

Abstract
Protein farnesyltransferase (FTase) and protein geranylgeranyltransferase-I (GGTase-I) add 15- or 20-carbon lipids, respectively, to proteins that terminate with a CaaX motif. These posttranslational modifications of proteins with lipids promote protein interactions with membrane surfaces in cells, but the in vivo importance of the CaaX prenyltransferases and the protein lipidation reactions they catalyze remain incompletely defined. One study concluded that a deficiency of FTase was inconsequential in adult mice and led to little or no tissue pathology. To assess the physiologic importance of the CaaX prenyltransferases, we used conditional knockout alleles and an albumin–Cre transgene to produce mice lacking FTase, GGTase-I, or both enzymes in hepatocytes. The hepatocyte-specific FTase knockout mice survived but exhibited hepatocellular disease and elevated transaminases. Mice lacking GGTase-I not only had elevated transaminases but also had dilated bile cannaliculi, hyperbilirubinemia, hepatosplenomegaly, and reduced survival. Of note, GGTase-I–deficient hepatocytes had a rounded shape and markedly reduced numbers of actin stress fibers. Hepatocyte-specific FTase/GGTase-I double-knockout mice closely resembled mice lacking GGTase-I alone, but the disease was slightly more severe. Our studies refute the notion that FTase is dispensable and demonstrate that GGTase-I is crucial for the vitality of hepatocytes.
Keywords: protein farnesyltransferase, protein geranylgeranyltransferase, hyperbilirubinemia, hepatic steatosis, prelamin A, actin stress fibers
Protein farnesyltransferase (FTase) and protein geranylgeranyltransferase-I (GGTase-I) add 15-carbon farnesyl or 20-carbon geranylgeranyl lipids, respectively, to the cysteine in proteins that terminate with a CaaX motif (“C” is a cysteine; “a” is often an aliphatic amino acid; the “X” can be one of many residues). This posttranslational modification is generally called “protein prenylation” (1, 2). Cell culture studies have demonstrated that protein prenylation promotes the ability of proteins to associate with membrane surfaces (2). For example, when the farnesylation of H-RAS (a proto-oncogene implicated in the pathogenesis of human cancers) is eliminated, the protein loses its ability to reach the plasma membrane and relay the signals that promote cell growth (3–5). These studies prompted the pharmaceutical industry to develop FTase inhibitors (FTIs) for the treatment of cancer (6–9). GGTase-I inhibitors (GGTIs) and dual FTase/GGTase-I inhibitors have also been developed as potential anticancer therapies (10, 11).
Despite considerable interest in FTase as a therapeutic target, the in vivo importance of FTase for the vitality of mammalian cells and tissues has never been clearly defined. One group developed a knockout allele for Fntb (encoding the β-subunit of FTase) and concluded that FTase is required for embryonic development but that FTase deficiency is associated with little or no pathology in adult tissues (12). The authors suggested that the dispensability of FTase in the tissues of adult mice might relate, in part, to alternate prenylation of FTase substrates by GGTase-I (12). The conclusion that FTase is dispensable in adult tissues was somewhat surprising, considering the importance of this enzyme in yeast (13, 14). Nevertheless, the notion that FTase was not essential for mammalian cells and tissues seemed consistent with preclinical studies of FTIs, which were reported to have few adverse side effects (15, 16). But newer studies raised uncertainties about the notion that FTase is dispensable. A deficiency of FTase in keratinocytes led to severe alopecia (17). The fact that FTase deficiency caused significant pathology in one study (17) but not in another (12) indicated a need for additional studies on the physiologic importance of FTase in mammalian tissues.
The physiologic consequences of GGTase-I deficiency are also poorly defined. Early studies with GGTIs indicated that they were quite toxic (11). That finding, together with the observation that GGTase-I deficiency is lethal in budding yeast (13), led many to suspect that GGTase-I would be an essential protein in mammalian cells. However, more recent studies of GGTase-I inhibitors have found less toxicity (18, 19), and genetic models of GGTase-I deficiency (with a conditional knockout allele for Pggt1b, which encodes the β-subunit of GGTase-I) suggested that mammalian cells can survive in the absence of GGTase-I (20, 21). For example, inactivating GGTase-I in mouse pneumocytes retarded the growth of lung tumors without toxicity to normal lung tissue (20). Although GGTase-I–deficient lung tumors grew at a slower rate, they ultimately became quite large and caused the mice to die, implying that GGTase-I–deficient cells can survive in vivo (20). More recently, Khan et al. (22) created mice lacking GGTase-I in macrophages and found that GGTase-I–deficient macrophages were activated, leading to erosive arthritis. These genetic studies, along with conflicting information on the properties of GGTIs, underscored a need for a fresh look at the functional relevance of GGTase-I in mammalian cells.
Given the uncertainties about the functional importance of both FTase and GGTase-I in mammals, we decided to assess the impact of deficiencies of the two protein prenyltransferases, alone and in combination, in hepatocytes. We chose hepatocytes for three reasons. The first is that one can obtain very efficient recombination—and efficient gene inactivation—with the albumin–Cre transgene (23, 24). The second is that abnormalities in hepatocyte function can be readily detected with simple blood tests and histological studies. Third, it is straightforward to isolate and analyze hepatocytes from mouse models.
MATERIALS AND METHODS
Hepatocyte-specific Fntb and Pggt1b knockout mice
A conditional knockout allele for Fntb, Fntbfl, was recently generated by Liu et al. (21). Cre-mediated excision of the “floxed” segment of DNA removes the Fntb promoter, the translational start site, and exon 1. Fntb+ and Fntbfl alleles were identified by PCR with forward primer 5′–AGGATTTGGGTGAGGGAA–3′ and reverse primer 5′–AGCAGCCACCTGGAGACT–3′; Fntb+ and Fntbfl alleles yielded 250- and 320-bp amplicons, respectively. Mice harboring a Pggt1b conditional knockout allele, Pggt1bfl (20), were also used. Both Fntbfl/fl and Pggt1bfl/fl mice were bred with albumin–Cre (AlbCre) transgenic mice (24). In this article, hepatocyte-specific Fntb knockout mice (Fntbfl/flAlbCre) were designated “FntbΔ/Δ mice”; hepatocyte-specific Pggt1b knockout mice (Pggt1bfl/flAlbCre) were designated “Pggt1bΔ/Δ mice.” The AlbCre transgene was detected by PCR with forward primer 5′–GCATTACCGGTCGATGCAACGAGTGATGAG–3′ and reverse primer 5′–GAGTGAACGAACCTGGTCGAAATCAGTGCG–3′.
Mice were weaned at 21 days of age, housed in a virus-free barrier facility with a 12-h light-dark cycle, and fed a chow diet containing 4.5% fat. All experiments were approved by UCLA's Animal Research Committee.
Mouse primary hepatocytes
Primary hepatocytes were isolated by a two-step perfusion protocol based on previously described methods (25, 26). Briefly, mice were anesthetized by intraperitoneal injection of pentobarbitone (50 mg/kg). The abdominal cavity was opened and the liver was perfused via the portal vein, first with 37°C 10 mM HEPES-buffered saline for 5 min, and subsequently with 37°C 0.05% type I collagenase (Worthington Biochemical Corp., Lakewood, NJ) in 10 mM HEPES-buffered saline for 7 min at 5 ml/min. After perfusion, the liver was excised and chopped with scissors in Hepato-STIM medium (BD Biosciences, San Jose, CA) to disperse the cells. A series of five low-speed (50 g) centrifugation steps was performed to enrich the population of hepatocytes (and limit numbers of Kupffer and endothelial cells). The resulting cell pellet was resuspended, and an aliquot was taken to assess cell number and viability (by trypan blue exclusion). Hepatocyte preparations were then plated at a density of 2.0 × 106 cells on 60-mm collagen-coated plates (BD Biosciences) in Hepato-STIM medium containing 10% FBS (ATCC, Manassas, VA) containing pen/strep (ATCC) and Fungizone (Invitrogen, Carlsbad, CA) and allowed to adhere for 4 h. The medium was then removed; the cells washed with PBS and fresh medium was added.
Extraction of RNA, cDNA synthesis, and RT-PCR
Total RNA was extracted from freshly prepared primary hepatocytes with TRI reagent (Molecular Research Center), treated with DNase I (Ambion, Applied Biosystems), and reverse-transcribed with SuperScript III (Invitrogen). qRT-PCR reactions were performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems) with 50 ng cDNA, 200 nM oligonucleotides, and 10 μl SensiMixPlus SYBR (Bioline). Expression levels were calculated with the comparative cycle threshold method (27) and normalized to β2-microglobulin. To assess Fntb expression, we used oligonucleotide primers 5′–GACGGCTGCTACTCCTTCTG–3′ and 5′–TGCTGATGGAACATCCAATG–3′; for Pggt1b expression, we used primers 5′–GGGCATGGATATGAAGAAAGC–3′ and 5′–AAGGTGGATCCTCCATGAGAC–3′; for β2-microglobulin, we used primers 5′–TGGTGCTTGTCTCACTGACC–3′ and 5′–TATGTTCGGCTTCCCATTCT–3′.
Histology and immunohistochemistry
Livers were fixed in 10% paraformaldehyde (PFA), dehydrated for 24 h in 70% ethanol, and embedded in paraffin. Hematoxylin and eosin, Masson's trichrome, Sirius red, Periodic acid-Schiff (PAS), and reticulin stains were performed by UCLA's Department of Pathology. Oil Red O staining for neutral lipids was performed on frozen sections. For immunofluorescence studies, livers were embedded in Sakura Tissue-Tec OCT compound, frozen on dry ice, and 8-μm thick sections were cut with a cryostat. Sections were fixed in 3% PFA for 10 min, permeabilized with PBS containing 0.1% Tween-20, and incubated in blocking buffer (PBS containing 10% fetal bovine serum, 0.2% BSA, 1 mM CaCl2, and 1 mM MgCl2) for 1 h. Sections were then incubated for 1 h at 4°C with Alexa Fluor 488–labeled phalloidin (1:200, Invitrogen) diluted in blocking buffer containing 0.2% Triton X-100. After washing the slides, the tissue sections were postfixed with 3% PFA for 10 min, incubated in PBS/DAPI for 10 min, washed twice with PBS, and then mounted with Prolong-Gold Antifade reagent (Invitrogen).
For immunocytochemical studies on primary hepatocytes, cells were grown on collagen I–coated cover slips (BD Biosciences), fixed in 3% PFA, permeabilized with 0.2% Triton X-100, and blocked with PBS containing 10% fetal bovine serum and 0.2% BSA (28). Cells were incubated for 2 h with primary antibodies. After washing, cells were incubated with secondary antibodies for 1 h. After washing, slides were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen). The following antibodies and probes were used: lamin A (rabbit, 1:200, Santa Cruz Biotechnology); prelamin A (7G11, 1:50) (29); Alexa Fluor α-tubulin (1:400, Invitrogen); and Alexa Fluor 488–phalloidin (1:200, Invitrogen). Alexa Fluor 488– and Alexa Fluor 568–conjugated secondary antibodies were used at a 1:200 dilution.
Images were recorded with an Axiovert 200M microscope equipped with an AxioCam MRm and an ApoTome (all from Zeiss). Nuclear shape (2,000 cells/genotype) was assessed in a blinded fashion as described (29–32).
Protein extraction and Western blots
Protein extracts were prepared from primary hepatocytes with a urea-containing solubilization buffer (28, 33). Proteins were size-fractionated on 4–12% polyacrylamide Bis-Tris gels (Invitrogen) and then transferred to nitrocellulose for Western blotting. We used antibodies for the following proteins: lamin A/C (goat, 1:400, Santa Cruz Biotechnology); FTaseβ (rabbit, 1:200, Santa Cruz Biotechnology); HDJ-2 (mouse, 1:400, NeoMarkers); actin (goat, 1:1000, Santa Cruz Biotechnology); RAP1A (goat, 1:200, Santa Cruz Biotechnology); and prelamin A (29). After washing, the membranes were incubated with 1:5000 IRDye secondary antibodies (Rockland). Signals were detected with an Odyssey infrared imaging scanner (Li-Cor Biosciences).
Flow cytometry and cell sorting
Freshly prepared primary hepatocytes were analyzed by flow cytometry in UCLA's Flow Cytometry Laboratory (http://cyto.mednet.ucla.edu/). Briefly, cells were stained with propidium iodide (PI) and analyzed on a FACScan flow cytometer (BD Biosciences). Cell cycle analysis was performed with ModFit LT software (Verity Software House, Topsham, ME). To segregate cells based on DNA content, samples were sorted on a BD FACSAriaII sorter (BD Biosciences), and PI fluorescence was collected after a 670 LP filter with a 695/40-nm BP filter.
In vivo detection of apoptosis
Active caspases 3 and 7 in hepatocytes were detected with an in vivo technique (FLIVO, Immunochemistry Technologies). FLIVO is a fluorescently labeled inhibitor of caspases 3 and 7 that binds to active caspases. Mice were injected via the tail vein with the fluorescent probe (FAM-FLIVO), and livers were excised and frozen 1 h later. Then 8-µm sections were cut and fixed for 10 min in 3% PFA. The slides were incubated in PBS/DAPI for 10 min, washed twice with PBS, and mounted with Prolong-Gold Antifade reagent (Invitrogen). The fluorescent signal (green) was imaged with an Axiovert 200M microscope equipped with an AxioCam MRm (Zeiss).
Liver cholesterol and triglyceride measurements
Neutral lipids were extracted from livers of Fntb+/+ and FntbΔ/Δ mice as described (34). Livers were weighed and homogenized in a 2:1 solution of chloroform and methanol. After 2-h incubation, 0.5 ml of 0.1 M NaCl was added, and the samples were mixed. After centrifugation, the organic phase was collected and dried. Samples were solubilized in ethanol containing 1% NP-40, and triglyceride and cholesterol levels were measured with enzymatic kits (Sigma Triglyceride kit and the Wako Cholesterol kit).
Statistical analyses
Body weight curves were compared with repeated-measures ANOVA and the log-rank test. Differences in the percentages of misshapen nuclei were assessed with a χ2 test. Survival differences were assessed with a Kaplan-Meier test.
RESULTS
Hepatocyte-specific Fntb knockout mice
We used an Fntb conditional knockout allele, Fntbfl, and an albumin–Cre (AlbCre) transgene to produce mice lacking Fntb in hepatocytes (Fntbfl/flAlbCre+, designated FntbΔ/Δ mice). Cre-mediated recombination was efficient; the β-subunit of FTase (FTβ) was undetectable in hepatocytes of FntbΔ/Δ mice, as judged by Western blots with an FTβ-specific antibody (Fig. 1A). Also, protein farnesylation in hepatocytes was strongly inhibited, as judged both by an accumulation of nonfarnesylated HDJ-2 (Fig. 1A) and the appearance of prelamin A in cells (Fig. 1A, B). Consistent with these findings, Fntb transcript levels in FntbΔ/Δ livers and isolated hepatocytes were very low (Fig. 1C).
Fig. 1.

Inactivating FTase in the liver. (A) Western blot of extracts from primary hepatocytes from Fntb+/+ and Fntbfl/flAlbCre+ (FntbΔ/Δ) mice with antibodies against the β-subunit of FTase (FTβ), prelamin A, lamin A/C, and HDJ-2. Actin was used as a loading control. (B) Immunohistochemical staining of primary hepatocytes from Fntb+/+ and FntbΔ/Δ mice with antibodies against lamin A (green) and prelamin A (red). DNA was visualized with DAPI (blue). Scale bar, 20 µm. (C) Quantitative RT-PCR analysis of Fntb expression with RNA prepared from whole liver or primary hepatocytes from Fntb+/+, Fntbfl/+AlbCre+ (Fntb+/Δ), and FntbΔ/Δ mice (n = 4 mice/group). Data were normalized to β2-microglobulin, and expression levels were compared with those of Fntb+/+ mice (set at 1.0). Error bars indicate SEM.
FntbΔ/Δ mice were born at the expected Mendelian frequency, appeared normal at birth, were fertile, and remained alive after 1 year of observation. However, FntbΔ/Δ mice grew slower than wild-type littermates (supplementary Fig. I-A, B), and the liver/body weight ratio was increased (supplementary Fig. I-C). Histological examination of livers from 12-month-old FntbΔ/Δ mice revealed steatosis, increased collagen staining, apoptosis, and loss of hepatocytes (Fig. 2). Steatosis in livers from FntbΔ/Δ mice was confirmed by Oil Red O staining (Fig. 3A, B) and electron microscopy (Fig. 3C, D). Liver triglycerides were increased in FntbΔ/Δ mice (Fig. 3E), but there were no significant changes in liver cholesterol levels (Fig. 3F). The serum alanine transaminase (ALT) and aspartate aminotransferase (AST) levels in FntbΔ/Δ mice were markedly elevated, but bilirubin and albumin levels were within normal limits (Table 1).
Fig. 2.

Fntb deficiency in the liver causes steatosis, fibrosis, and apoptosis. (A, B) Hematoxylin and eosin–stained sections of liver from 12-month-old Fntb+/+ and FntbΔ/Δ mice, revealing disordered hepatic lobules and steatosis in FntbΔ/Δ mice. (C, D) Masson's trichrome stain revealing increased collagen (blue) in the liver of the FntbΔ/Δ mouse, along with steatosis and ballooning of hepatocytes. (E, F) Reticulin stain (black) revealing a disorganized liver architecture and regions with collapse of reticulin fibers (black arrow), corresponding to areas of hepatocyte loss. Images in A–F were recorded with a 20× objective. Scale bar, 50 µm. (G, H) Fluorescence microscopy revealing active caspases 3 and 7 in the livers of 6-month-old Fntb+/+ and FntbΔ/Δ mice (n = 3 mice/group were examined). Scale bar, 10 µm.
Fig. 3.

Increased lipids in the liver of FntbΔ/Δ mice. (A, B) Oil Red O–stained liver sections of 6-month-old Fntb+/+ and FntbΔ/Δ mice (n = 3 mice/group were examined). Sections were counterstained with Mayer's hematoxylin (blue). Scale bar, 100 µm. (C, D) Electron micrographs of livers from Fntb+/+ and FntbΔ/Δ mice. L, lipid droplets; N, nucleus. Magnification, ×14,000; scale bar, 2.0 μm. (E, F) Percentage triglycerides (E) and cholesterol (F) mass in livers of Fntb+/+ and FntbΔ/Δ mice (n = 8 mice/group). Error bars indicate SEM.
TABLE 1.
Serum chemistry studies in wild-type and Fntb mice
| Male |
Female |
||||||
| Wild-type | Fntb | P | Wild-type | Fntb | P | Normal Range | |
| ALT (U/L) | 39 ± 5 | 531 ± 79 | <0.0001 | 45 ± 6 | 462 ± 64 | <0.0001 | 7–227 |
| AST (U/L) | 159 ± 36 | 585 ± 60 | <0.0001 | 104 ± 20 | 546 ± 84 | <0.0001 | 37–329 |
| ALP (U/L) | 37 ± 14 | 152 ± 17 | <0.0001 | 68 ± 13 | 185 ± 28 | <0.0001 | 13–291 |
| CHOL (mg/dl) | 80 ± 16 | 92 ± 24 | NS | 94 ± 8 | 102 ± 10 | NS | 34–219 |
| GLU (mg/dl) | 296 ± 27 | 183 ± 27 | NS | 232 ± 13 | 190 ± 14 | NS | 46–279 |
| TRIG (mg/dl) | 99 ± 10 | 89 ± 13 | NS | 153 ± 18 | 83 ± 7 | NS | 16–193 |
| DBILI (mg/dl) | 0.3 ± 0.01 | 0.2 ± 0.03 | NS | 0.3 ± 0.01 | 0.3 ± 0.01 | NS | 0–0.3 |
| TBILI (mg/dl) | 0.4 ± 0.04 | 0.3 ± 0.01 | NS | 0.3 ± 0.01 | 0.4 ± 0.02 | NS | 0.1–1.1 |
| ALB (g/dl) | 2.8 ± 0.11 | 2.7 ± 0.31 | NS | 3.1 ± 0.12 | 3.0 ± 0.10 | NS | 2.5–5.4 |
n = 12 mice/group, including 6 males and 6 females; 6 months of age. Mean ± SEM.
ALB, albumin; CHOL, cholesterol; DBILI, direct bilirubin; GLU, glucose; NS, not significant; TBILI, total bilirubin.
Several abnormalities were readily apparent in hepatocytes isolated from the livers of FntbΔ/Δ mice. First, we observed an increased frequency of nuclei with nuclear blebs (Fig. 4). Second, we observed a reduced number of binuclear cells (supplementary Fig. II). Third, many nuclei in FntbΔ/Δ hepatocytes were larger than those in wild-type hepatocytes (supplementary Fig. II). Flow cytometry studies (supplementary Fig. III) revealed a reduced percentage of FntbΔ/Δ cells in the “G2 phase” (i.e., fewer FntbΔ/Δ hepatocytes had a twice-normal amount of DNA, consistent with the reduced numbers of binuclear hepatocytes).
Fig. 4.
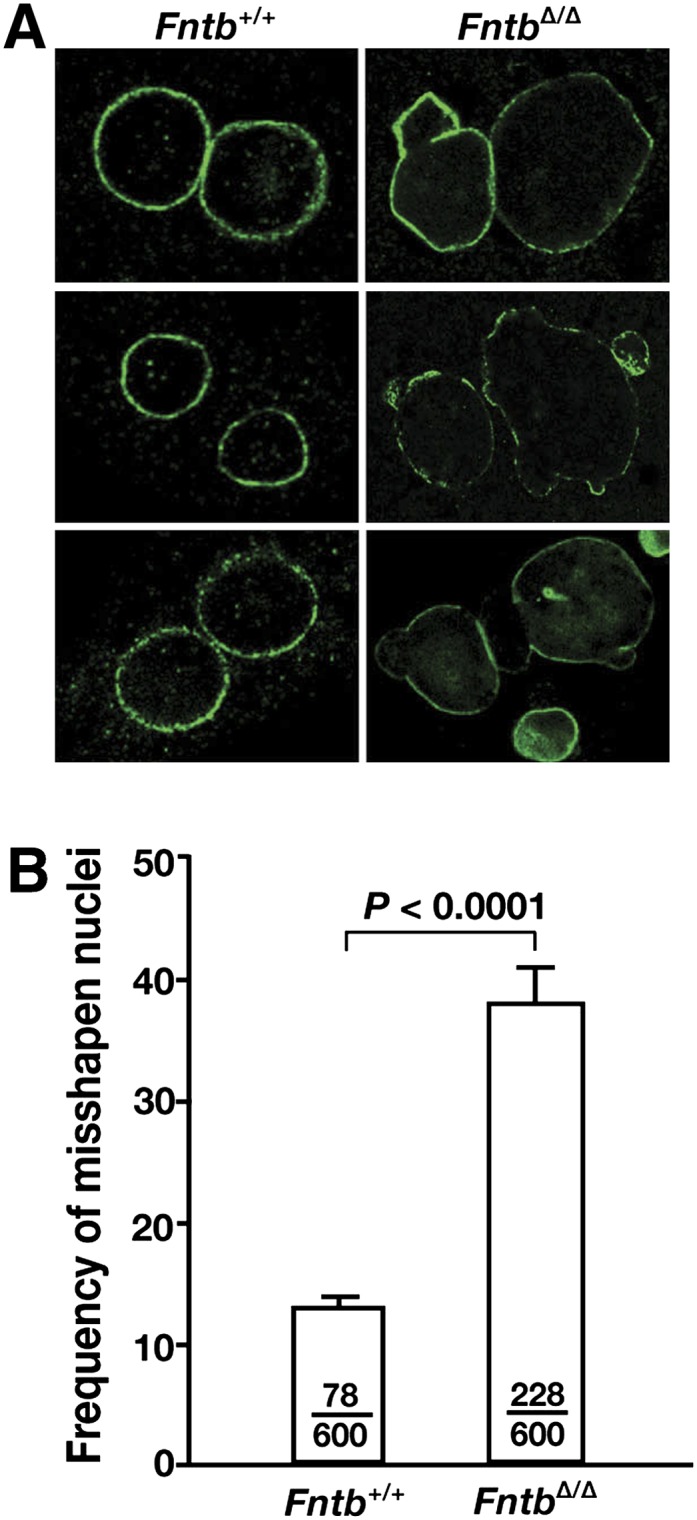
Misshapen nuclei in FntbΔ/Δ hepatocytes. (A) Immunohistochemical staining of primary hepatocytes from Fntb+/+ and FntbΔ/Δ mice with an antibody against lamin A (green). (B) Frequency of misshapen nuclei in Fntb+/+ and FntbΔ/Δ hepatocytes. Bars indicate the percentage of cells with misshapen nuclei; the number of cells harboring nuclear blebs and the total number of cells examined are recorded within each bar. Error bars indicate SEM for results with different cell preparations of the same genotype (n = 4/group).
Hepatocyte-specific Pggt1b knockout mice
A conditional knockout allele for Pggt1b, Pggt1bfl (20), and the AlbCre transgene were used to produce hepatocyte-specific Pggt1b knockout mice (Pggt1bfl/flAlbCre+, designated Pggt1bΔ/Δ mice). As expected, nonprenylated RAP1A (a protein that is normally geranylgeranylated) could be detected in Pggt1bΔ/Δ hepatocytes (Fig. 5A). Pggt1bΔ/Δ hepatocytes contained very low levels of Pggt1b transcripts (Fig. 5B).
Fig. 5.
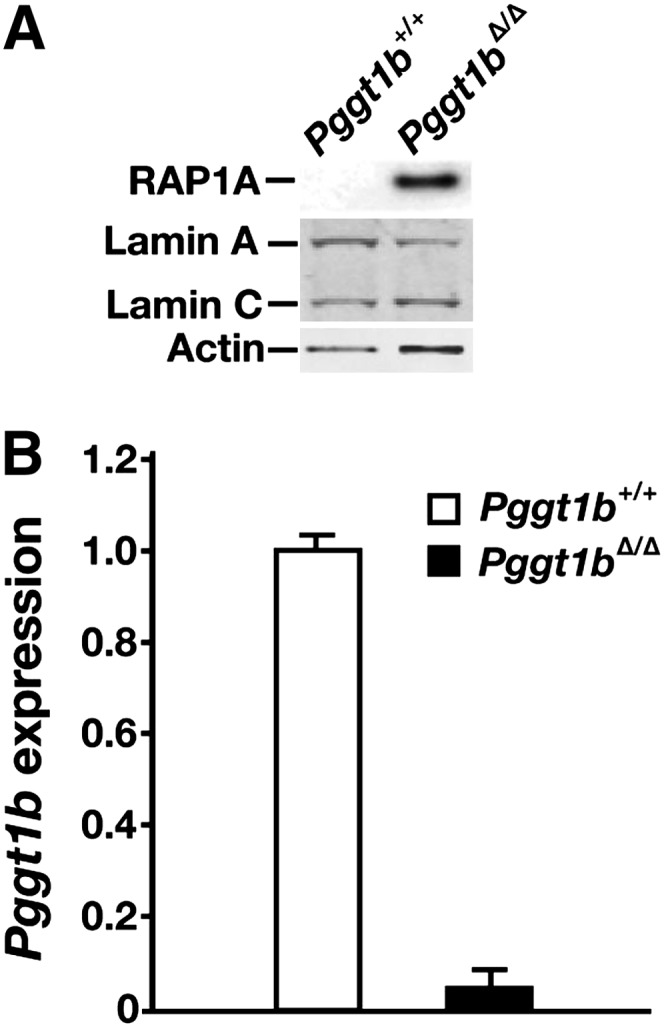
Inactivating GGTase-I in mouse hepatocytes. (A) Western blot of extracts from primary hepatocyte preparations from Pggt1b+/+ and Pggt1bΔ/Δ mice with antibodies against nonprenylated RAP1A and lamin A/C. Actin was used as a loading control. (B) Quantitative RT-PCR analysis of Pggt1b expression in hepatocytes from Pggt1b+/+ and Pggt1bΔ/Δ mice (n = 4 mice/group were examined). Data were normalized to β2-microglobulin, and expression levels were compared with levels observed in Pggt1b+/+ mice (which was set at 1.0). Error bars indicate SEM.
Pggt1bΔ/Δ mice survived development and were born at the expected Mendelian ratios. All Pggt1bΔ/Δ mice had a distended abdomen, beginning at 1 month of age, and were jaundiced. The body weight of Pggt1bΔ/Δ mice tended to be lower than wild-type mice (supplementary Fig. IV-A, B). The liver and spleen of Pggt1bΔ/Δ mice were massively enlarged (supplementary Fig. IV-C, D), and serum ALT, AST, and alkaline phosphatase (ALP) levels were markedly elevated, as were total and direct bilirubin levels (Table 2). Not surprisingly, the partial thromboplastin and prothrombin times in Pggt1bΔ/Δ mice were prolonged (Table 3). Unlike FntbΔ/Δ mice (which appeared healthy at 1 year of age), approximately 50% of Pggt1bΔ/Δ mice died within 9 weeks of age and all died by 26 weeks of age (supplementary Fig. IV-E).
TABLE 2.
Serum chemistry studies in wild-type and Pggt1b mice
| Wild-type | Pggt1b | P | |
| ALT (U/L) | 39 ± 5 | 517 ± 91 | <0.0001 |
| AST (U/L) | 159 ± 36 | 321 ± 64 | <0.0001 |
| ALP (U/L) | 51 ± 4 | 350 ± 34 | <0.0001 |
| CHOL (mg/dl) | 80 ± 16 | 121 ± 23 | NS |
| GLU (mg/dl) | 296 ± 27 | 211 ± 21 | NS |
| TRIG (mg/dl) | 99 ± 10 | 87 ± 10 | NS |
| DBILI (mg/dl) | 0.1 ± 0.01 | 32 ± 3 | <0.0001 |
| TBILI (mg/dl) | 0.2 ± 0.02 | 35 ± 7 | <0.0001 |
n = 8 mice/group, 8 weeks of age. Mean ± SEM.
CHOL, cholesterol; DBILI, direct bilirubin; GLU, glucose; NS, not significant; TBILI, total bilirubin; TRIG, triglyceride.
TABLE 3.
Blood coagulation studies in wild-type and Pggt1b mice
| Wild-type | Pggt1b | Normal Range | |
| PTT (second) | 8.2 ± 0.21 | 23.3 ± 0.34 | 7–19 |
| PT (second) | 21.2 ± 1.1 | 67.6 ± 0.62 | 18.5–33.4 |
| Fibrinogen (mg/dl) | 1.6 ± 0.04 | 2.1 ± 0.19 | 1.4–2.1 |
n = 4 mice/group, 8 weeks of age. Mean ± SEM.
PT, activated prothrombin time; PTT, activated partial thromboplastin time.
Hepatocytes from Pggt1bΔ/Δ mice quickly attached to plastic and ∼70% were viable, as judged by trypan blue exclusion, but the cells failed to spread and they manifested an abnormal, rounded shape (supplementary Fig. V-A, B). The nuclei of Pggt1bΔ/Δ hepatocytes tended to be slightly smaller than those in wild-type hepatocytes (supplementary Fig. V-C, D). The pattern of actin stress filaments in Pggt1bΔ/Δ hepatocytes was abnormal, as judged by phalloidin staining (supplementary Fig. V-E, F), consistent with the fact that the formation of actin stress fibers depends on geranylgeranylation of RHO GTPases (35). The pattern of tubulin staining within Pggt1bΔ/Δ cells was also abnormal, likely due to the striking abnormalities in cell shape (supplementary Fig. V-G, H).
The livers of Pggt1bΔ/Δ mice had necrotic blood-filled cavities, and there appeared to be more glycogen, as judged by a PAS staining (Fig. 6C, D). We also observed evidence of apoptosis (caspases 3 and 7 activation) (Fig. 6E, F). In addition, the pattern of actin stress fibers was markedly abnormal (Fig. 6G, H), consistent with findings in isolated hepatocyte preparations (supplementary Fig. V-E, F). Electron microscopy showed markedly dilated bile cannaliculi in livers of Pggt1bΔ/Δ mice (Fig. 7), along with increased amounts of glycogen, smaller and more osmophilic mitochondria, and reduced amounts of rough endoplasmic reticulum. Some of the dilated bile cannaliculi were in direct continuity with the space of Disse (Fig. 7B).
Fig. 6.

Severe liver disease in hepatocyte-specific Pggt1b knockout mice (Pggt1bΔ/Δ). (A, B) Hematoxylin and eosin–stained liver sections from 4-month-old Pggt1b+/+ and Pggt1bΔ/Δ mice, showing disordered hepatic lobules with dilated sinusoids and multiple blood-filled cavities. (C, D) PAS stain revealing increased glycogen (magenta) in the liver of a Pggt1bΔ/Δ mouse. Images in panels A–D were recorded with a 20× objective. Scale bar, 50 µm. (E, F) In vivo detection of active caspases 3 and 7 (green) in the livers of 4-month-old Pggt1b+/+ and Pggt1bΔ/Δ mice (n = 3 mice/group were examined). DNA was visualized with DAPI (blue). (G, H) Abnormal organization of actin fibrils in the liver of Pggt1bΔ/Δ mice, as judged by phalloidin staining (green). DNA was visualized with DAPI (blue). Scale bar for images in panels E–H, 10 µm.
Fig. 7.

Electron micrographs of livers from Pggt1b+/+ and Pggt1bΔ/Δ mice (A–F). Pggt1bΔ/Δ livers exhibit dilated bile canaliculi (B), increased amounts of glycogen (D), smaller and darker-staining mitochondria (D, F), and reduced amounts of rough endoplasmic reticulum (F). BC, bile canaliculus; D, space of Disse; Gl, glycogen; M, mitochondria; Nu, nucleus; RER, rough endoplasmic reticulum. Magnification in panels A–B, ×14,000; scale bar, 2.0 μm. Magnification in panels C–D, ×23400; scale bar, 1.0 μm. Magnification in panels E–F, ×37,400; scale bar, 1.0 μm.
Hepatocyte-specific Fntb/Pggt1b double-knockout mice
We also bred mice lacking both Fntb and Pggt1b in hepatocytes (FntbΔ/ΔPggt1bΔ/Δ). As expected, hepatocytes from FntbΔ/ΔPggt1bΔ/Δ mice exhibited an accumulation of nonprenylated RAP1A, nonprenylated HDJ-2, and prelamin A (Fig. 8A, B). Transcript levels for Fntb and Pggt1b in FntbΔ/ΔPggt1bΔ/Δ hepatocytes were very low (Fig. 8C, D).
Fig. 8.

Inactivating both FTase and GGTase-I in mouse hepatocytes. (A) Western blots of hepatocytes from wild-type and FntbΔ/ΔPggt1bΔ/Δ mice with antibodies against the β-subunit of FTase (FTβ), nonprenylated RAP1A, and HDJ-2. (B) Western blots of hepatocyte extracts from wild-type, FntbΔ/Δ, Pggt1bΔ/Δ, and FntbΔ/ΔPggt1bΔ/Δ mice with antibodies against lamin A/C and prelamin A (n = 3 hepatocyte preparations/group). Actin was used as loading control. (C, D) Quantitative RT-PCR analysis of Fntb (C) and Pggt1b (D) expression in hepatocytes from wild-type and FntbΔ/ΔPggt1bΔ/Δ mice (n = 4 hepatocyte preparations/group). Data were normalized to β2-microglobulin, and gene-expression levels were compared with those of wild-type mice (set at 1.0). Error bars indicate SEM.
Like Pggt1bΔ/Δ mice, the FntbΔ/ΔPggt1bΔ/Δ mice had markedly enlarged abdomens, and their body weights were lower than in wild-type mice (supplementary Fig. VI). The lifespan of FntbΔ/ΔPggt1bΔ/Δ mice was slightly shorter than in Pggt1bΔ/Δ mice (supplementary Fig. IV-E).
Hepatocytes from FntbΔ/ΔPggt1bΔ/Δ mice exhibited the same rounded shape as Pggt1bΔ/Δ hepatocytes (supplementary Fig. VII-A, B). Also, compared with wild-type hepatocytes, there were fewer numbers of binuclear hepatocytes and increased numbers of cells with enlarged nuclei (supplementary Fig. VII-C–E). Liver histology in FntbΔ/ΔPggt1bΔ/Δ mice was markedly abnormal, with areas of necrosis and increased collagen staining (Fig. 9A–D). The reticulin stain revealed multiple areas of hepatocyte loss (Fig. 9E, F). The serum ALT, AST, and ALP levels in FntbΔ/ΔPggt1bΔ/Δ mice (Table 4) were higher than we observed in FntbΔ/Δ or Pggt1bΔ/Δ mice (Tables 1 and 2), but we did not find extremely high levels of bilirubin in FntbΔ/ΔPggt1bΔ/Δ mice.
Fig. 9.

Inactivation of both Fntb and Pggt1b in liver causes severe hepatocellular disease. (A, B) Hematoxylin and eosin–stained sections of liver from 3-month-old wild-type and FntbΔ/ΔPggt1bΔ/Δ mice, revealing disordered hepatic lobules and areas of necrosis (black asterisks) in FntbΔ/ΔPggt1bΔ/Δ mice. (C, D) Sirius Red–stained liver sections, revealing increased amounts of collagen in the liver of FntbΔ/ΔPggt1bΔ/Δ mouse. Black asterisks in panels B and D indicate areas of necrosis. (E, F) Liver sections from 4-month-old wild-type and FntbΔ/ΔPggt1bΔ/Δ mice stained for reticulin, revealing disorganized liver architecture, lipid globules, and areas of hepatocyte loss. Images were recorded with a 20× objective. Scale bars, 50 µm.
TABLE 4.
Serum chemistry studies in wild-type and FntbPggt1b mice
| Wild-type | Fntb Pggt1b | P | |
| ALT (U/L) | 39 ± 5 | 1201 ± 48 | <0.0001 |
| AST (U/L) | 159 ± 36 | 1487 ± 72 | <0.0001 |
| ALP (U/L) | 51 ± 4 | 1501 ± 47 | <0.0001 |
| CHOL (mg/dl) | 80 ± 16 | 69 ± 8 | NS |
| GLU (mg/dl) | 296 ± 27 | 87 ± 11.3 | NS |
| TRIG (mg/dl) | 99 ± 10 | 52 ± 7.6 | NS |
| DBILI (mg/dl) | 0.1 ± 0.02 | 9.45 ± 3.62 | <0.0001 |
| TBILI (mg/dl) | 0.2 ± 0.01 | 13.7 ± 3.41 | <0.0001 |
CHOL, cholesterol; GLU, glucose; DBILI, direct bilirubin; NS, not significant; TBILI, total bilirubin; TRIG, triglyceride.
n = 8 mice/group, 6–8 weeks of age. Mean ± SEM.
DISCUSSION
The importance of protein isoprenylation for targeting CaaX proteins to membrane surfaces has been well documented in cell culture studies (2), but the physiologic relevance and importance of these posttranslational modifications for the vitality and viability of mammalian tissues has remained unclear. In the current study, we show that inactivation of either Fntb or Pggt1b in hepatocytes causes severe hepatocellular disease, with elevated plasma transaminases and histological evidence of hepatocyte damage and hepatocyte loss. But while hepatocellular disease was a shared feature of the two knockout models, several phenotypes were distinct. Most impressively, the Pggt1b knockout led to markedly elevated bilirubin and alkaline phosphatase levels, along with severe hepatosplenomegaly. The Fntb knockout also had unique features—hepatic steatosis, reduced numbers of binuclear hepatocytes, and more misshapen cell nuclei. These studies show that FTase-deficient cells are not free of pathology and that FTase is not dispensable in adult mice, as was suggested earlier (12), and they further demonstrated that GGTase-I deficiency is devastating for liver function. The knockout of both CaaX prenyltransferases resulted in more severe hepatocellular disease, as judged by reduced survival and exaggerated increases in serum transaminases. The fact that the liver in FntbΔ/ΔPggt1bΔ/Δ mice develops and functions (albeit at a minimal level) in the absence of both FTase and GGTase-I was remarkable, given that the loss of both prenyltransferases in yeast is lethal (13, 36).
From a technical standpoint, the knockouts of Fntb and Pggt1b in the liver worked well. The β-subunit of the FTase was undetectable in livers of FntbΔ/Δ mice, and most of the HDJ-2 in FntbΔ/Δ hepatocytes was nonprenylated. The levels of Fntb transcripts in livers of FntbΔ/Δ mice were extremely low, as were Pggt1b transcripts levels in livers of Pggt1bΔ/Δ mice. However, the transcripts in the knockout models were not completely eliminated, and that was not surprising. First, the albumin–Cre transgene inactivates floxed genes within hepatocytes, but not other liver cell types. Also, the Fntb and Pggt1b knockouts caused apoptosis, which likely caused an influx of inflammatory cells with normal levels of Fntb and Pggt1b expression.
In an earlier study, Mijimolle et al. (12) concluded that a generalized deficiency in FTase caused few or no histological abnormalities, but in the current study, we found severe hepatocellular disease. What explains this discrepancy? We suspect that Mijimolle and coworkers were simply unsuccessful in inactivating FTase. Their mutant allele yielded a short in-frame deletion within the Fntb transcript—not the frameshift that they had hoped for (37); hence, it seems possible that their mutant allele may not have inactivated enzyme function. In support of this explanation, they found that a large percentage of the HDJ-2 in cloned “knockout” fibroblasts was prenylated. With our mutant allele, the recombination event eliminated the promoter and exon 1, and the vast majority of the HDJ-2 in hepatocytes was nonprenylated. Also, in their studies, Mijimolle and coworkers relied on a ubiquitously expressed inducible Cre transgene, and it seems possible that their recombination rates were far lower than they expected—and far lower than we obtained with the albumin–Cre transgene.
In the current studies, the knockout of Fntb in the liver led to an accumulation of prelamin A, in agreement with cell culture studies where protein prenylation was inhibited with statins (38), FTIs (31, 39–41), or bisphophonates (30). We also observed reduced levels of mature lamin A in the setting of the Fntb knockout—a finding that did not surprise us. In earlier studies (30), we found a striking decrease in mature lamin A levels in mouse fibroblasts that had been treated with an FTI. We did not observe an accumulation of prelamin A in Pggt1bΔ/Δ hepatocytes, consistent with the fact that prelamin A is normally farnesylated. The prelamin A Western blot on independent preparations of hepatocytes from FntbΔ/ΔPggt1bΔ/Δ mice were interesting, particularly in light of the suggestion by Varela et al. (42) that prelamin A is geranylgeranylated by GGTase-I in the setting of FTase inhibition. In support of their idea, they reported that a FTI/GGTI combination led to more prelamin A accumulation than an FTI alone, but the specificity of their drugs was not well documented. In our studies, the levels of prelamin A accumulation were no greater in FntbΔ/ΔPggt1bΔ/Δ hepatocytes than in FntbΔ/Δ hepatocytes, providing little support for the idea that prelamin A is alternately prenylated. However, caution is warranted in the interpretation of these experiments because we detected mature lamin A, as well as small amounts of prenylated HDJ-2, in the FntbΔ/ΔPggt1bΔ/Δ hepatocytes, indicating that the hepatocyte preparations may have been contaminated with other cell types.
Previous studies have shown that geranylgeranylation of RHO GTPases is critical for the formation of actin stress fibers within cells (35). Our studies support that finding. Cultured GGTase-I–deficient hepatocytes exhibited a marked depletion of actin stress fibers, as judged by phalloidin staining, and they had a markedly abnormal, rounded shape. Abnormal organization of actin stress fibers was also evident in the livers of Pggt1bΔ/Δ mice. We suspect that the defects in the hepatocyte cytoskeleton may have contributed to the hyperbilirubinemia in Pggt1bΔ/Δ mice. In Pggt1bΔ/Δ livers, adjacent hepatocytes were often separated, and the cannaliculi between cells were frequently enlarged. Occasionally, bile cannaliculi were in direct continuity with the space of Disse, providing a likely explanation for the elevated levels of bilirubin in the plasma.
In summary, we found that deficiencies in either FTase or GGTase-I lead to severe hepatocellular disease. Our studies lay to rest the earlier notion that FTase is dispensable in adult tissues. Our studies do not speak to the potential utility of FTIs and GGTIs as anticancer agents, either alone or in combination with each other or other agents. However, our findings imply that pharmacological inhibition of FTase and GGTase-I, if it were sustained and complete, could cause significant injury to the liver and other tissues.
Supplementary Material
Acknowledgments
The authors thank Drs. Pinchas Cohen (UCLA) for the albumin–Cre transgenic mice, Larry Castellani (UCLA) for some of the hepatocyte preparations, Ingrid Schmid (UCLA) for flow cytometry analysis, and Ms. Jinny Wong (UCSF) for electron microscopy.
Footnotes
Abbreviations:
- ALP
- alkaline phosphatase
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- Fntb
- mouse gene for the β-subunit of protein farnesyltransferase
- FTase
- protein farnesyltransferase
- FTI
- protein farnesyltransferase inhibitor
- GGTase-I
- protein geranylgeranyltransferase-I
- GGTI
- protein geranylgeranyltransferase inhibitor
- Pggt1b
- mouse gene for the β-subunit of protein geranylgeranyltransferase-I
This work was supported by National Institutes of Health Grants HL-76839, CA-099506-07, HL-086683, and HL-089781; Ellison Medical Foundation Senior Scholar Program; March of Dimes Grant 6-FY2007-1012; American Heart Association, Western States Affiliate, Beginning Grant-in-Aid 0865262F; and Vascular Biology Program, UCLA, Fellowship 2 T32 HL069766:06. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of seven figures.
REFERENCES
- 1.Casey P. J., Seabra M. C. 1996. Protein prenyltransferases. J. Biol. Chem. 271: 5289–5292. [DOI] [PubMed] [Google Scholar]
- 2.Zhang F. L., Casey P. J. 1996. Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 65: 241–269. [DOI] [PubMed] [Google Scholar]
- 3.Casey P. J., Solski P. A., Der C. J., Buss J. E. 1989. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. USA. 86: 8323–8327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hancock J. F., Magee A. I., Childs J. E., Marshall C. J. 1989. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 57: 1167–1177. [DOI] [PubMed] [Google Scholar]
- 5.Cox A. D., Hisaka M. M., Buss J. E., Der C. J. 1992. Specific isoprenoid modification is required for function of normal, but not oncogenic, Ras protein. Mol. Cell. Biol. 12: 2606–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lerner E. C., Hamilton A. D., Sebti S. M. 1997. Inhibition of Ras prenylation: a signaling target for novel anti-cancer drug design. Anticancer Drug Des. 12: 229–238. [PubMed] [Google Scholar]
- 7.Gibbs J. B., Oliff A., Kohl N. E. 1994. Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell. 77: 175–178. [DOI] [PubMed] [Google Scholar]
- 8.Tamanoi F. 1993. Inhibitors of Ras farnesyltransferases. Trends Biochem. Sci. 18: 349–353. [DOI] [PubMed] [Google Scholar]
- 9.James G. L., Goldstein J. L., Brown M. S., Rawson T. E., Somers T. C., McDowell R. S., Crowley C. W., Lucas B. K., Levinson A. D., Marsters J. C., Jr 1993. Benzodiazepine peptidomimetics: potent inhibitors of Ras farnesylation in animal cells. Science. 260: 1937–1942. [DOI] [PubMed] [Google Scholar]
- 10.Sebti S. M., Hamilton A. D. 2000. Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and bench-to-bedside translational studies. Oncogene. 19: 6584–6593. [DOI] [PubMed] [Google Scholar]
- 11.Lobell R. B., Omer C. A., Abrams M. T., Bhimnathwala H. G., Brucker M. J., Buser C. A., Davide J. P., deSolms S. J., Dinsmore C. J., Ellis-Hutchings M. S., et al. 2001. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 61: 8758–8768. [PubMed] [Google Scholar]
- 12.Mijimolle N., Velasco J., Dubus P., Guerra C., Weinbaum C. A., Casey P. J., Campuzano V., Barbacid M. 2005. Protein farnesyltransferase in embryogenesis, adult homeostasis, and tumor development. Cancer Cell. 7: 313–324. [DOI] [PubMed] [Google Scholar]
- 13.He B., Chen P., Chen S-Y., Vancura K. L., Michaelis S., Powers S. 1991. RAM2, an essential gene of yeast, and RAM1 encode the two polypeptide components of the farnesyltransferase that prenylates a-factor and Ras proteins. Proc. Natl. Acad. Sci. USA. 88: 11373–11377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallim M. A., Fernandes L., Alspaugh J. 2004. The RAM1 gene encoding a protein-farnesyltransferase β-subunit homologue is essential in Cryptococcus neoformans. Microbiology. 150: 1925–1935. [DOI] [PubMed] [Google Scholar]
- 15.Hanrahan E. O., Kies M., Glisson B., Khuri F., Feng L., Tran H., Ginsberg L., Truong M., Hong W., Kim E. 2009. A phase II study of Lonafarnib (SCH66336) in patients with chemorefractory, advanced squamous cell carcinoma of the head and neck. Am. J. Clin. Oncol. 32: 274–279. [DOI] [PubMed] [Google Scholar]
- 16.Santucci R., Mackley P., Sebti S., Alsina M. 2003. Farnesyltransferase inhibitors and their role in the treatment of multiple myeloma. Cancer Control. 10: 384–387. [DOI] [PubMed] [Google Scholar]
- 17.Lee R., Chang S. Y., Trinh H., Tu Y., White A. C., Davies B. S., Bergo M. O., Fong L. G., Lowry W. E., Young S. G. 2010. Genetic studies on the functional relevance of the protein prenyltransferases in skin keratinocytes. Hum. Mol. Genet. 19: 1603–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kazi A., Carie A., Blaskovich M., Bucher C., Thai V., Moulder S., Peng H., Carrico D., Pusateri E., Pledger W., et al. 2009. Blockade of protein geranylgeranylation inhibits cdk2-dependent p27Kip1 phosphorylation on thr187 and accumulates p27Kip1 in the nucleus: implications for breast cancer therapy. Mol. Cell. Biol. 29: 2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J., Chan L., Fiji H., Dahl R., Kwon O., Tamanoi F. 2009. In vivo antitumor effect of a novel inhibitor of protein geranylgeranyltransferase I. Mol. Cancer Ther. 8: 1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sjogren A. K., Andersson K. M., Liu M., Cutts B. A., Karlsson C., Wahlstrom A. M., Dalin M., Weinbaum C., Casey P. J., Tarkowski A., et al. 2007. GGTase-I deficiency reduces tumor formation and improves survival in mice with K-RAS-induced lung cancer. J. Clin. Invest. 117: 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu M., Sjogren A., Karlsson C., Lbrahim M., Andersson K., Olofsson F., Wahistrom A., Dalin M., Yu H., Chen Z., et al. 2010. Targeting the protein prenyltransferases efficiently reduces tumor development in mice with K-RAS-induced lung cancer. Proc. Natl. Acad. Sci. USA. 107: 6471–6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan O. M., X. Ibrahim M., Jonsson I., Karlsson C., Liu M., Sjogren A., Olofsson F., Brisslert M., Andersson S., Ohlsson C., et al. 2011. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J. Clin. Invest. 121: 628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weisend C. M., Kundert J., Suvorova E., Prigge J., Schmidt E. 2009. Cre activity in fetal albCre mouse hepatocytes: utility for developmental studies. Genesis. 47: 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anzo M., Cobb L., Hwang D., Mehta H., Said J., Yakar S., LeRoith D., Cohen P. 2008. Targeted deletion of hepatic Igf1 in TRAMP mice leads to dramatic alterations in the circulating insulin-like growth factor axis but does not reduce tumor progression. Cancer Res. 68: 3342–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seglen P. O. 1976. Preparation of isolated rat liver cells. Methods Cell Biol. 13: 29–83. [DOI] [PubMed] [Google Scholar]
- 26.Klaunig J. E., Goldblatt P., Hinton D., Lipsky M., Chacko J., Trump B. 1981. Mouse liver cell culture. I. Hepatocyte isolation. In Vitro. 17: 913–925. [DOI] [PubMed] [Google Scholar]
- 27.Livak K. J., Schmittgen T. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 28.Fong L. G., Ng J. K., Meta M., Cote N., Yang S. H., Stewart C. L., Sullivan T., Burghardt A., Majumdar S., Reue K., et al. 2004. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA. 101: 18111–18116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fong L. G., Ng J. K., Lammerding J., Vickers T. A., Meta M., Cote N., Gavino B., Qiao X., Chang S. Y., Young S. R., et al. 2006. Prelamin A and lamin A appear to be dispensable in the nuclear lamina. J. Clin. Invest. 116: 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toth J. I., Yang S. H., Qiao X., Beigneux A. P., Gelb M. H., Moulson C. L., Miner J. H., Young S. G., Fong L. G. 2005. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc. Natl. Acad. Sci. USA. 102: 12873–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang S. H., Bergo M. O., Toth J. I., Qiao X., Hu Y., Sandoval S., Meta M., Bendale P., Gelb M. H., Young S. G., et al. 2005. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutation. Proc. Natl. Acad. Sci. USA. 102: 10291–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang S. H., Andres D., Spielmann H., Young S., Fong L. 2008. Progerin elicits disease whether or not it is farnesylated. J. Clin. Invest. 118: 3291–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinert P., Zackroff R., Aynardi-Whitman M., Goldman R. D. 1982. Isolation and characterization of intermediate filaments. Methods Cell Biol. 24: 399–419. [DOI] [PubMed] [Google Scholar]
- 34.Folch J., Lees M., Stanley G. H. S. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226: 497–509. [PubMed] [Google Scholar]
- 35.Hall A. 1998. Rho GTPases and the actin cytoskeleton. Science. 279: 509–514. [DOI] [PubMed] [Google Scholar]
- 36.Powers S., Michaelis S., Broek D., Anna-A S. S., Field J., Herskowitz I., Wigler M. 1986. RAM, a gene of yeast required for a functional modification of RAS proteins and for production of mating pheromone a-factor. Cell. 47: 413–422. [DOI] [PubMed] [Google Scholar]
- 37.Yang S. H., Bergo M. O., Farber E., Qiao X., Fong L. G., Young S. G. 2009. Caution! Analyze transcripts from conditional knockout alleles. Transgenic Res. 18: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinensky M., Beck L. A., Leonard S., Evans R. 1990. Differential inhibitory effects of lovastatin on protein isoprenylation and sterol synthesis. J. Biol. Chem. 265: 19937–19941. [PubMed] [Google Scholar]
- 39.Yang S. H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., Majumdar S., Bergo M. O., Young S. G., Fong L. G. 2006. Treatment with a protein farnesyltransferase inhibitor improves disease phenotypes in mice with a targeted Hutchinson-Gilford progeria syndrome mutation. J. Clin. Invest. 116: 2115–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fong L. G., Frost D., Meta M., Qiao X., Yang S., Coffinier C., Young S. 2006. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 311: 1621–1623. [DOI] [PubMed] [Google Scholar]
- 41.Young S. G., Meta M., Yang S. H., Fong L. G. 2006. Prelamin A farnesylation and progeroid syndromes. J. Biol. Chem. 281: 39741–39745. [DOI] [PubMed] [Google Scholar]
- 42.Varela I., Pereira S., Ugalde A. P., Navarro C. L., Suarez M. F., Cau P., Cadinanos J., Osorio F. G., Foray N., Cobo J., et al. 2008. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat. Med. 14: 767–772. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.