

Background: p21Cip1 is controlled by both a transcriptional and post-translational mechanism during cell cycle progression and differentiation.
Results: CDK2 blocks APC/CCdc20-mediated ubiquitylation of p21 at the N terminus, causing G2 arrest in mitogen-stimulated cardiomyocytes.
Conclusion: The regulation of p21 ubiquitylation is required to maintain a terminal differentiation state of cardiomyocytes.
Significance: This provides a novel insight on the control of cell cycle arrest in terminal differentiation.
Keywords: Cardiac Muscle, Cell Cycle, Cyclins, Ubiquitin Ligase, Ubiquitylation, APC/Cdc20, Cardiomyocyte, Cyclin-dependent Kinase 2, p21Cip1
Abstract
Cyclin-dependent kinase inhibitor p21Cip1 plays a crucial role in regulating cell cycle arrest and differentiation. It is known that p21Cip1 increases during terminal differentiation of cardiomyocytes, but its expression control and biological roles are not fully understood. Here, we show that the p21Cip1 protein is stabilized in cardiomyocytes after mitogenic stimulation, due to its increased CDK2 binding and inhibition of ubiquitylation. The APC/CCdc20 complex is shown to be an E3 ligase mediating ubiquitylation of p21Cip1 at the N terminus. CDK2, but not CDC2, suppressed the interaction of p21Cip1 with Cdc20, thereby leading to inhibition of anaphase-promoting complex/cyclosome and its activator Cdc20 (APC/CCdc20)-mediated p21Cip1 ubiquitylation. It was further demonstrated that p21Cip1 accumulation caused G2 arrest of cardiomyocytes that were forced to re-enter the cell cycle. Taken together, these data show that the stability of the p21Cip1 protein is actively regulated in terminally differentiated cardiomyocytes and plays a role in inhibiting their uncontrolled cell cycle progression. Our study provides a novel insight on the control of p21Cip1 by ubiquitin-mediated degradation and its implication in cell cycle arrest in terminal differentiation.
Introduction
Cell cycle inhibitor p21Cip1 (p21) plays a crucial role in several biological aspects, including cell cycle, differentiation, senescence, and apoptosis (1). It is well established that p21 protein binds to cyclin-dependent kinases (CDKs)2 through its N-terminal domain to function as a stoichiometric inhibitor, thereby regulating cell cycle progression. Furthermore, the C-terminal domain of p21 binds to proliferating cell nuclear antigen and inhibits the processivity of DNA polymerase.
The expression of p21 protein during the cell cycle is regulated at levels of both transcription and protein stability. Notably, p21 mRNA is induced at the transcriptional level (2). For example, p21 is one of the target genes of the tumor suppressor p53 and is involved in p53-dependent DNA damage-induced G1 arrest of the cell cycle (3, 4). In contrast, down-regulation of p21 protein during cell cycle progression is mainly exerted by post-translational regulation, such as ubiquitin-dependent or -independent degradation by the proteasome (5–11). Among the E3 ligase complex, SCFSkp2 complex is implicated in degrading p21 at the G1/S transition and during S phase by ubiquitylating four distinct lysine residues located in the C-terminal region (12, 13), and CRLCdt2 targets the degradation of p21 binding to proliferating cell nuclear antigen in the S phase (14, 15). Amador et al. (19) showed that anaphase-promoting complex/cyclosome and its activator Cdc20 (APC/CCdc20), which targets mitotic proteins for degradation by proteasome (16–18), controls the ubiquitin-mediated degradation of p21 during G2/M. The protein degradation of p21 is regulated by interaction with CDKs. Notably, the CDK2-cyclin E complex binds to p21 and phosphorylates its Ser-130 residue, thereby facilitating the degradation of p21 during S phase of the cell cycle (12, 13, 20). This accelerated degradation of p21 by CDK2 is crucial for cell cycle progression in proliferating cells.
p21 is also involved in differentiation of cells, such as intestinal epithelial cells, keratinocytes, PC12 cells, glioma cells, skeletal muscle cells, and cardiomyocytes (21). Mammalian cardiomyocytes irreversibly withdraw from the cell cycle soon after birth and enter terminal differentiation (22–24). Several studies have reported that the expression of p21 protein increases significantly throughout cardiac development in mouse and human (25, 26). Recently, Di Stefano et al. (27) reported that triple transfection with p21, p27Kip1, and p57Kip2 small interfering RNAs (siRNAs) induces cardiomyocyte to enter S phase. Thus, p21 plays an important role for withdrawal of cardiomyocytes from the cell cycle. Although the transcriptional regulation of p21 is reported throughout cardiac development (28), detailed mechanism by which p21 protein is controlled has remained elusive.
We have previously shown that cyclin D1 is induced by mitogenic stimuli (29, 30), but its nuclear import is impaired in terminally differentiated cardiomyocytes. The expression of the nuclear localization signal (NLS)-tagged cyclin D1 (D1NLS) and CDK4 triggers the cell cycle, indicating that the impairment of nuclear expression of cyclin D1 is one of the barriers of the cell cycle in cardiomyoyctes (31). Furthermore, we found that Skp2 is actively ubiquitylated to be degraded, and this leads to accumulation of CDK inhibitor p27, one of the targets of SCFSkp2 ubiquitin ligase. Thus, combined expression of Skp2 and D1NLS/CDK4 promotes cardiomyocyte proliferation both in vitro and in vivo (32, 33). However, the proliferation of these cardiomyocytes is still limited. Thus, it is expected that p21 is another important key factor of the maintenance of cardiomyocyte cell cycle arrest.
In this study, we investigated the post-transcriptional control of p21 in terminally differentiated cardiomyocytes. We show here that CDK2 is implicated in increased stabilization of p21 protein after mitogenic stimulation by inhibiting APC/CCdc20 ubiquitylation. It is further demonstrated that p21 protein causes G2 arrest in cardiomyocytes that had been forced to re-enter the cell cycle. Our findings provide a novel insight into the control mechanism of p21 by which mammalian cardiomyocytes cease to proliferate during terminal differentiation.
EXPERIMENTAL PROCEDURES
Plasmids and Reagents
Wild type rat p21 was PCR-amplified using pCRW0.8 that was provided by Dr. Vogelstein. The primer set was as follow: 5′-ATGTCCGATCCTGGTGATGT-3′ (forward) and 5′-TCAGGGCTTTCTCTTGCAGA-3′ (reverse). Mutant p21(1–140) lacking the C terminus was generated by PCR using the primers 5′-ATGTCCGATCCTGGTGATGT-3′ (forward) and 5′-TTATCGGCCCTGAGATGTCC-3′ (reverse). p21 CK− (R18A, L20A, and F50A) and K3R(1–140) mutants were generated by site-directed mutagenesis as described (Stratagene) using p21 (WT) and p21(1–140) mutant as templates, respectively. N-terminally HA-tagged p21 was generated by PCR using the forward primer that encoded HA-precision protease sequence, 5′-ATGTATCCATACGATGTGCCAGATTACGCACTGGAAGTTCTGTTCCAGGGGCCCATGTCCGATCCTGGTGATGT-3′ (forward) and 5′-TTATCGGCCCTGAGATGTCC-3′ (reverse). All the PCR products were subcloned into pGEMT vector (Promega). Lactacystin was purchased from Peptide Institute Inc., and cycloheximide was from Sigma. Other chemicals were reagent grade.
Antibodies
The following antibodies were used in this study: mouse monoclonal anti-Cdc27 (AF3.1, Sigma); anti-cyclin D1 (CC12, Oncogene Research Products); anti-CDC2 (sc-54, Santa Cruz Biotechnology); anti-glutathione S-transferase (GS019, Nacalai Tesque, Inc.); anti-tropomyosin (clone CH1, Lab Vision Corp.); anti-HA tag (2367, Cell Signaling Technology); anti-p55CDC/Cdc20 (SC-5296, Santa Cruz Biotechnology); anti-sarcomeric actin (M0874, DAKO); anti-β-actin (A2228, Sigma); anti-ubiquitin (P4D1, Cell Signaling); rabbit polyclonal anti-phosphohistone H3 (Ser-10) (06-570, Upstate); anti-glutathione S-transferase (A5800, Molecular Probes); anti-Skp1 (2156, Cell Signaling Technology); anti-Cul1 (Zymed Laboratories Inc.); anti-p21 (SC-756, Santa Cruz Biotechnology); anti-p21 (SC-397, Santa Cruz Biotechnology); anti-Ub (SC-9133, Santa Cruz Biotechnology); anti-CDK2 (SC-163, Santa Cruz Biotechnology); anti-CDK4 (SC-260, Santa Cruz Biotechnology); and anti-cyclin E (SC-481, Santa Cruz Biotechnology).
Adenoviruses
Adenoviruses encoding nuclear localization signal-tagged cyclin D1 (Ad-D1NLS) and CDK4 (Ad-CDK4) were as described previously (32). The cDNA corresponding to His-Myc-ubiquitin K48R mutant (34), CDK2, CDK2 dominant negative (CDK2DN) mutant (35), and cyclin E were ligated into the SwaI site of the E1-deleted region of cassette cosmid vector pAxCAwt (TaKaRa). The DNA containing the siRNA sequence that targets rat p21 or CDK2 inserted into pcPURU6β were obtained by TaKaRa and subsequently ligated into the SwaI site of pAxcw (TaKaRa). Oligonucleotide sequences specific for rat p21 from 79 to 97 were 5′-GGGGTAAAGTATGCTGTCAACGTGTGCTGTCCGTTGACGGCATACTTTGCTCCTTTTT-3′ (sense) and 5′-AAAAAGGAGCAAAGTATGCCGTCAACGGACAGCACACGTTGACAGCATACTTTACCCC-3′ (antisense); sequences specific for rat CDK2 from 1011 to 1029 were 5′-GGGTGTGATTAAACTAGTAACGTGTGCTGTCCGTTACTGGTTTAGTCACATCCTTTTT-3′ (sense) and 5′-AAAAAGGATGTGACTAAACCAGTAACGGACAGCACACGTTACTAGTTTAATCACACCC-3′ (antisense), respectively.
Cell Culture and Treatment
Rat neonatal cardiomyocytes were isolated and cultured as described previously (31). Briefly, heart ventricles were isolated from 3-day-old postnatal Sprague-Dawley rats, trisected, and then digested four times with 1 mg/ml collagenase type II (Worthington) at 37 °C for 20 min. The dispersed cells were purified by centrifugation through a discontinuous Percoll gradient of 1.050, 1.060, and 1.082 g/ml. The cardiac cells were collected and plated on a 60-mm dish (2 × 106 cells) or on 25-mm collagen-coated coverslips (2 × 105 cells) in minimum essential medium supplemented with 5% calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cardiomyocytes were cultured at 37 °C for 24 h in humidified air with 5% carbon dioxide, after which the medium was changed to MEM containing 0.5% calf serum and further incubated for 36 h. Cells were then treated with medium containing 10% fetal bovine serum or 10 ng of bFGF (Sigma) and further incubated for another 24 h. Cells were infected with Ad-D1NLS, Ad-CDK4, and His-Myc-ubiquitin K48R at a multiplicity of infection of 20 and Ad-CDK2, Ad-CDK2DN, Ad-cyclin E, Ad-CDC2 (1764, Vector Biolabs, The Gene Delivery Co.), CDK2 siRNA, and p21 siRNA at a multiplicity of infection of 20 or 80 for 48 h. Use of rats in this study was approved by the Institutional Animal Care and Use Committee of Tokyo Medical and Dental University.
Real Time Reverse Transcription-PCR
Total RNA was isolated by the acid-guanidinium method using a kit from Nacalai Tesque Inc. Isolated total RNA was reverse-transcribed using the PrimeScript reverse transcriptase kit (TaKaRa). Primers were used as follows: rat GAPDH, 5′-AAGATGGTGAAGGTCGGTGT-3′ (forward) and 5′-TTCCCATTCTCAGCCTTGAC-3′ (reverse); and rat p21, 5′-TCTTGCACTCTGGTGTCTCA-3′ (forward) and 5′-GGGCTTTCTCTTGCAGAAGA-3′ (reverse). PCRs were performed using ABI PRISM 7900HT sequence detection systems (Applied Biosystems) and Power SYBR Green PCR master mix (Applied Biosystems). The thermal cycling conditions were composed of an initial denaturation step at 50 °C for 2 min and 95 °C for 10 min followed by 50 cycles of PCR under the following conditions: 95 °C for 15 s, 60 °C for 20 s, and 72 °C for 30 s.
Whole Cell Extract and Western Blot
Cells treated as indicated were washed in phosphate-buffered saline, lysed in ice-cold lysis buffer (20 mm HEPES (pH 7.9), 420 mm NaCl, 0.5% Nonidet P-40, 1.5 mm MgCl2, 0.2 mm EDTA, 25% glycerol, 1 mm DTT, and phosphatase and a mixture of protease inhibitors) for 30 min. Lysates were then sonicated, and insoluble matter was removed by centrifugation. Whole cell extracts were standardized for protein content using the protein assay kit (Bio-Rad). Cell extracts (usually 12 μg of protein) were separated by SDS-PAGE, transferred onto an Immobilon-P membrane (Millipore), and subjected to Western blot with indicated antibodies. Quantitative analysis was performed with the gel analysis component of ImageJ software.
In Vivo Ubiquitylation Assay
Cardiomyocytes were infected with various adenoviruses as indicated for 42 h and treated with 20 μm lactacystin for 6 h and harvested. Whole cell extracts (50 μg of protein) in 150 μl of IP buffer (50 mm HEPES (pH 7.6), 150 mm NaCl, 1 mm EDTA, 2.5 mm EGTA, 10 mm glycerophosphate, 1 mm NaF, 0.1 mm Na2VO4, 10% glycerol, 0.1% Tween 20, 1 mm DTT, and a mixture of protease inhibitors) were mixed with 30 μl of agarose beads bound to the UBA domains from Rad23 (ubiquitinated protein enrichment kit, Calbiochem) at 4 °C for 3 h. After the beads were washed, bound protein was subjected to SDS-PAGE and Western blot.
In Vitro Ubiquitylation Assay
Whole cell extracts (40 μg of protein) were obtained from cells infected with D1NLS/CDK4 for 48 h and mixed with 15 μl of ExactaCruz C (Santa Cruz Biotechnology) that had been incubated with anti-Cdc27 antibody (3 μg) and further incubated for 2 h, followed by washing four times in buffer (50 mm HEPES (pH 7.6), 100 mm NaCl, 1 mm EDTA, 2.5 mm EGTA, 10 mm glycerophosphate, 1 mm NaF, 0.1 mm Na2VO4, 10% glycerol, 0.1% Tween 20, 1 mm DTT, and a mixture of protease inhibitors), and two times in reaction buffer (40 mm Tris (pH 7.5), 60 mm NaCl, 1 mm DTT, and a mixture of protease inhibitors). The immunoprecipitates (immunopurified APC/CCdc20 complex) or whole cell extracts from D1NLS/CDK4 incubated with 2 μl of in vitro transcribed/translated p21 in 30 μl of reaction buffer containing 5 mm MgCl2, 1.5 mm ATP, 50 ng of Uba1, 200 ng of UbcH10, 3 μg of glutathione S-transferase (GST)-ubiquitin, and/or GST-CDK2/FLAG-cyclin E (kindly provided by Dr. Kamura, Nagoya University, Japan) at 26 °C for 1 h. As substrates, various p21 mutants were prepared in vitro using 1 μg each of pGEMT vector encoding p21 in 25 μl of rabbit reticulocyte lysate (TnT kit from Promega) according to the protocol. Ubiquitylated p21 was determined by autoradiography of 35S labeled p21. To detect ubiquitylated p21 by immunoblotting, in vitro ubiquitylation assay reaction samples were immunoprecipitated with anti-p21 antibody followed by immunoblotting with anti-GST antibody. To detect p21 ubiquitylated sites, in vitro ubiquitylation assay reaction samples using HA-p21 substrates were immunoprecipitated with anti-HA antibody. The beads were washed four times in Wash buffer and two times in PreScission protease cleavage buffer (50 mm Tris (pH 7.0), 150 mm NaCl, 1 mm DTT) and incubated with 80 units/ml Precision protease (GE Healthcare) overnight at 4 °C. After incubation, the beads and the supernatants were subjected to SDS-PAGE and immunoblotted with anti-GST antibody. To specify which lysine residue is linked to ubiquitin chains, GST-p21 (506104, Calbiochem) as substrate and mutant types of ubiquitin were used, and reaction samples were purified by GST pulldown assay and immunoblotted with anti-Ub (P4D1, Cell Signaling) antibody.
Cell Cycle Analysis and Immunohistochemistry
For cell cycle analysis, cells were treated with propidium iodide (50 μg/ml) and RNase A (500 μg/ml). The DNA content of cells was analyzed using a laser scanning cytometer (LSC 101, Olympus). For analysis of cell number, cardiomyocytes were untreated or infected with D1NLS expressing adenoviruses and cultured. Cell numbers were counted at 2–6 days after infection, and the relative cell proliferation was expressed as mean ± S.E. of three independent experiments. For immunohistochemistry, cells cultured on glass coverslips were fixed with 4% paraformaldehyde and stained with primary antibodies as indicated and Alexa 488 or Alexa 568 secondary antibodies (Invitrogen). Nuclei were stained with DAPI. Images were obtained with the laser scanning confocal image system (LSM510, ZEISS, Jena, Germany).
In Vitro CDK-Associated Kinase Assay
Whole cell extracts were immunoprecipitated with anti-CDK2 or anti-CDC2 antibody, and the complex was assayed for CDK kinase activity as described (32). The immunoprecipitated complexes were incubated with 2 μg of histone H1, 60 μm cold ATP, and 5 μCi [γ-32P]ATP (6000 Ci/mmol) for 30 min at 30 °C. Phosphorylated proteins were subjected to autoradiography.
Statistical Analysis
Quantitative data were expressed as mean ± S.E. Statistical analysis was performed with the Student's t test.
RESULTS
p21 Is Stabilized in Cardiomyocytes in Response to Mitogenic Stimuli
Some of the cell cycle regulators are known to be increased with several mitogenic stimuli in primary cardiomyocytes (23, 24, 29, 36, 37). As shown in Fig. 1A, the expression of p21 was increased in cardiomyocytes treated with bFGF and serum. In particular, adenoviruses encoding NLS-tagged cyclin D1 and CDK4 caused a robust induction of p21 expression in the nuclei of the cells stained with tropomyosin, one of the cardiac specific markers (Fig. 1, A and B). Although p21 is known to be controlled by transcriptional regulation, it was shown that p21 mRNA level had no apparent change and 1.4-, 0.8-, and 2.3-fold change compared with the control for bFGF, serum, or D1NLS-CDK4-treated cardiomyocytes, respectively (supplemental Fig. S1A). We further treated cardiac cells with mutant ubiquitin K48R (UbK48R) in which lysine residue 48 was changed to arginine, proteasome inhibitor lactacystin, or both. p21 protein was remarkably increased with these treatments, and the level of p21 was almost similar among them, indicating that p21 is actively degraded via ubiquitin-dependent proteasome system. The percentage of p21 levels in cells treated with bFGF, serum, and D1NLS/CDK4 compared with those in the presence of a proteasome inhibitor was up-regulated from 8% (nontreated) to 30% (bFGF), 64% (serum), and 64% (D1NLS/CDK4) (Fig. 1C and supplemental Fig. S1B). As expected from the result that p21 was stabilized, Ub (K48R), lactacystin, and both decreased CDK2 kinase activity (Fig. 1C and supplemental Fig. S1C). Furthermore, they also reduced D1NLS/CDK4-induced proliferation of cardiomyocytes to the same extent as control (supplemental Fig. S1D). In contrast, p21 was not clearly stabilized in proliferating rat embryonic fibroblasts compared with in serum-starved cells (supplemental Fig. S2). These data, taken together, strongly support the notion that p21 protein is induced through increased stabilization in response to mitogenic stimulation especially in cardiomyocytes.
FIGURE 1.

p21 is stabilized in cardiomyocytes in response to mitogenic stimuli. Cardiomyocytes were treated with 10 ng of bFGF, 10% FBS for 24 h or infected with a combination of adenoviruses encoding D1NLS and CDK4 for 48 h. A, Western blotting of whole cell extracts for p21, CDK2, cyclin D1, CDK4, sarcomeric actin, and CDK2 in immunoprecipitates (IP) with anti-CDK2 antibody and kinase activity of CDK2. B, immunostaining for p21 in D1NLS/CDK4-expressing cardiomyocytes. Green, p21; red, tropomyosin; white, DAPI. Arrowheads and arrows indicate cardiomyocytes and cardiac fibroblasts, respectively. Scale bar, 50 μm. C, Western blotting of whole cell extracts for p21, ubiquitin, sarcomeric actin, and CDK2 in immunoprecipitates with anti-CDK2 antibody and kinase activity of CDK2. Cells were treated with adenovirus for His-Myc-tagged ubiquitin (K48R) for 48 h, and with 20 μm lactacystin for 6 h before harvest.
CDK2 Binds to and Stabilizes p21 Protein
As shown in Fig. 1A, cardiomyocytes treated with bFGF, serum, or D1NLS/CDK4 all induced the expression of CDK2 on parallel with the up-regulation of p21. The activity of CDK2 was also up-regulated in bFGF-, FBS-, and D1NLS/CDK4-treated cells (Fig. 1A). Thus, we examined the effect of CDK2 on p21. Overexpression of wild type CDK2 caused the increase of p21. Furthermore, the loss of function approach was employed using gene delivery of specific siRNA against CDK2. Knockdown of CDK2 significantly decreased the p21 protein in D1NLS/CDK4 cells (Fig. 2A), suggesting that CDK2, which is capable of binding of p21, controls the stability of the p21 protein. Intriguingly, the dominant negative mutant type of CDK2 (CDK2DN), which is defective in CDK2 kinase activity in D1NLS/CDK4-infected cells (supplemental Fig. S3), also elevated p21 to the same level as wild type of CDK2 (Fig. 2A), indicating the catalytic activity of CDK2 is dispensable. Consistent with this, cyclin E, the regulatory cyclin for CDK2, had no effect on the increase of p21 (supplemental Fig. S4, A and B). In contrast, p21 was not elevated by overexpression of CDC2 (supplemental Fig. S5A). Thus, the relationship of p21 binding to CDK2 and its stability were examined. Fig. 2B showed that whole amount of p21 protein in the cells was rapidly degraded with a half-life less than 1 h. In contrast, overexpression of CDK2 prolonged the half-life of p21 (Fig. 2B). Furthermore, p21 protein in the complex of CDK2 exhibited a more prolonged half-life (Fig. 2B). Collectively, these data clearly demonstrate that p21 protein is stabilized by CDK2 independently of its kinase activity.
FIGURE 2.

p21 stabilization via CDK2 binding. A, Western blotting of whole cell extracts for p21, CDK2, and sarcomeric actin. Cells were infected with adenoviruses encoding D1NLS/CDK4, wild type CDK2, dominant negative mutant type of CDK2 (CDK2DN), or siRNA for CDK2 for 48 h. B, Western blotting of whole cell extracts for p21, CDK2, and sarcomeric actin. Cells were infected with adenoviruses for D1NLS/CDK4 and wild type CDK2. 50 μg/ml cycloheximide (Sigma) was added 48 h after infection, and cells were harvested at the indicated time. To determine the p21 protein in the complex of CDK2, whole cell extracts were immunoprecipitated (IP) with anti-CDK2 antibody, followed by immunoblotting with anti-p21 antibody. Right panel is quantitative measurements. p21 in immunoprecipitates with anti-CDK2 antibody are normalized to CDK2 level, and p21 in others are normalized to actin level. C, in vivo ubiquitylation analysis of p21. Cardiomyocytes were infected with adenoviruses for His-Myc-tagged ubiquitin, D1NLS/CDK4, and wild type CDK2. Ubiquitylated proteins were isolated from total cell lysates using the Rad23 UBA domains as under “Experimental Procedures.” Bound proteins were subjected to an immunoblotting with anti-p21 antibody. WB, Western blot. D, in vitro ubiquitylation analysis of p21. WCE (40 μg of protein) from cells infected with D1NLS/CDK4 for 48 h were reacted with 35S-labeled p21 protein as a substrate in the absence or presence of GST-CDK2/FLAG-cyclin E (CDK2/CycE). The reaction products were analyzed by SDS-PAGE followed by autoradiography. E, in vitro ubiquitylation analysis of p21. WCE (40 μg of protein), obtained from cells infected with D1NLS/CDK4 for 48 h, were incubated with 0.5 μg of recombinant human p21, GST fusion (506104, Calbiochem) as a substrate, and 5 μg of ubiquitin (WT, R48, R63, K48, K63, or K0, kindly provided from Dr. Baer, Columbia University) in the reaction buffer. To detect ubiquitylated p21, ubiquitylation assay reaction samples were purified by GST pulldown assay, fractionated by SDS-PAGE, and detected by immunoblotting with mouse monoclonal anti-ubiquitin antibody (P4D1, Cell Signaling). Reactions in the presence of wild type ubiquitin (lanes 1 and 5), Ub (R48) mutant ubiquitin (lane 2), Ub (R63) mutant ubiquitin (lane 3), lysine-less Ub-K0 mutant ubiquitin (lanes 4 and 8), Ub (K48) mutant ubiquitin (lane 6), or Ub (K63) mutant ubiquitin (lane 7).
CDK2 Suppresses Polyubiquitylation of p21
We next addressed if ubiquitin-mediated pathway is involved in the p21 stabilization by CDK2. Fig. 2C showed that p21 was actively polyubiquitylated in untreated cardiomyocytes, and its activity was significantly reduced in cells treated with D1NLS/CDK4. Overexpression of CDK2 further suppressed the polyubiquitylation of p21. Ubiquitylation assay in vitro using cell extract showed p21 protein was actively ubiquitylated in cardiomyocytes, and this activity was suppressed by addition of recombinant CDK2 in a dose-dependent manner (Fig. 2D). In contrast, CDC2 had no effect on p21 ubiquitylation (supplemental Fig. S5B). Polyubiquitin chains linked through the Lys-48 residue of ubiquitin are most commonly associated with proteins targeted for proteosomal degradation; in contrast, polyubiquitylation through Lys-63 is associated with nonproteolytic functions such as signal transduction (38–40). Therefore, to examine which lysine residue is linked to ubiquitin chains, we performed in vitro ubiquitylation assay using ubiquitin mutants, Ub(R48) and Ub(R63), in which arginine was substituted for Lys-48 or Lys-63, respectively, and Ub(K48) and Ub(K63), in which all the lysines are changed to arginine with the exception of residue Lys-48 or Lys-63, respectively, and lysine-less ubiquitin, Ub-K0 (41). As shown in Fig. 2E (lanes 1–4), the levels of polyubiquitylation were remarkably reduced in Ub(R48) and Ub-K0 reactions. In contrast, the reaction in Ub(R63) was retained at the same level of Ub (WT). Furthermore, the robust ubiquitylation was observed in Ub(K48) reaction as much as in Ub (WT), but not in Ub(K63) (Fig. 2E, lanes 5–8). These data indicate that Lys-48-linked chains are conjugated to p21 during polyubiquitylation. Our results confirm that the p21 protein is actively polyubiquitylated followed by degradation in cardiomyocytes, and the activity is inhibited by CDK2.
p21 Is Polyubiquitylated by the APC/CCdc20 Complex and CDK2 Blocks the Interaction of p21 with Cdc20 and Suppresses Its Ubiquitylation
To determine the E3 ligase complex(es) responsible for active ubiquitylation of p21 in cardiomyocytes, we purified the p21 complex by immunoprecipitation and examined E3 ligase components in the complex. Fig. 3A showed that both the Cdc20 and Cdc27 subunits of APC/C complex were bound to p21. In contrast, it was observed that there was no significant binding of Skp1 and Cul1 in the p21 complex. Next, the interaction of p21 with Cdc20 was examined in cells overexpressing CDK2. The amount of Cdc20 binding to p21 was remarkably reduced by overexpression of CDK2 (Fig. 3B). The in vitro binding assay showed that adding recombinant GST-CDK2/cyclin E (CycE) reduced binding of p21 to Cdc20 in cell lysates from D1NLS/CDK4 cells (Fig. 3C). These suggest that CDK2 antagonizes interaction of p21 with Cdc20. We next investigated if the APC/CCdc20 complex has an activity to ubiquitylate p21 protein. To address this, we used whole cell extract from cardiomyocytes expressing D1NLS/CDK4 (WCE) or anti-cdc27 immunoprecipitates from WCE as the APC/CCdc20 complex (immunopurified APC/CCdc20 complex) for the in vitro ubiquitylation assay of p21. It was confirmed that the APC/CCdc20 complex included cdc20 and p21 (Fig. 3D). Fig. 3E showed that p21 was polyubiquitylated in the presence of the APC/CCdc20 complex at a comparable level to that in WCE. Moreover, the activity in the immunopurified APC/CCdc20 complex was remarkably inhibited by CDK2/cyclin E as in WCE. In contrast, a p21 mutant (p21CK−), which is defective in its binding to CDK2, was resistant to inhibition by CDK2/cyclin E, despite being ubiquitylated at the same level to wild type of p21 (p21WT) (Fig. 3, F and G).
FIGURE 3.

CDK2 blocks the p21-cdc20 interaction and prevents p21 ubiquitylation mediated by APC/CCdc20. A, interaction of p21 with components of various E3 ligase. Whole cell extracts of D1NLS/CDK4-infected cardiomyocytes were immunoprecipitated (IP) with anti-p21 antibody or control IgG, and the immunocomplexes were assayed by Western blotting (WB). B, Western blotting of immunoprecipitates using anti-p21 antibody. Cardiomyocytes were infected with adenoviruses for D1NLS/CDK4 and wild type CDK2. Whole cell extracts were immunoprecipitated with anti-p21 antibody and immunoblotted with indicated antibodies. C, in vitro binding analysis of p21 with Cdc20. Whole cell extracts (WCE) (40 μg of protein) from D1NLS/CDK4 cells were incubated with or without GST-CDK2/FLAG-cyclin E (CDK2/CycE) at 4 °C for 2 h and immunoprecipitated using anti-p21 antibody. Bound Cdc20 and GST-CDK2 proteins were assayed by Western blot. Right panel represents quantitative data of Cdc20 expression in anti-p21 immunoprecipitates. D, immunopurification of APC/C complex. Whole cell extracts were immunoprecipitated with anti-Cdc27 antibody and immunoblotted with indicated antibodies. E, in vitro ubiquitylation analysis using immunopurified APC/C complex (D) or whole cell extracts (WCE) in the absence or presence of GST-CDK2/FLAG-cyclin E (CDK2/CycE). In vitro translated p21 protein was used as a substrate. The reaction products were immunoprecipitated with anti-p21 antibody, followed by immunoblotting with anti-GST antibody. Asterisk indicates light chain of IgG. F, inability of p21CK− to bind to CDK2. GST-CDK2/cyclin E complex was incubated with p21WT or p21CK− at 4 °C for 30 min. The protein lysates were immunoprecipitated with anti-p21 antibody (left panel) or incubated with glutathione-Sepharose beads (middle panel), and the binding of p21 and GST-CDK2 was assayed by Western blot. G, in vitro ubiquitylation analysis using in vitro translated p21 or p21CK− as a substrate and immunopurified APC/C complex represented in D.
These data clearly indicate that p21 is polyubiquitylated, at least in part, by the APC/CCdc20 complex in cardiomyocytes, and CDK2 counteracts p21 binding to the E3 ligase complex, resulting in inhibiting its polyubiquitylation via CDK/cyclin binding domain of p21.
APC/CCdc20-mediated Ubiquitylation of p21 Occurs at the N Terminus and Is Inhibited by CDK2
Specific lysine residue(s) of target protein is often ubiquitylated by distinct E3 ligase, and this association is considered to be one of the determinants that control stability. To determine amino acid residue(s) of p21 ubiquitylated by the APC/CCdc20 complex in cardiomyocytes, we first generated two recombinant p21 mutant proteins, a mutant deleted C-terminal cluster of lysine residues (WT(1–140)), and a mutant with Lys-Arg mutations at 15, 74, and 91 lysines in WT(1–140) (K3R residues 1–140) (supplemental Fig. S6A). These two types of mutant p21 were ubiquitylated equally to wild type of p21, and its activity was inhibited by CDK2 (supplemental Fig. S6B). This raised a possibility that other lysine residue(s) might be a target of modification by the APC/CCdc20 complex. Because it has been reported that p21 is specifically ubiquitylated at the N terminus (42, 43), we sought to examine the possibility that the N terminus of p21 is the target for ubiquitylation. We generated a p21 fusion protein that is N-terminally fused with HA and a recognition sequence for precision protease (Fig. 4A) and did ubiquitylation assay with this fusion protein and WCE from D1NLS/CDK4 cardiomyocytes. The product was divided in 2 aliquots. One aliquot was treated with precision protease. We then performed anti-HA immunoprecipitation with the cleaved and uncleaved forms of p21 and immunoblotted with an antibody to GST, because a recombinant ubiquitin was tagged with GST. After immunoprecipitation, we observed ubiquitylated species of both uncleaved p21 and the cleaved N-terminal HA tag (beads). In contrast, very little ubiquitylation was detected in the released p21 fraction (sup) (Fig. 4B). This strongly indicated that the ubiquitylation of p21 occurred at the N terminus much more efficiently compared with several other lysine residues. Furthermore, the similar result was obtained from ubiquitylation assay with immunopurified APC/CCdc20 instead of WCE. When the reaction was performed in the presence of the recombinant CDK2, most of the ubiquitylation in the bead fractions was abolished (Fig. 4C). Taken together, these data indicate that the N terminus of p21 protein is an efficient target of ubiquitylation by APC/CCdc20, and its activity was inhibited by CDK2.
FIGURE 4.
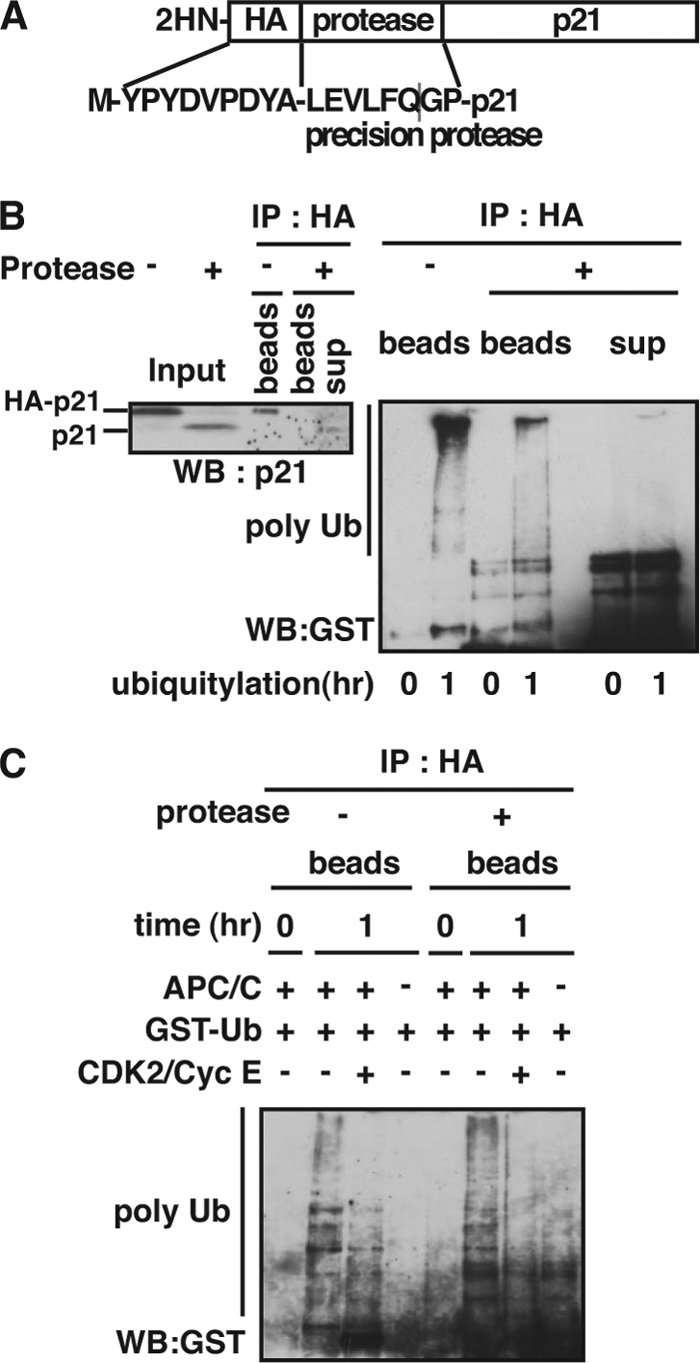
N terminus of p21 is a target of ubiquitylation by APC/CCdc20 in cardiomyocytes. A, p21 protein was N-terminally tagged with a peptide of HA and precision protease recognition consensus sequence. B, in vitro ubiquitylation analysis using translated HA-tagged p21 and whole cell extracts of D1NLS/CDK4 cardiomyocytes. After the reaction, the product was divided in 2 aliquots. One aliquot was incubated with PreScission protease at 4 °C overnight. An equivalent amount of the beads and supernatant fractions of the cleaved sample and the uncleaved sample were immunoprecipitated with anti-HA antibody and then immunoblotted with anti-p21 antibody (left panel) or anti-GST antibody (right panel). C, in vitro ubiquitylation analysis using translated HA-tagged p21 and immunopurified APC/C in the absence or presence of GST-CDK2/FLAG-cyclin E (CDK2/CycE). The reaction products were incubated with PreScission protease at 4 °C overnight and then immunoprecipitated with anti-HA antibody. The polyubiquitylated p21 was detected by immunoblotting with anti-GST antibody.
Accumulation of p21 Causes G2 Arrest of the Cell Cycle in Cardiomyocytes
We next addressed the biological role of p21 stabilization by CDK2 in cardiomyocytes. To this end, we examined cell cycle progression in cells whose p21 protein was down-regulated by gene transfer of adenovirus encoding specific siRNA against p21. As shown in Fig. 5A, down-regulation of p21 correlated with the increase of both CDK2 and CDC2 kinase activity. Consistent with the biochemical activation of these kinases, proliferation of D1NLS/CDK4 cells was significantly increased when p21 protein was knocked down (Fig. 5B), strongly supporting that p21 accumulation plays a negative role in proliferation of cardiomyocytes in vivo. We next examined the effect of CDK2. Fig. 5C showed that overexpression of CDK2 caused a significant increase in the percentage of cells at G2/M phase, and this increase was almost abolished by knockdown of p21 protein. The role of p21 in causing G2/M arrest was further examined by counting cells positive for phosphorylated histone H3 (H3P), a marker for cells with M phase progression. As shown in Fig. 5D, the rate of H3P-positive cells in D1NLS/CDK4 cells was remarkably suppressed by overexpressing CDK2, and this suppression was reversed by siRNA against p21. Down-regulation of p21 protein in D1NLS/CDK4 cells moderately increased the appearance of H3P-positive cells, consistent with accelerated cell proliferation by p21-specific siRNA (Fig. 5B). All these data strongly support that CDK2-mediated stabilization of p21 protein is implicated in G2 arrest of cardiomyocytes.
FIGURE 5.

Accumulation of p21 protein causes G2 arrest of cell cycle in cardiomyocytes. A, down-regulation of p21 in D1NLS/CDK4 cardiomyocytes infected with adenovirus encoding siRNA specific for p21. At 48 h post-infection, the cells were harvested and extracted for CDK2 and CDC2 kinase assay. B, measurement of cell number of cardiomyocytes infected with adenoviruses for D1NLS/CDK4 and p21 siRNA. At each day indicated, cell number was counted, and the relative cell proliferation was expressed as mean ± S.E. of three independent experiments. C, cell cycle analysis of cardiomyocytes infected with a combination of adenoviruses for D1NLS/CDK4, CDK2, and p21 siRNA as indicated using a laser scanning cytometer. D, quantitative measurement of H3P-positive cardiomyocytes. Cells were treated as in C. At 48 h post-infection, cells were fixed and immunostained with anti-phospho-histone H3 (H3P) and anti-tropomyosin antibodies. Cardiomyocytes positive for H3P were counted and expressed as the percent of total. Data are the means with S.E. of three independent experiments.
DISCUSSION
Expression of the cell cycle regulator p21, a short lived protein, is controlled by both transcriptional and post-translational mechanisms during cell cycle progression and differentiation. In this study, we demonstrate for the first time that p21 is stabilized in mitogen-stimulated cardiomyocytes through antagonizing effect of CDK2 on APC/CCdc20-mediated ubiquitylation. Furthermore, it is shown that this negative regulation plays a role in causing G2 arrest of the cell cycle in terminally differentiated cardiomyocytes.
SCFSkp2, CRL4Cdt2, and APC/CCdc20 are reported to be involved in the degradation of p21 during the cell cycle (12–15, 19, 44). We have reported that Skp2 is actively ubiquitylated and degraded in cardiomyocytes, and its overexpression induced degradation of p27 (32), but not p21 (data not shown). In addition, we showed that p21 interacted with neither Skp1 nor Cul1, components of SCFSkp2 complex (Fig. 3A). Thus, SCFSkp2 does not represent a major ubiquitin ligase responsible for p21 degradation in differentiated cardiomyocytes. In contrast, p21 interacted with Cdc20 and Cdc27 (Fig. 3A), and immunopurified APC/Ccdc20 actively ubiquitylated p21 at the same level of WCE (Fig. 3E). It is thus evident that ubiquitylation of p21 in cardiomyocytes expressing D1NLS/CDK4 is mainly due to APC/CCdc20, although we did not analyze about CRL4Cdt2. Furthermore, we showed that ubiquitylation of a p21 mutant missing all lysines was detected (supplemental Fig. S6), and the N terminus of p21 was ubiquitylated (Fig. 4). It was reported that p21 is ubiquitylated at not only internal lysines but also at the N terminus (42, 43). Based on in vitro ubiquitylation assay using extracts from cardiomyocytes, p21 is thus ubiquitylated by APC/CCdc20 complex at the N terminus.
It is well established that CDK promotes the degradation of p21 protein. For example, the p21 mutant deficient for interaction with CDKs greatly reduces sensitivity to proteasome-mediated degradation (45). CDK2 phosphorylates p21 at Ser-130 and facilitates the ubiquitin-dependent degradation of p21 by SCFSkp2 both in vitro and in vivo (12, 13, 19, 20). In contrast, in this study, CDK2 efficiently inhibited the p21 ubiquitin-mediated degradation, counteracting p21 ubiquitylation by APC/CCdc20, possibly because of its interaction with p21. Cdc20 contains at its C terminus a WD40 repeat domain that directly contacts the destruction box (D box) motif of APC/C substrates (46). The APC/CCdc20-mediated ubiquitylation and degradation of p21 require an intact D box (RXXL) motif within 84–87 amino acids of p21 (19). Interestingly, the binding site of p21 for various CDKs, including CDK2, has been mapped to within 28–82 amino acid residues (47). Tyr-76 of p21, which corresponds to Tyr-88 of p27, a closely related CDK inhibitor (48, 49), is considered to be required for CDK-p21 interaction. Therefore, the close location of the CDK2 binding domain and the D box motif of p21 may interfere with the interaction of p21 with Cdc20 upon CDK2 binding, thereby leading to inhibition of p21 ubiquitylation. Additionally, it is also possible that specific modification of p21 by other protein kinases or interaction between CDK2 and p21 through other protein factor(s) are involved in CDK2-induced p21 stability. Ono et al. (50) have reported that TOK-1 binds to the C terminus of p21 and enhances its interaction with CDK2. This ternary complex may regulate CDK2-mediated stability of p21, because TOK-1 is expressed in tissues such as skeletal muscle and heart. Thus, more study is required to understand the regulation of CDK2 interaction with p21, such as the protein modification of p21 and modulatory protein factor(s), and tissue-specific regulation.
p21 is also implicated in cell differentiation. For instance, p21 triggers megakaryocytic maturation in K562, a multipotent human leukemia cell line (28), and the inactivation of p21 by the adenovirus E1A protein leads to the induction of DNA replication in differentiated muscle cells (51). In cardiomyocyte differentiation, the loss of proliferative capacity correlates an increased expression of p21, which inhibits CDK2 activity and down-regulates proliferating cell nuclear antigen expression (26, 52). Consistent with this, CDK2-induced accumulation of p21 caused a G2 arrest of the cell cycle in cardiomyocytes (Fig. 5), suggesting that p21 plays a role in withdrawing the cells from the cell cycle to maintain terminal differentiation. In contrast, the loss of p21 caused no significant abnormality of cardiac development in mice (53). Thus, p21 alone is not sufficient for withdrawing cardiomyocytes from the cell cycle during differentiation. We propose CDK2-mediated inhibition of p21 degradation represents one mechanism that tightly maintains cardiomyocytes in the terminally differentiated state.
It has recently been shown that the delivery of cell cycle regulators induces myocardial regeneration and enhances cardiac function (24, 54–56). We have recently shown that a combination of cell cycle drivers (cyclin D1/CDK4) and removal of a cell cycle brake (Skp2) induces efficient and stable proliferation of adult cardiomyocytes in situ and prevents development of ischemic heart failure (33). However, the proliferative capacity of cardiomyocytes is still limited. Thus, it is possible that accumulation of p21 through CDK2-mediated suppression of APC/CCdc20 is implicated in withdrawal from the cell cycle after forced cell proliferation.
Finally, this study demonstrates for the first time that the N terminus of p21 is ubiquitylated by APC/CCdc20 E3 ligase, and this activity is inhibited by CDK2 in cardiomyocytes. This provides not only a novel control mechanism of p21 stability in cardiomyocytes associated with terminal differentiation but a therapeutic target for improving cardiac repair following myocardial injury.
Supplementary Material
Acknowledgments
We thank Dr. Y. Tanaka for critical discussion. We are also grateful to Dr. R. Baer, Dr. B. Vogelstein, Dr. S. van den Heuvel, and Dr. R. Kopito for materials.
This work was supported in part by a grant-in-aid for research on priority areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, a grant from the Japan Society for the Promotion of Science, a grant from the Japan Space Forum, a grant from Takeda Science Foundation, and a grant from the Atsuko Ouchi Memorial Fund.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
- CDK
- cyclin-dependent kinase
- APC/CCdc20
- anaphase-promoting complex/cyclosome and its activator Cdc20
- NLS
- nuclear localization signal
- WCE
- whole cell extract
- Ub
- ubiquitin
- H3P
- phosphorylated histone H3.
REFERENCES
- 1. Sherr C. J., Roberts J. M. (1999) Genes Dev. 13, 1501–1512 [DOI] [PubMed] [Google Scholar]
- 2. Gartel A. L., Tyner A. L. (1999) Exp. Cell Res. 246, 280–289 [DOI] [PubMed] [Google Scholar]
- 3. Duli V., Kaufmann W. K., Wilson S. J., Tlsty T. D., Lees E., Harper J. W., Elledge S. J., Reed S. I. (1994) Cell 76, 1013–1023 [DOI] [PubMed] [Google Scholar]
- 4. el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B. (1993) Cell 75, 817–825 [DOI] [PubMed] [Google Scholar]
- 5. Bendjennat M., Boulaire J., Jascur T., Brickner H., Barbier V., Sarasin A., Fotedar A., Fotedar R. (2003) Cell 114, 599–610 [DOI] [PubMed] [Google Scholar]
- 6. Chen X., Barton L. F., Chi Y., Clurman B. E., Roberts J. M. (2007) Mol. Cell 26, 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coleman M. L., Marshall C. J., Olson M. F. (2003) EMBO J. 22, 2036–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin Y., Lee H., Zeng S. X., Dai M. S., Lu H. (2003) EMBO J. 22, 6365–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li X., Amazit L., Long W., Lonard D. M., Monaco J. J., O'Malley B. W. (2007) Mol. Cell 26, 831–842 [DOI] [PubMed] [Google Scholar]
- 10. Sheaff R. J., Singer J. D., Swanger J., Smitherman M., Roberts J. M., Clurman B. E. (2000) Mol. Cell 5, 403–410 [DOI] [PubMed] [Google Scholar]
- 11. Touitou R., Richardson J., Bose S., Nakanishi M., Rivett J., Allday M. J. (2001) EMBO J. 20, 2367–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bornstein G., Bloom J., Sitry-Shevah D., Nakayama K., Pagano M., Hershko A. (2003) J. Biol. Chem. 278, 25752–25757 [DOI] [PubMed] [Google Scholar]
- 13. Wang W., Nacusi L., Sheaff R. J., Liu X. (2005) Biochemistry 44, 14553–14564 [DOI] [PubMed] [Google Scholar]
- 14. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., Dutta A. (2008) Genes Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim Y., Starostina N. G., Kipreos E. T. (2008) Genes Dev. 22, 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harper J. W., Burton J. L., Solomon M. J. (2002) Genes Dev. 16, 2179–2206 [DOI] [PubMed] [Google Scholar]
- 17. Peters J. M. (2002) Mol. Cell 9, 931–943 [DOI] [PubMed] [Google Scholar]
- 18. Peters J. M. (2006) Nat. Rev. Mol. Cell Biol. 7, 644–656 [DOI] [PubMed] [Google Scholar]
- 19. Amador V., Ge S., Santamaría P. G., Guardavaccaro D., Pagano M. (2007) Mol. Cell 27, 462–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu H., Nie L., Maki C. G. (2005) J. Biol. Chem. 280, 29282–29288 [DOI] [PubMed] [Google Scholar]
- 21. Studzinski G. P., Harrison L. E. (1999) Int. Rev. Cytol. 189, 1–58 [DOI] [PubMed] [Google Scholar]
- 22. Ahuja P., Sdek P., MacLellan W. R. (2007) Physiol. Rev. 87, 521–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bicknell K. A., Coxon C. H., Brooks G. (2007) J. Mol. Cell. Cardiol. 42, 706–721 [DOI] [PubMed] [Google Scholar]
- 24. Pasumarthi K. B., Field L. J. (2002) Circ. Res. 90, 1044–1054 [DOI] [PubMed] [Google Scholar]
- 25. Burton P. B., Yacoub M. H., Barton P. J. (1999) Eur. Heart. J. 20, 604–611 [DOI] [PubMed] [Google Scholar]
- 26. Poolman R. A., Gilchrist R., Brooks G. (1998) Int. J. Cardiol. 67, 133–142 [DOI] [PubMed] [Google Scholar]
- 27. Di Stefano V., Giacca M., Capogrossi M. C., Crescenzi M., Martelli F. (2011) J. Biol. Chem. 286, 8644–8654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans-Anderson H. J., Alfieri C. M., Yutzey K. E. (2008) Circ. Res. 102, 686–694 [DOI] [PubMed] [Google Scholar]
- 29. Tamamori-Adachi M., Ito H., Nobori K., Hayashida K., Kawauchi J., Adachi S., Ikeda M. A., Kitajima S. (2002) Biochem. Biophys. Res. Commun. 296, 274–280 [DOI] [PubMed] [Google Scholar]
- 30. Tamamori-Adachi M., Goto I., Yamada K., Kitajima S. (2008) Cell Cycle 7, 3768–3774 [DOI] [PubMed] [Google Scholar]
- 31. Tamamori-Adachi M., Ito H., Sumrejkanchanakij P., Adachi S., Hiroe M., Shimizu M., Kawauchi J., Sunamori M., Marumo F., Kitajima S., Ikeda M. A. (2003) Circ. Res. 92, e12–19 [DOI] [PubMed] [Google Scholar]
- 32. Tamamori-Adachi M., Hayashida K., Nobori K., Omizu C., Yamada K., Sakamoto N., Kamura T., Fukuda K., Ogawa S., Nakayama K. I., Kitajima S. (2004) J. Biol. Chem. 279, 50429–50436 [DOI] [PubMed] [Google Scholar]
- 33. Tamamori-Adachi M., Takagi H., Hashimoto K., Goto K., Hidaka T., Koshimizu U., Yamada K., Goto I., Maejima Y., Isobe M., Nakayama K. I., Inomata N., Kitajima S. (2008) Cardiovasc. Res. 80, 181–190 [DOI] [PubMed] [Google Scholar]
- 34. Yu H., Kopito R. R. (1999) J. Biol. Chem. 274, 36852–36858 [DOI] [PubMed] [Google Scholar]
- 35. van den Heuvel S., Harlow E. (1993) Science 262, 2050–2054 [DOI] [PubMed] [Google Scholar]
- 36. Engel F. B. (2005) Cell Cycle 4, 1360–1363 [DOI] [PubMed] [Google Scholar]
- 37. Parker T. G., Packer S. E., Schneider M. D. (1990) J. Clin. Invest. 85, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ikeda F., Dikic I. (2008) EMBO Rep. 9, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newton K., Matsumoto M. L., Wertz I. E., Kirkpatrick D. S., Lill J. R., Tan J., Dugger D., Gordon N., Sidhu S. S., Fellouse F. A., Komuves L., French D. M., Ferrando R. E., Lam C., Compaan D., Yu C., Bosanac I., Hymowitz S. G., Kelley R. F., Dixit V. M. (2008) Cell 134, 668–678 [DOI] [PubMed] [Google Scholar]
- 40. Stewart G. S., Panier S., Townsend K., Al-Hakim A. K., Kolas N. K., Miller E. S., Nakada S., Ylanko J., Olivarius S., Mendez M., Oldreive C., Wildenhain J., Tagliaferro A., Pelletier L., Taubenheim N., Durandy A., Byrd P. J., Stankovic T., Taylor A. M., Durocher D. (2009) Cell 136, 420–434 [DOI] [PubMed] [Google Scholar]
- 41. Wu-Baer F., Lagrazon K., Yuan W., Baer R. (2003) J. Biol. Chem. 278, 34743–34746 [DOI] [PubMed] [Google Scholar]
- 42. Bloom J., Amador V., Bartolini F., DeMartino G., Pagano M. (2003) Cell 115, 71–82 [DOI] [PubMed] [Google Scholar]
- 43. Coulombe P., Rodier G., Bonneil E., Thibault P., Meloche S. (2004) Mol. Cell. Biol. 24, 6140–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu Z. K., Gervais J. L., Zhang H. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 11324–11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cayrol C., Ducommun B. (1998) Oncogene 17, 2437–2444 [DOI] [PubMed] [Google Scholar]
- 46. Yu H. (2007) Mol. Cell 27, 3–16 [DOI] [PubMed] [Google Scholar]
- 47. Fotedar R., Fitzgerald P., Rousselle T., Cannella D., Dorée M., Messier H., Fotedar A. (1996) Oncogene 12, 2155–2164 [PubMed] [Google Scholar]
- 48. Pavletich N. P. (1999) J. Mol. Biol. 287, 821–828 [DOI] [PubMed] [Google Scholar]
- 49. Russo A. A., Jeffrey P. D., Patten A. K., Massagué J., Pavletich N. P. (1996) Nature 382, 325–331 [DOI] [PubMed] [Google Scholar]
- 50. Ono T., Kitaura H., Ugai H., Murata T., Yokoyama K. K., Iguchi-Ariga S. M., Ariga H. (2000) J. Biol. Chem. 275, 31145–31154 [DOI] [PubMed] [Google Scholar]
- 51. Mal A., Chattopadhyay D., Ghosh M. K., Poon R. Y., Hunter T., Harter M. L. (2000) J. Cell Biol. 149, 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Engel F. B., Hauck L., Boehm M., Nabel E. G., Dietz R., von Harsdorf R. (2003) Mol. Cell. Biol. 23, 555–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deng C., Zhang P., Harper J. W., Elledge S. J., Leder P. (1995) Cell 82, 675–684 [DOI] [PubMed] [Google Scholar]
- 54. Pasumarthi K. B., Nakajima H., Nakajima H. O., Soonpaa M. H., Field L. J. (2005) Circ. Res. 96, 110–118 [DOI] [PubMed] [Google Scholar]
- 55. Woo Y. J., Panlilio C. M., Cheng R. K., Liao G. P., Atluri P., Hsu V. M., Cohen J. E., Chaudhry H. W. (2006) Circulation 114, I206–213 [DOI] [PubMed] [Google Scholar]
- 56. Engel F. B., Hsieh P. C., Lee R. T., Keating M. T. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15546–15551 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.