

Abstract
Background:
Cisplatin (CDDP) is a drug used for treatment of many types of malignancy but pancreatic cancer is relatively resistant to it. This study aims to determine whether and how cyclin D1 (D1) and c-Myc influence the response of pancreatic cancer cells to CDDP.
Materials and Methods:
Ela-mycPT mouse pancreatic cancer cells were transfected with D1 or c-myc cDNA and treated with CDDP alone or together with NPCD, an inhibitor of cyclin dependent ckinase (CDK) 4 and 6. Reverse transcription followed by polymerase chain reaction (RT-PCR) and western blot assays were used to determine the mRNA and protein levels of interested genes. Cell viability was determined using 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay.
Results:
Treatment of Ela-mycPT1 cells with CDDP caused an increase in c-myc expression but a slightly latent decrease in D1 expression, whereas D1 and c-Myc proteins repressed each other. D1 or c-Myc rendered Ela-mycPT1 cells resistant or sensitive, respectively, to CDDP. D1 induced the expression of several members of the NF-κB family, including RelA, RelB, Nfκb1 and Nfκb2. D1 also induced BIRC5 and several pro-survival members of the Bcl-2 gene family, including Bcl-2 , Mcl-1 and Bad while it decreased the level of the pro-apoptotic Noxa. Inhibition of CDK4 or CDK6 kinase activity by NPCD did not affect these effects of D1. In contrast, c-Myc in Ela-mycPT1 and Ela-mycPT4 cells has the opposite effects to D1 on the expression of most of these apoptosis regulating genes.
Conclusion:
Our results suggest that induction of c-Myc and inhibition of D1 may be mechanisms for CDDP to elicit cytotoxicity. On the other hand, D1 induces whereas c-Myc represses the expression of key NF-κB family members to induce and repress, respectively, the expression of BIRC5 and several Bcl-2 family members, in turn inhibiting or enhancing the response to CDDP.
Keywords: Bcl-2, chemotherapy, c-myc, cyclin D1, NF-κB, pancreatic cancer
INTRODUCTION
Pancreatic cancer is one of the few most lethal malignant diseases. It accounts for 6-7% of all cancer deaths and is the fourth leading course of cancer death in the United States and many other western countries.[1] Many patients are dead within 6 months of the first presentation, and the 5-year survival rate is less than 5% even after surgical resection. This dismal prognosis is mainly because the tumors are usually diagnosed at an advanced stage and are resistant to various treatments, including chemotherapy.[1,2] Cisplatin (CDDP) is a potent chemotherapeutic agent used for many types of malignancy. It is also commonly used in pancreatic cancer treatment, but the cancer is relatively resistant to it, compared with other malignancies.[3,4] Several mechanisms have been suggested to explain the CDDP resistance, whose molecular details are still not fully understood.
Cyclin D1 (D1) protein encoded by the CCND1 gene is a critical regulator of cell cycle progression. As reviewed by one of us previously,[5,6] D1 also has many other functions, such as transcriptional control. Rearrangement, amplification and increased expression of CCND1 are common in various malignancies.[5] We have previously shown that D1 can render pancreatic cancer cells resistant to chemotherapy,[7] which is in line with some clinical studies that associate D1 overexpression with chemoresistance of pancreatic cancer.[5]
The c-Myc protein is a master transcription factor that is estimated to regulate, directly or indirectly, the expression of 10-15% of the genes in human genome.[5,6,8] As a consequence, c-Myc mediates most cellular functions including cell proliferation, apoptosis, differentiation and metabolism [5,6,8] Translocation, amplification or elevated expression of the c-myc gene occurs very frequently in virtually all types of malignancy, including pancreatic cancer.[5,9] Although classified as an oncoprotein, c-Myc expressed ectopically is also capable of inducing apoptosis or sensitizing cells to various apoptotic stimuli.[8] We have previously shown that opposite to D1, ectopic c-Myc renders pancreatic cancer cells sensitive to several chemotherapeutic agents, which occurs in part via inhibition of D1.[10]
Nuclear factor (NF)-κB is a family of proteins, many members of which are ubiquitously expressed and inducible by a variety of extracellular growth stimuli.[11] Key members of the genes in this family include RelA, RelB, Nfκb1, Nfκb2, and IκBα. Protein products of the RelA, Nfκb1 and Nfκb2 genes are processed to p65, p50 and p52, respectively, which then form p65/p50 and p52/RelB dimmers to regulate the transcription of genes that mediate a variety of cellular functions, including cell proliferation, differentiation, apoptosis, inflammation, etc.[11–13] These transcriptional activities can be modulated by a number of events, including changes in the status of the inhibitory protein IκBα, the cellular localization of the NF-κB protein complex, and different protein modifications of NF-κB members such as phosphorylation, acetylation, and S-nitrosylation.[11–13] NF-κB has been found to be constitutively active in pancreatic cancer and many other malignancies.[11] NF-κB is also often activated in response to chemotherapeutic agents and thus augments chemoresistance of many malignancies, [11–14] including pancreatic cancer.[15] The mechanisms for the constitutive activation are still not well understood, but known activating factors include mutation of IκBα, proteosomal degradation of IκBα, and enhanced inflammatory cytokine expression.[11–14] Because of the pivotal role of NF-κB in chemoresistance, inhibition of its activity has become one of the major strategies to sensitize cancer cells to chemotherapeutic agents.[14,16]
Of the many transcriptional targets of NF-κB are CCND1, [17,18] BIRC5 that encodes survivin proteins,[19] and several members of the Bcl-2 family.[20,21] NF-κB activates the expression of several pro-survival members[20] but represses several other pro-apoptotic members of the Bcl-2 family,[21] which collectively augments the cell survival or chemoresistance. However, the effects of NF-κB on Bcl-2 members may be cell-type specific, and thus not all of its target genes are regulated in a given cell type.[20,21] Moreover, most Bcl-2 members undergo alternative splicing to produce different mRNA variants. While some of these splice variants remain elusive for their functions, such as Bcl-2β and Bcl-xβ,[22] some other variants of the same Bcl-2 members are known to function oppositely. For instance, the exon 2 of the Bcl-x is alternatively spliced to produce a short (Bcl-xS) and a long (Bcl-xL) variant. While Bcl-xL is pro-survival, Bcl-xS is pro-apoptotic.[22]
In this communication, we confirm the previous findings that D1 and c-Myc render pancreatic cancer cells resistant and sensitive, respectively, to CDDP. We also found that CDDP might repress D1 but induce c-Myc to elicit cytotoxicity, whereas D1 and c-Myc may repress each other. D1 induces the expression of RelA, RelB, Nfkb1 and Nfkb2 as well as BIRC5, Bcl-2, Bcl-2β, Bad and Mcl-1 that are pro-survival but represses the expression of Noxa that is pro-apoptotic, whereas c-Myc has the opposite effects on these genes or their mRNA variants. The effects may be a significant part of the mechanism for the influence of D1 and c-Myc in the response to CDDP.
MATERIALS AND METHODS
Reagents
Naphtho [2, 1-α] pyrrolo [3, 4-c] carbazole-5, 7 (6H, 12H)-dione (NPCD) was synthesized and purified by us at a purity of over 99% proved by high performance liquid chromatography (HPLC).[23] It was dissolved with dimethyl sulfoxide (DMSO) and kept at −20°C until use. Primary antibodies used in this study were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA), including a mouse monoclonal anti-β-actin (sc-47778), a rabbit polyclonal anti-cyclin D1 (sc-718), and a rabbit polyclonal anti-c-Myc (c-19). Peroxidase-conjugated anti-mouse (NA931) and anti-rabbit (NA934) secondary antibodies were purchased from Amersham Biosciences (Piscataway, NJ, USA).
Transient transfection
Cells were grown as monolayer culture in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were incubated at 37°C in a humidified atmosphere of 5% CO 2 and 95% air. Ela-mycT1, MDA-MB-231 (MB231) and D1.5 cells were transiently transfected with pcDNA3.1-cyclin D1 (D1), pcDNA3.1-neo vector, pcDNA3.1-c-myc, pcDNA3.1-hygro vector, pSiD1shRNA and pSishRNA plasmids using LipofectAMINE reagent (www.Invitrogen.com) according to the manufacturer's protocol. In cases when cells were treated with CDDP after transfection, the medium was replaced with a fresh one containing the indicated concentration of CDDP every 24 hours. A pcDNA3.1neo vector containing green fluorescent protein (GFP) cDNA was co-transfected in indicated experiment to show the transfection efficiency.
DNA ladder assay
Ela-myc-PT1 cells treated with 2 μM CDDP for 48 hours were collected, washed in phosphate buffered saline (PBS), resuspended in lysis buffer containing 0.5% Triton X-100, 10 mM ethylenediaminetetraacetic acid (EDTA) and 10 mM Tris-HCl (pH 8.0), and incubated for 30 min on ice, followed by centrifugation for 30 min at 12,000 rpm. The supernant was collected, added with sodium dodecyl sulfate (0.5%) and 0.4 μg/μl RNase A, followed by incubation at 37°C for 90 min to get rid of RNA. The samples were then digested with 1.5 μg/μl Proteinase K at 56°C for another 90 min to remove the proteins. An equal amount of phenol solution (phenol:chloroform:isoamylalcohol = 25:24:1) was added followed by centrifugation for 1 min at 12,000 rpm. The phenol/chloroform extraction was repeated three times. The DNA was precipitated with 2.5 volumes of cold ethanol and 0.1 volume of 3 M sodium acetate overnight at −70°C, centrifuged at 12,000 rpm for 10 min, and washed with 70% ethanol. The precipitated DNA was dissolved in 20 μl of TE buffer, fractioned by electrophoresis on 2% agarose gel, and visualized with ethidium bromide staining and a UV transilluminator.
MTT assay
Cells were seeded in a 96-well microplate at 4000 cells per well, five wells per dose of to-be-tested drug and per time point, and cultured at 37°C with 5% CO2. CDDP or NPCD was added 24 hours later at the indicated concentrations, with the solvent (saline for CDDP and DMSO for NPCD) as non-treated control. The culture was continued for the indicated time period. At the end of the treatment, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was added into each well at a final concentration of 0.5 mg/ml, followed by incubation at 37°C for 3 hours in dark. The culture medium containing MTT was discarded and the dye crystals were dissolved in DMSO. The viable cells were detected by reading the absorbance of the metabolized MTT at a wavelength of 570 nm using the Beckman Coulter AD340 absorbance detector (Beckman Coulter Inc., Fullerton, CA, USA).
Protein extraction and western blot assay
Total protein from the samples was extracted as described by Andrew and Faller[24] with inhibitors of proteases, kinases and phosphatases, followed by determination of protein concentration using Bradford assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). An equal amount of proteins from each sample was fractionated in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto an Immobilon-P Nylon membrane (Millipore, Bedford, MA, USA) in a tank transfer system. After being blocked with 5% milk, the membrane was incubated with a specific primary antibody at an optimized concentration (1:1000) and then with horseradish peroxidase-conjugated secondary antibody, with three washes between each antibody. The signal was visualized with enhanced chemiluminescent (ECL) substrates (Pierce, Rockford, IL, USA) on X-ray film (ISC BioExpress, Kaysville, UT, USA).
RT-PCR assay and densitometry quantification
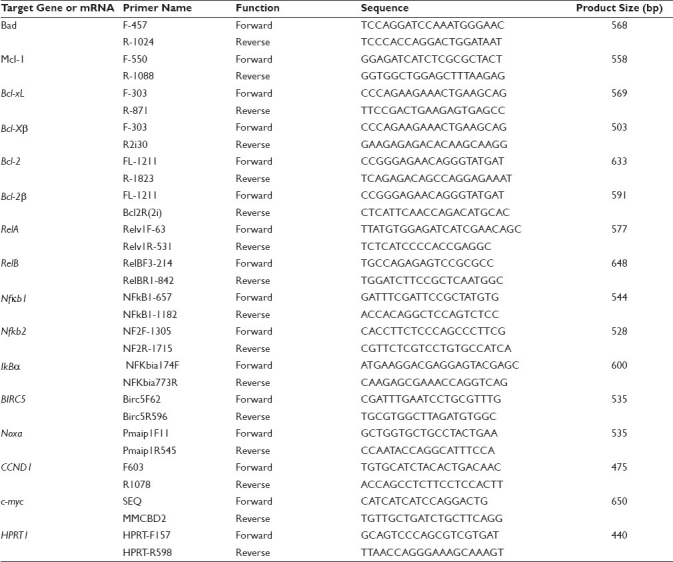
Total RNA of the samples was extracted from cultured cells using TRIzol (Invitrogen, Cat. 15596-026) by following the manufacturer's instruction. To digest out genomic DNA residuals, 5 units of RNase-free DNase (http://www.promega.com/) were added, followed by incubation at 37°C for 30 min. The DNase was inactivated by heating the sample to 99°C for 5 min. An aliquot (2.5 μg) of the RNA samples was then reverse transcribed (RT) in a 25-μl reaction solution to the first strand of cDNA using random hexamer primer. The RT products were diluted with water to a final volume of 50 μl. For polymerase chain reaction (PCR) to amplify cDNA, 1 μl of the diluted RT products was used as template and hypoxanthine-guanine phosphoribosyl transferase-1 (HPRT1) was used as the loading control as its mRNA is less abundantly expressed than β-actin. PCR was performed in an Eppendorf AG Mastercycler (Hamburg, Germany) and with Hot-Start Taq Mastermix™ (Denville Scientific Inc., Cat. CB4030-4, Metuchen, NJ, USA). PCR was started with an initial denaturation step at 95°C for 5 min. Each cycle included a denaturation step at 95°C for 30 seconds, a primer-annealing step at a temperature optimized for each pair of primers, and an elongation step at 72°C for a time period optimal for the size of the PCR product. The PCR condition was optimized for each gene and was stopped within the linear portion of the amplification. The forward and reverse primers for each gene, listed in Table 1, were designed in such a way that they were localized at two different exons of the given gene with one or several large introns in between. In this way, if there still is a traceable amount of DNA residual, it either cannot be amplified due to large intron(s) or is amplified as a molecule larger than the expected size indicated in Table 1. PCR products were separated in 1% agarose gel and visualized with ethidium bromide staining. The expression levels of D1 and c-myc mRNA were quantified as the band density on the agarose gel, calculated as the ratio to the HPRT1 expressed by the same sample.
Table 1.
List of the primers and the expected size of PCR products with these primer pairs
DNA sequencing
Several PCR products manifested as multiple bands in agarose gel. In this case, each band was cut out and purified. In this case, each band was cut out and purified with UltraClean Gel DNA Extraction Kit (ISC BioExpress, Kaysville, UT), following the manual. The purified DNA sample was sequenced by Genewiz, Inc. (South Plainfield, NJ, USA). The sequences were analyzed with DNAStar software and were compared with the NCBI database via Blast and with UCSC genome via Blat (http://genome.ucsc.edu/) to obtain the identity of the band.
Statistical analyses
Statistical comparisons of MTT data between groups were made with the student's t-test. A P value <0.05 was considered as significant.
RESULTS
CDDP decreases D1 but increases c-myc expression
We had previously established a cell line from a pancreatic tumor developed in an Ela-myc transgenic mouse.[7] This cell line, named Ela-mycPT1, is wild type for the p53 and k-ras genes. Its level of the c-myc mRNA is not very high, presumably because the Elastase-1 gene promoter used to drive the c-myc transgene is not active in cell culture. We first treated this cell line with escalating concentrations (0.5, 1, 2 and 4 μM) of CDDP or vehicle (saline) for 48 hours. MTT assay showed that CDDP decreased the cell viability with an IC50 at about 2 μM [Figure 1a]. We then determined the time course of the response and observed that the cell viability at 12, 24, 36, 48 and 60 hours post CDDP treatment was about 90, 80, 60, 45 and 20%, respectively, of the non-treated counterparts [Figure 1b]. CDDP-induced apoptosis was confirmed by DNA ladder assay of the cells treated with 2 μM CDDP for 48 hours [Figure 1c].
Figure 1.
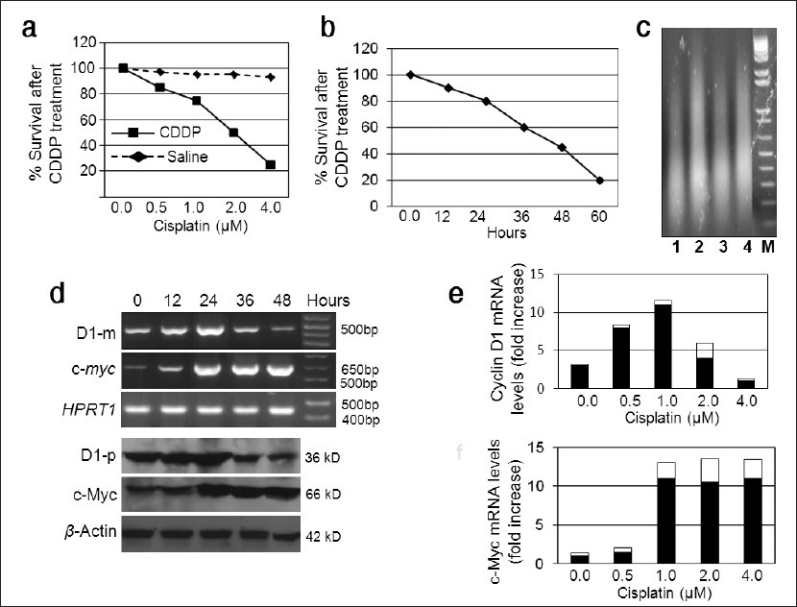
Response of Ela-mycPT1 cells to CDDP treatment. (a) Dose response curve of Ela-mycPT1 cells to a 48-hour treatment with CDDP or saline (as vehicle). Note that IC50 was at about 2 μM. (b) Time course of 2 μM CDDP treatment. (c) DNA ladder assay showing smear DNA fragments stained by ethidium bromide in four (lanes 1-4) different experiments of Ela-mycPT1 cells treated with 2μM CDDP for 48 hours. “M” is a maker of DNA molecular weights. (d) D1 and c-myc expression in the cells treated with 2μM CDDP for the indicated time period. The top panel is the mRNA level detected by RT-PCR with HPRT1 as the loading control whereas the bottom panel is the protein level detected by western blot with β-actin as the loading control. (e and f) Densitometry quantification of the D1 (e) and c-myc (f) mRNA levels, which quantifies the band density in the agarose gel and then is calculated as the ratio to the density of the HPRT in the same sample
Treatment of the Ela-mycPT1 cells with CDDP at the concentration of IC50, i.e. 2 μM, transiently increased the D1 mRNA level 12 and 24 hours after the treatment, as determined by RT-PCR assay, but the D1 level gradually declined at later time points, with a level lower than that of the non-treated control at 48 hours [Figure 1d]. The mRNA level of c-myc was also increased at 12 hours; however, when it peaked at 24 hours, it sustained at the peak for a period of time, unlike D1 [Figure 1d]. Densitometry quantification of the bands on agarose gel clearly showed a reciprocal relationship between D1 and c-Myc at the later time points [Figure 1e and 1f]. At 60 hours post treatment, the expression levels of both genes were very low (data not shown), likely because most cells were already dead. The protein levels of the D1 and c-Myc, determined by western blot assay [Figure 1d], showed similar changes as the mRNA.
Cyclin D1 imposes resistance to CDDP
To confirm our previous report that D1 renders Ela-mycPT1 cells resistant to CDDP, we transfected the cells with a D1 construct, pcDNA3.1-D1, or the empty vector as control. Co-transfection with a green fluorescent protein (GFP) construct resulted in many GFP expressing cells seen under a fluorescent microscope [Figure 2a], indicating that this cell line could be transfected at a high efficiency. RT-PCR and western blot assays also confirmed higher levels of D1 mRNA and protein in the D1 transfectants, compared with the vector counterparts [Figure 2b]. As shown in [Figure 2c], the D1 cells survived better than the vector cells when treated with 2 μM CDDP, which was manifested as early as 24 hours post treatment and became more evident at later time points.
Figure 2.
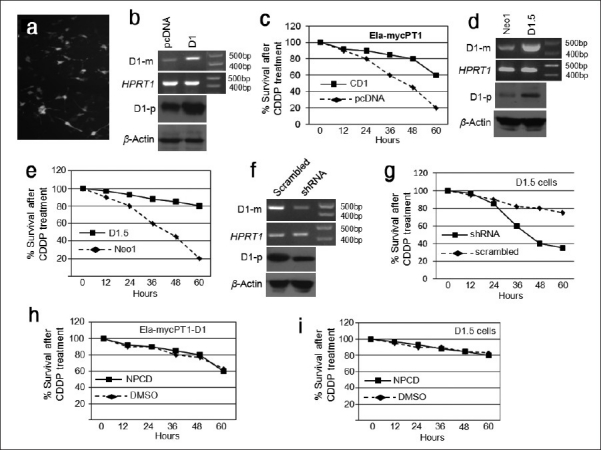
Effects of D1 on the CDDP cytotoxicity of Ela-mycPT1 cells. (a) Many cells co-transfected with not only D1 construct but also a GFP cDNA construct show GFP positive under fluorescent microscope. (b) RT-PCR and western blot assays show a higher D1 mRNA (D1-m) and protein (D1-p) level in the cells transfected with the D1 cDNA construct, relative to the pcDNA3.1-neo vector transfectants. HPRT1 and β-actin are used as the loading control for the mRNA and protein, respectively. (c) D1 cDNA transfected cells survive better than the pcDNA3.1-neo vector transfectants treated with 2μ CDDP for a period of 60 hours. (d) D1.5 clone expresses a higher D1 mRNA (D1-m) and protein (D1-p) level than the Neo1 clone. (e) D1.5 clone survives better than the Neo1 clone when treated with 2μ CDDP for a period of 60 hours. (f) D1.5 clone transfected with a D1 shRNA construct shows a decreased D1 mRNA (D1-m) and protein (D1-p) level than the same clone transfected with a scrambled shRNA construct. (g) The D1-shRNA transfected D1.5 cells show a much less cell viability than the scrambled shRNA transfectants. (h) D1 cDNA transfected cells treated with CDDP together with NPCD or DMSO manifest similar cell viability in a period of 60 hours. (i) D1.5 cells treated with CDDP together with NPCD or DMSO show similar cell viability in a period of 60 hours
We also determined the effects of D1 in the D1.5 and Neo1 clones of Ela-mycPT1 cells that were previously established by us to stably express D1 and the pcDNA3.1-neo empty vector, respectively.[7] RT-PCR and western blot assays confirmed that the D1.5 cells expressed higher levels of D1 mRNA and protein, respectively, than the Neo1 cells [Figure 2d]. Again, the D1.5 cells survived better than the Neo1 cells when treated with 2 μM CDDP [Figure 2e]. To further confirm the D1 relevance, we transfected the D1.5 cells with a CCND1 shRNA construct (pSiD1shRNA) or a scrambled shRNA construct (pSishRNA) as control. RT-PCR and western blot assays confirmed that the pSiD1shRNA downregulated the D1 mRNA and protein levels, compared with the scrambled shRNA [Figure 2f]. The D1shRNA expressing cells showed a decreased cell viability when treated with 2 μM CDDP, compared with the scrambled counterparts [Figure 2g], thus confirming that a decrease in the D1 level restored the sensitivity of the cells to CDDP.
Because many, but not all, functions of D1 are elicited via its binding to CDK4 or CDK6,[6] we wonder whether the kinase activity of these CDKs is involved in the D1-imposed resistance to CDDP. We transfected Ela-mycPT1 cells with the pcDNA3.1-D1 construct and treated the cells with 2 μM CDDP alone or together with 1 μM NPCD, which is an indolocarbazole-derived inhibitor of CDK4 and CDK6 that has an IC50 on the kinase activity of these CDKs at the nanomolar range and requires at least several micromoles to elicit cytotoxicity, as reported by us recently.[23] Surprisingly, NPCD at this dose and time period did not affect the viability of the D1 transfected cells [Figure 2h] and the D1.5 clone cells [Figure 2i] treated with CDDP. It seems that the kinase activity of CDK4/6 may not be involved in D1-imposed resistance to CDDP.
c-Myc enhances the response to CDDP
We transfected Ela-mycPT1 cells with a c-myc cDNA construct or with the pcDNA3.1-hygro empty vector as control and performed RT-PCR assay to confirm the increased expression of c-myc [Figure 3a]. When treated with 2 μM CDDP, the c-myc transfectants showed a much less cell viability than the vector counterparts, manifested as 65 versus 80%, 30 versus 60% and 20 versus 45% at 24, 36 and 48 hours after the treatment, respectively [Figure 3b]. Both transfectants died dramatically at 60 hours, which narrowed the difference between the two. Thus, ectopic c-Myc enhanced the cytotoxicity of CDDP at certain time points.
Figure 3.

Effects of c-Myc on the cyotoxicity of CDDP. (a) Ela-mycPT1 cells and D1.5 cells transfected with a c-myc cDNA construct show higher levels of c-myc mRNA detected by RT-PCR assay, compared with the corresponding pcDNA3.1-hygro vector transfectants. HPRT1 is included as a loading control. (b and c) c-myc transfected Ela-mycPT1 cells (b) and D1.5 cells (c) show a less cell viability during 12-60 hours of CDDP treatment, compared with the corresponding vector transfected cells
Since c-Myc elicited opposite effects to D1 on the response to CDDP, we transfected the D1.5 cells with the c-myc construct and carried out RT-PCR assay to confirm the increased c-myc expression in the transfected cells, compared with the vector transfectants [Figure 3a]. When treated with 2 μM CDDP, the c-myc transfectants showed a decreased cell viability between 24 and 48 hours post treatment, compared with the vector counterparts [Figure 3c], which shows for the first time that c-Myc imposed sensitivity to CDDP occurs also in cells ectopically expressing D1.[10]
D1 inhibits c-myc but induces several members of the NF-κB and Bcl-2 families
Since we previously observed that D1 imposed chemoresistance by increasing NF-κB activity, we wonder whether it is due to increased expression of NF-κB members. We transfected the Ela-mycPT1 cells with the pcDNA3.1-D1 construct and performed RT-PCR and western blot assays to confirm the increased mRNA and protein levels of D1, relative to the levels in the empty vector transfectants [Figure 4a]. When treated with 2 μM CDDP for 48 hours, the D1 transfected cells manifested increased mRNA levels of the RelA, RelB, Nfκb1 and Nfκb2 genes, compared with the vector counterparts, while the level of the IκBα was not obviously affected [Figure 4b]. The BIRC5 mRNA level was also increased [Figure 4b].
Figure 4.
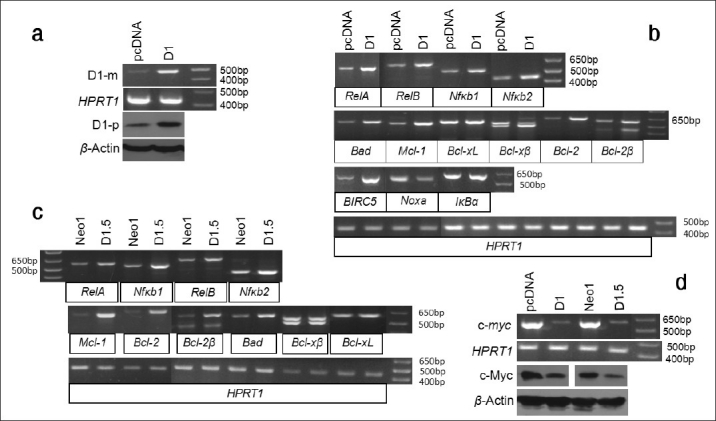
Effects of D1 on BIRC5 and several members of the NF-κ B and Bcl-2 families. (a) RT-PCR and western blot assays show that Ela-mycPT1 cells transfected with D1 cDNA express increased D1 mRNA (D1-m) and protein (D1-p) levels, compared with the cells transfected with the pcDNA3.1-neo empty vector. HPRT1 or β-actin is used as a loading control. (b) RT-PCR detection of the mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, IκBα , Bad, MCl-1, Bcl-xL, Bcl-xβ , Bcl-2, Bcl-2β , BIRC5 and Noxa in the D1-transfected Ela-mycPT1 cells and the vector-transfected counterparts, with HRPT1 as the loading control. (c) RT-PCR detection of the mRNA levels of the RelA, RelB, Nfκb1, Nfκ b2, Mcl-1, Bcl-2, Bcl-2β , Bad, Bcl-xL and Bcl-xβ in the D1 expressing D1.5 cells and the vector expressing Neo1 cells, with HPRT1 as the loading control. (d) RT-PCR or western blot detection of the c-myc mRNA or c-Myc protein level in the D1 cDNA or pcDNA3.1-neo vector transfected Ela-mycPT1 cells as well as in the D1.5 and Neo1 clones, with HPRT1 or β-actin as the corresponding loading control.
In the D1 transfected cells, the mRNA levels of Bad, Mcl-1, Bcl-2 and one of its variants, Bcl-2β,[22] were increased while Noxa was decreased and the Bcl-XL was not affected [Figure 4b]. Bxl-xS was not included because it was not detectable in Ela-mycPT1 cells in our previous study (data not shown). A third Bcl-x splice variant, Bcl-xβ,[22,25] was detected as two bands in agarose gel [Figure 4b]. Purifying and then sequencing these two bands revealed that the faster migrating (the bottom) band was Bcl-xβ, the level of which was higher in the D1 transfectants than the vector counterparts, while the slower migrating (the top) band was the musculus retinoic acid induced 14 (Mrai-14) transcript detected by mis-priming in PCR. Bcl-2β was also detected as two bands, both of which showed a higher level in the D1 transfectants than in the vector counterparts. Sequencing these two bands revealed that the slower migrating (the top) band is Bcl-2β while the faster migrating (the lower) band is a chimeric RNA formed between Nek9 and Bcl-2 due to mis-priming of the reverse primer to the Nek9 mRNA. Thus, the higher level of this chimera in the D1 transfectants is likely because of a higher Bcl-2β level. The intron sequences of Bcl-x and Bcl-2 are too short for us to design a better reverse primer to improve the PCR specificity.
We also determined expression of these genes in the D1.5 and Neo1 clones and obtained similar results [Figure 4c]. Moreover, we determined the c-myc level in the D1 transfected Ela-mycPT1 cells and the D1.5 cells, and surprisingly found that it was decreased compared with the corresponding vector counterparts [Figure 4d].
We also transfected MDA-MB231 human breast cancer cells with the D1 cDNA construct and performed RT-PCR and western blot assays to confirm the increased mRNA and protein levels of D1 in the D1 transfectants, compared with the vector counterparts [Figure 5a]. When treated with 2 μM CDDP for 48 hours, the D1 transfected cells also manifested an upregulation of the mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, Bcl-2β, Mcl-1 and several BIRC5 variants [26] as well as a down-regulation of Noxa and c-myc [Figure 5b]. Thus, the effects of D1 on these apoptosis-regulating genes are not specific to the mouse pancreatic cancer cell line, but rather they occur in human and in other types of cancer cells as well.
Figure 5.
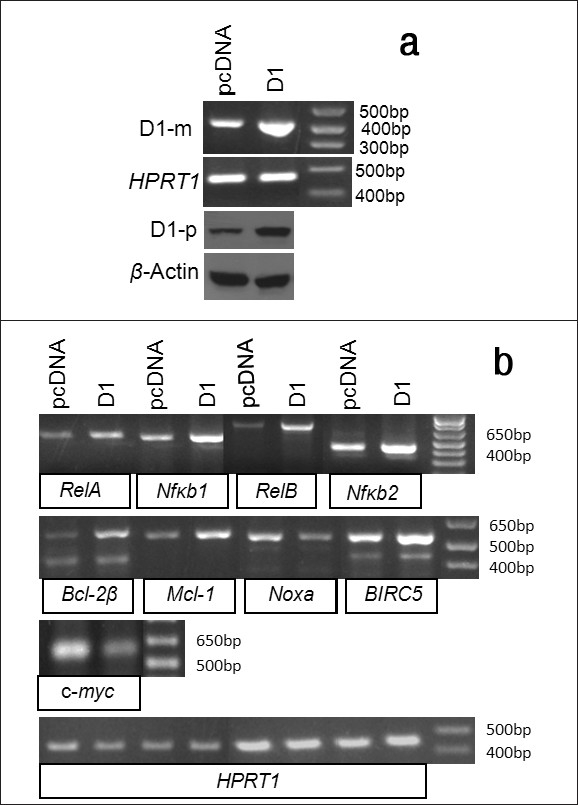
The effects of D1 on the expression of apoptosis regulating genes in MDA-MB231 human breast cancer cells. (a)RT-PCR or western blot detection of the D1 mRNA (D1-m) or protein (D1-p) level in the MDA-MB231 cells transfected with a D1 cDNA construct or the pcDNA3.1-neo vector, with HPRT1 or β-actin as the corresponding loading control. (b) RT-PCR detection of the mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, Bcl-2μ, Mcl-1, Noxa, BIRC5 and c-myc genes in the D1 cDNA or vector transfected MDA-MB-231 cells, with HPRT1 as the loading control
Wondering whether the effect of D1 on the expression of these genes involves CDK4 or CDK6 kinase activity, we determined the expression of these genes in the cells treated simultaneously with 2 μM CDDP and 1 μM NPCD for 48 hours. Similar to what is described above for the cells treated with only CDDP [Figure 5], the D1 transfected Ela-mycPT1 cells [Figure 6a] or the D1.5 cells [Figure 6b] still manifested increased mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, Bad, Mcl-1, Bcl-2 and BIRC5 as well as decreased levels of Noxa and c-myc, compared with the responding vector counterparts. Wondering whether NPCD alone affects the expression of these genes, we also treated the D1 transfected Ela-mycPT1 cells and the D1.5 cells with 1 μM NPCD alone for 48 hours (without concomitant CDDP) and determined the gene expression. The results showed that the mRNA levels of most of these genes were not obviously changed by NPCD, except Bad that was slightly induced [Figure 6c and d]. Collectively, these results showed that inhibition of CDK4 or CDK6 activity by a low concentration of NPCD, with or without a concomitant CDDP treatment, did not obviously influence the effects of D1 on the mRNA levels of most apoptosis regulating genes studied in the Ela-mycPT1 cells.
Figure 6.
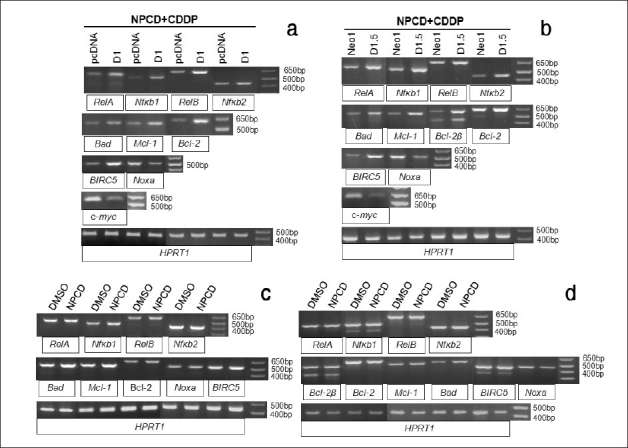
Effects of CDK4 and CDK6 inhibitor NPCD on the regulation of the apoptosis regulating genes by D1. (a) RT-PCR detection of the mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, Bad, Mcl-1, Bcl-2, Noxa, BIRC5 and c-myc in the D1 cDNA or vector transfected Ela-mycPT1 cells treated with both CDDP and NPCD for 48 hours. (b) RT-PCR detection of the mRNA levels of the above mentioned genes in D1.5 and Neo1 clones treated with both CDDP and NPCD for 48 hours. (c and d) RT-PCR detection of the mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, Bad, Mcl-1, Bcl-2, Noxa and BIRC5 in the D1 cDNA transfected Ela-mycPT1 (c) and D1.5 cells (d) treated with NPCD alone. HPRT1 is used as the loading control for all the above RT-PCR assays
c-Myc inhibits D1 and several members of the NF-κB and Bcl-2 families
To examine whether c-myc imposed chemosensitivity is associated with regulation of the above-studied apoptosis regulating genes, we transfected the Ela-mycPT1 cells with a c-myc cDNA construct or the pcDNA3.1-hygro vector as control. RT-PCR and western blot assays confirmed increased mRNA and protein levels of the c-myc, compared with the vector transfectants [Figure 7a]. When treated with 2 μM CDDP for 48 hours, the c-myc transfected cells manifested decreased mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, Mcl-1, Bcl-2 and Bcl-2β, compared with the vector transfectants, while the levels of Bcl-xL and Bel-xβ remained unchanged [Figure 7b].
Figure 7.
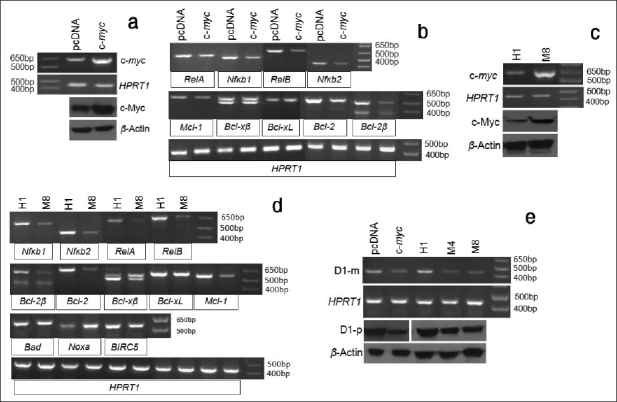
Effects of c-Myc protein on the expression of apoptosis regulating genes. (a) RT-PCR and western blot assays show a higher c-myc mRNA (c-myc) and protein (c-Myc) level in the c-myc cDNA transfected Ela-mycPT1 cells, compared with the pcDNA3.1-hygro vector transfected counterparts. HPRT1 or β-actin is used as a loading control. (b) RT-PCR detection of the mRNA levels of the RelA, RelB, Nfκb1, Nfκb2, Mcl-1, Bcl-xβ, Bcl-xL, Bcl-2, and Bcl-2β in c-myc cDNA or the empty pcDNA3.1-hygro vector transfected Ela-mycPT1 cells, with HPRT1 as the loading control. (c) the c-myc mRNA (c-myc) or protein (c-Myc) level in the H1 and M8 clones of the Ela-mycPT4 cells, with HPRT1 or β-actin as the corresponding loading control. (d) RT-PCR detection of the mRNA levels of the Nfκ b1, Nfκ b2, RelA, RelB, Bcl-2β , Bcl-2, Bcl-xβ, Bcl-xL, Mcl-1, Bad, Noxa and BIRC5 genes in the H1 and M8 clones. (e) The D1 mRNA (D1-m) and protein (D1-p) level is lower in the c-myc cDNA transfected Ela-mycPT1 cells as well as in M4 and M8 c-myc expressing cell clones, in relative to the corresponding vector (pcDNA3.1-hygro and H1) counterparts. HPRT1 or β-actin is used as a loading control.
We also determined the mRNA levels of these genes in M8 and H1 clones of the Ela-mycPT4 cell line that were previously established by us to stably express the c-myc and the pcDNA3.1-hygro empty vector, respectively.[10] The parental Ela-mycPT4 cell line was established by us from a different pancreatic tumor developed in a different Ela-myc transgenic mouse compared with the Ela-mycPT1 cell line.[10] As shown in Figure 7c, the M8 cells indeed expressed a higher level of c-myc mRNA and protein detected by RT-PCR and western blot assays, respectively, compared with the H1 cells. Relative to H1 cells, the M8 cells treated with 2 μM CDDP for 48 hours also showed lower mRNA levels of the Nfκb1, Nfκb2, RelA, RelB, Bcl-2, Bcl-2β , and Mcl-1 [Figure 7d]. Bcl-xL and Bcl-x β levels may also be slightly lower, considering that the HPRT1 used as the loading control for these two variants was slightly lower in the H1 cells. Bad and BIRC5 were similar between M8 and H1 cells while the level of Noxa was higher in M8 cells [Figure 7d]. Also interestingly, the mRNA and protein level of D1 was lower in the c-myc transfected Ela-mycPT1 cells and the M8 cells treated with 2 μM CDDP, compared with the corresponding vector controls [Figure 7e]. To further confirm this finding, another c-myc expression clone, M4, [10] was treated with CDDP and then determined for the D1 level. A lower mRNA and protein level of D1 was observed in M4 cells as well, compared with the H1 cells [Figure 7e]. Taken together, these results showed that c-Myc has opposite effects to D1 on these apoptosis regulating genes in the cells treated with CDDP. Moreover, c-myc expressing cells also manifested a lower D1 level than the vector counterparts when the cells were treated with CDDP, similar to our previous observation in the c-myc expressing cells before CDDP treatment.[10]
DISCUSSION
We have previously reported that high D1 and c-Myc levels render Ela-mycPT1 mouse pancreatic cancer cells resistant and sensitive, respectively, to several chemotherapeutic agents, including CDDP.[7,10] D1 acts via induction of NF-κB activity and stabilization of the Bcl-2 protein upon treatment with these chemotherapeutic agents,[7] whereas c-Myc acts by downregulation of NFκB activity and D1 expression.[10] The present study confirms these novel functions of D1 and c-Myc and discloses some underlying mechanisms.
Increased c-Myc and decreased D1 may be mechanisms of CDDP cytotoxicity
CDDP is known to elicit cytotoxicity by binding to DNA and then causing DNA damage.[27] A novel finding of the present study is that CDDP decreases the level of D1 after an initial increase on the first day. This transient increase is likely to be a cellular response to survive the assault by CDDP, but CDDP eventually decreases D1 as part of its killing mechanism. We also observe for the first time that CDDP increases the c-myc expression, which is sustained after D1 has leveled off. These findings are interesting for two reasons: 1) It is the first time to observe that cell demise, although induced by a drug, is associated with increased expression of the endogenous c-myc at both mRNA and protein levels, although ectopic c-Myc is well known to be capable of inducing apoptosis.[8] 2) Although ectopic c-Myc has been shown to inhibit D1, including in M4 and M8 cells shown by us previously,[10] there still is a dearth of evidence for such effect from the endogenous allele of c-myc. The reciprocal relationship between D1 and c-Myc observed in the present study suggests that the endogenous c-Myc may inhibit D1 and, vice versa, the endogenous D1 may also inhibit c-myc as discussed below. In short, an increase in c-Myc and a slightly latent decrease in D1 may be two previously unrecognized mechanisms for CDDP to elicit its cytotoxicity [Figure 8]. Increased c-Myc is known to be capable of inducing DNA damage under some situations.[28] It is thus possible that increased c-Myc may also contribute to the induction of DNA damage by CDDP in cancer cells.
Figure 8.
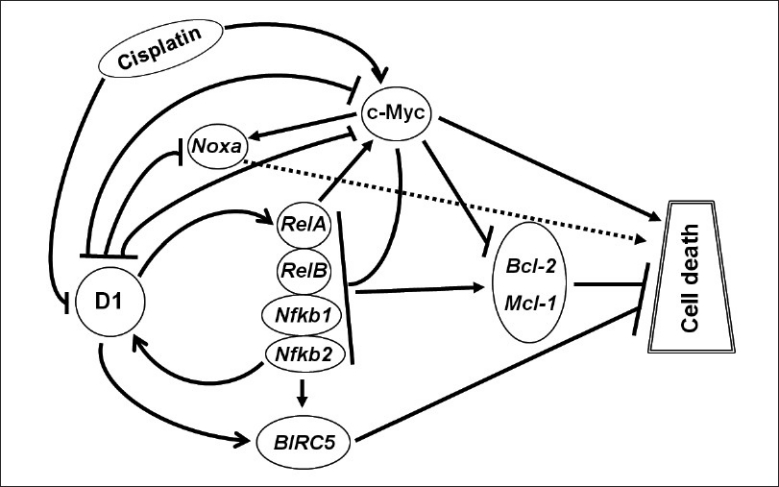
Depiction of how D1 and c-Myc proteins regulate CDDP-induced cell death, with arrow indicating induction and hyphen (-|) indicating inhibition. CDDP causes an increase in the c-Myc level but a slightly latent decrease in the D1 level. D1 induces but c-Myc inhibits RelA, RelB, Nfκb1 and Nfκb2 expression, resulting in an increased or decreased NF-κB activity, respectively. The expression of Bcl-2 and Mcl-1 as critical NF-κB targets is changed accordingly. D1 also induces BIRC5 but inhibits Noxa while c-Myc induces Noxa without affecting BIRC5 expression. c-Myc is known to have other molecular pathways to cause cell death but whether D1 also has other pathways is currently unclear. Moreover, D1 and c-Myc may repress each other, thus affecting the other's apoptotic pathway as a secondary (i.e. indirect) mechanism to elicit its effects on cell death
D1 regulates expression of some NF-κB and Bcl-2 family members during CDDP treatment
Although in the previous study we showed that D1 induced the NF-κB activity, what caused and what followed the induction were unknown.[7] We show in the present study that D1 increases the expression of several key NF-κB family members, including RelA, RelB, Nfκb1 and Nfκb2, which may be a significant part of the mechanism for the increased NF-κB activity during CDDP treatment. NF-κB is known to utilize multiple mechanisms to raise chemoresistance of cancer cells, the major one of which is to upregulate BIRC5 and several pro-survival members of the Bcl-2 family. Supporting this mechanism, we observe upregulation of BIRC5 as well as Bad, Mcl-1 and Bcl-2 in D1 expressing cells, compared with the vector expressing counterparts during CDDP treatment. Bcl-2β is found to be increased as well, but its meaning is currently unclear because little has been known about this mRNA variant.[22] Nevertheless, the fact that both Bcl-2 and Bcl-2β splice variants are induced by D1 suggests that the increase may occur at the transcriptional level, since splicing immediately follows transcription. Bcl-xL is not obviously affected by D1 although Bcl-xβ, the function of which is basically unknown,[22] is transiently increased in the D1 transfectants. D1 also decreases Noxa, a pro-apoptotic member in the Bcl-2 family. A collective outcome of these changes in BIRC5 and Bcl-2 family members, presumably due in part to increased NF-κB activity, should be a strong resistance to apoptosis. It needs to be pointed out that NF-κB and BIRC5 (survivin) proteins have also been shown to activate D1. Therefore, a mutual activation may constitute a positive feedback loop to further augment the chemoresistant effect of D1. A caveat needs to be given that Bcl-2 is a huge family of genes, and many of its members are not studied in the present study but some of them may be regulated by D1 to mediate cheomoresistance as well.
As reviewed by one of us recently,[6] D1 functions via three different mechanisms as follows: 1) binding to CDK4/6 to form a holoenzyme to phosphorylate pRB or other substrates, 2) forming a complex with CDK4/6 without involving their kinase activity, or 3) acting via a CDK4/6-independent mechanism. The third mode of action has so far been observed only in regulation of genes' transcription, which is achieved by binding of D1 to some transcription factors such as steroid hormone receptors. Few, if any, of these transcriptional regulations involve CDK4/6, to our knowledge. NPCD is an indolocarbazole derivative that inhibits CDK4 and CDK6 at tens of nanomoles and causes cytotoxicity only when the concentration is higher than several micromoles and the treatment is longer than 3 days, as reported by us recently.[23] In the present study, we carefully set the concentration at 1 μM and the duration at 2 days to avoid its cytotoxicity. Under this condition, NPCD does not seem to mediate the effects of D1 on the cytotoxicity of CDDP and on the expression of the apoptosis regulating genes studied. Therefore, these effects of D1 may not involve the kinase activity of CDK4/6, thus in line with the third mechanism of D1 described above, although it is unclear from our data whether the regulation by D1 occurs at the transcriptional level. We also do not know whether these CDK proteins per se are involved via the second mechanism aforementioned, but it has been reported that depletion of the endogenous CDK4 in human oral squamous cell carcinoma cells did not affect the IC50 of CCDP in cdk4-depleted cells.
c-Myc has the opposite effects to D1 on the NF-κB and Bcl-2 family members
Opposite to D1, ectopic c-Myc is shown in this study to decrease the mRNA levels of RelA, RelB, Nfκb1 and Nfκb2, which is in line with several reports that c-Myc suppresses the expression of several components of the NF-κB family.[29–32] We also observe that c-Myc downregulates Bcl-2 and Mcl-1 but induces Noxa. It has been reported that c-Myc selectively suppresses the expression of Bcl-2 but not Bcl-xL or Mcl-1 in immortal myeloid cells[33] while it suppresses Bcl-xL in mouse embryonic fibroblasts (MEF) cells.[34] Consistent with the notion that the effect of c-Myc on Bcl-2 members is cell type specific, we do not observe obvious effect of c-Myc on Bcl-xL and Bad. Nevertheless, the effects of c-Myc observed herein may constitute another mechanism for the cytotoxicity of CDDP and for D1-imposed resistance to CDDP, since D1 downregulates the c-myc [Figure 8]. Therefore, the induction of the NF-κB family members by D1 may also be secondary via the inhibition of the c-myc expression [Figure 8].
CONCLUSIONS
In this study, we confirm the previous findings that D1 renders pancreatic cancer cells resistant to CCDP, whereas c-Myc imposes sensitivity. We show for the first time that CDDP may elicit its cytotoxicity via induction of c-Myc and repression of D1. The effects of D1 and c-Myc on the chemoresponse are elicited via regulation of expression of several members of the NF-kB and Bcl-2 families as well as BIRC5. Moreover, D1 and c-Myc may repress each either, which constitutes another mechanism.
AUTHOR'S PROFILE
Dr. Dezhong Joshua Liao, Dr. Joshua Liao is an associate professor at Hormel Institute and the corresponding author of this manuscript
Dr. Ying-xia Li, Dr. Ying-xia Li is a professor at Fudan University, China
Dr. Ayman El-Kady, Dr. Ayman Ei-Kady was a postdoctoral fellow at Hormel Institute but recently returned back to his home country, Egypt, as a faculty in the Department of Zoology, Faculty of Science, Alexandria University, Alexandria, Egypt
Dr. Yuan Sun, Dr. Yuan Sun is a postdctoral fellow at Hormel Institute
ACKNOWLEDGMENTS
The study was supported by a grant from NIH RO1CA100864 to D. J. Liao. We thank Dr. Fred Bogott at Austin Medical Center, Austin of Minnesota, USA, for his excellent English editing of the revised manuscript.
Contributor Information
Ying-xia Li, Email: liyx417@fudan.edu.cn.
D Joshua Liao, Email: djliao@hi.umn.edu.
REFERENCES
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 3.Brown DB, Gonsalves CF, Yeo CJ, Witkiewicz AK, Carr BI. One year survival with poorly differentiated metastatic pancreatic carcinoma following chemoembolization with gemcitabine and cisplatin. Oncol Rep. 2010;24:767–9. doi: 10.3892/or_00000919. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez SE, Trevino JG. Current adjuvant and targeted therapies for pancreatic adenocarcinoma. Curr Med Chem. 2008;15:1674–83. doi: 10.2174/092986708784872348. [DOI] [PubMed] [Google Scholar]
- 5.Liao DJ, Thakur A, Wu J, Biliran H, Sarkar FH. Perspectives on c-Myc, Cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit Rev Oncog. 2007;13:93–158. doi: 10.1615/critrevoncog.v13.i2.10. [DOI] [PubMed] [Google Scholar]
- 6.Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57–67. doi: 10.4161/cc.10.1.14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biliran H, Jr, Wang Y, Banerjee S, Xu H, Heng H, Thakur A, et al. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin Cancer Res. 2005;11:6075–86. doi: 10.1158/1078-0432.CCR-04-2419. [DOI] [PubMed] [Google Scholar]
- 8.Wang C, Tai Y, Lisanti MP, Liao DJ. c-Myc induction of programmed cell death may contribute to carcinogenesis: a perspective inspired by several concepts of chemical carcinogenesis. Cancer Biol Ther. 2011;11:615–26. doi: 10.4161/cbt.11.7.14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao JD, Adsay NV, Khannani F, Grignon D, Thakur A, Sarkar FH. Histological complexities of pancreatic lesions from transgenic mouse models are consistent with biological and morphological heterogeneity of human pancreatic cancer. Histol Histopathol. 2007;22:661–76. doi: 10.14670/hh-22.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biliran H, Jr, Banerjee S, Thakur A, Sarkar FH, Bollig A, Ahmed F, et al. c-Myc-induced chemosensitization is mediated by suppression of cyclin D1 expression and nuclear factor-kappa B activity in pancreatic cancer cells. Clin Cancer Res. 2007;13:2811–21. doi: 10.1158/1078-0432.CCR-06-1844. [DOI] [PubMed] [Google Scholar]
- 11.Prasad S, Ravindran J, Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Mol Cell Biochem. 2010;336:25–37. doi: 10.1007/s11010-009-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-kappaB addiction and its role in cancer: ‘one size does not fit all’. Oncogene. 2011;30:1615–30. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng C, Yin Q, Wu H. Structural studies of NF-kappaB signaling. Cell Res. 2011;21:183–95. doi: 10.1038/cr.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799:775–87. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sebens S, Arlt A, Schafer H. NF-kappaB as a molecular target in the therapy of pancreatic carcinoma. Recent Results Cancer Res. 2008;177:151–64. doi: 10.1007/978-3-540-71279-4_17. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki J, Ogawa M, Muto S, Itai A, Isobe M, Hirata Y, et al. Novel IkB kinase inhibitors for treatment of nuclear factor-kB-related diseases. Expert Opin Investig Drugs. 2011;20:395–405. doi: 10.1517/13543784.2011.559162. [DOI] [PubMed] [Google Scholar]
- 17.Witzel II, Koh LF, Perkins ND. Regulation of cyclin D1 gene expression. Biochem Soc Trans. 2010;38:217–22. doi: 10.1042/BST0380217. [DOI] [PubMed] [Google Scholar]
- 18.Diehl JA, Benzeno S. Cyclin D1 and pancreatic carcinoma: a proliferative agonist and chemotherapeutic antagonist. Clin Cancer Res. 2005;11:5665–7. doi: 10.1158/1078-0432.CCR-05-1016. [DOI] [PubMed] [Google Scholar]
- 19.Papanikolaou V, Iliopoulos D, Dimou I, Dubos S, Kappas C, Kitsiou-Tzeli S, et al. Survivin regulation by HER2 through NF-kappaB and c-myc in irradiated breast cancer cells. J Cell Mol Med. 2011;15:1542–50. doi: 10.1111/j.1582-4934.2010.01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luqman S, Pezzuto JM. NFkappaB: a promising target for natural products in cancer chemoprevention. Phytother Res. 2010;24:949–63. doi: 10.1002/ptr.3171. [DOI] [PubMed] [Google Scholar]
- 21.Sarnico I, Lanzillotta A, Benarese M, Alghisi M, Baiguera C, Battistin L, et al. NF-kappaB dimers in the regulation of neuronal survival. Int Rev Neurobiol. 2009;85:351–62. doi: 10.1016/S0074-7742(09)85024-1. [DOI] [PubMed] [Google Scholar]
- 22.Akgul C, Moulding DA, Edwards SW. Alternative splicing of Bcl-2-related genes: functional consequences and potential therapeutic applications. Cell Mol Life Sci. 2004;61:2189–99. doi: 10.1007/s00018-004-4001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y, Li YX, Wu HJ, Wu SH, Wang YA, Luo DZ, et al. Effects of an Indolocarbazole-Derived CDK4 Inhibitor on Breast Cancer Cells. J Cancer. 2011;2:36–51. doi: 10.7150/jca.2.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ban J, Eckhart L, Weninger W, Mildner M, Tschachler E. Identification of a human cDNA encoding a novel Bcl-x isoform. Biochem Biophys Res Commun. 1998;248:147–52. doi: 10.1006/bbrc.1998.8907. [DOI] [PubMed] [Google Scholar]
- 26.Sampath J, Pelus LM. Alternative splice variants of survivin as potential targets in cancer. Curr Drug Discov Technol. 2007;4:174–191. doi: 10.2174/157016307782109652. [DOI] [PubMed] [Google Scholar]
- 27.Koberle B, Tomicic MT, Usanova S, Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta. 2010;1806:172–82. doi: 10.1016/j.bbcan.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Prochownik EV. c-Myc: linking transformation and genomic instability. Curr Mol Med. 2008;8:446–58. doi: 10.2174/156652408785747988. [DOI] [PubMed] [Google Scholar]
- 29.Keller U, Nilsson JA, Maclean KH, Old JB, Cleveland JL. Nfkb 1 is dispensable for Myc-induced lymphomagenesis. Oncogene. 2005;24:6231–40. doi: 10.1038/sj.onc.1208779. [DOI] [PubMed] [Google Scholar]
- 30.Keller U, Huber J, Nilsson JA, Fallahi M, Hall MA, Peschel C, et al. Myc suppression of Nfkb2 accelerates lymphomagenesis. BMC Cancer. 2010;10:348. doi: 10.1186/1471-2407-10-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klapproth K, Sander S, Marinkovic D, Baumann B, Wirth T. The IKK2/NF-{kappa}B pathway suppresses MYC-induced lymphomagenesis. Blood. 2009;114:2448–58. doi: 10.1182/blood-2008-09-181008. [DOI] [PubMed] [Google Scholar]
- 32.Schlee M, Hölzel M, Bernard S, Mailhammer R, Schuhmacher M, Reschke J, et al. C-myc activation impairs the NF-kappaB and the interferon response: implications for the pathogenesis of Burkitt's lymphoma. Int J Cancer. 2007;120:1387–95. doi: 10.1002/ijc.22372. [DOI] [PubMed] [Google Scholar]
- 33.Eischen CM, Packham G, Nip J, Fee BE, Hiebert SW, Zambetti GP, et al. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene. 2001;20:6983–93. doi: 10.1038/sj.onc.1204892. [DOI] [PubMed] [Google Scholar]
- 34.Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol Cell Biol. 2003;23:7256–70. doi: 10.1128/MCB.23.20.7256-7270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]