroxyceramides, which compose part of the external layer of the CCE that replaces the plasma membrane in terminally differentiated keratinocytes, are cross-linked by TGase-1 to CCE precursor proteins, including involucrin [Wertz et al., 1989; Steinert and Marekov, 1995; Nemes et al., 1999b; Candi et al., 2005].
ARCI is a genetically heterogenous disease. Germline mutations in at least five other genes have been implicated in ARCI [Lefèvre et al., 2003; Jobard et al., 2002; Lefèvre et al., 2004; Lefèvre et al., 2006]. ALOX12B and ALOXE3, both located at chromosome 17p13.1, encode for arachidonate 12(R)-lipoxygenase and arachidonate lipoxygenase-3, respectively The protein product of CYP4F22, a cytochrome P450 protein, along with arachindonate 12(R)-lipoxygenase and arachidonate lipoxgenase-3 are part of the lipid metabolism pathway involved in the formation of ω-hydroxyceramides from arachidonic acid [Brash et al., 2007]. Mutations ALOX12B, ALOXE3 and CYP4F22 have been reported associated with NBCIE [Jobard et al., 2002;Lefèvre et al., 2006]. ABCA12 on chromosome 2q34 encodes a keratinocyte ATP-binding transporter involved in lipid trafficking [Lefèvre et al., 2003]. Truncating ABCA12 mutations tend to cause harlequin ichthyosis, while missense mutations in ABCA12 are associated with with LI or NBCIE [Lefèvre et al., 2003; Kelsell et al., 2005; Akiyama et al., 2005]. Germline mutations in ichthyin, a gene encoding a protein of unknown function have also been reported to be associated with ARCI [Lefèvre et al., 2004]. ARCI patients with mutations in ichthyin show specific ultrastructural abnormalities of the epidermis characterized by abnormal lamellar bodies and perinuclear elongated membranes in stratum granulosum classified as ARCI EM type III [Dahlqvist et al., 2007]. Additional loci for ARCI have been suggested to reside at chromosome bands 3p21, 19p12-q21, 19p13.1-p13.2 and 12p11.2-q13 [Virolainen et al., 2000; Fischer et al., 2000; Mizrachi-Koren et al., 2005].
TGM1 MUTATION ANALYSES
Novel Mutations
We report 23 novel TGM1mutations identified among 25 ARCI patients including: 15 (65%) missense (V209F, R225P, E285K, F293V, I304F, R323W, W342R, V359M, L366P, H405N, M421V, F435V, A560G, R689C, R689H), four (17%) putative splice-site (c.1-1G>C, c.984+1G>A, c.1159+1G>T, c.2226-2A>G), two (9%) frameshift (c.802delG, c.1223_1227delACACA), and two (9%) nonsense (R54X, Q124X). We found that TGase-1 residues affected by novel missense mutations were conserved among species including Pan troglodytes (chimpanzee), Bos Taurus (cow), Mus musculus (mouse), and Rattus norvegicus (rat) [Altschul et al., 1997]. Missense mutations were the most common mutation type, affecting 76% (19/25) of patients. Seventy-three percent (11/15) of novel missense mutations were within the catalytic core domain. Of the other four missense mutations, two occurred each in the β sandwich and β-barrel 2 protein domain, respectively.
Exon six, which codes for part of the catalytic core domain, was the exon mutated most often accounting for 15 % (7/47) of the mutated alleles. The most common mutation detected was c.877-2A>G, which affected 15% (7/47) of mutated alleles and 28% (7/25) of patients. Patients with the c.877-2A>G mutation were all compound heterozygous; 71% (5/7) possessed another missense mutation, and 29% (2/7) had another putative splice-site mutation. R323W, the second most commonly detected mutation, was present in 8.5% (4/47) of mutated alleles and 12% (3/25) of patients. Five mutations (R54X, R225P, R323W, R689C, R689H) occurred at four different arginine codons that contained CpG dinucleotides. None of the 23 novel mutations reported here were observed in at least 100 CEPH DNA samples analyzed in our laboratory.
Review of TGM1 Mutations Reported in the Literature
Including this study, a total of 115 different TGM1 mutations has been reported in 234 individuals with ARCI from diverse racial/and ethnic backgrounds (Caucasion Americans, Norwegians, Swedish, Finnish, German, Swiss, French, Italian, Dutch, Portuguese, Hispanics, Iranian, Tunisian, Moroccan, Egyptian, Afghani, Hungarian, African-Americans, Korean, Japanese and South African) (Supp. Table S1). Seventy-one (62%) mutations were missense; twenty (17%) nonsense; nine (8%) deletion; eight (7%) splice-site and seven (6%) insertion (Figure 1). Fourty-four of the mutations are predicted to lead to truncated proteins [Huber et al., 1997; Shevchenko et al., 2000]. Nine of the twenty nonsense mutations (45%) have been found within the first five translated exons. Only three nonsense mutations occurred within exons 7–10, while eight occurred within exons 11–15. All translated exons (2–15) displayed at least one mutation. Overall, 85% (98/115) of the mutations were located in the first two-thirds of TGM1. The most 5′ mutation (c.1-1G>C) occurred in intron 1 and the most 3′ mutation (Q774X) occurred in exon 15. Exon three had the most identified mutations, accounting for 14% (16/115) of all mutations, followed by exon eight with 12% (14/115), exon seven with 11% (13/115), and exons four, five and six, each with 10% (11/115) of all mutations (Figure 2). Recently, we reported that 28% of patients with ARCI had TGM1 mutations in exon 3 and that the R142H mutation was the most frequent mutation in exon 3 and second most common in the cohort overall [Farasat et al., 2008].
Figure 1. TGM1 mutations associated with ARCI.

A, TGM1 mutations reported in this study and the literature. B, TGM1 gene structure. C, TGAse-1 protein domains. Mutations reported in the present study (novel) are in red font and mutations in the literature are in blue.
1Mutations found in patients diagnosed with ichthyosis with sparing of the lims/bathing suit ichthyosis.
2Mutations found in patients diagnosed with self-healing collodion baby.
*indicates novel missense mutations.
Figure 2. Distribution of all arginine and non-arginine TGM1 mutations reported in the literature including this report.

A, Structure of TGM1 with exons (black boxes) drawn to scale and the distribution of all reported TGM1 mutations. B, TGase-1 protein domains and the distribution of all reported TGM1 mutations.
TGase-1 domain shown are based on the amino acid: Anchoring domain (1–92), β-sandwich domain (94–246), catalytic core domain (247–572), β-barrel 1 (573–688), β-barrel 2 (689–817). Triangles indicate active site residues C377, H436, and D459. “M” indicates myristolation motif of amino acids 47–54 (CCGCCSCR), and “P” indicates phosphorylation site S82. “X” indicates cleavage sites between amino acids S92 and R93 and N572 and R573. 5′, 124bp, and 3′, 197bp, untranslated regions are shown in grey.
The most commonly reported TGM1 mutation has been the c.877-2A>G splice-site mutation. It has been identified in ARCI patients from a variety of ethnic and racial backgrounds, including Caucasian Americans, African-Americans, Germans, Norwegians, and Egyptians [Petit et al., 1997; Huber et al., 1997; Hennies et al., 1998a; Hennies et al., 1998b; Pigg et al., 1998; Pigg et al., 2000; Shevchenko et al., 2000; Esposito et al., 2001; Shawky et al., 2004; Oji et al., 2006; Farasat et al., 2008;]. Including this study, 34 % (147/435) of all TGM1 mutated alleles reported to date carry the c.877-2A>G mutation. Recently, using data from the National Ichthyosis and Related Diseases Registry in the US, we showed that the c.877-2A>G was the most common TGM1 mutation among North Americans, accounting for a 28% mutation allele frequency [Farasat et al. 2008]. That is very similar to the worldwide frequency in this study (34%). It had been shown that this splice-site mutation is common among North American and Norwegian patients due to a founder effect [Pigg et al., 1998; Shevchenko et al., 2000]. It has been estimated that c.877-2A>G was introduced in the Norwegian population around 1000–1100 AD [Shevchenko et al., 2000]. It was also hypothesized that this mutation originated in Westphalia, a region in Northern Germany, and that the immigration of German families introduced this mutation into the Finnish and North American populations, respectively [Shevchenko et al., 2000].
Reverse transcription polymerase chain reaction (RT-PCR) analysis of mRNA extracted from purified blood cells of a patient homozygous for the mutation c.877-2A>G showed that the splice-site mutation leads to two different splice variants, both of which result in a premature stop codon [Shevchenko et al., 2000]. One variant results in an insertion of a G before nucleotide c.877T [Pigg et al., 1998], while the other causes an insertion of intron 5 in between exons 5 and 6 leading to a frameshift [Huber et al., 1997]. Keratinocytes homozygous for the c.877-2A>G mutation have membrane TGAse-1 enzyme levels 6.3–6.5% of control [Huber et al., 1997] (Supp. Table S2). Previously, RT-PCR analysis of mRNA from two LI patients showed that 10/14 of the cDNA clones with the splice-site mutation c.2088+1G>T lacked fifty-four nucleotides (c.2035–2088) [Huber et al., 1997]. This mutation seems to cause a misread during splicing that identifies c.2035–36 as the new GT splice donor site [Huber et al., 1997]. The effect on the translated protein is the deletion of amino acids 679–696. Keratinocytes compound heterozygous for c.2088+1G>T and S358R the mutations have TGase-1 enzyme activity levels 2.2–4.0% of control [Huber et al., 1997] (Supp. Table S2). Including this work, five additional putative splice-site mutations (c.1-1G>C, c.984+1G>A, c.1159+1G>T, c.1645+1G>A, c.2226-2A>G) have been reported but are yet to be fully characterized [Farasat et al., 2008]. We did not find these nucleotide changes upon our examination of 137 CEPH DNA samples supporting that they are disease causing mutations.
CpG Dinucleotides
Of the 817 amino acids in TGase-1, 54 (6.6%) are arginines of which 49 (91%) of their codons begin with a CpG dinucleotide. Mutations have been found in 37% (20/54) of arginine codons in TGase-1, and all of these mutated arginine codons begin with a CpG dinucleotide. Thirty-one percent (36/115) of all TGM1 mutations and 41% (28/71) of missense mutations occurred in arginine residues in TGase-1 (Figure 2). Forty-nine percent (35/71) of missense mutations were within CpG dinucleotides, and 74% (26/35) of these mutations were C> T or G>A transitions. 5′ methylation and demethylation of cytosines that are part of CpG dinucleotides are often associated with the transcriptional inactivation and activation of genes [Robertson and Wolffe, 2000]. Arginine is the most frequently mutated amino acid accounting for 15% of all mutations in genetic diseases [Vitkup et al. 2003]. Four of the six codons that encode arginine contain CpG dinculeotides. The high frequency of mutated arginine codons in TGM1 may be due to methyl-induced deamination of CpG dinucleotides [Coulondre et al., 1978; Cooper and Youssoufian, 1988; Magewu and Jones, 1994; Andrews et al., 1996].
Polymorphisms
The c.726G>A variant was originally reported in a patient with sporadic LI whose keratinocytes did not show a significant decrease in membrane TGase-1 activity (<75%), when compared with control keratinocytes [Huber et al.1995a; Huber et al., 1995b]. c.726G>A changes codon 242 from GAG to GAA, both encoding glutamic acid. c.726G>A is a polymorphism [Huber et al.1995a] since we also found this transition in 4 of 130 CEPH DNA samples.
The G382R (c.1144G>A) change was first reported as a GGA>AGA (4294G>A) transition in codon 382 [Hennies et al., 1998b]. However, the codon G382 published in GenBank (NM_000359.2) is GGC and a G>A transition from that sequence cannot produce an arginine codon. Our sequence analysis of a panel of 116 CEPH sequences, showed an A at nucleotide c.1146 in 46 cases. Therefore, the patient in whom the G382R mutation was originally described must have had the C>A nucleotide change in the third base of codon 382 (GGC>GGA), and this change represents a common SNP.
The c.1552G>A (V518M) variant was first reported as a disease causing mutation [Hennies et al., 1998b]. In that original report, a second TGM1 mutation could not be found in one family with the V518M variant, while another family was homozygous for the variant, and none of 100 German controls’ DNA had this variant [Hennies et al., 1998b]. However, in another report 7 of 100 Italian controls had the c.1552 G>A transition. Furthermore, an unaffected father who possessed the c.1552G>A variant and the mutation E520G did not pass the c.1552G>A variant to either of his two affected children who also received the E520G mutation from their mother [Esposito et al., 2001]. In addition, we identified this variant in 2 of 88 CEPH DNA sequences, supporting that c.1552G>A is a SNP.
BIOLOGICAL RELEVANCE
Transglutaminase-1 has been associated with the development of the cornified cell envelope (CCE) [Rice and Green, 1978; Thacher and Rice, 1985], and increased TGase-1 activity coincides with the terminal differentiation of keratinocytes [Steinert et al., 1996a]. CCE is a physical and water impermeable barrier that replaces the plasma membrane in mature skin cells [Candi et al., 2005]. Ultrastructural investigations revealed that the CCE was missing in patients with LI, and keratinocytes of ARCI patients have decreased TGase-1 enzyme activity levels [Kanerva et al., 1983; Hohl et al., 1998]. Human epidermal scales and nails of ARCI patients with TGM1 mutations have shown a structurally perturbed or attenuated CCE [Rice et al., 2005]. ARCI with mutation in TGM1 have defects in skin-barrier function, thus TGase-1 seems to be essential for normal epidermal barrier function [Huber et al., 1995a; Elias et al., 2002]. The inner layer of the human CCE contains, loricrin, involucrin and small proline rich proteins cross-linked together by TGase-1 [Steinert and Marekov, 1997; Robinson, 1997; Rice and Green 1979; Nemes et al., 1999a]. TGase-1, like other transglutaminases, is thought to create isodipeptide Nε-(γ-glutamyl) lysine cross-links [Lorand and Conrad, 1984]. TGase-1 has also been shown to form ester bonds between involucrin and an analog of ω-hydroxyceramides in vitro [Nemes et al., 1999b]. The ω-hydroxyceramides are attached to the outer surface of the CCE, probably by involucrin and other proteins, and form a water retaining layer known as the “lipid envelope.” TGM1 has been reported to be down regulated by the transcription factor HOXA7, and evidence exists for non-sense mediated mRNA decay (NMD) to regulate TGM1 post-transcriptionally [Huber et al., 1995a; Huber et al., 1997; La Celle and Polakowska, 2001; Maquat, 2004]. TGM1 promoter is targeted by several transcription factors such as p63, grainheadlike 3. TGase-1 membrane-cytosolic partitioning favors the membrane upon myristation and palmitoylation [Steinert et al., 1996b].
Furthermore, three uncommon cis peptide bonds are thought to be critical to the enzymatic reaction mediated by TGase-1 [Boeshans et al., 2007]. Calcium ions ubiquitously lead to the activation of transglutaminases, including TGase-1 [Green and Rice, 1979; Lorand and Conrad, 1984; Gibson et al., 1996]. TGase-1 is thought to be regulated by three Ca2+binding sites that promote cis to trans isomerization of those peptide bonds leading to increased enzyme activity [Boeshans et al., 2007]. The catalytic triad of TGase-1, C377, H436 and D459, is higly conserved [Kim et al., 1991;Huber et al., 1997; Altschul et al., 1997]. Further regulation from an inactive to an active form is achieved by proteolytic cleavage after amino acids S92 and N572, possibly by cathepsin D [Kim et al., 1995; Steinert et al., 1996a; Egberts et al., 2004]. Serine residues S24, S85, S92, and especially S82 have been identified as locations for phosphorylation in human TGase 1 [Rice et al., 1996]. Nitric oxide has also shown to inhibit TGase-1 activity and CCE formation in human epidermal keratinocytes [Rossi et al., 2000].
Previously, using 32P labeled TGM1 probes, mRNA was shown to be absent from cultured keratinocytes taken from a patient homozygous for the nonsense mutation R127X [Huber et al., 1997]. Northern blotting of tissue taken from a skin biopsy and cultured keratinocytes from a patient homozygous for the truncating mutation c.1297delT showed absent TGase-1 mRNA in vitro [Huber et al., 1995a]. NMD, which decreases the amount of cellular mRNA with premature termination codons (PTCs), could be the mechanism that explains these observations [Maquat, 2004]. In addition, other mutations in TGM1 that also lead to PTCs have demonstrated decreased enzyme expression in the skin [Akiyama et al., 2001a; Akiyama et al., 2003]. Epidermis from a patient with the TGM1 mutations c.2114delA and R389H showed reduced TGase-1 immunoreactivity, while TGase-2 and TGase-3 showed normal expression [Akiyama et al., 2001a]. Similarly, immunofluorescence labeling detected TGase-3, but not TGase-1, in the granular layer from a patient homozygous for c.374delA [Akiyama et al., 2003].
Keratinocytes from affected patients have been analyzed for TGase-1 enzyme activity based on in vitro 3H-labeled putrescine incorporation into dimethylcaseine [Yuspa et al., 1980; Lichti et al., 1985]. The results of the measured TGase-1 enzyme activities identified in the literature are summarized in Supp. Table S2. Cultured keratinocytes from unaffected TGM1 mutation heterozygotes (R307W//WT and D102V/WT had TGase-1 membrane activities 48–52% those of controls. In comparison, cultured keratinocytes from compound heterozygous or homozygus patients (n=14 patients) had TGase-1 membrane activities 0.05–6% those of controls. TGase-1 membrane activities from cultured keratinocytes of patients with exclusively truncating or missense mutations were similar (Supp. Table S2). Keratinocytes from patients homozygous for TGM1 deletion (c.1279delT), splice-site (c.8772G>A), or nonsense (R127X) mutations have TGase-1 membrane activity levels ranging from 0.3–3% those of controls. Similarly, those with only missense mutations (n=7 patients) had membrane activity levels of TGase-1 that ranged from 0.2–6% those of controls. Given the small number of samples tested, it cannot be determined whether missense mutations in the β-sandwich domain or the catalytic core are more detrimental to TGase-1 membrane activity.
Another assay to determine cytosolic and membrane enzyme activities uses cDNA cloned into a pVL1392 baculovirus vector and then transfected into Sf9 insect cells, where the protein is expressed [Candi et al., 1998; Raghunath etl al., 2003]. Half (4/8) of the missense mutations (R142C, R142H, R143H, G278R) led to no detectable membrane activity, but two missence mutations (S42Y and R315L) had protein activities at least twice those of controls [Candi et al., 1998]. Sf9 cells expressing the TGase-1 protein with the truncating mutation c.1294delT had virtually no enzyme activity (Supp. Table S3) [Huber et al., 1995a; Huber et al., 1997; Candi et al., 1998]. In the absence of a functional TGase-1 protein, increased TGase-3 activity has been shown, but not without the development of scaling [Oji et al., 2006].
We performed a search for amino-acid sequence similar to transglutaminase-1 using the program BlastP 2.218 in the RefSeq protein database on the NCBI website [Altschul et al., 1997]. TGase-1 shared 52–60% amino acid sequence homology with the other known human transglutaminase enzymes, including factor XIIIa and transglutaminases-2, 3, 4, 5, 6, and 7. Human coagulation factor XIIIa shows the highest homology (60%) and identity (43%) to TGase-1.
Molecular Modeling
Since the crystal structure of TGase-1 has not been determined, other transglutaminases, of known structure such as human coagulation factor XIIIa [Huber et al., 1997; Altschul et al., 1997; Candi et al., 1998; Yang et al., 2001; Kon et al., 2003 ; Raghunath et al., 2003], TGase-3 [Oji et al., 2006] or combination of TGases 2, 3 and 5 have also been used to model TGase-1 [Boeshans et al., 2007]. To date 23 TGM1 mutations have been studied with molecular modeling: 6 mutations have been studied using TGase-3 as a template structure [Oji et al., 2006], while 17 mutations have been studied using human factor XIIIa [Huber et al., 1997; Candi et al., 1998; Yang et al., 2001; Kon et al., 2003; Raghunath et al., 2003]. Candi and co-workers proposed that the R315L mutation interferes with N-terminal proteolytic activation, based on that the S42Y mutation disrupts an inhibitory function of the 10kB anchoring region [Candi et al., 1998].
We used the SegMod algorithm [Levitt, 1992] implemented in the program GeneMine to build a model of TGase-1 (sequence: GI 4507475), based on its homology to factor XIII (template structure: 1ex0.pdb) [Fox et al. 1999]. Residues 106 to 788 of TGase-1 were modeled. Within this range, identity between the template and model sequences is 44 %. Conservation is highest within the catalytic-core domain (53 %), as compared to 37 % in the β-sandwich domain, 32 % in β-barrel 1, and 35 % in β-barrel 2. The TGase-1 model includes eighteen of the twenty arginines observed to be mutated in patients with ARCI, and eight of these eighteen (44%) are in the catalytic core (Figure 3).
Figure 3. Wall-eyed stereo view of TGase-1 model.
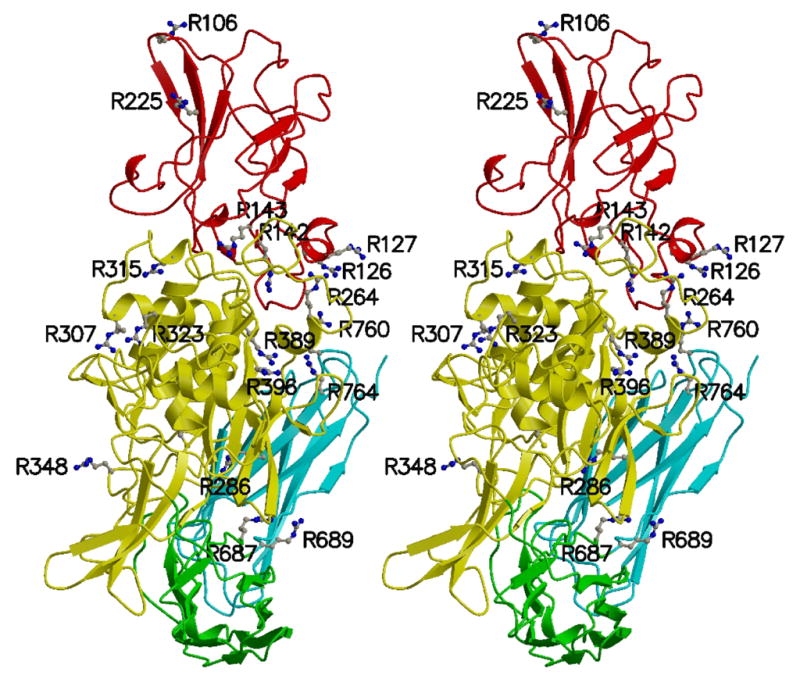
The 18 arginine residues, mutations of which have been associated with ARCI, are shown as ball-and-stick. All arginine residues shown in the two beta-barrel domains as well as R142 and R143 of the beta-sandwich domain, are located at domain boundaries. To indicate the active site, C377 is also shown as ball-and-stick (to the left of R286). The protein main chain is colored by domain: beta-sandwich (red), catalytic core (yellow), beta-barrel 1 (green), and beta-barrel 2 (cyan). Figures 3, 4, and 5 were prepared with the programs MOLSCRIPT [Kraulis, 1991] and Raster3D [Merritt and Bacon, 1997].
Figure 4a shows clusters of charged residues (arginines, aspartic acids, glutamatic acids) at the domain interfaces. Two (R142 and R143) of the six mutated arginines in the β-sandwich domain, are located at domain interfaces in the modeled structure. Note that R143 and D254 form a salt bridge, and Y134 forms a hydrogen bond with R142. These two interactions are present in the template crystal structure of factor XIIIa. The amino-acid composition of the interface between the β-sandwich and catalytic core domains is very similar in factor XIIIa and TGase-1 (Figure 4b). In addition, mutations at residues R142, R143, and Y134 have been identified in patients with ARCI [Huber et al., 1995; Laiho et al 1997; Farasat et al. 2008], suggesting that weakening interactions at this interface can have serious consequences.
Figure 4. Arginine residues located at domain interfaces in TGase-1 model.

a) The arginines are shown as space-filling, and nearby acidic residues are shown as sticks. The protein main chain is colored by domain: beta-sandwich (orange), catalytic core (yellow), beta-barrel 1 (green), and beta-barrel 2 (cyan). b) Interface between the beta-sandwich and catalytic-core domains of TGase-1, colored by the sequence identity between the model and the template structure (red where identical, blue where different). The amino-acid composition of this interface is highly conserved between model and template. Six residues found mutated in patients with ARCI are indicated with spheres.
All of the mutated arginines in the two β-barrel domains (R687, R689, R760, and R764) appear at domain interfaces (Figure 4a). R764 and R760 in β-barrel 2 domain are proximal to E271 in the catalytic core domain and to D132, E133 and E135 in the β-sandwich domain. Displacement of these three acidic residues could weaken the nearby interaction of Y134 with R142. Elsewhere, R687 and R689 reside at the interface of the β-barrel 1, β-barrel 2, and catalytic-core domains. We report two novel mutations (R689H, R689C) at residue R689. R689C changes a positively charged arginine to a neutral cysteine. The 687H mutation was previously reported [Oji et al., 2006]. We also identify a novel mutation (E285K) that changes this acidic residue to a basic residue. In our model, E285 resides across the domain boundary from R687 and R689.
The catalytic triad of TGase-1, along with eight neighboring residues mutated in ARCI patients, is depicted in Figure 5. Four of these mutations are novel (R323W, W342R, H405N, and F435V). R323W and W342R both alter the total charge near the active site, as does R323Q reported previously [Huber et al., 1995a]. Interestingly, mutations F401V and F435V are both found in ARCI patients [Tok et al. 1999], strongly suggesting that the interaction of these two phenylalanine residues is important for the positioning of H436 relative to the other catalytic resides, C377 and D459. It is worth noting that there is a cis peptide bond between G473 and P474 and that the mutation G473S has been reported in an ARCI patient [Huber et al., 1997]. G473S would be expected to introduce strain in the TGase-1 structure near the active site, since the main-chain atoms of glycine are more flexible than those of serine.
Figure 5. Wall-eyed stereo view of the catalytic triad and neighboring residues in TGase-1 model.

Side chains are shown as ball-and-stick, with the catalytic residues (Cys 377, His 436, and Asp 459) denoted with darker carbon atoms. Residues associated with missense mutations observed in ARCI patients and the catalytic residues are labeled. For clarity, the side chains of Trp 378, Phe 380, and Ala 381 are not shown. The protein backbone (alpha-carbon trace) is colored according to the sequence identity between the TGase-1 model and coagulation factor XIII template (red where identical, blue where different).
Inspection of the TGase-1 model also provides insight into other novel mutations reported here. The replacement of valine with a larger phenylalanine side chain (V209F) presumably disrupts the hydrophobic packing with nearby residues in the β-sandwich domain: F147, L165, L207, I183, W193, and A195. The mutation A560G may also destabilize hydrophobic interactions with neighboring residues, e.g., F495 and V485. A560 resides in a helix located in a highly conserved region (relative to factor XIIIa) near the putative calcium binding sites [Boeshans et al., 2007].
Other novel mutations were observed at the protein surface or at domain interfaces. R225 resides at the surface of the β-sandwich domain adjacent to both E166 and E232. The R225P mutation may cause an unfavorable imbalance of charge at the protein surface. R225 has been reported previously mutated (R225H) [Hennies et al., 1998b]. The mutation M421V occurs on the surface of β-barrel 1 such that the methionine sulfur atom is accessible to solvent and other proteins. The mutation V359M is located at the interface between the catalytic-core and β-barrel 2 domains. L366P is nearby, also on the surface of the catalytic-core domain.
Mouse Models
Knockout mice have been constructed by deleting exons 1–3 of the TGM1 gene by homologous recombination in R1 embryonic stem (ES) cells [Matsuki et al., 1998]. There was no evidence of embryonic lethality in F2 mice, but neonatal TGase-1 −/− mice died within 4–5 hours of birth [Matsuki et al., 1998]. TGase-1 −/− mice were often born with a rigid translucent membrane and neonates’ skin was wrinkled, taught, shiny, and erythematous [Matsuki et al., 1998]. Ultrastructurally, TGase-1 −/− skin lacked a cornified cell envelope and displayed a somewhat swollen stratum corneum that became more compact over time [Matsuki et al., 1998]. In addition, the TGase-1 −/− mice did not feed, weighed less, and were smaller and less active than wild-type littermates [Matsuki et al., 1998]. Their tails and extremities became waxy and shriveled due to dehydration [Matsuki et al., 1998]. The transepidermal water loss (TEWL) of TGase-1 −/− mouse skin was increased by a factor of 100, when compared to normal mouse skin [Matsuki et al., 1998].
Using the same TGase-1 knockout mouse developed by Matsuki et al., Kuratomo and coworkers grafted dorsal skin of TGase-1 +/− and TGase-1 −/− mice, to athymic “nude” mice [Kuramoto et al., 2002]. The TGase-1 +/− grafted skin grew hair. In contrast, TGase-1 −/−grafted skin showed erythema, thick scales and immature hair follicles. Histologically, it showed acanthosis, hyperkeratosis, lacked a cornified envelope and displayed electron dense granules close to the plasma membrane [Kuramoto et al., 2002]. The grafted TGase-1 −/− mouse skin with scales displayed control level TEWL values, while grafted skin of TGase-1 −/− mice without scales showed high TEWL values as seen in TGase-1 −/−neonates [Kuramoto et al., 2002].
Northern and Western blotting have been used to show that TGM1 was expressed in the epithelial cells of the liver, lungs and kidneys of mice [Hiiragi et al., 1999]. It has been identified that TGase-1, phosphorylated at tyrosine residues, was associated with β-catenin, radixin, and N-Cadherin in the junctional fraction of mouse epithelial liver cells in vitro. In the presence of CaCl2, but not EDTA, junctional cross-linking in adherens junctions was increased in incubated mouse liver fractions [Hiiragi et al., 1999]. The group suggested that TGase-1 cross-linking might be important for epithelial cell structure at adherens junctions.
Yamada et al. constructed transgenic mice with a lac Z region controlled by the 5′ 2.5kb upstream region of tgm1 [Yamada et al., 1997]. Transgene positive offspring were subsequently mated. Strong β-gal staining was observed in the skin, tongue, esophagus, stomach and vaginal tissue from adult transgenic mice and not in similar tissues in adult non-transgenic mice. Some β-gal staining was also seen in tissue from the oral mucosa of adult transgenic mice [Yamada et al., 1997]. After 18 days post-conception through the neonatal period, β-gal staining was identified in the upper spinous and lower granular layers of mouse epidermis. The authors suggested that this was evidence for a promoter function in the 2.5kb region 5′ of TGM1 [Yamada et al., 1997].
CLINICAL AND DIAGNOSTIC RELEVANCE
Although originally it was thought that LI and NBCIE were distinct disease entities, it has since been shown that both conditions can be caused by mutations in TGM1, including the same compound heterozygous mutations [Williams and Elias, 1985; Laiho et al., 1997]. Currently, it is generally recognized that there is a variable phenotypic expression among and within ARCI families and patients. Neonates with ARCI are often born with a collodion membrane [Shwayder, 1999]. Those with LI often show ectropion and eclabium, which are less common in neonates with NBCIE [Williams and Elias, 1985; Shwayder, 1999]. Neonates with ARCI have a high risk of systemic infection, dehydration, respiratory problems and temperature dysregulation [Sandberg, 1981; Shwayder, 1999].
Patients with TGM1 mutations can also present with uncommon clinical variants of ARCI, including self-healing collodion baby (SHCB) and ichthyosis with sparing of the limbs (also designated by some investigators as bathing suit ichthyosis). SHCB is inherited in an autosomally recessive manner and patients typically exhibit a collodion membrane at birth, but after desquamination of the collodion membrane, many show considerable improvement leading to healthy-looking skin in as little as three months [Frenk and de Techtermann, 1992]. Three Swiss SHCB patients, each born in a collodion membrane and one with ectropion, all developed completely normal-looking skin after variable periods of time [Frenk and de Techtermann, 1992]. Some patients who developed only minor scaling, including two Albanian siblings both compound heterozygous for the D490G and G278R TGM1 mutations, have also been described as SHCBs [Raghunath et al., 2003; Harting et al., 2008]. The TGM1 mutation D490G has only been associated with (SHCB) [Raghunath et al., 2003]. After resolution of the collodion membrane, one Albanian infant had complete improvement of scaling on the extremities and face by four weeks of age, and the other presented with only mild scaling of the trunk at four years of age [Raghunath et al., 2003]. In addition, patient ARCI1 in this report is compound heterozygous (G291D/V359M) and has been diagnosed with SHCB (Table 1). Recently, a white and a Hispanic patient, both SHCBs, have been reported to have mutations in the ALOX12B gene [Harting et al., 2008].
Table 1.
Novel TGM1 Germline Mutations Associated with ARCI
| Number | Nucleotide changea NM_000359.2 | Amino acid changeb NP_000350 | Exon/intron | Mutation type | References | Allelesc |
|---|---|---|---|---|---|---|
| 1 | c.1-1G>C | ND | Intron 1 | Splice Site | This report | Compound heterozygous (c.1-1G>C/c.877-2A>G) |
| 2 | c.160C>T | p.Arg54X | Exon 2 | Nonsense | This report | Homozygous |
| 3 | c.370C>T | p.Gln124X | Exon 3 | Nonsense | This report | Compound heterozygous (Q124X/R126C) |
| 4 | c.625G>T | p.Val209Phe | Exon 4 | Missense | This report | Compound heterozygous (R143C/V209F) |
| 5 | c.674G>C | p.Arg225Pro | Exon 4 | Missense | This report | Compound heterozygous (R225P/c.877-2A>G) |
| 6 | c.802delG | p.Val268PhefsX63 | Exon 5 | Frameshift | This report | Compound heterozygous (c.802delG/R323Q) |
| 7 | c.853G>A | p.Glu285Lys | Exon 5 | Missense | This report | Compound heterozygous (E285K/A560G) |
| 8 | c.877T>G | p.Phe293Val | Exon 6 | Missense | This report | Compound heterozygous (c.877-2A>G/F293V) |
| 9 | c.910A>T | p.Ile304Phe | Exon 6 | Missense | This report | Homozygous |
| 10 | c.967C>T | p.Arg323Trp | Exon 6 | Missense | This report | Compound heterozygous (c.877-2A>G/R323W) Heterozygous |
| 11 | c.984+1G>A | ND | Intron 6 | Splice Site | This report | Compound heterozygous with (c.882_888delCCACGGG/c.984+1G>A/E520G) |
| 12 | c.1024T>C | p.Trp342Arg | Exon 7 | Missense | This report | Compound heterozygous (c.877-2A>G/W342R) |
| 13 | c.1075G>A | p.Val359Met | Exon 7 | Missense | This report | Compound heterozygous (V359M/G291D) |
| 14 | c.1097T>C | p.Leu366Pro | Exon 7 | Missense | This report | Compound heterozygous (G291D/L366P) |
| 15 | c.1159+1G>T | ND | Intron 7 | Splice Site | This report | Homozygous |
| 16 | c.1213C>A | p.His405Asn | Exon 8 | Missense | This report | Compound heterozygous (R389P/H405N) |
| 17 | c.1223_1227delACACA | p.Asp408ValfsX21 | Exon 8 | Frameshift | This report | Homozygous Compound heterozygous (R106X/c.1223_1227delACACA) |
| 18 | c.1261A>G | p.Met421Val | Exon 8 | Missense | This report | Compound heterozygous (c.877-2A>G/M421V) |
| 19 | c.1303T>G | p.Phe435Val | Exon 9 | Missense | This report | Homozygous |
| 20 | c.1679C>G | p.Ala560Gly | Exon 12 | Missense | This report | Compound heterozygous (E285K/A560G) |
| 21 | c.2065C>T | p.Arg689Cys | Exon 13 | Missense | This report | Heterozygous |
| 22 | c.2066G>A | p.Arg689His | Exon 13 | Missense | This report | Compound heterozygous (R348X/R689H) |
| 23 | c.2226-2A>G | ND | Intron 14 | Splice Site | This report | Compound heterozygous (c.877-2A>G/c.2226-2A>G) |
The nucleotide numbering is based on the published GenBank sequence NM_000359.2, and nucleotide +1 is the adenine of the ATG initiation codon.
The amino acid sequence is taken from the published GenBank sequence NP_000350 and begins with the translation initiation codon as 1.
TGM1 alleles of affected patients. ND, not determined
In 1994 Schulz first described South African patients with large darkly colored scales on the trunk sparing the face and extremities that he called bathing suit ichthyosis (BSI)[Schulz 1994]. In 1997, Petit and co-workers described a child homozygous for TGM1 mutation V382M and reduced TGase-1 enzyme activity who presented with large brown scales distributed over his thorax and abdomen that spared the limbs [Petit et al. 1997]. This clinical variant was referred as ichthyosis with sparing of the limbs. In essence ichthyosis with sparing of the limbs and BSI are different names to describe the same clinical variant. Ichthyosis with sparing of the limbs/BSI has also been reported in families from Germany, France, Turkey, the Netherlands and Morocco [Jacyk, 2005; Oji et al., 2006; Arita et al., 2007; Petit et al. 1997]. Fifteen different mutations in TGM1 have been reported in nineteen patients [Oji et al., 2006; Arita et al., 2007 Petit et al. 1997]. Turk, Morrocan, German and South African patients were reported homozygous for Y276N, R315H, R687H, and R315L, and V382M [Arita et al., 2007; Oji et al., 2006; Petit et al. 1997]. Three German patients had the common splice-site mutation c.877-2A>G, and all three were compound heterozygous each with either R264W, or R307G, or R315C [Oji et al., 2006]. Compound heterozygous patients included those with the mutations: (R264Q/c.1074delC), (R126C/R142H), (R307G/R389P) and (R307G/W263X) [Oji et al., 2006].
Ichthyosis fetalis or harlequin ichthyosis (HI) is the most severe form of ARCI [Sandberg, 1981]. We contend that HI represents a third major subtype of ARCI different from LI and NBCIE based on its dissimilar genetic and clinical presentation [Craig et al., 1970; Kelsell et al., 2005]. Truncating mutations in ABCA12 have been demonstrated in patients with HI from diverse ethnic backgrounds [Akiyama et al 2005, Kelsell et al 2005, Thomas et al 2006], but, to date, no TGM1 mutations have been reported to cause HI. Patients with HI are often born prematurely (34–36 gestational weeks) [Kelsell et al., 2005]. Neonates consistently present with hyperkeratonic thick scales that cover nearly the whole body with deep fissures as well as with malformed digits, ears and nose, severe eclabium and ectropion [Craig et al., 1970; Lawlor, 1988; Dale et al., 1990; Dahlstrom et al., 1995]. Hair has been observed to grow between the cracks in the plate-like scales but not elsewhere [Craig et al., 1970; Lawlor, 1988]. Histologically, the thickening of the neonate’s stratum corneum has been observed [Dale et al., 1990; Akiyama et al., 2005]. In addition to the health-risks outlined above, HI neonates are prone to sepsis [Lawlor, 1988; Dahlstrom et al., 1995; Kelsell et al., 2005] and often have difficulty breathing [Craig et al., 1970] and feeding [Craig et al., 1970; Lawlor, 1988; Dahlstrom et al., 1995]. Surviving patients have been shown to develop thinning scales, hair-growth, and erythema [Lawlor, 1988].
Mutation Detection Rate
Identification of a large number of individuals with ARCI without mutations in the TGM1 gene suggested that ARCI is genetically heterogenous [Paramentier et al., 1995] and contribute to the challenge in confirming the clinical diagnosis. Recently, we demonstrated that the TGM1 gene was mutated in 55% of patients with cases of ARCI in North America [Farsat et al., 2008]. The other 45% of patients might possess undetected large, partial or complete deletions of TGM1 or mutations deep within an intron or the promoter region. In addition, ARCI patients without TGM1 mutations may have mutations in any of the five other known ARCI causing genes or at any of the four uncharacterized loci that have been linked to ARCI [Virolainen et al., 2000; Fischer et al., 2000; Jobard et al., 2002; Lefèvre et al., 2003; Lefèvre et al., 2004; Mizrachi-Koren et al., 2005; Lefèvre et al., 2006].
Genotype-Phenotype Correlations
Many studies have attempted to show genotype-phenotype correlations between mutations in the TGM1 gene and clinical and/or epidermal ultrastructural findings. In some previous studies, patients with TGM1 mutations were classified with LI and NBCIE [Laiho et al., 1997; Hennies et al., 1998b; Laiho et al., 1999; Shawky et al., 2004], or with and without plate-like scales [Pigg et al., 1998; Shevchenko et al., 2000]. These clinical case series, which investigated between 9 and 55 patients, did not report statistically significant genotype-phenotype correlations [Laiho et al., 1997; Hennies et al., 1998b; Pigg et al., 1998; Laiho et al., 1999; Shevchenko et al., 2000; Shawky et al., 2004]. However, most of these studies were under powered to detect potential genotype-phenotype correlations.
In a recent study of 104 ARCI patients in the US, we found that patients with TGM1 mutations were more likely to report the presence of a collodion membrane at birth (p=0.006), ectropion (p=0.001), and plate-like scales (p=0.005) than were patients without TGM1 mutations [Farasat et al., 2008]. These results support the findings of a previous study of eighty-three Swedish and Estonian ARCI patients by Ganemo and colleagues that also reported ectropion (p<0.001) and collodion membrane at birth (p<0.001) associated with TGM1 mutations [Ganemo et al., 2003]. In addition, we found that patients with TGM1 truncating mutations were also associated with moderate or severe hypohidrosis (p=0.001) and worst overheating (p=0.0007), compared to patients with only TGM1 missense mutations. [Farasat et al., 2008]. In agreement with the Ganemo study [Ganemo et al., 2003], ARCI patients with TGM1 mutations reported alopecia more often than patients without TGM1 mutations [Farasat et al., 2008]. However, we found a strong correlation between mutations in TGM1 and alopecia (p=0.001) [Farasat et al., 2008]. TGase-1 is expressed in the cortex and medulla cells as well as the outer and inner root sheath cells of normal hair follicles [Yoneda et al., 1998]. The expression of TGase-1 suggests a direct involvement of this enzyme in hair formation [Yoneda et al., 1998]. Therefore, mutations in TGM1 may explain the alopecia present in patients with ARCI [Farasat et al., 2008].
Despite the genetic heterogeneity of ARCI [Lefevre et al., 2003], TGM1 has been the causative gene identified most often with this disease [Farasat et al., 2008], and, consequently, TGM1 should be the first gene to be analyzed to screen for mutations. We recently developed a logistic model that predicted that ARCI patients with a history of collodion membrane at birth, alopecia, and eye problems are, respectively, 4.24, 4.13, and 3.6 times more likely to have mutations in TGM1 than ARCI patients without TGM1 mutations [Farasat et al., 2008]. These results could be used to identify ARCI patients who would be more likely to have mutations in TGM1 upon mutation analysis.
Prenatal Diagnosis
Prenatal diagnosis for pregnancies at an increased risk for ARCI is very useful in families who have a previous child affected with ARCI and in which TGM1 mutations or other ARCI causing mutations were identified in the parents or a sibling [Schorderet et al., 1997; Bichakjian et al., 1998; Pigg et al., 2000]. Since ARCI is genetically heterogenous, a prenatal test not showing mutations in TGM1 does not guarantee a neonate will be unaffected by ARCI. Similarly a prenatal test not showing mutations in CYP4F22, ALOX12B, ALOXE3 or ichthyin does not guarantee a neonate will be unaffected by ARCI. Both disease-causing alleles of an affected family member must be identified before prenatal testing can be performed. Prenatal diagnosis has been reported in patients with ARCI, usually by extracting DNA from a chorionic villus sample (CVS) obtained at either 10 or 13 weeks of gestation [Schorderet et al., 1997; Pigg et al., 2000] or by DNA extraction from fetal cells obtained by amniocentesis performed at approximately 15–18 weeks gestation. TGM1 mutation analysis of DNA from a CVS at 13 weeks of gestation revealed mutations in the TGM1, indicating a fetus at risk for ARCI [Schorderet et al., 1997]. In another report, the haplotyping and subsequent sequencing of TGM1 from two CVSs taken at 10 weeks gestation, did not reveal TGM1 mutations, thus excluding LI in two at-risk fetuses [Pigg et al., 2000]. Amniocentesis at 14 weeks of gestation and the subsequent genotyping and sequencing of TGM1 in DNA extracted from fetal cells have been used to exclude TGM1 associated ARCI [Bichakjian et al., 1998]. Preimplantation genetic diagnosis (PGD) may be available for families in which the disease-causing mutations have been identified.
FUTURE PROSPECTS
Future characterization of TGM1mutations and genotype–phenotype correlations in ARCI may provide valuable insights into the molecular pathogenesis of ARCI. Future clinical studies and laboratory investigations using in vitro systems and animal models may help us to elucidate the sequence of pathogenetic events that lead to the clinical findings and the organ preference of involvement that we observe in ARCI. A crystal structure of TGase-1 would also increase the understanding of the molecular effects associated with ARCI. Characterization of mutations and their outcomes would provide more accurate prenatal genetic counseling for parents of at-risk individuals. A more extensive understanding of the genetic, molecular, and patho-physiological aspects of ARCI would hopefully lead to novel treatments.
Supplementary Material
Acknowledgments
We would like to thank the for, the patients with ichthyosis their families, and the physicians who referred them. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. This research was supported in part by the Intramural Research Program of DCEG, NCI, NIH.
References
- Akiyama M, Takizawa Y, Kokaji T, Shimizu H. Novel mutations of TGM1 in a child with congenital ichthyosiform erythroderma. Br J Dermatol. 2001a;144:401–407. doi: 10.1046/j.1365-2133.2001.04037.x. [DOI] [PubMed] [Google Scholar]
- Akiyama M, Takizawa Y, Suzuki Y, Ishiko A, Matsuo I, Shimizu H. Compound heterozygous TGM1 mutations including a novel missense mutation L204Q in a mild form of lamellar ichthyosis. J Invest Dermatol. 2001b;116:992–995. doi: 10.1046/j.0022-202x.2001.01367.x. [DOI] [PubMed] [Google Scholar]
- Akiyama M, Takizawa Y, Suzuki Y, Shimizu H. A novel homozygous mutation 371delA in TGM1 leads to a classic lamellar ictiosis phenotype. Br J Dermatol. 2003;148:149–153. doi: 10.1046/j.1365-2133.2003.05041.x. [DOI] [PubMed] [Google Scholar]
- Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, Tsuji-Abe Y, Tabata N, Matsuoka K, Sasaki R, Sawamura D, Shimizu H. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115:1777–1784. doi: 10.1172/JCI24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews JD, Mancini DN, Singh SM, Rodenhiser DI. Site and sequence specific DNA methylation in the neurofibromatosis (NF1) gene includes C5839T: the site of the recurrent substitution mutation in exon 31. Hum Mol Genet. 1996;5:503–507. doi: 10.1093/hmg/5.4.503. [DOI] [PubMed] [Google Scholar]
- Arita K, Jacyk WK, Wessagowit V, van Rensburg EJ, Chaplin T, Mein CA, Akiyama M, Shimizu H, Happle R, McGrath JA. The South African “bathing suit ichthyosis” is a form of lamellar ichthyosis caused by a homozygous missense mutation, p.R315L, in transglutaminase 1. J Invest Dermatol. 2007;127:490–493. doi: 10.1038/sj.jid.5700550. [DOI] [PubMed] [Google Scholar]
- Bale SJ, Doyle SZ. The genetics of ichthyosis: a primer for epidemiologists. J Invest Dermatol. 1994;102:49S–50S. doi: 10.1111/1523-1747.ep12388591. [DOI] [PubMed] [Google Scholar]
- Bale SJ, Compton JG, Russell LJ, DiGiovanna JJ. Genetic heterogeneity in lamellar ichthyosis. J Invest Dermatol. 1996;107:140–141. doi: 10.1111/1523-1747.ep12298430. [DOI] [PubMed] [Google Scholar]
- Becker K, Csikós M, Sárdy M, Szalai ZS, Horváth A, Kárpáti S. Identification of two novel nonsense mutations in the transglutaminase 1 gene in a Hungarian patient with congenital ichthyosiform erythroderma. Exp Dermatol. 2003;12:324–329. doi: 10.1034/j.1600-0625.2003.120313.x. [DOI] [PubMed] [Google Scholar]
- Bichakjian CK, Nair RP, Wu WW, Goldberg S, Elder JT. Prenatal exclusion of lamellar ichthyosis based on identification of two new mutations in the transglutaminase 1 gene. J Invest Dermatol. 1998;110:179–182. doi: 10.1046/j.1523-1747.1998.00104.x. [DOI] [PubMed] [Google Scholar]
- Boeshans KM, Mueser TC, Ahvazi B. A three-dimensional model of the human transglutaminase 1: insights into the understanding of lamellar ichthyosis. J Mol Model. 2007;13:233–246. doi: 10.1007/s00894-006-0144-9. [DOI] [PubMed] [Google Scholar]
- Brash AR, Zheyong Y, Boeglin WE, Schneider C. The hepoxilin connection in the epidermis. FEBS J. 2007;274:3494–3502. doi: 10.1111/j.1742-4658.2007.05909.x. [DOI] [PubMed] [Google Scholar]
- Candi E, Melino G, Lahm A, Ceci R, Rossi A, Kim IG, Ciani B, Steinert PM. Transglutaminase 1 mutations in lamellar ichthyosis. Loss of activity due to failure of activation by proteolytic processing. J Biol Chem. 1998;273:13693–13702. doi: 10.1074/jbc.273.22.13693. [DOI] [PubMed] [Google Scholar]
- Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–340. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- Craig JM, Goldsmith LA, Baden HP. An abnormality of keratin in the harlequin fetus. Pediatrics. 1970;46:437–440. [PubMed] [Google Scholar]
- Cserhalmi-Friedman PB, Milstone LM, Christiano AM. Diagnosis of autosomal recessive lamellar ichthyosis with mutations in the TGM1 gene. Br J Dermatol. 2001;144:726–730. doi: 10.1046/j.1365-2133.2001.04126.x. [DOI] [PubMed] [Google Scholar]
- Dahlstrom JE, McDonald T, Maxwell L, Jain S. Harlequin Ichthyosis—a case report. Pathology. 1995;27:289–292. doi: 10.1080/00313029500169143. [DOI] [PubMed] [Google Scholar]
- Dale BA, Holbrook KA, Fleckman P, Kimball JR, Brumbaugh S, Sybert VP. Heterogeneity in harlequin ichthyosis, an inborn error of epidermal keratinization: variable morphology and structural protein expression and a defect in lamellar granules. J Invest Dermatol. 1990;94:6–18. doi: 10.1111/1523-1747.ep12873301. [DOI] [PubMed] [Google Scholar]
- Dahlqvist J, Klar J, Hausser I, Anton-Lamprecht I, Pigg MH, Gedde-Dahl T, Jr, Gånemo A, Vahlquist A, Dahl N. Congenital ichthyosis: mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. J Med Genet. 2007;44:615–20. doi: 10.1136/jmg.2007.050542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egberts F, Heinrich M, Jensen JM, Winoto-Morbach S, Pfeiffer S, Wickel M, Schunck M, Steude J, Saftig P, Proksch E, Schütze S. Cathepsin D is involved in the regulation of transglutaminase 1 and epidermal differentiation. J Cell Sci. 2004;117:2295–2307. doi: 10.1242/jcs.01075. [DOI] [PubMed] [Google Scholar]
- Elias PM, Schmuth M, Uchida Y, Rice RH, Behne M, Crumrine D, Feingold KR, Holleran WM, Pharm D. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol. 2002;11:258–256. doi: 10.1034/j.1600-0625.2001.110308.x. [DOI] [PubMed] [Google Scholar]
- Esposito G, Auricchio L, Rescigno G, Paparo F, Rinaldi M, Salvatore F. Transglutaminase 1 gene mutations in Italian patients with autosomal recessive lamellar ichthyosis. J Invest Dermatol. 2001;116:809–812. doi: 10.1046/j.1523-1747.2001.01314.x. [DOI] [PubMed] [Google Scholar]
- Esposito G, Tadini G, Paparo F, Viola A, Ieno L, Pennacchia W, Messina F, Giordano L, Piccirillo A, Auricchio L. Transglutaminase 1 deficiency and corneocyte collapse: an indication for targeted molecular screening in autosomal recessive congenital ichthyosis. Br J Dermatol. 2007;157:799–846. doi: 10.1111/j.1365-2133.2007.08070.x. [DOI] [PubMed] [Google Scholar]
- Farasat S, Wei MH, Toure O, Herman ML, Liewehr DJ, Steinberg SM, Bale SJ, Fleckman P, Toro JR. Transglutaminase-1 genotype phenotype investigations of 104 North American patients with autosomal recessive congenital ichthyosis. Submitted J Med Genet. 2008 doi: 10.1136/jmg.2008.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Faure A, Bouadjar B, Blanchet-Bardon C, Karaduman A, Thomas I, Emre, Cure S, Özgüc M, Weissenbach J, Prud’homme Two new loci for autosomal recessive ichthyosis on chromosomes 3p21 and 19p12-q12 and evidence for further genetic heterogeneity. Am J Hum Genet. 2000;66:904–913. doi: 10.1086/302814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenk E, de Techtermann F. Self-healing collodion baby: evidence for autosomal recessive inheritance. Pediatr Dermatol. 1992;9:95–97. doi: 10.1111/j.1525-1470.1992.tb01221.x. [DOI] [PubMed] [Google Scholar]
- Ganemo A, Pigg M, Virtanen M, Kukk T, Raudsepp H, Rossman-Ringdahl I, Westermark P, Niemi KM, Dahl N, Vahlquist A. Autosomal recessive congenital ichthyosis in Sweden and Estonia: Clinical, genetic and ultrastructural findings in eighty-three patients. Acta Derm Venereol. 2003;83:24–30. doi: 10.1080/00015550310002666. [DOI] [PubMed] [Google Scholar]
- Gibson DFC, Ratnam AV, Bikle DD. Evidence for separate control mechanisms at the message, protein, and enzyme activation levels for transglutaminase during calcium-induced differentiation of normal and transformed human keratinocytes. J Invest Dermatol. 1996;106:154–161. doi: 10.1111/1523-1747.ep12329856. [DOI] [PubMed] [Google Scholar]
- Harting M, Brunetti-Pierri N, Chan CS, Kirby J, Dishop MK, Richard G, Scaglia F, Yan AC, Levy ML. Self-healing collodion membrane and mild nonbullous congenital ichthyosiform erythroderma due to 2 novel mutations in the ALOX12B gene. Arch Dermatol. 2008;144:351–356. doi: 10.1001/archderm.144.3.351. [DOI] [PubMed] [Google Scholar]
- Hennies HC, Raghunath M, Wiebe V, Vogel M, Velten F, Traupe H, Reis A. Genetic and immunohistochemical detection of mutations inactivating the keratinocyte transglutaminase in patients with lamellar ichthyosis. Hum Genet. 1998a;102:314–318. doi: 10.1007/s004390050697. [DOI] [PubMed] [Google Scholar]
- Hennies HC, Küster W, Wiebe V, Krebsová A, Reis A. Genotype/phenotype correlation in autosomal recessive lamellar ichthyosis. Am J Hum Genet. 1998b;62:1052–1061. doi: 10.1086/301818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiiragi T, Sasaki H, Nagafuchi A, Sabe H, Shen SC, Matsuki M, Yamanishi K, Tsukita S. Transglutaminase type 1 and its cross-linking activity are concentrated at adherens junctions in simple epithelial cells. J Biol Chem. 1999;274:34148–34154. doi: 10.1074/jbc.274.48.34148. [DOI] [PubMed] [Google Scholar]
- Hohl D, Huber M, Frenk E. Analysis of the cornified cell envelope in lamellar ichthyosis. Arch Dermatol. 1993;129:618–623. [PubMed] [Google Scholar]
- Hohl D, Aeschlimann D, Huber M. In vitro and rapid in situ transglutaminase assays for congenital ichthyosis-a comparative study. J Invest Dermatol. 1998;110:268–271. doi: 10.1046/j.1523-1747.1998.00132.x. [DOI] [PubMed] [Google Scholar]
- Huber M, Rettler I, Bernasconi K, Frenk E, Lavrijsen SPM, Ponec M, Bon A, Lautenschlager S, Schorderet DF, Hohl D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995a;267:525–528. doi: 10.1126/science.7824952. [DOI] [PubMed] [Google Scholar]
- Huber M, Rettler I, Bernasconi K, Wyss M, Hohl D. Lamellar ichthyosis is genetically heterogenous-cases with normal keratinocyte transglutaminase. J Invest Dermatol. 1995b;105:653–654. doi: 10.1111/1523-1747.ep12324122. [DOI] [PubMed] [Google Scholar]
- Huber M, Yee VC, Burri N, Vikerfors E, Lavrijsen APM, Paller AS, Hohl D. Consequences of seven novel mutations on the expression and structure of keratinocyte transglutaminase. J Biol Chem. 1997;272:21018–21026. doi: 10.1074/jbc.272.34.21018. [DOI] [PubMed] [Google Scholar]
- Jacyk WK. Bathing-suit ichthyosis. A peculiar phenotype of lamellar ichthyosis in South African blacks. Eur J Dermatol. 2005;15:433–436. [PubMed] [Google Scholar]
- Jessen BA, Phillips MA, Hovnanian A, Rice RH. Role of Sp1 response element in transcription of the human transglutaminase 1 gene. J Invest Dermatol. 2000;115:113–117. doi: 10.1046/j.1523-1747.2000.00027.x. [DOI] [PubMed] [Google Scholar]
- Jobard F, Lefèvre C, Karaduman A, Blanchet-Bardon C, Emre S, Weissenbach J, Özgüc M, Lathrop M, Prud’homme JF, Fischer J. Lipoxygenase-3 (ALOXE3) and 12(R)-lipogenase (ALOX12b) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum Mol Genet. 2002;11:107–113. doi: 10.1093/hmg/11.1.107. [DOI] [PubMed] [Google Scholar]
- Kaneko KJ, Rein T, Guo ZS, Latham K, DePamphilis ML. DNA methylation may restrict but does not determine differential gene expression at the Sgy/Tead2 locus during mouse development. Mol Cell Biol. 2004;24:1968–1982. doi: 10.1128/MCB.24.5.1968-1982.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanerva L, Niemi K, Lauharanta J, Lassus A. New observations of the fine structure of lamellar ichthyosis and the effect of treatment with etretinate. Am J Dermatopathol. 1983;5:555–568. doi: 10.1097/00000372-198312000-00006. [DOI] [PubMed] [Google Scholar]
- Kalinin AE, Kajava AV, Steinert PM. Epithelial barrier function: assembly and structural features of the cornified cell envelope. Bioessays. 2002;24:789–800. doi: 10.1002/bies.10144. [DOI] [PubMed] [Google Scholar]
- Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, Dopping-Hepenstal PJ, Dale BA, Tadini G, Fleckman P, Stephens KG, Sybert VP, Mallory SB, North BV, Witt DR, Sprecher E, Taylor AE, Ilchyshyn A, Kennedy CT, Goodyear H, Moss C, Paige D, Harper JI, Young BD, Leigh IM, Eady RA, O’Toole EA. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet. 2005;76:794–803. doi: 10.1086/429844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HC, Idler WW, Kim IG, Han JH, Chung SI, Steinert PM. Complete amino acid sequence of the human transglutaminase K enzyme deduced from the nucleic acid sequences of cDNA clones. J Biol Chem. 1991;266:536–539. [PubMed] [Google Scholar]
- Kim IG, McBride OW, Wang M, Kim SY, Idler WW, Steinert PM. Structure and organization of the human transglutaminase 1 gene. J Biol Chem. 1992;267:7710–7717. [PubMed] [Google Scholar]
- Kim SY, Chung SI, Steinert PM. Highly active soluble processed forms of the transglutaminase 1 enzyme in epidermal keratinocytes. J Biol Chem. 1995;270:18026–18035. doi: 10.1074/jbc.270.30.18026. [DOI] [PubMed] [Google Scholar]
- Kon A, Takeda H, Sasaki H, Yoneda K, Nomura K, Ahvazi B, Steinert PM, Hanada K, Hashimoto I. Novel transglutaminase 1 gene mutations (R348X/Y365D) in a Japanese family with lamellar ichthyosis. J Invest Dermatol. 2003;120:170–2. doi: 10.1046/j.1523-1747.2003.19522.x. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Cryst. 1991;24:946–950. [Google Scholar]
- Kuramoto N, Takizawa T, Takizawa T, Matsuki M, Morioka H, Robinson JM, Yamanishi Development of ichthyosiform skin compensates for defective permeability barrier function in mice lacking transglutaminase 1. J Clin Invest. 2002;109:243–250. doi: 10.1172/JCI13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Celle PT, Polakowska RR. Human homoebox HOXA7 regulates keratinocyte tranglutaminase type 1 and inhibits differentiation. J Biol Chem. 2001;276:32844–32853. doi: 10.1074/jbc.M104598200. [DOI] [PubMed] [Google Scholar]
- Laiho E, Ignatius J, Mikkola H, Yee VC, Teller DC, Niemi KM, Saarialho-Kere U, Kere J, Palotie A. Transglutaminase 1 mutations in autosomal recessive congenital ichthyosis: private and recurrent mutations in an isolated population. Am J Hum Genet. 1997;61:529–538. doi: 10.1086/515498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiho E, Niemi KM, Ignatius J, Kere J, Palotie A, Saarialho-Kere U. Clinical and morphological correlations for transglutaminase 1 gene mutations in autosomal congenital ichthyosis. Eur J Hum Genet. 1999;7:625–632. doi: 10.1038/sj.ejhg.5200353. [DOI] [PubMed] [Google Scholar]
- Lawlor F. Progress of a harlequin fetus to nonbullous ichthyosiform erythroderma. Pediatrics. 1988;82:870–873. [PubMed] [Google Scholar]
- Lefèvre C, Audebert S, Jobard F, Bouadjar B, Lakhdar H, Boughdene-Stambouli O, Blanchet-Bardon C, Heilig R, Foglio M, Weissenbach J, Lathrop M, Prud’homme JF, Fischer J. Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum Mol Genet. 2003;12:2369–2378. doi: 10.1093/hmg/ddg235. [DOI] [PubMed] [Google Scholar]
- Lefèvre C, Bouadjar B, Karaduman A, Jobard F, Saker S, Özguc M, Lathrop M, Prud’homme JF, Fischer J. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum MolGenet. 2004;13:2473–2482. doi: 10.1093/hmg/ddh263. [DOI] [PubMed] [Google Scholar]
- Lefèvre C, Bouadjar B, Ferrand V, Tadini G, Mégarbané A, Lathrop M, Prud’homme JF, Fischer J. Muations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet. 2006;15:767–776. doi: 10.1093/hmg/ddi491. [DOI] [PubMed] [Google Scholar]
- Levitt M. Accurate modeling of protein conformation by automatic segment matching. J Mol Biol. 1992;226:507–533. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- Lichti U, Ben T, Yuspa SH. Retinoic acid-induced transglutaminase in mouse epidermal cells is distinct from epidermal transglutaminase. J Biol Chem. 1985;260:1422–1426. [PubMed] [Google Scholar]
- Lorand L, Conrad SM. Transglutaminases. Mol Cell Biochem. 1984;58:9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- Lugassy J, Hennies HC, Indelman M, Khamaysi Z, Bergman R, Sprecher E. Rapid detection of homozygous mutations in congenital recessive ichthyosis. Arch Dermatol Res. 2008;300:81–85. doi: 10.1007/s00403-007-0815-0. [DOI] [PubMed] [Google Scholar]
- Magewu AN, Jones PA. Ubiquitous and tenacious methylatioin of the CpG site in codon 248 of the p53 gene may explain its frequent appearance as a mutational hot spot in human cancer. Mol Cell Biol. 1994;14:4225–4232. doi: 10.1128/mcb.14.6.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki M, Yamashita F, Ishida-Yamamoto A, Yamada K, Kinoshita C, Fushiki S, Ueda E, Morishima Y, Tabata K, Yasuno H, Hashida M, Iizuka H, Ikawa M, Okabe M, Kondoh G, Kinoshita T, Takeda J, Yamanishi K. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase) Proc Natl Acad Sci USA. 1998;95:1044–1049. doi: 10.1073/pnas.95.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- Merritt Ethan A, Bacon David J. Raster3D: Photorealistic molecular graphics. Method Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Mizrachi-Koren M, Geiger D, Indelman M, Bitterman-Deutsch O, Bergman R, Sprecher E. Identification of a novel locus associated with congenital recessive ichthyosis on 12p11.2-q13. J Invest Dermatol. 2005;125:456–462. doi: 10.1111/j.0022-202X.2005.23777.x. [DOI] [PubMed] [Google Scholar]
- Mizrachi-Koren M, Shemer S, Morgan M, Indelman M, Khamaysi Z, Petronius D, Bitterman-Deutsch O, Hennies HC, Bergman R, Sprecher E. Homozygosity mapping as a screening tool for the molecular diagnosis of hereditary skin diseases in consanguineous populations. J Am Acad Dermatol. 2006;55:393–401. doi: 10.1016/j.jaad.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Muramatsu S, Suga Y, Kon J, Matsuba S, Hashimoto Y, Ogawa H. A Japanese patient with a mild form of lamellar ichthyosis harbouring two missense mutations in the core domain of the transglutaminase 1 gene. Br J Dermatol. 2004;150:390–392. doi: 10.1111/j.1365-2133.2003.05803.x. [DOI] [PubMed] [Google Scholar]
- Nemes Z, Marekov LN, Steinert PM. Involucrin cross-linking by transglutaminase 1. Binding to membranes directs residue specificity. J Biol Chem. 1999a;274:11013–11021. doi: 10.1074/jbc.274.16.11013. [DOI] [PubMed] [Google Scholar]
- Nemes Z, Marekov LN, Fésüs L, Steinert PM. A novel function for transglutaminase 1: attachment of long-chain ω-hydroxyceramides to involucrin by ester bond formation. Proc Natl Acad Sci USA. 1999b;96:8402–8407. doi: 10.1073/pnas.96.15.8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oji V, Hautier JM, Ahvazi B, Hausser I, Aufenvenne K, Walker T, Seller N, Steijlen PM, Küster W, Hovnanian A, Hennies HC, Traupe H. Bathing suit ichthyosis is caused by transglutaminase-1 deficiency: evidence for a temperature-sensitive phenotype. Hum Mol Genet. 2006;15:3083–3097. doi: 10.1093/hmg/ddl249. [DOI] [PubMed] [Google Scholar]
- Parmentier L, Blanchet-Bardon C, Nguyen S, Prud’homme JF, Dubertret L, Weissenbach J. Autosomal recessive lamellar ichthyosis: identification of a new mutation in transglutaminase 1 and evidence for genetic heterogeneity. Hum Mol Genet. 1995;4:1391–1395. doi: 10.1093/hmg/4.8.1391. [DOI] [PubMed] [Google Scholar]
- Petit E, Huber M, Rochat A, Bodemer C, Teillac-Hamel D, Müh JP, Revuz J, Barrandon Y, Lathrop M, de Prost Y, Hohl D, Hovnanian A. Three novel point mutations in the keratinocyte transglutaminase (TGK) gene in lamellar ichthyosis: significance for mutant transcript level, TGK immunodetection and activity. Eur J Hum Genet. 1997;5:218–228. [PubMed] [Google Scholar]
- Pigg M, Gedde-Dahl T, Jr, Cox D, Hauβer I, Anton-Lamprecht I, Dahl N. Strong founder effect for a transglutaminase 1 gene mutation in lamellar ichthyosis and congenital ichthyosiform erythroderma from Norway. Eur J Hum Genet. 1998;6:589–596. doi: 10.1038/sj.ejhg.5200224. [DOI] [PubMed] [Google Scholar]
- Pigg M, Gedde-Dahl T, Jr, Cox DW, Haugen G, Dahl N. Haplotype association and mutation analysis of the transglutaminase 1 gene for prenatal exclusion of lamellar ichthyosis. Prenat Diagn. 2000;20:132–137. doi: 10.1002/(sici)1097-0223(200002)20:2<132::aid-pd765>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Raghunath M, Hennies HC, Ahvazi B, Vogel M, Reis A, Steinert PM, Traupe H. Self-healing collodion baby: a dynamic phenotype explained by a particular transglutaminase-1 mutation. J Invest Dermatol. 2003;120:224–228. doi: 10.1046/j.1523-1747.2003.12032.x. [DOI] [PubMed] [Google Scholar]
- Rice RH, Greeen H. Relation of protein synthesis and transglutaminase activity to formation of the cross-linked envelope during terminal differentiation of the cultured human epidermal keratinocyte. J Cell Bio. 1978;76:705–711. doi: 10.1083/jcb.76.3.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RH, Green H. Presence in human epidermal cells of a soluble protein precursor of the cross-linked envelope: activation of the cross-linking by calcium ions. Cell. 1979;18:681–694. doi: 10.1016/0092-8674(79)90123-5. [DOI] [PubMed] [Google Scholar]
- Rice RH, Mehrpouyan M, Qin Q, Phillips MA, Lee YM. Identification of phosphorylation sites in keratinocyte transglutaminase. Biochem J. 1996;320:547–550. doi: 10.1042/bj3200547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RH, Crumrine D, Uchida Y, Gruber R, Elias PM. Structural changes in epidermal scale and appendages as indicators of defective TGM1 activity. Arch Dermatol Res. 2005;297:127–133. doi: 10.1007/s00403-005-0591-7. [DOI] [PubMed] [Google Scholar]
- Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- Robinson NA, Lapic S, Welter JF, Eckert RL. S100A11, S100A10, annexin I, desmosal proteins, small proline-rich proteins, plasminogen activator inhibitor-2, and involucrin are components of the cornified envelope of cultured human epidermal keratinocytes. J Biol Chem. 1997;272:12035–12036. doi: 10.1074/jbc.272.18.12035. [DOI] [PubMed] [Google Scholar]
- Rossi A, Catani MV, Candi E, Bernassola F, Puddu P, Melino G. Nitric oxide inhibits cornified envelope formation in human keratinocytes by inactivating transglutaminases and activating protein 1. J Invest Dermatol. 2000;115:731–739. doi: 10.1046/j.1523-1747.2000.00116.x. [DOI] [PubMed] [Google Scholar]
- Rothnagel JA, Rogers GE. Transglutaminase-mediated cross-linking in mammalian epidermis. Mol Cell Biochem. 1984;58:113–119. doi: 10.1007/BF00240610. [DOI] [PubMed] [Google Scholar]
- Russell LJ, DiGiovanna JJ, Hashem N, Compton JG, Bale SJ. Linkage of autosomal recessive lamellar ichthyosis to chromosome 14q. Am J Hum Genet. 1994;55:1146–1152. [PMC free article] [PubMed] [Google Scholar]
- Russell LJ, DiGiovanna JJ, Rogers GR, Steinert PM, Hashem N, Compton JG, Bale SJ. Mutations in the gene for transglutaminase 1 in autosomal recessive lamellar ichthyosis. Nat Genet. 1995;9:279–283. doi: 10.1038/ng0395-279. [DOI] [PubMed] [Google Scholar]
- Sandberg NO. Lamellar ichthyosis. Pediatr Rev. 1981;2:213–216. [Google Scholar]
- Schorderet DF, Huber M, Laurini RN, Von Moos G, Gianadda B, Délèze G, Hohl D. Prenatal diagnosis of lamellar ichthyosis by direct mutational analysis of the keratinocyte transglutaminase gene. Prenat Diagn. 1997;17:483–486. doi: 10.1002/(sici)1097-0223(199705)17:5<483::aid-pd80>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Shawky RM, Sayed NS, Elhawary NA. Mutations in transglutaminase 1 gene in autosomal recessive congenital ichthyosis in Egyptian families. Dis Markers. 2004;20:325–332. doi: 10.1155/2004/965968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko YO, Compton JG, Toro JR, DiGiovanna JJ, Bale SJ. Splice-site mutation in TGM1 in congenital recessive ichthyosis in American families: molecular, genetic, genealogic, and clinical studies. Hum Genet. 2000;106:492–499. doi: 10.1007/s004390000284. [DOI] [PubMed] [Google Scholar]
- Schulz EJ. Genodermatoses. Dermatol Clin. 1994;12:787–796. [PubMed] [Google Scholar]
- Shwayder T. Ichthyosis in a nutshell. Pediatr Rev. 1999;20:5–12. doi: 10.1542/pir.20-1-5. [DOI] [PubMed] [Google Scholar]
- Steinert PM, Marekov LN. The proteins elafin, filaggrin, keratin intermediate filaments, loricrin, and small proline-rich proteints 1 and 2 are isodipeptide cross-linked components of the human epidermal cornified cell envelope. J Biol Chem. 1995;270:17702–17711. doi: 10.1074/jbc.270.30.17702. [DOI] [PubMed] [Google Scholar]
- Steinert PM, Chung SI, Kim SY. Inactive zymogen and highly active proteolytically processed membrane-bound forms of the transglutaminase 1 enzyme in human epidermal keratinocytes. Biochem Biophys Res Commun. 1996a;221:101–106. doi: 10.1006/bbrc.1996.0552. [DOI] [PubMed] [Google Scholar]
- Steinert PM, Kim SY, Chung SI, Marekov LN. The transglutaminase 1 enzyme is variably acylated by myristate and palmitate during differentiation in epidermal keratinocytes. J Biol Chem. 1996b;271:26242–26250. doi: 10.1074/jbc.271.42.26242. [DOI] [PubMed] [Google Scholar]
- Steinert PM, Marekov LN. Direct evidence that involucrin is a major early isopeptide cross-linked component of the keratinocyte cornified cell envelope. J Biol Chem. 1997;272:2021–2030. doi: 10.1074/jbc.272.3.2021. [DOI] [PubMed] [Google Scholar]
- Thacher SM, Rice RH. Keratinocyte-specific transglutaminase of cultured human epidermal cells: relation to cross-linked envelope formation and terminal differentiation. Cell. 1985;40:685–695. doi: 10.1016/0092-8674(85)90217-x. [DOI] [PubMed] [Google Scholar]
- Tok J, Garzon MC, Cserhalmi-Friedman P, Lam HM, Spitz JL, Christiano M. Identification of mutations in the transglutaminase 1 gene in lamellar ichthyosis. Exp Dermatol. 1999;8:128–133. doi: 10.1111/j.1600-0625.1999.tb00360.x. [DOI] [PubMed] [Google Scholar]
- Virolainen E, Wessman M, Hovatta I, Niemi KM, Ignatius J, Kere J, Peltonen L, Palotie A. Assignment of a novel locus for autosomal recessive congenital ichthyosis to chromosome 19p13.1-p13.2. Am J Hum Genet. 2000;66:1132–1137. doi: 10.1086/302813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkup D, Sander C, Church GM. The amino-acid mutational spectrum of human genetic disease. Genome Biol. 2003;4:R72. doi: 10.1186/gb-2003-4-11-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz PW, Madison KC, Downing DT. Covalently bound lipids of human stratum corneum. J Invest Dermatol. 1989;92:109–111. doi: 10.1111/1523-1747.ep13071317. [DOI] [PubMed] [Google Scholar]
- Williams ML, Elias PM. Heterogeneity in autosomal recessive ichthyosis. Clinical and biochemical differentiation of lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch Dermatol. 1985;121:477–488. doi: 10.1001/archderm.121.4.477. [DOI] [PubMed] [Google Scholar]
- Yamada K, Matsuki M, Morishima Y, Ueda E, Tabata K, Yasuno H, Suzuki M, Yamanishi K. Activation of the human transglutaminase 1 promoter in transgenic mice: Terminal differentiation-specific expression of the TGM1-lacZ transgene in keratinized stratified squamous epithelia. Hum Mol Genet. 1997;6:2223–2231. doi: 10.1093/hmg/6.13.2223. [DOI] [PubMed] [Google Scholar]
- Yang JM, Ahn KS, Cho MO, Yoneda K, Lee CH, Lee JH, Lee ES, Candi E, Melino G, Ahvazi B, Steinert PM. Novel mutations of the transglutaminase 1 gene in lamellar ichthyosis. J Invest Dermatol. 2001;117:214–218. doi: 10.1046/j.0022-202x.2001.01429.x. [DOI] [PubMed] [Google Scholar]
- Yoneda K, Akiyama M, Morita K, Shimizu H, Imamura S, Kim SY. Expression of transglutaminase 1 in human hair follicles, sebaceous glands and sweat glands. Br J Dermatol. 1998;138:37–44. doi: 10.1046/j.1365-2133.1998.02024.x. [DOI] [PubMed] [Google Scholar]
- Yotsumoto S, Akiyama M, Yoneda K, Fukushige T, Kobayashi K, Takeyori S, Kanzaki T. Analyses of the transglutaminase 1 gene mutation and ultrastructural characteristics in a Japanese patient with lamellar ichthyosis. J Dermatol Sci. 2000;24:119–125. doi: 10.1016/s0923-1811(00)00087-6. [DOI] [PubMed] [Google Scholar]
- Yuspa SH, Ben T, Hennings H, Lichti U. Phorbol ester tumor promotors induce epidermal transglutaminase activity. Biochem Biophys Res Commun. 1980;97:700–708. doi: 10.1016/0006-291x(80)90321-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.