

Background: The mechanism of anion selectivity of Cl− transporting CLC proteins is not fully understood.
Results: Mutational analysis of the extracellularly exposed CLC-5 residue Lys210 indicates its essential role in anion discrimination.
Conclusion: A positive charge at position 210 is crucial to confer NO3− over Cl− preference in coupled anion/proton transport.
Significance: These findings suggest a novel mechanism responsible for anion discrimination in CLC proteins.
Keywords: Anion Transport, Chloride Transport, Membrane Biophysics, Membrane Transport, Proton Transport, Transport, Transporters
Abstract
Mammalian CLC proteins comprise both Cl− channels and Cl−/H+ antiporters that carry out fundamental physiological tasks by transporting Cl− across plasma membrane and intracellular compartments. The NO3− over Cl− preference of a plant CLC transporter has been pinpointed to a conserved serine residue located at Scen and it is generally assumed that the other two binding sites of CLCs, Sext and Sin, do not substantially contribute to anion selectivity. Here we show for the Cl−/H+ antiporter CLC-5 that the conserved and extracellularly exposed Lys210 residue is critical to determine the anion specificity for transport activity. In particular, mutations that neutralize or invert the charge at this position reverse the NO3− over Cl− preference of WT CLC-5 at a concentration of 100 mm, but do not modify the coupling stoichiometry with H+. The importance of the electrical charge is shown by chemical modification of K210C with positively charged cysteine-reactive compounds that reintroduce the WT preference for Cl−. At saturating extracellular anion concentrations, neutralization of Lys210 is of little impact on the anion preference, suggesting an important role of Lys210 on the association rate of extracellular anions to Sext.
Introduction
The central role of CLC proteins deduced by studies in mammals is the transport of Cl− ions across the plasma membrane and intracellular compartments (1–4). This is critical for muscle excitability, transepithelial transport, endocytosis, lysosomal function, and synaptic activity as proved by the involvement of several CLC human isoforms in hereditary diseases (5, 6).
Interestingly, CLC proteins include both Cl− channels and Cl−/H+ antiporters. In the channels, Cl− ions passively diffuse down their electrochemical gradient, whereas in the transporters Cl− and H+ are coupled with a stoichiometry of 2 Cl−:1 H+ (7–9). Despite the different thermodynamic transport mechanisms, the overall architecture is conserved across the protein family (10–14). Structures of the bacterial and eukaryotic homologues CLC-ec1 (11) and CLC-Cm (15) showed that the protein assembles as a homodimer with each subunit harboring a narrow permeation pathway containing 3 anion binding sites (Fig. 1, A and B). At the innermost binding site (Sin)2 the anion is still partially exposed to the intracellular solution. The central binding site (Scen) is located approximately halfway across the membrane, and is mostly contributed by conserved Ser and Tyr residues. The third and outermost binding site (Sext) is present in the structure of CLC-Cm in which the side chain of a critical glutamate residue, Gluext (Glu148 in CLC-ec1, Glu210 in CLC-Cm, and Glu211 in CLC-5), occupies Scen and in the structure of CLC-ec1 when Gluext is neutralized (11). In WT CLC-ec1 the side chain of Glu148 occupies Sext and isolates the anion at Scen from the extracellular space. In the E148Q mutant, an anion occupies Sext, whereas the Gln side chain is displaced from the pore (Fig. 1B). Gluext is crucial for gating and in CLC channels its mutations induce a constitutively open phenotype (11, 12, 16, 17). In the transporters Gluext is required for anion/proton coupling and its substitution transforms the antiporters into permanently open and pH independent anionic conductances (9, 18, 19).
FIGURE 1.

Location of residue Lys210 mapped on the structure of the CLC-ec1 mutant E148Q (PDB entry 1OTU). A, dimeric structure of CLC-ec1 viewed from the membrane plane (extracellular side above and cytoplasmic side below). The two subunits are shown in green and cyan. Residue Gln148 is colored in red, Arg147, corresponding to Lys210 of CLC-5, is shown in blue, and Ser107 in orange. Chloride anions bound to Sext, Scen, Sint are shown in magenta. B, expanded representation of the anion permeation pathway for one of the subunits. The positions of the three binding sites are also indicated by horizontal dashed lines. C, alignment of the sequence stretch comprising Lys210 (shown in bold) for several CLC proteins.
The three anion binding sites provide a structural framework to understand the mechanism of anion selectivity of CLC proteins. Interestingly, the plant AtCLC-a is not a Cl−/H+ exchanger but rather a coupled 2 NO3−:1 H+ antiporter responsible for NO3− accumulation into the vacuoles (20). The different anion selectivity of AtCLC-a is surprising because polyatomic anions like NO3− and SCN− lead to uncoupling of anion and H+ flux in CLC-ec1 and CLC-5 probably by slippage (18, 21).
The molecular determinant responsible for the different selectivity for NO3− in coupled proton antiport activity has been pinpointed to a single residue in the central binding site. The conserved serine residue present in Cl−/H+ antiporters (Ser107 in CLC-ec1, Ser168 in CLC-5) (Fig. 1B) corresponds to a Pro in the NO3−/H+ antiporter. Substitution of the WT Ser with a Pro is sufficient to convert CLC-5 from a Cl−/H+ to a NO3−/H+ antiporter and confers higher NO3− permeability to CLC-ec1 and CLC-0 (7, 22, 23), whereas the opposite substitution endowed AtCLC-a with coupled Cl−/H+ exchange activity (23, 24). Consistent with an important role of Scen in the coupling mechanism, structural analysis showed that anion occupancy of the central binding site of CLC-ec1 correlates with the degree of coupling with H+ countertransport (21, 25).
These findings clearly indicate that the central binding site is critical for anion selectivity and anion/H+ coupling efficiency. However, the question remains as to whether other protein regions are also involved in anion selectivity.
Several lines of evidence obtained from CLC-ec1 suggest that the internal binding site is not a likely candidate. The fact that anions at this position are still partially hydrated argues against a role in anion selectivity (10, 11). Consistent with this, it has been suggested that Cl− binding to Sin is weak (22) and that Sin is poorly selective between Cl− and SCN− (21).
The external binding site is in principle another site potentially involved in anion selectivity but its role has been investigated only sporadically and importantly, for CLC transporters it has been probed only by mutating the conserved Gluext (18, 19, 26, 27). From these studies it was concluded that Sext does not have a substantial role in anion selectivity (22, 28). However, considering the complex role of Gluext, it is conceivable that its substitutions, besides modifying Sext, have drastic effects on general transport properties that can potentially hinder the detection of selectivity changes. Here we investigate the role of the external binding site of CLC-5 in anion discrimination by a different approach.
A conserved positive residue (Arg147 in CLC-ec1 and Lys210 in CLC-5, but Trp209 in CLC-Cm) is adjacent to the important Gluext (Fig. 1, A and B) and is part of a sequence stretch highly conserved in CLCs from different organisms (Fig. 1C). The structure of the E148Q mutant of CLC-ec1 (11) shows that Arg147 is in close proximity to the outermost Cl− ion with its side chain directed toward the extracellular space and its backbone probably participating in anion coordination at Sext (Fig. 1B). These elements potentially suggest a role for this residue in the discrimination between transported anions, a hypothesis that we tested on CLC-5 by substituting Lys210 with several amino acids.
Our results indicate that a positive charge at position 210 is responsible for the NO3− over Cl− preference of WT CLC-5. Detailed analysis of the anion concentration dependence of transport suggests that Lys210 is primarily involved in regulating the voltage-dependent association of anions from the extracellular medium.
EXPERIMENTAL PROCEDURES
Molecular Biology
Oocytes were obtained by dissection and collagenase treatment of ovaries from Xenopus laevis frogs. Mutations were introduced by recombinant PCR as previously described (29). All constructs were in the pTLN vector (30) and were expressed by injecting 25–50 ng of cRNA transcribed from linearized cDNA from the Ambion mMessage mMachine SP6 kit. Oocytes were kept in a solution containing (in mm) 90 NaCl, 10 HEPES, 2 KCl, 1 MgCl2, 1 CaCl2, pH 7.5, for 3–6 days at 18 °C.
Two-electrode Voltage Clamp
Two-electrode voltage clamp was performed with a Turbotec 03 amplifier (npi, Tamm, Germany) and the custom acquisition program GePulse. Measurements were performed at room temperature (20–25 °C). Unless otherwise stated, the voltage protocol consisted of voltage steps from 120 to −60 mV in decrements of 20 mV from a holding potential corresponding to the resting potential of the oocytes (between −15 and −30 mV, a range that is typical for CLC-5 expression). Measurements of stationary current were performed by applying voltage steps of 20 ms to 80 mV every 2 s. The standard bath solution contained (in mm): 100 NaCl, 4 MgCl2, 10 HEPES, pH 7.3. For measurements of anion selectivity, NaCl was substituted with equimolar amounts of NaNO3, NaBr, NaSCN, or NaI.
For some batches of oocytes Br− and NO3− gave rise to small current increases in uninjected oocytes compared with Cl−. This small contribution did not affect the results obtained for the WT and the mutants with the highest expression, K210A, K210C, K210R, and K210Q, but had to be considered for K210E and K210H. On the other hand, in some batches, I− and SCN− significantly increased the currents elicited in uninjected oocytes. Therefore the results for I− and SCN− on all the constructs were corrected for their effect on uninjected oocytes in the following manner. The contribution of endogenous currents in the different measuring solutions was estimated for at least 3 noninjected oocytes. The average difference between the current in the different anions and the current in Cl− was then subtracted from the measurements in oocytes of the same batch expressing CLC-5.
Solutions with the methanethiosulfonate (MTS) reagents, MTSEH (2-hydroxyethyl methanethiosulfonate), MTSES (2-sulfonatoethyl methanethiosulfonate), MTSET (2-(trimethylammonium)ethyl methanethiosulfonate)), and with DTT (dithiothreitol) were freshly prepared the day of the experiment by dissolving in Cl− or NO3− bath solutions the appropriate amount of substance from stock solutions stored at −80 or −20 °C. The final concentration was 1 mm for MTS reagents (unless otherwise stated) and 10 mm for DTT, pH 7.3, adjusted with H2SO4 or NaOH. MTS reagents and DTT were purchased from Biotum (Biotium Inc., Hayward, CA).
For measurements of [Cl−]ext dependence the extracellular solution with the highest [Cl−] contained (in mm) 300 NaCl, 4 MgSO4, 10 HEPES, pH 7.3. Lower [Cl−] was obtained by substituting in a bath solution similar to the standard one (4 mm MgSO4 instead of 4 mm MgCl2) equimolar amounts of NaCl with NaAsp.
For a quantitative analysis of the anion concentration dependence we assumed a simple saturating behavior (in agreement with the data) described by the following equation,
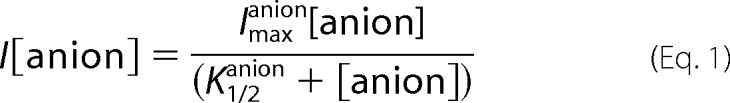 |
where anion can be chloride or nitrate, Imax is the current at saturating [anion], and K1/2 the concentration of half-maximal current. Thus, the dependence of the normalized currents on [Cl−]ext was fitted at each potential with Equation 2,
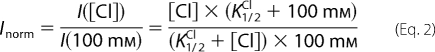 |
providing an estimate of K1/2Cl.
The dependence of the macroscopic currents on [NO3−]ext was analyzed by normalizing to the currents in the presence of 100 mm Cl−. Thus, these normalized currents were fitted with Equation 3,
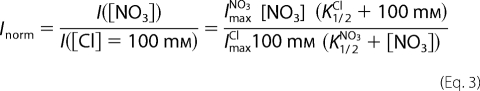 |
using the value of K1/2Cl obtained from the fit of Equation 2. The fit of Equation 3 provides two important parameters: K1/2Cl, the concentration of half-maximal current in NO3−, and ImaxNO3/ImaxCl, the ratio of currents in NO3− and Cl− in saturating concentrations of both anions.
The voltage dependence of K1/2 for Cl− and NO3− was fitted with a Woodhull model (31),
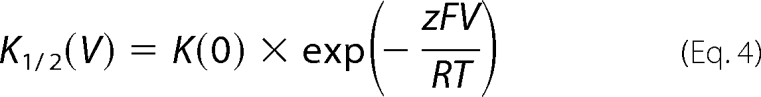 |
where K(0) is the apparent binding constant for the anion at 0 mV, z is the apparent electrical distance of a hypothetical anion binding site, V is the voltage, F, R, and T have their usual meanings (F, Faraday constant; R, gas constant; T, absolute temperature).
Data analysis was performed with a custom software (written in Visual C++, Microsoft) and the Origin program (OriginLab Corporation). Data are presented as mean ± S.E. unless otherwise stated. Statistical differences were assessed using Student's t test.
Extracellular pH Measurements Using a pH-sensitive Microelectrode
Proton transport was qualitatively measured by monitoring the acidification of the extracellular solution close to the oocyte using a pH-sensitive microelectrode as described previously (19). Briefly, a silanized microelectrode was back-filled with a proton ionophore (Mixture B, Fluka) and a solution containing phosphate-buffered saline, and connected to a custom high-impedance amplifier. The electrodes were routinely checked and responded consistently with a slope of 57–61 mV/pH unit. The pH-sensitive microelectrode was gently pushed onto the vitelline membrane without rupturing the plasma membrane. Application of a series of pulses to a positive test potential under voltage-clamp induced acidification of the extracellular solution. Solutions contained 0.5 mm HEPES buffer (pH 7.3).
Determination of the Relative Coupling Efficiency
The microelectrode method cannot be used to determine absolute coupling coefficients but can provide an estimate of the relative coupling efficiency of anion/proton exchange comparing measurements in the presence of different anions performed on the same oocyte with the pH-sensitive microelectrode in the same position (7). We applied a train of defined length of activating voltage pulses and measured the final change in extracellular pH, ΔpH, and the net current during the voltage pulses, Inet, in the presence of Cl− or NO3− from the same oocyte. The current is the sum of the proton current, IH and the anion current, Ianion, and the latter is equal to ranion × IH, where ranion is the number of anions transported for each proton.
As a first approximation, the acidification is proportional to the proton current,
and thus Equation 7.
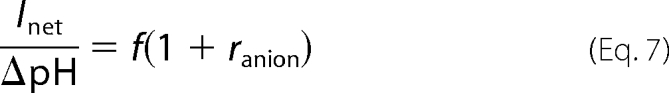 |
The proportionality factor, f, depends on the distance of the microelectrode from the oocyte surface, the local density of membrane expression, and other unknown factors. For the same oocyte and microelectrode positions, however, f can be considered identical for Cl− and NO3−. Therefore, the ratio of these quantities in NO3− and Cl−, respectively, defining the relative coupling efficiency, erel, as,
is a measure of the relative efficiency of the Cl−/H+ exchange compared with the NO3−/H+ exchange. A large number of erel indicates that NO3− transport is less coupled than Cl− transport, i.e. rNO3 > rCl.
RESULTS
Mutational Analysis
To test the possible role of Sext in substrate specificity of CLC-5 we mutated the conserved lysine residue at position 210 to Ala, Cys, Glu, His, Gln, and Arg.
All mutants gave rise to measurable currents (Fig. 2A) that, taking into account the variability of protein expression in oocytes and possibly a different turnover rate, had a magnitude similar to WT, except K210E and K210H for which the current magnitude was reduced (Fig. 2, A and B).
FIGURE 2.

Expression and rectification of the currents for WT CLC-5 and Lys210 mutants. A, representative current recordings for WT and the indicated mutants. B, average of the ratio between the current of WT and the indicated mutants at 100 mV. In each batch of oocytes, the ratio was calculated by dividing the average current of the mutants and the WT. Data were obtained for at least three batches (n ≥ 7). Error bars are S.D. The average current of the WT was 5.7 ± 1.8 μA (n = 38). C, average values of the ratio of currents at 120 and 80 mV calculated for each individual oocyte for WT (Lys210) and the indicated mutants (the number of experiments is the same as in panel B, error bars are S.D.). The difference with the WT value is statistically significant for K210R (p < 10−6) and K210C and K210E (p < 0.05).
Comparison of the macroscopic currents (Fig. 2A) shows that in general the behavior of the mutants is similar to WT. They display the strong outward rectification typical of WT (note the lack of current at negative voltages) (18, 26, 32) and the characteristic quasi-instantaneous activation with an additional, residual, slow component (Fig. 2A). The voltage dependence in the positive voltage range also appears very similar with the surprising exception of the conservative mutation K210R for which the voltage dependence is decreased compared with WT (Fig. 2A). To obtain a more quantitative description of the voltage dependence we estimated the degree of rectification by calculating the ratio of the current amplitudes at 120 and 80 mV. The results reported in Fig. 2C confirm that K210R is the only mutant with substantially altered voltage dependence.
To test whether the mutants have normal Cl−/H+ exchange activity, we assessed proton transport activity by measuring the degree of extracellular acidification produced by the activation of the constructs in Cl− containing extracellular solution at 100 mV and found that it is qualitatively very similar to WT (data not shown). Importantly, control experiments on noninjected oocytes did not elicit any detectable proton transport activity (data not shown). In conclusion, despite some minor differences, all mutants retained the basic transport properties of WT CLC-5.
We next asked whether the mutations affected the anion specificity of CLC-5. The strong rectification of macroscopic currents of CLC-5 precludes an estimation of anion selectivity based on measurements of the reversal potential, which is traditionally the method of choice to investigate selectivity of ion channels (33) and has been applied also to transporters that mediate macroscopic current at both positive and negative potentials, including CLC-ec1 (27). However, as detailed under “Discussion,” the selectivity of transport proteins can be measured by different methods, in particular by comparing the current amplitude in different ionic conditions (33, 34). The various methods, assessing different thermodynamic properties, are not expected to provide the same results (34) and therefore great attention has to be taken in comparing the selectivity obtained with different techniques. In this work, the selectivity of WT and mutant CLC-5 was assessed in terms of the relative macroscopic current that allows comparison of different anions according to the relative efficiency with which they are transported. To avoid confusion between this type of measurement and the selectivity obtained from permeability ratios (or other methods), we will refer to the anion discrimination measured in this work as substrate specificity for transport activity.
Supplemental Fig. S1 shows representative current recordings of an oocyte expressing WT CLC-5 in extracellular solutions containing different anions (note the different current scale for SCN−). In agreement with previous reports (7, 32) we found that Br− slightly decreases the currents compared with Cl−, whereas the reduction produced by I− is much more pronounced. NO3− and SCN− increase the currents (18). The ratio Ianion/ICl− of the current magnitude in the presence of a particular anion with the current in the presence of Cl− at 100 mV (for SCN− at 80 mV) and at a concentration of 100 mm, is shown in Fig. 3, B and C.
FIGURE 3.

Selectivity of WT CLC-5 and Lys210 mutants. A, representative current recordings in Cl− solution (black trace) and NO3− solution (red trace) at 100 mV for two oocytes expressing WT CLC-5 and the mutant K210A, respectively. The ratio INO3−/ICl− was 1.61 for WT and 0.67 for K210A. Traces for K210A were scaled to obtain the same current level in Cl− solution for WT and K210A. B, average currents in Br−, NO3−, and I− relative to Cl− at 100 mV (n ≥ 7). The difference with the WT value of INO3−/ICl− is statistically significant for all the mutants (p < 0.01), for IBr−/ICl− and IIodide−/ICl− is statistically significant only for K210H (p < 0.05). C, average of the currents in SCN− relative to Cl− at 80 mV (n ≥ 7). The difference with the WT value is statistically significant for K210A, K210E, and K210Q (p < 0.05).
In performing these experiments several problems have to be taken into account. First of all, for SCN−, due to the large increase in current magnitude, the ratio ISCN−/ICl− was calculated at 80 mV to avoid errors due to series resistance (Fig. 3C). Moreover, to compensate for possible drifts of the currents during the perfusion with different anions, measurements with Cl− solution were repeated after each anion tested. Finally, if endogenous currents were present their contribution was subtracted (see “Experimental Procedures”).
In agreement with previous results (7, 32) the transport activity of WT followed the substrate specificity sequence, SCN− > NO3− > Cl− > Br− > I− (Fig. 3). Fig. 3B shows that with the exception of K210H none of the mutations significantly affected the relative specificity of Br− and I− compared with WT. However, considering the small effect we did not investigate this aspect in further detail. The effect of the mutations on relative specificity of NO3− was in contrast much more consistent and robust. In fact, with the exception of the conservative mutation K210R, all mutants decreased the relative specificity of NO3− (Fig. 3, A and B).
At 100 mV, transport activity of WT CLC-5 is higher in NO3− than in Cl− with INO3−/ICl− = 1.6. In contrast, INO3-/ICl− is only 1.1 for K210H, whereas K210A, K210E, K210C, and K210Q reverse the preference of NO3− and Cl− (INO3−/ICl− < 1) with the strongest effect produced by the mutant K210A (INO3−/ICl− = 0.7). The strong effect of K210A on the relative NO3−/Cl− preference is also illustrated in Fig. 3A, which shows representative macroscopic currents in NO3− and Cl− for WT and K210A.
SCN− robustly increases the WT current compared with Cl−, (ISCN−/ICl− at 80 mV is 2.8). However, ISCN−/ICl− is strongly reduced for K210A, K210C, and K210E and for K210Q transport activity is similar in the presence of SCN− and Cl− (Fig. 3C). The mutations with the largest impact on NO3− specificity, K210A, K210E, and K210Q, are the ones that also affect more strongly SCN− specificity (Fig. 3, B and C). Considering the strong effect of K210A on NO3− specificity, we felt compelled to provide additional evidence that this mutation preserved the transport properties of the WT.
First of all, we analyzed the dependence of macroscopic currents of WT and K210A on [Cl−]ext. Fig. 4A shows the dependence on [Cl−]ext for WT and K210A at 100 mV. At a concentration of 3 mm currents are just above background, but raising [Cl−]ext leads to a progressive increase of current magnitude for both WT and K210A. The data points follow a Michaelis-Menten behavior and the fit with Equation 1 allows estimating the apparent K1/2 for Cl− (Fig. 4A). K1/2 decreases with increasing voltage (Fig. 4B), consistent with the idea that inward Cl− movement is favored at positive voltages. The K1/2 voltage dependence could be described by a Woodhull model (Equation 4), with K(0) values of 1.1 and 0.9 m and values for the apparent electric distance z of 0.50 and 0.67, respectively, for WT and K210A. This result indicates that K210A produces at most a small effect on Cl− transport further supporting the notion that this mutant retains the basic properties typical of the WT.
FIGURE 4.

Dependence of WT and K210A on [Cl−]ext. A, dependence on [Cl−]ext of the normalized currents for WT (n = 5, filled squares) and K210A (n = 3, open circles) at 100 mV (error bars are smaller than symbols). Normalization was performed with the current at 100 mm. Curves are fits to Equation 2, with K1/2 as the fitted parameter resulting in values of 71 ± 6 and 116 ± 10 mm for WT and K210A, respectively. B, voltage dependence of K1/2. Symbols are the same as panel A. Lines are fit with Equation 4 with K(0) and z as the fitted parameters resulting in values of 1064 ± 27 and 0.67 ± 0.09 mm for WT and 860 ± 15 and 0.50 ± 0.06 mm for K210A, respectively.
To gain some insight into the mechanism responsible for the decreased NO3− permeability of K210A, we investigated whether for this mutant NO3− transport is uncoupled from proton flux as for WT, or if the change in selectivity was due to a modified coupling with proton countertransport. To this aim we compared proton transport activity of WT and K210A in Cl− and NO3− solutions. Fig. 5A shows representative experiments in which we measured the level of extracellular acidification produced by activation of WT and K210A at 80 mV in Cl− and NO3− solutions. They clearly show that like for the WT, NO3− transport by mutant K210A is less coupled to proton flux compared with Cl− transport. To have a quantitative estimate of the coupling efficiency in Cl− and NO3− we calculated the ratio between the macroscopic current and the associated pH change, I/ΔpH, for the two anions obtaining the relative coupling efficiency, erel (see “Experimental Procedures”). The values of this parameter for WT and K210A at both 80 and 100 mV are not significantly different (p > 0.05) (Fig. 5B). This indicates that the degree of uncoupling produced by NO3− is similar in WT and K210A.
FIGURE 5.

Proton transport properties of WT and K210A. A, representative recordings of the extracellular acidification produced by the activation of WT and K210A. Horizontal gray bars indicate stimulation with trains of voltage pulses to 80 mV (400 ms duration) in Cl− (filled squares) and NO3− (open circles) solutions. B, average coupling efficiency of Cl− and NO3− with H+ transport, erel (Equation 8), at 80 and 100 mV measured for WT (n = 4) and K210A (n = 6) (errors bars are S.D.). The difference is not statistically significant (p > 0.05).
In summary, so far, our experiments indicate that the inversion of the NO3− and Cl− specificity observed for the mutant K210A (at 100 mV and 100 mm extracellular anion concentrations) is not due to a substantial modification of the Cl− and NO3−/proton coupling mechanism and it is not accompanied by changes in the voltage dependence of Cl− currents, nor in the mechanism of Cl− permeation compared with WT.
Modification of the Mutant K210C with MTS Reagents
The change in NO3− specificity produced by substitution of Lys210 with residues of different charge and properties is quite surprising. However, this result suggests that a positive charge at position 210 is critical to confer NO3− over Cl− preference to CLC-5. This interpretation makes a strong prediction. If it would be possible to modify K210C with MTS reagents of positive charge, we should be able to restore the preference for NO3− over Cl− of the WT that is inverted in this mutant.
Application of the positively charged MTSET, at a concentration of 1 mm, produced a marked increase of the stationary current when applied in NO3− solution for 2 min (Fig. 6A). The increase of stationary current was accompanied by an alteration of the NO3−/Cl− specificity (assessed from the ratio INO3−/ICl− at 100 mV) as illustrated in Fig. 6, B and C. Before modification, K210C passes larger currents in Cl− than in NO3− (Fig. 6B), similar to K210A (Fig. 3B). Modification with 1 mm MTSET produces an inversion of specificity (Fig. 6B) that becomes similar to WT (Fig. 3, A and B). Both the stationary current increase and the inversion of selectivity produced by MTSET could be reversed by washout with 10 mm DTT for 10 min (Fig. 6B). A similar conclusion is suggested by I-V relationships recorded before and after MTSET application and after DTT washout (supplemental Fig. S2).
FIGURE 6.

MTSET modification of K210C. A, representative current recordings showing the effect on the stationary current level of 1 mm MTSET (application indicated by a black bar) and of the washout with 10 mm DTT (indicated by a gray bar). Both reagents were dissolved in measuring solution containing 100 mm NO3−. Voltage protocol consisted of a 20-ms pulse to 80 mV applied every 2 s. After MTSET application the experiment was interrupted to record I-V relationships in Cl− and NO3− solutions. The dashed line represents the 0 current level. B, representative current recordings in Cl− (solid trace) and NO3− (dashed trace), at 100 mV from the same oocyte as shown in panel A (before and after MTSET application and after washout with DTT). In these conditions the value of INO3−/ICl− was, respectively, 0.89, 1.50, and 0.91. C, average value of INO3−/ICl− before and after MTSET application (n = 15) and after DTT washout (n = 6). The increase of INO3−/ICl− after MTSET modification is statistically significant (p < 10−6).
Although MTSET had only a minor effect on the stationary current amplitude of K210C when applied in Cl− solution (supplemental Fig. S3A) it induced the same inversion in selectivity upon application in either NO3− or Cl− solutions (Fig. 6 and supplemental Fig. S3B) and in both cases did not produce other major alterations of the current properties (supplemental Figs. S2 and S4). Importantly, MTSET modification had no effect on WT as assessed from the stationary current level (supplemental Fig. S5A), the anion specificity (supplemental Fig. S5B), and the general current properties (supplemental Fig. S6).
Application of 1 mm of the neutral reagent MTSEH did not have any effect on the stationary current level (Fig. 7A), on NO3−/Cl− specificity (Fig. 7B), or on general current properties (supplemental Fig. S7) of K210C. Importantly, application of MTSEH prevented subsequent modification by MTSET (Fig. 7 and supplemental Fig. S7) proving that MTSEH reacted with K210C such that the cysteine residue was not available for subsequent modifications. Therefore modification of K210C with a neutral charge does not alter the preference of K210C for Cl− over NO3−.
FIGURE 7.

MTSEH modification of K210C. A, representative current recordings showing the effect of 1 mm MTSEH on the stationary current level followed by application of 1 mm MTSET. Both reagents were dissolved in measuring solution containing 100 mm NO3−. Application of MTSEH is indicated by a gray bar and application of MTSET by a black bar. After MTSEH application the experiment was interrupted to record I-V relationships in Cl− and NO3− solutions. The dashed line represents the 0 current level. B, representative current recordings in Cl− (solid trace) and NO3− (dashed trace), at 100 mV from the same oocyte shown in panel A (before and after MTSEH application and after application of MTSET). In these conditions the value of INO3−/ICl− was, respectively, 0.91, 0.91, and 0.88. C, average value of INO3−/ICl− before and after MTSEH application (n = 6) and after application of MTSET (n = 3). The differences of the values of INO3−/ICl− in these three different conditions were not statistically significant (p > 0.8).
The negatively charged reagent MTSES even when applied at a concentration of 4 mm for 11 min did not have any effect on the stationary current level, on NO3−/Cl− specificity, or on general current properties (supplemental Figs. S8 and S9). However, unlike MTSEH, MTSES was not able to react with K210C as it did not prevent or alter the subsequent modification by MTSET (supplemental Figs. S8 and S9). These results support the essential role of the positive charge at position 210 for NO3−/Cl− specificity.
The Mechanism of Anion Preference
The method we used to assess anion specificity of the Cl−/H+ antiporter CLC-5 is based on the relative current level (in other words the relative transport rate) at 100 mV in the presence of different anions (at a concentration of 100 mm) compared with Cl−. In these conditions, the anion specificity can be determined by both the anion accessibility to the protein interior and the turnover rate of the anions. However, considering that anion accessibility should be rate-limiting for transport only at subsaturating concentrations, an analysis of the concentration dependence of the specificity can potentially provide a manner to distinguish between the two contributions. To this aim, we tested the effect of [NO3−]ext on the relative specificity of Cl− and NO3− on WT and K210A.
Fig. 8A shows the dependence of WT and K210A currents on [NO3−]ext at 120 mV. The values are normalized to the current in 100 mm Clext− and were fitted with Equation 3 (see “Experimental Procedures”). Two important pieces of information can be obtained from this analysis. First, as for Cl−, the K1/2 voltage dependence for NO3− could be described by a Woodhull model (Equation 4), with K(0) values of 3.1 and 11.9 m and values for the apparent electric distance z of 0.74 and 0.62, respectively, for WT and K210A (Fig. 8B). K210A therefore has a significantly increased K1/2 for NO3− compared with WT (values at 120 mV) (Fig. 8B). Second, from the fit with Equation 3 the ratio between the maximal current in NO3− and Cl−, ImaxNO3-/ImaxCl, was found to be 2.9 for WT, and 2.0 for mutant K210A. Unfortunately, it was not possible to apply anion concentrations higher than 300 mm as this quickly damaged the oocytes. This possibly affects our estimates of ImaxNO3-/ImaxCl. However, despite possible errors in the exact values, this result strongly suggests that in saturating concentrations of NO3−, when NO3− access is not rate-limiting, the turnover in NO3− is higher than in Cl− for both WT and K210A. According to this view the difference between WT and K210A in the relative preference for NO3− and Cl− emerges only when the NO3− concentration becomes rate-limiting for transport.
FIGURE 8.

Dependence of WT and K210A on [NO3−]ext and voltage. A, dependence on [NO3−]ext of the normalized currents for WT (n ≥ 7, filled squares) and K210A (n ≥ 8, open circles) at 120 mV. Normalization was performed with the current in 100 mm Cl− at 120 mV. Solid and dashed curves are fits to Equation 3, with K1/2 and ImaxNO3−/ImaxCl− as the fitted parameters resulting in values of 171 ± 26 and 372 ± 74 for K1/2 and 2.9 ± 0.2 and 2.0 ± 0.2 mm for ImaxNO3−/ImaxCl−, for WT and K210A, respectively. B, voltage dependence of K1/2. Symbols are the same as in panel A. Lines are fit with Equation 4 with K(0) and z as the fitted parameters resulting in values of 3.2 ± 0.6 and 0.74 ± 0.07 m for WT and 11.9 ± 3.4 and 0.62 ± 0.05 m for K210A, respectively.
DISCUSSION
Substrate discrimination is one of the defining properties of transport proteins. CLC Cl− channels and the bacterial Cl−/H+ antiporter CLC-ec1 transport anions by intrinsically different thermodynamic mechanisms but are characterized by the same anion selectivity sequence SCN− > Cl− > Br− > NO3− > I−, that could be estimated from measurements of permeability ratios (i.e. measurements of the reversal potential of the currents) (27, 35, 36).
Due to the strong outward rectification, this method cannot be applied to the mammalian Cl−/H+ antiporter CLC-5. Therefore, in this work anion selectivity was estimated by comparing the magnitude of macroscopic currents. With this approach, the selectivity is assessed in terms of the transport activity, i.e. the turnover rate supported by the different anions compared with Cl−. This method has been already applied to investigate substrate specificity of CLC exchangers and other transporters (7, 26, 32, 37, 38) and is highly analogous to the assessment of selectivity of ion channels performed by measurements of conductance, a technique that has been widely used. In fact, according to Hille (33), the Goldman-Hodgkin-Katz theory defines two empirical measures of selectivity, permeability ratio and macroscopic current. These methods probe different thermodynamic properties and therefore are not expected to generate the same results. Importantly, whereas both types of measurements can be influenced by parameters like ion concentration, the permeability ratio method tends to be less sensitive to experimental conditions compared with the use of macroscopic currents (34). For this reason, in assessing selectivity with the macroscopic current method, it is important to define the dependence of the current on the experimental factors (most notably ion concentration). Conforming to the definition normally adopted for transporters and to avoid confusion with selectivity measured with permeability ratios, we defined the selectivity obtained in this work as substrate specificity.
In agreement with previous results (7, 32), we found that the CLC-5 specificity sequence is SCN− > NO3− > Cl− > Br− > I−. In particular, properties of the NO3− transport through CLC exchangers has recently received revived attention with the discovery that the plant AtCLC-a homologue is a 2 NO3−:1 H+ antiporter characterized by a low permeability to Cl− (anion selectivity assessed by reversal potential is NO3− = I− > Br− > Cl−) (20). This is in contrast with the uncoupling produced by NO3− in the mammalian CLC-5 and the bacterial CLC-ec1 exchangers and showed that the mechanism of anion selectivity and anion/H+ coupling deserved deeper scrutiny. The molecular determinant of the different selectivity for Cl− and NO3− in coupled proton antiport activity has been identified in a single residue in Scen, indicating that this site is important for anion selectivity of transport and for the coupling efficiency of anion flux with H+ countertransport (7, 22–24). Currently it is assumed that no other protein regions, in particular Sext, influence anion selectivity in CLC channels and transporters. This conclusion mostly derives from studies on CLC-ec1 for which, however, the role of Sext has been probed exclusively mutating the conserved Gluext (Glu148) (27). Based on measurements of the shift of the reversal potential (9) and anion binding affinity by isothermal calorimetry (22), it was generally concluded that mutation E148A did not influence anion selectivity (9, 22, 28). However, we think that conclusions on the anion selectivity based on comparison between WT and mutants of the critical Gluext should be interpreted with caution for CLC-ec1 and CLC transporters in general for the following reasons.
First and most importantly, WT CLC-ec1 is a Cl−/H+ exchanger with a 2:1 stoichiometry for which NO3− and SCN− induce slippage. Mutations of Gluext turn the transporter into pure anionic conductance with an intrinsically different transport mechanism.
Moreover, measurements of binding affinity by isothermal calorimetry (22) concur with structural data (11) in suggesting that in equilibrium conditions, in WT CLC-ec1, only Scen is significantly occupied by anions, whereas in mutant E148A both Scen and Sext present a stably bound anion. This suggests relevant differences in the transport mechanism in WT and E148A due to the altered occupancy of the different binding sites as indicated also by the surprisingly decreased transport activity of the E148A mutant compared with WT (39).
Although some of the aspects just discussed might simply reflect our limited understanding of the complicated mechanism linking anion binding affinity and permeation through CLC proteins, they also indicate that in general, a direct comparison between the selectivity of WT and mutation of Gluext can be misleading, not only for CLC-ec1 but for all CLC transporters. Therefore, we believe that a different and less invasive approach could be beneficial to assess the role of Sext in anion selectivity. Motivated by this idea we investigated whether mutations of residues potentially contributing to Sext would affect anion selectivity of CLC-5, whereas preserving general transport properties.
Here, we found that a conserved lysine residue at position 210, probably contributing to anion coordination at Sext, has a limited impact on general transport properties of CLC-5 but is critical for its anion specificity. The ability of CLC-5 to tolerate mutations of this residue preserving function is quite unique among the CLC proteins in which the corresponding residue has been analyzed. The lysine corresponding to Lys210 of CLC-5 has been mutated in the CLC channels CLC-0 (Lys165), CLC-1 (Lys231), and CLC-Ka (Lys165) (17, 40–42). In CLC-0, the K165R mutation drastically altered the gating properties conferring a pronounced inwardly rectifying phenotype (40). The K165C mutation did not yield functional channels, indicating, differently from our results, very strict requirements on the properties of the residue at position 165 of CLC-0 to preserve function. However, modification of K165C with positive MTS reagents MTSEA and 3-aminopropyl methanethiosulfate (but not with MTSET) restored channel function and modified the gating properties similarly to the K165R mutation (41). In any case, no specific conclusion could be drawn about the role of the charge of this residue on anion selectivity of CLC-0. For CLC-1 it has been suggested that substitutions of Lys231, besides modifying the gating properties, affected anion selectivity (17). However, these conclusions were based on measurements of the membrane potential of cells expressing currents with a very pronounced inward rectification, rendering the assessment of relative permeability to different anions practically impossible. In CLC-Ka, mutant K165R strongly reduced currents, whereas mutation to Gln led to a complete loss-of-function. Therefore also for CLC-Ka no information could be gained about the role of Lys165 on anion selectivity (42).
Two independent lines of evidence support the role of Lys210 in anion specificity of CLC-5. 1) Substitution of Lys210 with Ala, Cys, Glu, His, and Gln, but not with Arg, produce a strong decrease (K210H) or an inversion (K210A, K210C, K210E, and K210Q) of the relative Cl−/NO3− specificity compared with WT (assessed at 100 mV and 100 mm anion concentration) with the strongest effect observed for K210A. SCN− transport activity was also identical to WT for K210R but was strongly decreased by other mutations with the strongest effect observed for K210Q, which is equally permeable to Cl− and SCN−. Importantly, substitutions with the largest impact on NO3− specificity are the same as the one with the biggest effect on SCN− specificity. However, none of the mutations induced major alterations of the halide (i.e. Br− and I−) specificity compared with WT.
2) Similar to K210A, the K210C mutant has a higher specificity for Cl− than for NO3−, but its modification by the positively charged MTS reagent MTSET is able to restore the NO3− over Cl− preference of the WT in a reversible manner, whereas modification with the neutral MTSEH was not able to alter the anion preference of K210C. Thus, it appears that a positive electric charge at position 210 is critical for NO3− over Cl− specificity of CLC-5.
Several independent elements suggest that the general transport properties of Lys210 mutants, in particular K210A, are very similar to WT. First of all we found that all the mutants analyzed retain two basic properties of WT CLC-5, the strong outward rectification of currents and a similar proton transport activity in the presence of Cl−.
In particular, for the mutant with the largest effect on NO3− selectivity, K210A, we additionally probed other transport properties. The decreased permeability of K210A for NO3− is not due to a change in coupling stoichiometry with H+ compared with WT. It seems therefore that Sext controls anion specificity but does not impact anion coupling to H+ transport. This is different from the effect of the S168P mutation, located at Scen, which increased NO3− over Cl− selectivity but also endowed CLC-5 with coupled NO3−/H+ antiport activity (7). It would be interesting to check whether these features reflect general properties of Sext and Scen or are due to a specific role of residues Lys210 and Ser168.
Moreover, we compared another transport property of WT and K210A, investigating for the first time the dependence of CLC-5 activity on [Cl−]ext. The small difference in the values of K(0) and z for WT and K210A further argues for an intact mechanism of Cl− gating and permeation in K210A and further support the notion that K210A specifically alters anion specificity of CLC-5. It should be noted that the value of the K(0) obtained here for WT CLC-5 (∼1 m) is 1000 times higher than the Cl− binding constants measured for CLC-ec1 by isothermal calorimetry (22). However, considering that our estimate is based on transport measurements (i.e. a nonequilibrium process) a direct comparison of the two measurements is not warranted. In fact, estimates of ion affinity based on measurements of equilibrium and nonequilibrium processes provide results that can differ by several orders of magnitude as illustrated by analysis of the BK K+ channel performed by Neyton and Miller (43).
Further insight into the mechanism of ion transport of CLC-5 was obtained by the analysis of the concentration dependence of the NO3−/Cl− preference. In general, transport efficiency depends on both the access rate of the anions to the protein interior and their subsequent translocation. These contributions are difficult to dissect. However, the dependence of transport efficiency on extracellular anion concentrations provides a way to distinguish between the two regimens as the anion access rate can be expected to be rate-limiting for transport only at subsaturating anion concentrations. Interestingly, the preference for Cl− over NO3− introduced by the K210A mutation at anion concentrations of 100 mm was reversed in saturating conditions indicating that when NO3− availability is not rate-limiting, NO3− is preferred to Cl− as a substrate in both WT and K210A.
In summary, we showed that neutralization of the conserved positive charge at position Lys210 alters anion specificity of CLC-5 for polyatomic anions (i.e. NO3− and SCN−) but not for halides (i.e. Br− and I−). A detailed analysis of the K210A mutant indicated that the altered preference for NO3− over Cl− appears at subsaturating NO3− concentrations. Considering the extracellular localization of Lys210, this potentially suggests that the K210A mutation reduces NO3− access to a binding site located in the transmembrane electric field.
In conclusion, we believe this is the first study to unveil a role of Sext in the substrate specificity of CLC proteins. This is important to understand the molecular basis of anion discrimination in CLC proteins in general and is relevant to elucidate their role in physiological settings in which selection between different anions is critical; an example is provided by plants that grow in soils rich in both Cl− and NO3− and need to strictly discriminate between them; plant CLC homologues have been shown to play a major role in this process (24, 44).
Supplementary Material
Acknowledgments
We thank A. R. Murgia and F. Quartino for excellent technical assistance.
This work was supported by Telethon Italy Grant GGP08064, the Italian Institute of Technology (Progetto SEED), and the Compagnia San Paolo.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S9.
- Sin
- internal binding site
- Scen
- central binding site
- Sext
- external binding site
- MTS
- methanethiosulfonate
- MTSEH
- 2-hydroxyethyl methanethiosulfonate
- MTSES
- 2-sulfonatoethyl methanethiosulfonate
- MTSET
- 2-(trimethylammonium)ethyl methanethiosulfonate
- ITC
- isothermal calorimetry.
REFERENCES
- 1. Novarino G., Weinert S., Rickheit G., Jentsch T. J. (2010) Science 328, 1398–1401 [DOI] [PubMed] [Google Scholar]
- 2. Steinberg B. E., Huynh K. K., Brodovitch A., Jabs S., Stauber T., Jentsch T. J., Grinstein S. (2010) J. Cell Biol. 189, 1171–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weinert S., Jabs S., Supanchart C., Schweizer M., Gimber N., Richter M., Rademann J., Stauber T., Kornak U., Jentsch T. J. (2010) Science 328, 1401–1403 [DOI] [PubMed] [Google Scholar]
- 4. Jentsch T. J., Stein V., Weinreich F., Zdebik A. A. (2002) Physiol. Rev. 82, 503–568 [DOI] [PubMed] [Google Scholar]
- 5. Jentsch T. J., Poët M., Fuhrmann J. C., Zdebik A. A. (2005) Annu. Rev. Physiol. 67, 779–807 [DOI] [PubMed] [Google Scholar]
- 6. Zifarelli G., Pusch M. (2007) Rev. Physiol. Biochem. Pharmacol. 158, 23–76 [DOI] [PubMed] [Google Scholar]
- 7. Zifarelli G., Pusch M. (2009) EMBO J. 28, 175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graves A. R., Curran P. K., Smith C. L., Mindell J. A. (2008) Nature 453, 788–792 [DOI] [PubMed] [Google Scholar]
- 9. Accardi A., Miller C. (2004) Nature 427, 803–807 [DOI] [PubMed] [Google Scholar]
- 10. Dutzler R., Campbell E. B., Cadene M., Chait B. T., MacKinnon R. (2002) Nature 415, 287–294 [DOI] [PubMed] [Google Scholar]
- 11. Dutzler R., Campbell E. B., MacKinnon R. (2003) Science 300, 108–112 [DOI] [PubMed] [Google Scholar]
- 12. Estévez R., Schroeder B. C., Accardi A., Jentsch T. J., Pusch M. (2003) Neuron 38, 47–59 [DOI] [PubMed] [Google Scholar]
- 13. Engh A. M., Maduke M. (2005) J. Gen. Physiol. 125, 601–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin C. W., Chen T. Y. (2003) J. Gen. Physiol. 122, 147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feng L., Campbell E. B., Hsiung Y., MacKinnon R. (2010) Science 330, 635–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Traverso S., Elia L., Pusch M. (2003) J. Gen. Physiol. 122, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fahlke C., Yu H. T., Beck C. L., Rhodes T. H., George A. L., Jr. (1997) Nature 390, 529–532 [DOI] [PubMed] [Google Scholar]
- 18. Zdebik A. A., Zifarelli G., Bergsdorf E. Y., Soliani P., Scheel O., Jentsch T. J., Pusch M. (2008) J. Biol. Chem. 283, 4219–4227 [DOI] [PubMed] [Google Scholar]
- 19. Picollo A., Pusch M. (2005) Nature 436, 420–423 [DOI] [PubMed] [Google Scholar]
- 20. De Angeli A., Monachello D., Ephritikhine G., Frachisse J. M., Thomine S., Gambale F., Barbier-Brygoo H. (2006) Nature 442, 939–942 [DOI] [PubMed] [Google Scholar]
- 21. Nguitragool W., Miller C. (2006) J. Mol. Biol. 362, 682–690 [DOI] [PubMed] [Google Scholar]
- 22. Picollo A., Malvezzi M., Houtman J. C., Accardi A. (2009) Nat. Struct. Mol. Biol. 16, 1294–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bergsdorf E. Y., Zdebik A. A., Jentsch T. J. (2009) J. Biol. Chem. 284, 11184–11193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wege S., Jossier M., Filleur S., Thomine S., Barbier-Brygoo H., Gambale F., De Angeli A. (2010) Plant J. 63, 861–869 [DOI] [PubMed] [Google Scholar]
- 25. Accardi A., Lobet S., Williams C., Miller C., Dutzler R. (2006) J. Mol. Biol. 362, 691–699 [DOI] [PubMed] [Google Scholar]
- 26. Friedrich T., Breiderhoff T., Jentsch T. J. (1999) J. Biol. Chem. 274, 896–902 [DOI] [PubMed] [Google Scholar]
- 27. Accardi A., Kolmakova-Partensky L., Williams C., Miller C. (2004) J. Gen. Physiol. 123, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Accardi A., Picollo A. (2010) Biochim. Biophys. Acta 1798, 1457–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Accardi A., Pusch M. (2003) J. Gen. Physiol. 122, 277–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lorenz C., Pusch M., Jentsch T. J. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 13362–13366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woodhull A. M. (1973) J. Gen. Physiol. 61, 687–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steinmeyer K., Schwappach B., Bens M., Vandewalle A., Jentsch T. J. (1995) J. Biol. Chem. 270, 31172–31177 [DOI] [PubMed] [Google Scholar]
- 33. Hille B. (2001) Ion Channels of Excitable Membranes, 3rd Ed., Sinauer, Sunderland, MA [Google Scholar]
- 34. Eisenman G., Horn R. (1983) J. Membr. Biol. 76, 197–225 [DOI] [PubMed] [Google Scholar]
- 35. Pusch M., Ludewig U., Rehfeldt A., Jentsch T. J. (1995) Nature 373, 527–531 [DOI] [PubMed] [Google Scholar]
- 36. Rychkov G. Y., Pusch M., Roberts M. L., Jentsch T. J., Bretag A. H. (1998) J. Gen. Physiol. 111, 653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vandenberg R. J., Shaddick K., Ju P. (2007) J. Biol. Chem. 282, 14447–14453 [DOI] [PubMed] [Google Scholar]
- 38. Rosental N., Kanner B. I. (2010) J. Biol. Chem. 285, 21241–21248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jayaram H., Accardi A., Wu F., Williams C., Miller C. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 11194–11199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ludewig U., Jentsch T. J., Pusch M. (1997) J. Gen. Physiol. 110, 165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin C. W., Chen T. Y. (2000) J. Gen. Physiol. 116, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gradogna A., Babini E., Picollo A., Pusch M. (2010) J. Gen. Physiol. 136, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Neyton J., Miller C. (1988) J. Gen. Physiol. 92, 569–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jossier M., Kroniewicz L., Dalmas F., Le Thiec D., Ephritikhine G., Thomine S., Barbier-Brygoo H., Vavasseur A., Filleur S., Leonhardt N. (2010) Plant J. 64, 563–576 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.