

Background: Signaling mediated by serine/threonine phosphatases during bacterial pathogenesis is not completely understood.
Results: In Group B Streptococcus (GBS), Stp1 controls serine/threonine kinase function, post-transcriptional regulation of hemolysin, autolysis, and virulence.
Conclusion: Although not essential for growth, Stp1 is critical for GBS pathogenesis.
Significance: The importance of Stp1 in virulence and autolysis accentuates the possibility of using phosphatase inhibitors to decrease GBS infections.
Keywords: Bacterial Pathogenesis, Bacterial Protein Phosphatases, Phosphoproteomics, Protein Phosphorylation, Virulence Factors, Streptococcus agalactiae, Autolysis, Blood-Brain Barrier, Hemolysin, Serine/threonine Phosphatase, Phosphopeptides
Abstract
Elucidating how serine/threonine phosphatases regulate kinase function and bacterial virulence is critical for our ability to combat these infections. Group B streptococci (GBS) are β-hemolytic Gram-positive bacteria that cause invasive infections in humans. To adapt to environmental changes, GBS encodes signaling mechanisms comprising two component systems and eukaryotic-like enzymes. We have previously described the importance of the serine/threonine kinase Stk1 to GBS pathogenesis. However, how the presence or absence of the cognate serine/threonine phosphatase Stp1 affects Stk1 function and GBS virulence is not known. Here, we show that GBS deficient only in Stp1 expression are markedly reduced for their ability to cause systemic infections, exhibit decreased β-hemolysin/cytolysin activity, and show increased sensitivity to autolysis. Although transcription of genes important for β-hemolysin/cytolysin expression and export is similar to the wild type (WT), 294 genes (excluding stp1) showed altered expression in the stp1 mutant and included autolysin genes. Furthermore, phosphopeptide enrichment analysis identified that 35 serine/threonine phosphopeptides, corresponding to 27 proteins, were unique to the stp1 mutant. This included phosphorylation of ATP synthase, DNA and RNA helicases, and proteins important for cell division and protein synthesis. Collectively, our results indicate that Stp1 is important for appropriate regulation of Stk1 function, hemolysin activity, autolysis, and GBS virulence.
Introduction
Signaling systems are essential for organisms to adapt to dynamic changes in their environment. Signaling in prokaryotic organisms is primarily achieved by two-component signaling systems that regulate gene expression in response to external/environmental signals, such as chemical gradients, Mg2+ concentration, osmolarity, and the presence of autoinducing or antimicrobial peptides (1–5). A typical two-component system comprises a membrane-associated sensor histidine kinase that responds to an environmental signal and phosphorylates its cognate DNA binding response regulator at an aspartate residue. Phosphorylation often alters the affinity of the response regulator to its target promoters, thus regulating gene expression.
In eukaryotes, adaptive responses and regulation of gene expression rely heavily on signaling mediated by multiple serine/threonine and tyrosine kinases and phosphatases. More than 600 members of these kinase-phosphatase families are present in humans (6), and deviation from normal phosphorylation events increases the risk of disease (7). The existence of eukaryotic-like serine/threonine phosphatases (STP)3 and serine/threonine kinases (STK) in prokaryotic organisms was described recently (for reviews, see Refs. 8–10). STK have been shown to be important for virulence of pathogenic bacteria, including Yersinia, Mycobacteria, Streptococcus sp., Enterococcus faecalis, and Staphylococcus aureus (11–23). However, many of these studies have primarily focused on the role of the kinase (STK) in these bacterial pathogens. Consequently, the role of serine/threonine phosphatases in regulation of kinase activity and bacterial pathogenesis is not completely understood (9, 10).
Although a few studies have described the role of serine/threonine phosphatases in bacteria (22, 24, 25), the inability to derive strains deficient only in Stp1 in certain bacteria (e.g. Streptococcus sp.) also led to the notion that it may be essential (9, 10, 26). Recently, Agarwal et al. (27) described the importance of the Stp1 homologue (SP-STP) of Group A Streptococcus (GAS) to its pathogenesis. Although the amino acid sequence of GBS Stp1 is 73% homologous to GAS SP-STP, the GBS Stp1 enzyme is not secreted as observed in GAS (18, 27, 28), suggesting mechanistic differences in regulation of Stk1 by Stp1 in bacterial pathogens. This study focuses on the role of Stp1 in regulation of Stk1 function and pathogenesis of Group B Streptococcus, a human neonatal pathogen.
Group B Streptococcus (GBS) or Streptococcus agalactiae are β-hemolytic, Gram-positive cocci with marked clinical pathogenesis in humans. GBS are the most common cause of bacterial infections in human newborns and are emerging pathogens of adult humans (29). These bacteria reside as commensal organisms in the lower gastrointestinal and genital tracts of healthy adult women. Transmission of GBS to the newborn can occur in utero due to ascending infection or from aspiration of contaminated amniotic/vaginal fluids during birth. GBS disease in human newborns includes pneumonia, sepsis, and meningitis (for reviews, see Refs. 29–31). Ascending GBS infections are also linked to intraamniotic infection, preterm birth, and stillbirth (32–36). The diverse host niches encountered by GBS during its disease cycle indicate that the pathogen efficiently adapts to the host environment.
Our long term interest is to understand mechanisms that enable GBS to adapt during its transition from commensal environments to invasive niches (31). Signaling systems are critical for environmental adaptation of GBS. Previous studies from our laboratory have identified that GBS encodes two eukaryotic-like signaling enzymes known as a serine/threonine kinase, Stk1, and a serine/threonine phosphatase, Stp1 (18). We have extensively described the importance of Stk1 to virulence of GBS (18, 37–40). However, given that signaling via phosphorylation is exploited by organisms due to their reversible nature, the lack of information on the cognate phosphatase Stp1 has resulted in inadequate understanding of eukaryotic-like signaling in bacterial pathogens, such as GBS. Therefore, we constructed an Stp1 mutant (Δstp1) to evaluate its role in GBS pathogenesis. Our studies indicate that GBS deficient only in Stp1 expression are significantly attenuated for their ability to cause systemic infections. Consistent with this finding, we observed that activity of the pluripotent GBS toxin β-hemolysin/cytolysin (β-H/C) was severely decreased in the Δstp1 mutant, and complementation restored normal hemolytic activity. Notably, the decrease in (β-H/C) in the Δstp1 mutant did not correlate to decreased expression of cyl genes encoding β-H/C. However, in vivo phosphopeptide enrichment analysis indicated increased serine/threonine phosphorylation of several GBS proteins, including those involved in ATP synthesis, that are critical for the activity of ABC transporters, which mediate export of β-H/C (41). Microarray analysis revealed a marked increase in expression of phage-encoded autolysin genes that correlated with increased sensitivity of the Δstp1 mutant to autolysis in vitro. Collectively, these data indicate that Stp1 plays a critical role in regulation of kinase function and GBS virulence.
EXPERIMENTAL PROCEDURES
General Growth
The strains, plasmids, and primers used in this study are listed in supplemental Table S1. The wild type (WT) GBS strain A909 is a serotype 1a capsular polysaccharide clinical isolate (42). Strain LR128 is isogenic to A909 and is deficient in CovR expression as described previously (39). Routine cultures of GBS were performed in tryptic soy broth (TSB; Difco) in 5% CO2 at 37 °C. Routine cultures of Escherichia coli were performed in Luria-Bertani broth (LB; Difco) at 37 °C. All chemicals were purchased from Sigma-Aldrich unless mentioned otherwise. GBS cell growth was monitored at 600 nm after incubation in 5% CO2 at 37 °C unless otherwise indicated. Antibiotics were added at the following concentrations when necessary: for GBS, kanamycin (1000 μg/ml), erythromycin (1 μg/ml), chloramphenicol (5 μg/ml), and spectinomycin (300 μg/ml); for E. coli, erythromycin (300 μg/ml), spectinomycin (50 μg/ml), kanamycin (40 μg/ml), and chloramphenicol (10 μg/ml).
Construction of the GBS stp1 Mutant
Approximately 1 kb of DNA located upstream of stp1 was amplified using the primers PF3:5′ and PR1+:5′ and high fidelity PCR (Invitrogen). Likewise, 1 kb of DNA that encoded the last 10 amino acids of Stp1 and the region downstream was amplified using primers NEW PF2+:5′ and PR4:5′ as described above. The gene conferring kanamycin (Ωkm-2) resistance was amplified from pCIV2 (43) using primers PF1:5′ and NEW PR2:5′ for allelic replacement of stp1. Subsequently, strand overlap extension PCR (44) was performed to introduce the antibiotic resistance gene between the flanking regions of stp1 described above. Note that the Ωkm-2 gene was engineered to be in frame with the last 10 amino acids of Stp1 in the downstream flanking region to retain the 1-bp overlap between stk1 and the upstream gene. The PCR fragment was then ligated into the temperature-sensitive vector pHY304 (45), and the resulting plasmid pKJ2 (Δstp1::km) was electroporated into GBS A909 as described previously (18). Selection and screening for the double crossover (Δstp1::km) was performed as described (18). PCR was used to verify the presence of Ωkm-2 and the absence of stp1. The Δstp1 strain (LR154) was used as the parent strain to derive the double Δstp1ΔcovR mutant using methods described previously (39).
Construction of GBS Strains Deficient in DivIVA, DivIVA Domain, and FtsZ
Approximately 1 kb of DNA located upstream of SAK_0586 (DivIVA), SAK_0373 (DivIVA domain protein), and SAK_0581 (FtsZ) was amplified using the primers SAK_0586 5′ XhoI/SAK_0586 5′ + Kan 5′; SAK_0373 5′ XhoI/SAK_0373 5′ + Kan 5′; and SAK_0581 5′ XhoI/SAK_0581 5′ + Kan 5′ and high fidelity PCR (Invitrogen). Similarly, the following primers were utilized to amplify 1 kb of DNA downstream of SAK_0586, SAK_0373, and SAK_0581: SAK_0586 3′ + Kan 3′/SAK_0586 3′ SacII; SAK_0373 3′ + Kan 3′/SAK_0373 3′ SpeI; and SAK_0581 3′ + Kan 3′/SAK_0581 3′ XbaI, respectively. The gene conferring kanamycin (Ωkm-2) resistance was amplified from pCIV2 (43) using the following primers: Kan 5′ + SAK_0586 5′/Kan 3′ + SAK_0586 3′; Kan 5′ + SAK_0373 5′/Kan 3′ + SAK_0373 3′; and Kan 5′ + SAK_0581 5′/Kan 3′ + SAK_0581 3′, respectively. The PCR fragments were then ligated into the temperature-sensitive vector pHY304 (45), and the resulting plasmids pJC1–3 were electroporated into GBS A909 as described previously (18). Selection and screening for the double crossovers were performed as described (18). PCR was used to verify the presence of Ωkm-2 and the absence of SAK_0586 (DivIVA), SAK_0373 (DivIVA domain protein), or SAK_0581 (FtsZ).
Autophosphorylation Assay
GBS were grown to an A600 of 0.6, and membrane fractions were enriched from total cellular extracts using ultracentrifugation as described previously (18). Protein concentrations in membrane fractions were estimated using the Bradford reagent (Sigma). 10 μci of [γ-32P]ATP (PerkinElmer Life Sciences) was added to ∼50 μg of total membrane protein from each strain. The reactions were incubated at 37 °C for 15 min, and then the products were resolved on 10% SDS-PAGE and exposed to the PhosphorImager (Amersham Biosciences) as described previously (18, 38).
Virulence Analysis
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC protocol 13311), Seattle Children's Research Institute, and performed using accepted veterinary standards.
Neonatal Rat Model of GBS Infection
Virulence analysis using the neonatal rat sepsis model of infection was performed as described (46). Briefly, time-mated, female Sprague-Dawley rats were obtained from Charles River Laboratories. GBS strains were grown to an A600 of 0.3, washed, and resuspended in phosphate-buffered saline and used as the inoculum. Groups of six rat pups (24–48 h of age) were given 10-fold serial dilutions of each strain by intraperitoneal injection, and the pups were checked for signs of morbidity every 8 h for 72 h. The lethal dose 50 or moribund 50 estimates were derived using logistic regression models for the probability of death conditional on dose and strain as described (47). The experiment was repeated twice, and p values of <0.05 were considered significant.
Adult Murine Model of GBS Infection
The murine model of GBS sepsis was used to compare virulence potential of strains used in this study. Briefly, 6-week-old male CD-1 mice obtained from Charles River Laboratories were injected via the tail vein with 3 × 107 cfu of either WT A909 or isogenic Δstp1 mutant (n = 5 per group). Blood, spleens, and brains from infected mice were collected aseptically 48 h after inoculation. Bacterial counts in blood, spleen, and brain homogenates were determined by plating serial 10-fold dilutions on TSB agar.
Sialylation of GBS Capsular Polysaccharide
Sialic acid concentrations in capsular polysaccharide isolated from WT GBS and the Δstp1 mutant were estimated as described (48). To quantitate the amount of sialic acid associated with the CPS of the strains, purified CPS from each strain was hydrolyzed under mild acid conditions, and the amount of sialic acid released was quantitatively determined by HPLC as described (48).
Growth in Human Serum
Growth of GBS in normal human serum was examined as described (49). Briefly, fresh blood, obtained with the consent of human volunteers, and serum were prepared as described (50). GBS strains grown to an A600 of 0.3 were centrifuged, washed, and resuspended to the original volume in PBS. 5 μl of the GBS cell suspension containing ∼105 cfu was added to 1.5 ml of normal human serum and incubated at 37 °C for a period of 24 h. Aliquots were removed, serially diluted, and plated to estimate GBS cfu as described previously (49).
Adherence and Invasion of Human Brain Microvascular Endothelial Cells (hBMEC)
The hBMEC line, immortalized by transfection with the SV40 large T antigen (51), was used in these studies. Propagation of hBMEC and GBS adherence, invasion, and intracellular survival were performed as described previously (52). A 2-fold increase or decrease in adherence or invasion compared with the isogenic WT A909 was considered significant as described previously (52).
Barrier Integrity Analysis with hBMEC
Changes in transendothelial electrical resistance across hBMEC monolayers in real time were measured using electric cell-substrate impedance sensing (ECIS) (53–56) as described previously (21). Briefly, hBMEC monolayers were established on gold-plated electrodes in 8-well array slides attached to a computer-operated sensing apparatus to allow measurements in real time. The hBMEC monolayers were infected with GBS WT A909, isogenic Δstp1, ΔcovR, ΔcovRΔstp1, and control ΔcylE at 1 × 105 cfu/well as described (21) and followed for a period of 20 h. The ECIS system measures the cell membrane capacitance (Cm), the resistance from the cell-electrode interaction (α), and the barrier function properties of the cell monolayer (Rb). Deconvolution of the overall ECIS signal into these parameters was performed by the ECIS software by fitting the mathematical model derived in Ref. 54 to the experimental data by least-square optimization procedures. Data are represented as a decrease in resistance as a proportion of the control over time. Uninfected wells served as controls for background levels of electrical resistance as described (21).
hBMEC Microarrays
hBMEC microarrays were performed as described previously (57). Briefly, hBMEC monolayers were cultured, washed, and infected with GBS strains as described (57). Subsequently, total RNA was isolated from hBMEC monolayers, and cDNA synthesis and labeling was performed as described (57). Hybridization of cDNA to the Human WG-6 v3 array from Illumina Inc. was also performed as described previously (57). A statistical algorithm developed for high density oligonucleotide arrays was used for data analysis, and a 2-fold increase or decrease in gene expression was considered significant (57). hBMEC microarrays were performed with two independent biological replicates of each strain (WT A909, Δstp1) and medium (PBS)-only controls. cDNA synthesis and quantitative PCR for IL-6, IL-8, CCL20, and GAPDH were performed using primers and methods described previously (58).
β-Hemolysin/Cytolysin Assays
Hemolysin activity assays were performed on GBS strains using methods described previously (39, 59). Briefly, ∼109 cfu for each GBS strain was centrifuged and resuspended in one-tenth the original volume with PBSGS (PBS + 1% glucose + 2% starch) and incubated at 37 °C for 1 h. Subsequently, the cells were centrifuged, and supernatants containing starch-bound β-H/C were collected. 2-Fold serial dilutions of the β-H/C extract in PBSG (PBS + 0.2% glucose) from each strain were incubated with an equal volume of 1% sheep red blood erythrocytes in 96-well plates at 37 °C for 1 h. Subsequently, the plates were spun at 1,000 × g for 10 min to pellet unlysed sheep red blood erythrocytes. The supernatants were transferred to a replica 96-well plate, and hemoglobin release was measured by recording the absorbance at 420 nm. Positive and negative controls included wells that contained sheep red blood erythrocytes with 0.1% SDS or PBSG, respectively. The hemolytic titer for a given strain was determined as the reciprocal of the greatest dilution producing 50% hemoglobin release compared with the SDS control as described previously (39, 59).
Western Blots
GBS strains were grown to an A600 of 0.6 and centrifuged. For preparation of total cell lysates, the cell pellets from above were washed in PBS and resuspended in cell lysis buffer (20 mm Trizma base, 10 mm MgCl2, 50 units/ml DNase (Qiagen, Inc., Valencia, CA), 50 μg/ml RNase, and 5 μg/ml protease inhibitor mixture). Cells were disrupted using a FastPrep FP101 bead beater (Bio 101) with three 30-s bursts at a power setting of 6, followed by clarification of cell lysates by centrifugation. The supernatant containing total proteins was quantified using the Bradford total protein assay (60). An equal amount of protein from each strain (30 μg) was mixed with 5× SDS sample buffer, heated at 95 °C for 10 min, and subjected to 12% SDS-PAGE. Following electrophoresis, the proteins were transferred to either nitrocellulose or 0.45-μm pore size Immobilon-P polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA) by a wet transfer method in a buffer containing 20% methanol (v/v), 0.3% Trizma base (w/v), and 1.44% glycine (w/v) for 1.5 h at 90 V and 4 °C. The membrane was blocked in 1:1 Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) in PBS and incubated at room temperature for 1 h with a 1:2000 dilution of primary Stp1 antibody (28), respectively. Subsequently, Alexa Fluor 680 goat anti-rabbit (Invitrogen) secondary antibody was added at 1:10,000, and washes were performed following the Odyssey LI-COR infrared imager instructions (LI-COR Biosciences).
Phosphopeptide Enrichment and Mass Spectrometry
Total protein was isolated from WT A909 and Δstp1 mutant as described previously (22, 40) with a few modifications. Briefly, each sample was normalized to contain an equal amount of protein, and the proteins were denatured and reduced in 5 mm DTT containing 0.1% Rapigest (Waters) at 50 °C for 30 min. Subsequently, the reduced cysteines were alkylated with 15 mm iodoacetamide for 1 h in the dark, and the samples were digested overnight at 37 °C using sequencing grade trypsin (1:100, trypsin/total protein). Rapigest was removed, and samples were desalted using Sep-Pak C-18 columns according to the manufacturer's instructions (Waters) and dried using a SpeedVac. From the peptide samples of each strain (1 mg), phosphopeptides were enriched and captured using soluble nanopolymer (PolyMAC) as described (61). Unbound non-phosphopeptides were washed, and phosphopeptides were eluted as described (61). The samples were analyzed by capillary liquid chromatography-nanoelectrospray tandem mass spectrometry (μLC-nanoESI-MS/MS) using a high resolution hybrid linear ion trap (LTQ-Orbitrap Velos, Thermo Fisher) coupled with Eksigent Ultra2D nanoflow HPLC and methods described previously (40, 62). Data were searched using Proteome DiscovererTM software with the SequestTM algorithm at 10 ppm precursor mass accuracy cut-off, and MS/MS tolerance was set at 0.8 Da. The searches included static modification on cysteine residues (+57.0214) and variable modifications on methionine (+15.9949) and serine and threonine residues (+79.997) to identify phosphorylation as described (21, 40, 62). Spectra were searched against the GBS A909 genome database, NC_007432, with a 1% false discovery rate cut-off based on the reverse database decoy search. Proteome Discoverer generates a reverse “decoy” database from the same protein database, and any peptides passing the initial filtering parameters that were derived from this decoy database are defined as false positive identification. The minimum cross-correlation factor (Xcorr) filter was readjusted for each individual charge state separately to optimally meet the predetermined target false discovery rate of 1% based on the number of random false positive matches from the reverse decoy database. Thus, each data set had its own passing parameters. Unique phosphopeptides identified were then manually validated. Phosphorylation site localization from collision-induced dissociation mass spectra (63) was determined by SEQUEST Xcorr scores, and only one phosphorylation site was counted using the top scored phosphopeptide for any phosphopeptide with potential ambiguous phosphorylation sites as described (61).
Isolation and Purification of GBS RNA
Total RNA from GBS was isolated as described previously (37). In brief, GBS strains were grown to an A600 of 0.6, centrifuged, washed in 1:1 mixed RNA Protect (Qiagen, Inc.) and TE buffer, and resuspended in kit-supplied RLT buffer. The cell suspensions were lysed through the use of a FastPrep FP101 bead beater (Bio 101), followed by clarification of the lysates via centrifugation. The supernatants were then purified and DNase-digested (Qiagen, Inc.) as described by the RNeasy Minikit manufacturer's instructions. RNA integrity and concentration were determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) or a NanoDrop 1000 (NanoDrop, Wilmington, DE) for use in microarrays or qRT-PCR, respectively.
GBS Microarrays and qRT-PCR
RNA was isolated from three independent biological replicates for each strain and purified as described above. Purified RNA was sent to NimbleGen Systems, Inc. (Madison, WI) for full expression services. NimbleGen performed cDNA synthesis, labeling of the cDNA, and hybridization of the labeled cDNA to the chip S. agalactiae A909 (catalog no. A6703-00-01, NimbleGen Systems, Inc.) according to company protocols. The chips were composed of 18 probes/target sequence, and each probe was replicated two times (see the NimbleGen Systems Web site). Microarray data were interpreted and analyzed using the program GeneSpring GX version 7.3.1 (Agilent Technologies). Genes with statistically significant differences among groups were calculated using the Welch t test (parametric, with variances not assumed equal) with a p value cut-off of 0.05 and an associated Benjamini and Hochberg false discovery rate multiple testing correction (about 5.0% of the identified genes would be expected to pass the restriction by chance (64)). Standard error propagation was calculated using the delta method for ratios of means from the three independent biological replicates for strains A909 (WT) and LR154 (Δstp1). All -fold changes were defined as relative to A909 WT. The entire set of microarray data is deposited at the Gene Expression Omnibus (GEO), accession number GSE21564. qRT-PCR was performed using a one-step QuantiTect SYBR Green RT-PCR kit (Qiagen, Inc.) as described previously (37). The reference gene used for all runs was the housekeeping ribosomal protein S12 gene rpsL (47).
GBS Autolysis
Autolysis of GBS was determined using methods described previously (65) with the exception that Triton X-100 was not included. Briefly, GBS strains grown to an A600 = 0.7 at 37 °C were centrifuged, washed in PBS, and resuspended to the original volume in 0.05 m Tris-HCl (pH 7.5). The initial A600 was measured (T0). The cultures were then incubated at 30 °C without shaking, and the A600 was recorded every 30 min for a period of 3 h. Autolysis for each strain is shown as a percentage of the initial A600 reading at T0.
Statistical Analysis
Unless mentioned otherwise, the Mann-Whitney test or unpaired t test was used to estimate differences between GBS strains. These tests were performed using GraphPad Prism version 5.0 for Windows (GraphPad Software, San Diego, CA).
RESULTS
GBS Deficient in Stp1 Expression Exhibit Attenuated Virulence
The GBS stp1 gene encodes a 26.5-kDa cytoplasmic protein that is homologous to eukaryotic-type PP2C protein phosphatases (18). Biochemical analysis revealed that Stp1 is a manganese-dependent phosphatase that dephosphorylates Stk1 in vitro (18). In GBS, the stp1 gene has a 1-base overlap with the downstream gene encoding stk1. Our previous attempts to construct a GBS strain that was deficient only for Stp1 expression were unsuccessful due to the weak endogenous promoter, which did not permit antibiotic (i.e. chloramphenicol) selection (for details, see Ref. 18). To evaluate the role of Stp1 in GBS virulence, we utilized allelic replacement to replace only the gene encoding stp1 with a gene that conferred resistance to kanamycin (km; see “Experimental Procedures”). We then confirmed that Stk1 expression in the Δstp1 strain was similar to the WT using an Stk1 autophosphorylation assay (Fig. 1) and qRT-PCR (see supplemental Table S2A). These results confirm that Stk1 is functional in the Δstp1 mutant and indicate the absence of polar effects of the allelic replacement. The Δstp1 mutant was also similar to WT for growth in laboratory media, such as TSB (supplemental Fig. S1A).
FIGURE 1.

The Δstp1 mutant is similar to WT for Stk1 autophosphorylation. In vitro phosphorylation reactions were performed using equal amounts (50 μg) of membrane proteins isolated from WT (A909) and the Δstp1 mutant as described (38) (also see “Experimental Procedures”). As controls, equal amounts of membrane proteins isolated from the Stk1-deficient GBS strains (Δstk1 and Δstp1Δstk1) are included. Note that autophosphorylation of Stk1 is observed in membrane fractions of WT (A909) and Δstp1 (-fold difference <2) and that Stk1 autophosphorylation is not observed in strains deficient for Stk1.
We next examined whether the absence of Stp1 affects virulence of GBS. The neonatal rat sepsis model of GBS infection (18, 46) was used to compare virulence properties of the Δstp1 mutant with those of the isogenic WT strain, A909. Briefly, bacteria grown to early log phase (A600 = 0.3) were used as the inoculum (for details, see “Experimental Procedures” and Refs. 18 and 46). Moribund 50 (MD50; formerly lethal dose 50 or LD50) estimates of the Δstp1 mutant were significantly higher than the WT strain (>3 log cfu compared with WT; see Table 1), indicating that the Δstp1 mutant is significantly attenuated for GBS virulence. Because the Δstp1 strain demonstrated decreased ability to cause bloodstream infections in neonatal rats, we hypothesized that this strain would also be attenuated for its ability to survive in the bloodstream of adult mice. To test this hypothesis, we used the adult murine model of GBS bloodstream infections (57). CD-1 mice (n = 5) were infected with 3 × 107 cfu of either WT or the Δstp1 strain as described (21, 57). At 48 h postinfection, blood, spleen, and brain were harvested, homogenized, serially diluted, and plated on TSB agar for quantitative bacterial counts (cfu). As shown in Fig. 2, A–C, the number of cfu recovered from the blood, spleen, and brain of mice infected with the Δstp1 strain was significantly lower compared with mice infected with the WT strain.
TABLE 1.
The GBS Δstp1 mutant is attenuated for virulence
Moribund 50 (MD50) estimates and confidence intervals were calculated as described under “Experimental Procedures.” The p value compares the value of the mutant strain with the WT A909 (p values of <0.05 were considered significant). NA, not applicable.
| Strain | MD50 | 95% confidence interval | p value |
|---|---|---|---|
| cfu | |||
| A909 (WT) | 3.9 × 105 | 1.8 × 105 to 8.3 × 105 | NA |
| Δstp1 | 5.3 × 108 | 9.5 × 107 to 3 × 1010 | <0.001 |
FIGURE 2.

The Δstp1 mutant shows decreased survival during GBS systemic infections. Five 6-week-old female CD-1 mice were intravenously injected with 3 × 107 cfu of WT or the Δstp1 mutant. At ∼24–48 h postinfection, blood, spleen, and brain were harvested from the infected mice, and cfu were enumerated. A significant decrease in cfu was observed in the blood (see A; p < 0.05), spleen (see B; p < 0.05), and brain (see C; p < 0.05) of mice infected with the Δstp1 mutant when compared with mice infected with the WT strain A909.
Growth in human serum is often used as an ex vivo assay to evaluate GBS growth defects that can occur in vivo (49, 68, 69). The Δstp1 mutant was compared with WT GBS for its ability to grow/survive in normal human serum for a period of 24 h as described (49) (also see “Experimental Procedures”). These results indicate that the overall growth of the Δstp1 mutant in human serum was not significantly different from that of WT (supplemental Fig. S1B, p = 0.17). Although sialylation of the capsular polysaccharide is critical for GBS bloodstream infections and virulence (66, 67), the Δstp1 mutant was similar to WT for the sialic acid capsular polysaccharide levels (see supplemental Fig. S1C).
Δstp1 Mutant Shows Decreased Blood-Brain Barrier Activation and Barrier Disruption
The dramatic decrease in survival of the Δstp1 mutant during GBS systemic infection (Fig. 2, A–C) prompted us to examine if Stp1 affects GBS penetration of the blood-brain barrier (BBB). The tissue culture model consisting of immortalized hBMEC (51) has been extensively used to evaluate the ability of GBS to adhere to, invade, and penetrate the BBB (21, 52, 57). Adherence and invasion of WT GBS and the Δstp1 mutant to hBMEC were performed as described (52, 57). These studies indicated that adherence and invasion of the Δstp1 strain to hBMEC were similar to those with WT A909 (supplemental Fig. S2).
Studies have also shown that in response to GBS, the BBB endothelium elaborates changes in gene expression that promote the neutrophilic inflammatory response characteristic of acute bacterial meningitis (57, 70). To determine if Stp1 affects the hBMEC response to GBS infection, microarray analysis was used to compare the transcriptional response of hBMEC to infection with WT GBS and Δstp1 as described (57, 58). We observed an approximately 5-fold decrease in transcription of genes encoding the proinflammatory cytokines and chemokines (e.g. IL-8, CCL20, IL-6, and TNFα; see Table 2) in hBMEC infected with the Δstp1 strain when compared with hBMEC infected with WT. Furthermore, a significant decrease (5–20-fold) in transcription of genes encoding nuclear receptor family proteins, oncogenes, and transcriptional factors involved in cell growth and differentiation (FOS, FOSB, and ATF3; Table 2) was observed in hBMEC infected with Δstp1 compared with hBMEC infected with WT A909. Levels of mRNA corresponding to housekeeping genes, such as β-actin and GAPDH, were similar for samples infected with either GBS WT or Δstp1 (data not shown). Collectively, these data indicate that although the Δstp1 strain is proficient for invasion of hBMEC (supplemental Fig. S2), this strain is unable to elicit the characteristic proinflammatory signaling response to GBS infection. To confirm the results obtained from the microarray, qRT-PCR was used to compare the transcript abundance of genes involved in proinflammatory signaling pathways. Consistent with the microarray analysis, transcript levels of IL-6, IL-8, and CCL20 were significantly decreased in cells infected with Δstp1 compared with hBMEC infected with the WT A909 (Fig. 3A; p < 0.005). These data suggest that the absence of Stp1 affects the ability of GBS to modulate the BBB endothelial response during infection.
TABLE 2.
Decreased transcription of hBMEC genes after infection with Δstp1
Gene expression is denoted as -fold difference relative to the WT GBS strain A909. Only genes that showed a >2-fold change (p < 0.05) in expression compared with WT A909 are listed.
| Δstp1 | |
|---|---|
| Chemokines and cytokines | |
| Chemokine (C-C motif) ligand 20 (CCL20) | 0.246 |
| Interleukin-6 (IL-6) | 0.237 |
| Tumor necrosis factor (TNFα) | 0.191 |
| Interleukin-8 (IL-8) | 0.167 |
| Cardiotrophin-like cytokine factor 1 (CLCF1) | 0.038 |
| Colony-stimulating factor 2 (granulocyte-macrophage) (CSF2) | 0.030 |
| Transcriptional factors/signal transduction | |
| Baculoviral IAP repeat (BIRC3) | 0.278 |
| Serpin peptidase inhibitor, clade E (SERPINE1) | 0.156 |
| Dual specificity phosphatase 5 (DUSP5) | 0.123 |
| Leukemia inhibitory factor (cholinergic differentiation factor) (LIF) | 0.117 |
| Sprouty homolog 4 (SPRY4) | 0.109 |
| Protein phosphatase 1, regulatory subunit 15A (PPP1R15A) | 0.105 |
| Heparin-binding EGF-like growth factor (HBEGF) | 0.094 |
| Immediate early response 3 (IER3) | 0.094 |
| FBJ murine osteosarcoma viral oncogene homolog B (FOSB) | 0.094 |
| Jun oncogene (JUN) | 0.090 |
| Brain cytoplasmic RNA 1, (BCYRN1) | 0.078 |
| Tumor necrosis factor α-induced protein 3 (TNFAIP3) | 0.071 |
| Kruppel-like factor 6 (KLF6) | 0.070 |
| Basic helix-loop-helix domain (BHLHB2) | 0.067 |
| Angiopoietin-like 4 (ANGPTL4) | 0.059 |
| Phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1) | 0.058 |
| AXIN1-up-regulated 1 (AXUD1) | 0.054 |
| Nuclear receptor subfamily 4, group A, member 3 (NR4A3) | 0.052 |
| Myeloid cell leukemia sequence 1 (BCL2-related) (MCL1) | 0.045 |
| Chromosome 2 open reading frame 26 (c2orf26) | 0.040 |
| Pim-3 oncogene (PIM3) | 0.031 |
| RNA, 7SK, nuclear (RN7SK) | 0.030 |
| Baculoviral IAP repeat-containing 3 (BIRC3) | 0.025 |
| Cysteine-rich, angiogenic inducer, 61 (CYR61) | 0.014 |
| GTP-binding protein overexpressed in skeletal muscle (GEM) | 0.013 |
| Tribbles homolog 1 (TRIB1) | 0.011 |
| Nuclear factor of κ light polypeptide gene enhancer in B-cells inhibitor, ζ (NFKBIZ) | 0.010 |
| Down syndrome critical region gene 1 (DSCR1) | 0.010 |
| RAS, dexamethasone-induced 1 (RASD1) | 0.007 |
| Nuclear factor, interleukin-3-regulated (NFIL3) | 0.002 |
| Unknown Function | |
| Hypothetical protein LOC387763 | 0.041 |
FIGURE 3.

The Δstp1 mutant is attenuated for its ability to induce an inflammatory response (A) and decrease barrier integrity (B) of hBMEC. A, qRT-PCR was performed on RNA isolated from hBMEC cells infected with either WT A909 or the Δstp1 strain. Note that transcription of IL-6, IL-8, and CCL20 was significantly decreased in hBMEC that were infected with the Δstp1 mutant compared with the WT A909 (p < 0.005). Error bars, S.E. B, time-dependent decrease in hBMEC resistance due to infection with Δstp1 was compared with WT, ΔcovR, ΔcylE, and ΔcovRΔstp1. Resistance was assessed by ECIS and is normalized to the uninfected or medium-only control.
GBS penetration of the BBB involves a combination of events that lead to failure of barrier function. We recently showed that hypervirulent GBS strains (e.g. CovR mutant (ΔcovR)) elicit a significant inflammatory response that accelerates BBB failure (21). We hypothesized that the decrease in inflammatory response observed in hBMEC infected with Δstp1 may correlate with decrease of barrier function. Barrier resistance of hBMEC to Δstp1 infection was determined by measuring the transendothelial electrical resistance across hBMEC monolayers in real time using ECIS as described (21). The results shown in Fig. 3B indicate that the decrease in hBMEC resistance in response to GBS infection was markedly delayed with Δstp1 when compared with WT, ΔcovR, and the control ΔcylE. Collectively, our studies indicate that although the Δstp1 mutant is capable of hBMEC invasion, the strain is attenuated for its ability to induce an inflammatory response and compromise BBB integrity.
Stp1 Is Important for GBS β-H/C Activity
The surface-associated toxin known as β-hemolysin/cytolysin is critical for the induction of the BBB inflammatory response and GBS virulence (21, 57, 59, 71–74). We previously showed that the serine/threonine kinase Stk1 can phosphorylate the two-component response regulator CovR at a threonine residue in position 65 to relieve CovR repression of cylE encoding β-H/C (37, 39). We hypothesized that if Stk1 activity is enhanced in the absence of Stp1, this can lead to increased threonine phosphorylation of CovR, transcription of cylE (β-H/C), and virulence of GBS. However, the attenuated virulence of the Δstp1 mutant (Figs. 2 and 3 and Tables 1 and 2) suggests that β-H/C expression may be decreased. To test this hypothesis, we compared β-H/C activity between WT and the Δstp1 mutant using sheep blood agar, and the results are shown in Fig. 4. Consistent with its virulence properties, we observed that hemolytic activity was severely decreased in the Δstp1 mutant (Fig. 4A). β-H/C was also extracted from WT, Δstp1, and the control β-H/C-deficient strain (ΔcylE) (75), and hemolytic activity was quantified as described (39, 59). These results indicate that whereas the hemolysin activity for WT A909 was 4 units, both Δstp1 and the ΔcylE strain had a hemolytic titer of <1 (-fold decrease of >0.25; see Table 3).
FIGURE 4.

Stp1 is important for β-hemolysin/cytolysin (β-H/C) activity of GBS. The zone of clearing observed around the colonies on the sheep blood agar plate represents β-H/C activity. A, the Δstp1 mutant shows no hemolytic activity, unlike the isogenic WT strain A909. B, Western blots on total cell lysates were performed using antibodies to the Stp1 homologue of Group A Streptococcus (28). These confirm that the expression of Stp1 is absent in the GBS Δstp1 mutant and restored in the complemented strain. C, complementation restores normal β-H/C activity to the Δstp1 mutant. D, the decrease in β-H/C activity in Δstp1 is also seen in the absence of the β-H/C repressor, CovR (compare ΔcovR and ΔcovRΔstp1).
TABLE 3.
Stp1 regulates GBS hemolytic activity independent of cylE transcription
| β-H/C activity |
|||
|---|---|---|---|
| Hemolytic titera | Change in hemolytic titer | Change in cylE transcriptionb | |
| -fold | -fold | ||
| A909 (WT) | 4 | NAc | 1 |
| ΔcylE | <1 | >0.25 | NA |
| Δstp1 | <1 | >0.25 | 0.92 ± 0.1 |
| ΔcovR | 128 | 32 | 11 ± 0.2 |
| ΔcovRΔstp1 | 32 | 8 | 11 ± 0.13 |
a Hemolytic titers were estimated as described (39, 59). The experiment was performed in triplicate, and changes that are >2-fold compared with WT GBS A909 are considered significant.
b qRT-PCR was performed as described under “Experimental Procedures.” Gene expression is denoted as -fold difference relative to the WT GBS strain A909. S.E. is indicated.
c NA, not applicable.
To confirm that the loss of hemolytic activity in the Δstp1 mutant was due to the absence of Stp1, we performed complementation studies. The gene encoding stp1 was previously cloned in the complementation vector pDC123 to construct pStp1 (18). To generate complementing GBS strains, the plasmid encoding stp1 (pStp1) and the vector control (pDC123) (76) were introduced into the Δstp1 strain as described (18). Of note, the plasmid pDC123 is routinely used as a complementation vector in GBS (18, 52, 75–77). The vector pDC123 was derived from the broad host range, high copy number plasmid pJS3, and constitutive tetM and cat promoters of pJS3 drive the expression of genes inserted in the multiple cloning site (for details, see Ref. 76). Western blot analysis was performed on total cellular proteins using antibodies to the Stp1 homologue in Streptococcus pyogenes (SP-Stp) (28). These results confirmed that Stp1 expression was abolished in the GBS Δstp1 mutant and restored in the complementing strain (Δstp1/pStp1; Fig. 4B). We then compared hemolytic activity between these strains. As shown in Fig. 4C, hemolytic activity is not observed in the GBS Δstp1 strain containing only the vector pDC123 (see Δstp1/pDC123). In contrast, hemolytic activity is restored to WT levels in the Δstp1 mutant that is complemented or restored for Stp1 expression (compare Δstp1/pStp1 with A909/pDC123 in Fig. 4C). Hemolytic titer of the complemented Stp1 strain was similar to that of WT (titer = 4). These data confirm that the decrease in β-H/C in the Δstp1 mutant is due to the loss of Stp1. The complemented strain was not included in virulence studies due to the unstable nature of the complementation vector (pDC123) in the absence of antibiotic (chloramphenicol) selection (for details, see Ref. 18).
Post-transcriptional Regulation of β-H/C by Stp1
We next examined whether the decrease in hemolytic activity observed in the Δstp1 strain correlated with decreased transcription of the gene encoding hemolysin (cylE). To this end, qRT-PCR was performed on WT and isogenic Δstp1 grown to an optical density (A600) of 0.6 as described (37). These studies indicated that transcription of the cylE gene in the Δstp1 mutant was not significantly different when compared with the isogenic WT (-fold difference compared with WT = 0.92 ± 0.1; Table 3). In GBS, the two-component regulator CovR binds to the promoter of hemolysin (PcylX) and represses transcription of the cyl operon, including cylE (37, 47, 49, 78). Because Stk1 regulates cylE transcription through phosphorylation of CovR (37), it was of interest to examine whether Stp1 regulation of β-H/C required the hemolysin repressor, CovR. To this end, using Δstp1 as the parent strain, we derived GBS strains that were deficient in CovR expression as described (39, 47). We then compared hemolysin (cylE) transcription and expression between ΔcovR and ΔcovRΔstp1. Of note, covR mutants are hyperhemolytic due to an approximate 10-fold increase in transcription of cylE compared with the isogenic WT A909 (37, 39). As shown in Fig. 4D and Table 3, hemolytic activity of the ΔcovRΔstp1 strain, although greater than Δstp1, was significantly lower than ΔcovR. Despite the decrease in hemolytic activity, transcription of cylE (β-H/C) in ΔcovRΔstp1 was comparable with the isogenic ΔcovR strain (i.e. transcription of cylE in ΔcovRΔstp1 and ΔcovR was ∼11-fold greater when compared with WT A909; see Table 3). These results indicate that decreased β-H/C activity in Δstp1 is independent of transcriptional regulation of cylE and is observed even in the absence of CovR. Consistent with the above observations, we observed that the decrease in hBMEC resistance to GBS infection was slightly delayed with the double ΔcovRΔstp1 mutant when compared with the isogenic ΔcovR (Fig. 3B).
Increased Serine/Threonine Phosphorylation of GBS Proteins Are Observed in Absence of Stp1
We next hypothesized that the decrease in β-H/C activity in the Δstp1 mutant could be attributed to deregulated kinase (Stk1) activity, which can lead to increased and/or altered Ser/Thr phosphorylation in GBS. To test this hypothesis, we compared Ser/Thr phosphopeptides present in the Δstp1 mutant to WT A909 using phosphopeptide enrichment and mass spectrometry as described (22, 40) (also see “Experimental Procedures”). Of note, Ser/Thr phosphopeptides identified in the Δstk1 mutant were recently described by us (40). Consistent with our previous findings (40), phosphopeptides corresponding to Stk1 were only identified in strains expressing Stk1 (WT A909 and Δstp1; Table 4 (top)). Of interest, 35 Ser/Thr phosphopeptides corresponding to 27 proteins were uniquely identified in the Δstp1 strain (i.e. these phosphopeptides were not identified in WT A909; Table 4 (middle)). Phosphopeptides unique to the Δstp1 strain included proteins predicted to be involved in cell division, such as PcsB, FtsZ, DivIVA, and DivIVA domain (also known as GpsB); some of these phosphopeptides were also previously identified in the GBS double Δstp1Δstk1 mutant that overexpressed Stk1 on a plasmid (40). Additional Ser/Thr phosphopeptides identified in Δstp1 corresponded to proteins belonging to the F0F1-ATP synthase complex, ATP-dependent RNA and DNA helicases, elongation factor Tu (EF-Tu), signal recognition particle FtsY, regulator of cell septum formation EzrA, ribosomal proteins, a peptidase of the collagenase family, proteins of as yet unknown functions, and the molecular chaperone GroEL (see Table 4 (middle) and supplemental Table S3). Representative phosphopeptides from Δstp1 that showed neutral loss of phosphoric acid during collision-induced dissociation in the mass spectrometer are shown in Fig. 5, A–C.
TABLE 4.
Increased serine/threonine phosphorylation in Δstp1
Phosphopeptide enrichment was performed as described previously (22) (also see “Experimental Procedures”). Phosphopeptides indicated above showed neutral loss of phosphoric acid in the MS analysis. The phosphorylated threonine or serine residue is indicated with a lowercase letter “t” or “s”, respectively. SAK numbers correspond to the ORF of the gene in the GBS A909 genome (88).
| Protein | Description | |
|---|---|---|
| Phosphopeptides identified in both A909 (WT) and Δstp1 | ||
| VTSTVSSLTtEQLLR | SAK_0389 | Serine/threonine protein kinase Stk1 (Stk1) |
| sLGNGIDPMDVIEK | SAK_0547 | Valyl-tRNA synthetase (ValS) |
| VLDEDDALPVVDDTESFDAtR | SAK_0586 | Cell division protein DivIVA |
| LTHLIsQNEVNDD | SAK_0862 | HPr kinase/phosphorylase (HprK) |
| VSGQTILDQEtK | SAK_1559 | Conserved hypothetical protein |
| FSDQEtKEFASSLSK | SAK_1681 | Glutamyl-tRNA(Gln) amidotransferase, C subunit (GatC) |
| sVGGFVLAGASHDATK | SAK_2014 | Chaperonin GroES (GroES) |
| Phosphopeptides unique to Δstp1 | ||
| IAATDSVINtLSGQQAAAQK | SAK_0050 | PcsB protein (PcsB) |
| FQAAAGQLEKtAR | SAK_0099 | Ribosomal protein L29 (RpmC) |
| VNVNtEQLAFQATR | SAK_0178 | Fructose-1,6-bisphosphate aldolase, class II (Fba) |
| IYYtHSMYPGGLK | SAK_0276 | Ribosomal protein L13 (RplM) |
| LDtEMIGLVK | SAK_0361 | ATP-dependent RNA helicase, DEAD/DEAH box family |
| NSGtAMYNQKPIAQSATNFDILK | SAK_0373 | DivIVA domain protein (GpsB) |
| AGItEEDSILDK | SAK_0375 | Conserved hypothetical protein |
| SSDFANLDtASLDDFIK | SAK_0375 | Conserved hypothetical protein |
| LVFNDTEStKTLPK | SAK_0389 | Serine/threonine protein kinase Stk1 (Stk1) |
| DNISRPtEGELDSK | SAK_0581 | Cell division protein FtsZ (FtsZ) |
| KDKTNQVSGFtTSAPTNQAPSER | SAK_0581 | Cell division protein FtsZ (FtsZ) |
| TGQEtSFDFDK | SAK_0583 | Conserved hypothetical protein |
| ESLSQSVILAQEtAER | SAK_0586 | Cell division protein DivIVA |
| QLEESGLLDtNNFQMEEPINLGETQTFK | SAK_0586 | Cell division protein DivIVA |
| LEtGDVALEDAIAEFQK | SAK_0598 | Exodeoxyribonuclease VII, small subunit (XseB) |
| LGKtEDDIIVNK | SAK_0651 | Conserved hypothetical protein |
| AEEHtIALGQITEQIPAIVAK | SAK_0709 | Septation ring formation regulator EzrA (EzrA) |
| VTHALDLYEtLQK | SAK_0709 | Septation ring formation regulator EzrA (EzrA) |
| TsEVPVAEDDSFLELER | SAK_0853 | Signal recognition particle-docking protein FtsY (FtsY) |
| APEtKVEDIVIDYK | SAK_0865 | Conserved hypothetical protein |
| DKASEYsNLAVDTFK | SAK_0865 | Conserved hypothetical protein |
| FESGELTtEDIVSAVK | SAK_0865 | Conserved hypothetical protein |
| GLDtGFYDFDPSTVK | SAK_0867 | Peptidase, U32 (collagenase) family |
| DTDKPLLLPVEDVFSItGR | SAK_0887 | Translation elongation factor Tu (EF-Tu, Tuf) |
| REEELSNAKtEANQIIDNAK | SAK_0982 | ATP synthase F0, B subunit (AtpF) |
| ELEAFtQFGSDLDAATQAK | SAK_0984 | ATP synthase F1, α subunit (AtpA) |
| IGHtAYQVTQNSATEHAFTGK | SAK_1026 | Methionine-R-sulfoxide reductase (MsrB) |
| EQPtQFGQGMSLQQALQAR | SAK_1228 | ATP-dependent DNA helicase PcrA (PcrA) |
| SSEFRtTENVPDIDLK | SAK_1628 | Conserved hypothetical protein |
| tAQLMADYEAQR | SAK_1628 | Conserved hypothetical protein |
| MTtENLGEIVISPR | SAK_1706 | Conserved hypothetical protein |
| NTEItRLYEQLK | SAK_1774 | Conserved hypothetical protein |
| TILEEEPIDEEAsRR | SAK_1774 | Conserved hypothetical protein |
| sQMEATTSDFDREK | SAK_2013 | Chaperonin GroEL (GroEL) |
| LTAPSVQFDEtTGDYSR | SAK_2048 | Ribosomal protein L32 (RpmF) |
| Phosphopeptides unique to WT A909 | ||
| sIWESQKEPIQEAITSFK | SAK_0186 | IgA-binding β antigen (Bag) |
| sLQDFIPLNEGK | SAK_0270 | Cysteinyl-tRNA synthetase (CysS) |
| sQFLQGSWNYER | SAK_0433 | PTS system, IID component, mannose/fructose/sorbose family |
| sLQLLAQNYLHDR | SAK_0483 | R3H domain protein |
| sFEGLYDLHNK | SAK_0897 | Peptide chain release factor 3 (PrfC) |
| sFDFITK | SAK_1544 | Ribosomal protein L11 (RplK) |
FIGURE 5.

MS spectra of threonine-phosphorylated peptides corresponding to PcsB (A), hypothetical protein SAK_0375 (B), and ATP synthase F1, α-subunit, AtpA (C). The mass spectra show neutral loss of phosphoric acid from representative phosphopeptides that were identified uniquely to the Δstp1 strain (i.e. not detected in WT A909; also see Table 4 (middle)). A, the m/z of the doubly charged precursor ion corresponding to PcsB is 1034.01. Note that the peak depicting neutral loss of phosphoric acid (H3PO4) has an m/z of 984.89 due to the loss of 49 Da from the doubly charged peptide. B, the peak depicting neutral loss of phosphoric acid (H3PO4) from the precursor ion corresponding to SAK_0375 has an m/z of 921.25 due to the loss of 49 Da from the doubly charged peptide. C, the peak depicting neutral loss of phosphoric acid (H3PO4) has an m/z of 1013.43 due to the loss of 49 Da from the doubly charged peptide corresponding to ATP synthase F1, α subunit, AtpA.
The increase in phosphorylation of proteins predicted to be involved in cell division prompted us to examine their role in GBS hemolysin activity, cell segregation, and virulence. We hypothesized that enhanced Ser/Thr phosphorylation may lead to decreased function of GBS proteins associated with cell division (leading to phenotypes similar to mutations in these genes). The role of PcsB in GBS cell wall separation was described previously (79). To further understand the role of Stp1, we constructed GBS strains deficient in expression of its targets that regulate cell division (i.e. DivIVA, DivIVA domain (GpsB), and FtsZ) and evaluated their role in hemolysin expression and GBS virulence. Hemolytic activity of GBS mutants deficient in expression of DivIVA, DivIVA domain, and FtsZ was similar to that of WT A909 (Fig. 6A). Also, virulence of GBS lacking FtsZ, DivIVA, or DivIVA domain was not significantly different from that of WT A909 (p > 0.05; see supplemental Table S4). We predict that the decreased hemolysin expression observed in the Δstp1 mutant may be due in part to decreased activity of the essential F0F1-ATP synthase that is critical for the function of the ABC transporters associated with hemolysin export and/or other targets uniquely phosphorylated in Δstp1 (see “Discussion”). Because GBS deficient in expression of DivIVA, DivIVA domain, and FtsZ showed the presence of long chains, similar to the Δstp1 mutant (see Fig. 6B), these observations suggest that increased post-translational modification of the cell division proteins is responsible for the abnormal chaining phenotype of the Δstp1 mutant.
FIGURE 6.

Stp1 targets, such as FtsZ, DivIVA, and DivIVA domain, do not regulate GBS hemolytic activity (see A) but are important for normal cell segregation (see B). A, note that hemolytic activity of GBS deficient in expression of the cell division proteins (ΔftsZ, ΔdivIVA, and ΔdivIVA domain) is similar to WT A909, in contrast to Δstp1, which lacks hemolytic activity. B, Gram-stained images of GBS are at 100× magnification. Note that the Δstp1 mutant indicates the presence of long chains in contrast to WT A909 or the complemented strain. Increased chain length is also seen in GBS strains deficient in expression of FtsZ, DivIVA, and DivIVA domain.
Changes in GBS Gene Expression in Δstp1
Because the absence of Stp1 led to increased phosphorylation of a number of GBS proteins (Table 4 (middle)), we also tested the hypothesis that these can lead to changes in gene expression. Global transcriptional profiling analysis was performed using RNA isolated from WT A909 and the isogenic Δstp1 strain as described under “Experimental Procedures.” These results confirmed that transcription of stp1 (SAK_0388) was decreased in the Δstp1 strain (370.3-fold less than WT; see supplemental Table S2B) and that Stk1 transcription was similar to that of the WT (shown in supplemental Table S2A). The microarray analysis also revealed that 132 genes showed increased expression, and 162 genes (excluding stp1) showed decreased expression in the stp1 mutant (supplemental Table S2B). Genes that showed altered expression in Δstp1 are grouped into functional categories based on their predicted function in the GBS genome. Consistent with our previous observations (Table 3), transcription of the gene encoding hemolysin (cylE) and other genes of the cyl operon in the Δstp1 mutant were comparable with WT A909. Furthermore, a comparison of genes that showed altered expression in the Δstp1 mutant with those regulated by CovR in GBS A909 (21) indicated that only a few CovR-regulated genes (e.g. FbsA, FbsB, and SAK_0607 to SAK_0648) showed changes in expression in Δstp1 (see asterisks in supplemental Table S2B). These data further confirm that Stp1 regulation of hemolysin and GBS virulence is independent of CovR regulation of gene expression.
Genes that showed decreased expression in Δstp1 included a few known virulence factors, such as C protein α and the fibrinogen-binding proteins FbsA and FbsB. Other genes that showed decreased expression include a two-component system (SAK_0380/0381), the stand alone transcriptional regulator of the MarR family, the putative competence protein, ABC transporters implicated in metal transport, precursors of purine and pyrimidine biosynthesis, and proteins of as yet unknown function in GBS (supplemental Table S2B). Genes that showed increased expression included transcriptional regulators of the AraC and RpiR family, ABC transporters that are implicated in the uptake of amino acids and oligopeptides, metallopeptidases, and proteins important for carbohydrate metabolism and autolysis (see below for details on autolysis).
To determine if changes in gene expression can be correlated with increased phosphorylation observed in Δstp1, we compared genes that showed altered expression in Δstp1 (supplemental Table S2, A and B) with proteins that were uniquely phosphorylated in Δstp1 (Table 4, middle). These analyses indicated that only SAK_0651 (a hypothetical protein), which showed a 32-fold increase in expression in Δstp1 (supplemental Table S2B), was also uniquely phosphorylated (Table 4, middle). Increased expression of SAK_0651 in Δstp1 may, in part, contribute to its increased phosphorylation. However, expression of the remaining 26 proteins that were identified as uniquely phosphorylated in Δstp1 (Table 4, middle) was similar to that of WT. Thus, phosphorylation of these proteins in Δstp1 cannot be attributed to an increase (or decrease) in their expression.
Δstp1 Mutant Is More Sensitive to Autolysis
Interestingly, the microarray analysis indicated a 30–50-fold increase in expression of phage-encoded lysin and holin genes (SAK_0652 and SAK_0653) in the Δstp1 mutant (supplemental Table S2B). Therefore, we tested the hypothesis that increased expression of these genes may promote autolysis of the Δstp1 mutant. The programmed action of the two-component holin and lysin proteins regulate bacteriophage-mediated lysis of host cells (for reviews, see Refs. 80–82). Although not much is known about mechanisms that trigger autolysis in GBS, previous studies have indicated that the autolysin protein had the ability to lyse GBS and other streptococci (83, 84). To determine if increased expression of lysin and holin promote autolysis of Δstp1, we compared survival of GBS (WT, Δstp1) with autolysis as described under “Experimental Procedures.” These results indicate that the Δstp1 mutant is more sensitive to autolysis when compared with WT A909 (Fig. 7). As expected, the complemented strain is similar to WT for resistance to autolysis (see Δstp1/pStp1 in Fig. 7).
FIGURE 7.
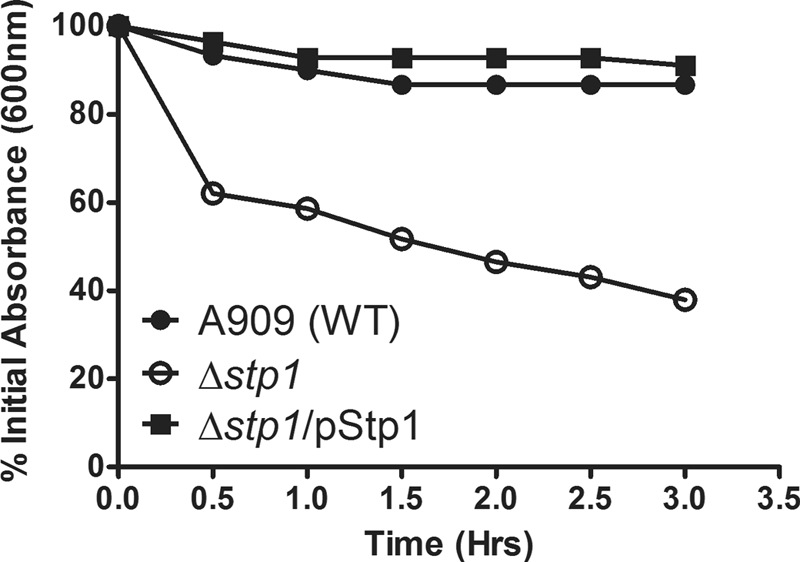
Increased autolysis of the Δstp1 mutant. The rate of GBS autolysis was determined as described under “Experimental Procedures.” Note that the stp1 mutant was more sensitive to autolysis when compared with WT A909 (p = 0.001). Autolysis of the complemented strain was similar to WT (p = 0.1).
Collectively, our studies indicate that Stp1 is important for regulation of Stk1 function and transcriptional and post-transcriptional processes that mediate GBS autolysis and virulence. Further studies will provide novel insight into how eukaryotic-like signaling systems regulate mechanisms important for bacterial pathogenesis. These will provide the basis for evaluation of Stp1 as an antimicrobial target in strategies to prevent bacterial infections in humans.
DISCUSSION
Signaling systems are essential for all living organisms to respond to their dynamic external environment. Signal transduction in bacteria typically involves reversible phosphorylation of histidine and aspartate residues. Although examples of serine/threonine phosphorylation are increasingly encountered in bacteria and other prokaryotes (for recent reviews, see Refs. 9 and 10), our understanding of the signal transduction mechanism is not complete. We have extensively described the role of the serine/threonine kinase Stk1 in regulation of GBS virulence (18, 21, 38–40). Because these studies have exclusively focused on the kinase (i.e. Stk1), the role of the cognate serine/threonine phosphatase Stp1 in regulation of Stk1 function and GBS virulence has remained unknown.
Our studies indicate that GBS strains deficient only in Stp1 expression are significantly attenuated for their ability to cause systemic infections. Furthermore, the Δstp1 mutant was attenuated for its ability to induce proinflammatory signaling pathways and disrupt barrier integrity of brain endothelium. The dramatic decrease in virulence of the Δstp1 mutant is consistent with the decrease in toxin (β-H/C) activity and increased sensitivity of the mutant to autolysis. The decrease in hemolytic activity observed in the Δstp1 mutant is independent of changes in transcription of the cylE gene encoding β-H/C and CovR/S-mediated transcriptional regulation of the cyl operon. Previous studies have indicated that components of an ABC transporter (i.e. the ATP binding protein CylA and the permease protein CylB) are required for β-H/C export in GBS (41). Because phosphopeptides corresponding to AtpA and AtpF (encoding α and B subunits of the F0F1-ATP synthase) were identified only in Δstp1, one possibility is that the decrease in hemolytic activity is, in part, due to by decreased function of F0F1-ATP synthase, leading to decreased CylA/CylB function. Evidence that protein phosphorylation inhibits the F0F1-ATP synthase is provided by recent studies that indicate that protein kinase Cζ (PKCζ) phosphorylation of AtpB inhibits the activity of the mitochondrial F0F1-ATP synthase (85, 86). Similarly, phosphorylation of AtpA by the serine/threonine protein kinase of Sulfolobus solfatarocus P2 is also suggested to decrease ATP synthase activity (87).4 Our efforts to construct GBS mutants deficient in the F0F1-ATP synthase subunits, AtpA and AtpF, were unsuccessful and may be due to the essential nature of these genes. Support for this conclusion is provided by observations that only a single operon encoding subunits of the F0F1-ATP synthase complex is present in the GBS A909 genome (SAK_0980–SAK_0987) (88), and these genes are essential in Streptococcus pneumoniae (89). Alternatively, it is also likely that the decrease in hemolytic activity observed in the Stp1 mutant can be attributed to a cumulative effect of increased phosphorylation and changes in gene expression that mediate protein synthesis and stability (Table 4 (middle)). Although we raised peptide antibodies to β-H/C (positions 2–19 (i.e. KDDNKLKISEASLEDYSE) and positions 644–662 (i.e. MFKRNSPYATNVSITEYR)), these antibodies failed to recognize β-H/C in starch extracts with hemolytic activity (e.g. GBS WT, hyperhemolytic ΔcovR; data not shown), which is consistent with previous conclusions that β-H/C may be non-immunogenic (74). The non-immunogenic nature of β-H/C poses an impediment on understanding mechanisms that regulate its secretion and development of vaccines.
Contrary to our hypothesis that the absence of Stp1 would enhance Stk1 (threonine) phosphorylation of CovR, leading to increased β-H/C (cylE) transcription and GBS virulence, our studies described here indicate the opposite. In S. pneumoniae, Stk1 and Stp1 homologues known as StkP and StpP form a ternary complex with their phosphorylated substrate, RitR (9, 90). Based on these observations and our findings, we predict that Stk1 phosphorylation and regulation of CovR function in vivo (i.e. in GBS) may require the interaction of CovR with both Stk1 and Stp1. In the absence of Stp1, it is likely that the substrate specificity of Stk1 is altered, leading to the phosphorylation of a target(s) that mediates post-transcriptional regulation of hemolysin and increased expression of autolysin genes. Support for this hypothesis is provided by our observations that phenotypes of the Δstp1 mutant (lack of hemolytic activity independent of changes in cylE transcription and increased sensitivity to autolysis) are not observed in the double Δstp1Δstk1 mutant (supplemental Fig. S3), suggesting that the phenotypic changes of Δstp1 are linked to the presence of Stk1. These observations implicate a key regulatory role for Stp1 in Stk1 function.
Changes in transcription of genes observed in the Δstp1 mutant can be attributed to altered expression of transcriptional regulators and phosphorylation of the ATP-dependent DNA and RNA helicases. Although the role of these proteins in GBS is not completely understood, helicases have an important role in unwinding DNA and RNA duplexes (91, 92) for transcriptional and post-transcriptional processes. Protein kinase-mediated phosphorylation had opposite effects on the activity of human DNA and RNA helicases (e.g. phosphorylation of the human DNA helicase II by the DNA-dependent serine/threonine protein kinase (DNA-PKcs) increased DNA unwinding (93), whereas phosphorylation of the human P68 RNA helicase by the serine/threonine protein kinase C abolished RNA binding (94)). Studies that elucidate the role of DNA and RNA helicases in transcription and post-transcriptional regulation of specific GBS genes and understanding of how Stp1 mediates repression of autolysin genes will provide novel information on GBS pathogenesis.
The abnormal chaining morphology observed with the Δstp1 mutant can be attributed to post-translational modifications of cell division proteins, such as FtsZ, DivIVA, DivIVA domain, and PcsB. Studies in other bacteria have indicated that serine/threonine phosphatases play a critical role in phosphorylation of cell division proteins (e.g. serine/threonine phosphatase PppL mutants of Streptococcus mutans show abnormal growth and irregular cell division (95)). In Mycobacteria, increased phosphorylation of DivIVA has distinct effects on cellular morphology (96), and serine/threonine kinase (PknA) phosphorylation of FtsZ is important for cell division during oxidative stress (97). Understanding the effect on Ser/Thr phosphorylation in the function of PcsB, FtsZ, DivIVA, and DivIVA domain will provide further insight into GBS cell division/cell separation.
It is also noteworthy that until recently, the inability to derive mutants deficient only in Stp1 expression, particularly in Streptococcus sp. (e.g. S. pneumoniae and previously in GBS and S. pyogenes), had led to the notion that Stp1 may be essential (10, 18, 26–28). Our studies of Stp1 in protein phosphorylation, autolysis, and GBS virulence may provide further insight into its role in other prokaryotes and will help determine if increased expression of autolysin genes is a conserved feature among Δstp1 mutants. Similar to our findings with GBS, Listeria monocytogenes deficient in Stp1 have been described to exhibit attenuated virulence and increased phosphorylation of EF-Tu; phosphorylation of EF-Tu is thought to regulate protein synthesis and impact virulence functions of Listeria (25). In GBS, Stp1 is important for regulation of Stk1 function and transcriptional and post-transcriptional processes that mediate autolysis, hemolysin activity, and virulence. Further studies on the role of Stp1 in bacteria will provide greater insight into signaling mechanisms that are conserved between prokaryotes and eukaryotes. These studies will be valuable for identification of specific inhibitors to the PP2C class of phosphatases (e.g. see Ref. 98) in treatment of bacterial infections.
Supplementary Material
Acknowledgments
We thank Donald Chaffin and Jessica Zbikowski Berry for help with the sialic acid capsular polysaccharide level analysis, Mason Craig Bailey for animal support, Sarah Collins for help with microscopy, and Chris Whidbey for critical reading of the manuscript. We thank Dr. Vijay Pancholi for the kind gift of the GAS Stp1 antibody, and we are grateful to Monique Stins and Kwang Sik Kim for providing hBMEC and to Christian Renken (Applied Biophysics) for assistance with ECIS. The host microarray analysis was performed at the Biogem Core Facility of the University of California San Diego, director Gary Hardiman.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Grant RO1AI070749 (to L. R.), NIH, NINDS, Grant RO1NS051247 (to K. S. D.), and NIH Grant GM088317 (to W. A. T.). This work was also supported by the National Science Foundation CAREER CHE-0645020 award (to W. A. T.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S4 and Figs. S1–S3.
R. A. Redbird and P. J. Kennedy, unpublished data.
- STP
- serine/threonine phosphatase(s)
- STK
- serine/threonine kinase(s)
- GAS
- Group A Streptococcus/streptococci
- GBS
- Group B Streptococcus/streptococci
- β-H/C
- β-hemolysin/cytolysin
- ECIS
- electric cell-substrate impedance sensing
- hBMEC
- human brain microvascular endothelial cell(s)
- EF-Tu
- elongation factor Tu
- qRT-PCR
- quantitative RT-PCR
- BBB
- blood-brain barrier.
REFERENCES
- 1. Hoch J., Silhavy T. (eds). (1995) Two-component Signal Transduction, American Society for Microbiology, Washington, D. C. [Google Scholar]
- 2. Beier D., Gross R. (2006) Curr. Opin. Microbiol. 9, 143–152 [DOI] [PubMed] [Google Scholar]
- 3. Calva E., Oropeza R. (2006) Microb. Ecol. 51, 166–176 [DOI] [PubMed] [Google Scholar]
- 4. Bassler B. L., Losick R. (2006) Cell 125, 237–246 [DOI] [PubMed] [Google Scholar]
- 5. Novick R. P. (2003) Mol. Microbiol. 48, 1429–1449 [DOI] [PubMed] [Google Scholar]
- 6. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 7. Cohen P. (2002) Nat. Rev. Drug Discov. 1, 309–315 [DOI] [PubMed] [Google Scholar]
- 8. Kennelly P. J. (2002) FEMS Microbiol. Lett. 206, 1–8 [DOI] [PubMed] [Google Scholar]
- 9. Pereira S. F., Goss L., Dworkin J. (2011) Microbiol. Mol. Biol. Rev. 75, 192–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Burnside K., Rajagopal L. (2011) Future Microbiol. 6, 747–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Håkansson S., Galyov E. E., Rosqvist R., Wolf-Watz H. (1996) Mol. Microbiol. 20, 593–603 [DOI] [PubMed] [Google Scholar]
- 12. Barz C., Abahji T. N., Trülzsch K., Heesemann J. (2000) FEBS Lett. 482, 139–143 [DOI] [PubMed] [Google Scholar]
- 13. Juris S. J., Rudolph A. E., Huddler D., Orth K., Dixon J. E. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 9431–9436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Madec E., Laszkiewicz A., Iwanicki A., Obuchowski M., Séror S. (2002) Mol. Microbiol. 46, 571–586 [DOI] [PubMed] [Google Scholar]
- 15. Koul A., Choidas A., Tyagi A. K., Drlica K., Singh Y., Ullrich A. (2001) Microbiology 147, 2307–2314 [DOI] [PubMed] [Google Scholar]
- 16. Chaba R., Raje M., Chakraborti P. K. (2002) Eur. J. Biochem. 269, 1078–1085 [DOI] [PubMed] [Google Scholar]
- 17. Cowley S., Ko M., Pick N., Chow R., Downing K. J., Gordhan B. G., Betts J. C., Mizrahi V., Smith D. A., Stokes R. W., Av-Gay Y. (2004) Mol. Microbiol. 52, 1691–1702 [DOI] [PubMed] [Google Scholar]
- 18. Rajagopal L., Clancy A., Rubens C. E. (2003) J. Biol. Chem. 278, 14429–14441 [DOI] [PubMed] [Google Scholar]
- 19. Débarbouillé M., Dramsi S., Dussurget O., Nahori M. A., Vaganay E., Jouvion G., Cozzone A., Msadek T., Duclos B. (2009) J. Bacteriol. 191, 4070–4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kristich C. J., Wells C. L., Dunny G. M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 3508–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lembo A., Gurney M. A., Burnside K., Banerjee A., de los Reyes M., Connelly J. E., Lin W. J., Jewell K. A., Vo A., Renken C. W., Doran K. S., Rajagopal L. (2010) Mol. Microbiol. 77, 431–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burnside K., Lembo A., de Los Reyes M., Iliuk A., Binhtran N. T., Connelly J. E., Lin W. J., Schmidt B. Z., Richardson A. R., Fang F. C., Tao W. A., Rajagopal L. (2010) PLoS ONE 5, e11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tamber S., Schwartzman J., Cheung A. L. (2010) Infect. Immun. 78, 3637–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaidenko T. A., Kim T. J., Price C. W. (2002) J. Bacteriol. 184, 6109–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Archambaud C., Gouin E., Pizarro-Cerda J., Cossart P., Dussurget O. (2005) Mol. Microbiol. 56, 383–396 [DOI] [PubMed] [Google Scholar]
- 26. Osaki M., Arcondéguy T., Bastide A., Touriol C., Prats H., Trombe M. C. (2009) J. Bacteriol. 191, 4943–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Agarwal S., Agarwal S., Pancholi P., Pancholi V. (September 14, 2011) J. Biol. Chem. 10.1074/jbc.M111.286690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jin H., Pancholi V. (2006) J. Mol. Biol. 357, 1351–1372 [DOI] [PubMed] [Google Scholar]
- 29. Baker C. J., Edwards M. W. (1995) in Infectious Diseases of the Fetus and Newborn Infant (Remington J. S., Klein J. O. eds) pp. 980–1054, W.B. Saunders, Philadelphia [Google Scholar]
- 30. Doran K. S., Nizet V. (2004) Mol. Microbiol. 54, 23–31 [DOI] [PubMed] [Google Scholar]
- 31. Rajagopal L. (2009) Future Microbiol. 4, 201–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hillier S. L., Martius J., Krohn M., Kiviat N., Holmes K. K., Eschenbach D. A. (1988) N. Engl. J. Med. 319, 972–978 [DOI] [PubMed] [Google Scholar]
- 33. Romero R., Sirtori M., Oyarzun E., Avila C., Mazor M., Callahan R., Sabo V., Athanassiadis A. P., Hobbins J. C. (1989) Am. J. Obstet. Gynecol. 161, 817–824 [DOI] [PubMed] [Google Scholar]
- 34. Hillier S. L., Krohn M. A., Kiviat N. B., Watts D. H., Eschenbach D. A. (1991) Am. J. Obstet. Gynecol. 165, 955–961 [DOI] [PubMed] [Google Scholar]
- 35. Verani J. R., McGee L., Schrag S. J. (2010) MMWR Recomm. Rep. 59, 1–36 [PubMed] [Google Scholar]
- 36. Weston E. J., Pondo T., Lewis M. M., Martell-Cleary P., Morin C., Jewell B., Daily P., Apostol M., Petit S., Farley M., Lynfield R., Reingold A., Hansen N. I., Stoll B. J., Shane A. L., Zell E., Schrag S. J. (2011) Pediatr. Infect. Dis. J. 30, 937–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin W. J., Walthers D., Connelly J. E., Burnside K., Jewell K. A., Kenney L. J., Rajagopal L. (2009) Mol. Microbiol. 71, 1477–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rajagopal L., Vo A., Silvestroni A., Rubens C. E. (2005) Mol. Microbiol. 56, 1329–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rajagopal L., Vo A., Silvestroni A., Rubens C. E. (2006) Mol. Microbiol. 62, 941–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Silvestroni A., Jewell K. A., Lin W. J., Connelly J. E., Ivancic M. M., Tao W. A., Rajagopal L. (2009) J. Proteome. Res. 8, 2563–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spellerberg B., Pohl B., Haase G., Martin S., Weber-Heynemann J., Lütticken R. (1999) J. Bacteriol. 181, 3212–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Madoff L. C., Michel J. L., Gong E. W., Kling D. E., Kasper D. L. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 4131–4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okada N., Geist R. T., Caparon M. G. (1993) Mol. Microbiol. 7, 893–903 [DOI] [PubMed] [Google Scholar]
- 44. Horton R. M. (1995) Mol. Biotechnol. 3, 93–99 [DOI] [PubMed] [Google Scholar]
- 45. Chaffin D. O., Beres S. B., Yim H. H., Rubens C. E. (2000) J. Bacteriol. 182, 4466–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jones A. L., Knoll K. M., Rubens C. E. (2000) Mol. Microbiol. 37, 1444–1455 [DOI] [PubMed] [Google Scholar]
- 47. Jiang S. M., Cieslewicz M. J., Kasper D. L., Wessels M. R. (2005) J. Bacteriol. 187, 1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chaffin D. O., Mentele L. M., Rubens C. E. (2005) J. Bacteriol. 187, 4615–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lamy M. C., Zouine M., Fert J., Vergassola M., Couve E., Pellegrini E., Glaser P., Kunst F., Msadek T., Trieu-Cuot P., Poyart C. (2004) Mol. Microbiol. 54, 1250–1268 [DOI] [PubMed] [Google Scholar]
- 50. Coligan J., Kruisbeek A., Marguiles D., Shevach E., Strober W. (eds) (1998) Current Protocols in Immunology, Vol. 1, John Wiley & Sons, Inc., New York [Google Scholar]
- 51. Stins M. F., Prasadarao N. V., Zhou J., Arditi M., Kim K. S. (1997) In Vitro Cell. Dev. Biol. Anim. 33, 243–247 [DOI] [PubMed] [Google Scholar]
- 52. Doran K. S., Engelson E. J., Khosravi A., Maisey H. C., Fedtke I., Equils O., Michelsen K. S., Arditi M., Peschel A., Nizet V. (2005) J. Clin. Invest. 115, 2499–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Giaever I., Keese C. R. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 3761–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Giaever I., Keese C. R. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 7896–7900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Giaever I., Keese C. R. (1993) Nature 366, 591–592 [DOI] [PubMed] [Google Scholar]
- 56. Lo C. M., Keese C. R., Giaever I. (1995) Biophys. J. 69, 2800–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Doran K. S., Liu G. Y., Nizet V. (2003) J. Clin. Invest. 112, 736–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Sorge N. M., Ebrahimi C. M., McGillivray S. M., Quach D., Sabet M., Guiney D. G., Doran K. S. (2008) PLoS ONE 3, e2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nizet V., Gibson R. L., Chi E. Y., Framson P. E., Hulse M., Rubens C. E. (1996) Infect. Immun. 64, 3818–3826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. (1997) Current Protocols in Molecular Biology, John Wiley & Sons, Inc., New York [Google Scholar]
- 61. Iliuk A. B., Martin V. A., Alicie B. M., Geahlen R. L., Tao W. A. (2010) Mol. Cell Proteomics 9, 2162–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Iliuk A., Tao W. A. (2009) Methods Mol. Biol. 527, 117–129, ix [DOI] [PubMed] [Google Scholar]
- 63. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 64. Benjamini Y., Hochberg Y. (1995) J. R. Stat. Soc. Ser. B 57, 289–300 [Google Scholar]
- 65. Mani N., Tobin P., Jayaswal R. K. (1993) J. Bacteriol. 175, 1493–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wessels M. R., Rubens C. E., Benedí V. J., Kasper D. L. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 8983–8987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Marques M. B., Kasper D. L., Pangburn M. K., Wessels M. R. (1992) Infect. Immun. 60, 3986–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Harris T. O., Shelver D. W., Bohnsack J. F., Rubens C. E. (2003) J. Clin. Invest. 111, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shelver D., Rajagopal L., Harris T. O., Rubens C. E. (2003) J. Bacteriol. 185, 6592–6599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Banerjee A., Kim B. J., Carmona E. M., Cutting A. S., Gurney M. A., Carlos C., Feuer R., Prasadarao N. V., Doran K. S. (2011) Nat. Commun. 2, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ring A., Braun J. S., Pohl J., Nizet V., Stremmel W., Shenep J. L. (2002) J. Infect. Dis. 185, 1745–1753 [DOI] [PubMed] [Google Scholar]
- 72. Hensler M. E., Liu G. Y., Sobczak S., Benirschke K., Nizet V., Heldt G. P. (2005) J. Infect. Dis. 191, 1287–1291 [DOI] [PubMed] [Google Scholar]
- 73. Doran K. S., Chang J. C., Benoit V. M., Eckmann L., Nizet V. (2002) J. Infect. Dis. 185, 196–203 [DOI] [PubMed] [Google Scholar]
- 74. Nizet V. (2002) Trends Microbiol. 10, 575–580 [DOI] [PubMed] [Google Scholar]
- 75. Pritzlaff C. A., Chang J. C., Kuo S. P., Tamura G. S., Rubens C. E., Nizet V. (2001) Mol. Microbiol. 39, 236–247 [DOI] [PubMed] [Google Scholar]
- 76. Chaffin D. O., Rubens C. E. (1998) Gene 219, 91–99 [DOI] [PubMed] [Google Scholar]
- 77. Quach D., van Sorge N. M., Kristian S. A., Bryan J. D., Shelver D. W., Doran K. S. (2009) J. Bacteriol. 191, 2023–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jiang S. M., Ishmael N., Dunning Hotopp J., Puliti M., Tissi L., Kumar N., Cieslewicz M. J., Tettelin H., Wessels M. R. (2008) J. Bacteriol. 190, 1956–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Reinscheid D. J., Gottschalk B., Schubert A., Eikmanns B. J., Chhatwal G. S. (2001) J. Bacteriol. 183, 1175–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang I. N., Smith D. L., Young R. (2000) Annu. Rev. Microbiol. 54, 799–825 [DOI] [PubMed] [Google Scholar]
- 81. Loessner M. J. (2005) Curr. Opin. Microbiol. 8, 480–487 [DOI] [PubMed] [Google Scholar]
- 82. Bayles K. W. (2007) Nat. Rev. Microbiol. 5, 721–726 [DOI] [PubMed] [Google Scholar]
- 83. Pritchard D. G., Dong S., Baker J. R., Engler J. A. (2004) Microbiology 150, 2079–2087 [DOI] [PubMed] [Google Scholar]
- 84. Cheng Q., Nelson D., Zhu S., Fischetti V. A. (2005) Antimicrob. Agents Chemother. 49, 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Guo H., Li D., Ling W., Feng X., Xia M. (2011) J. Lipid Res. 52, 908–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Struglics A., Fredlund K. M., Møller I. M., Allen J. F. (1998) Biochem. Biophys. Res. Commun. 243, 664–668 [DOI] [PubMed] [Google Scholar]
- 87. Redbird R. A., Ray K. W., Hite D., Dunham R., J K. P. (2007) FASEB J. 21, A986 [Google Scholar]
- 88. Tettelin H., Masignani V., Cieslewicz M. J., Donati C., Medini D., Ward N. L., Angiuoli S. V., Crabtree J., Jones A. L., Durkin A. S., Deboy R. T., Davidsen T. M., Mora M., Scarselli M., Margarit y Ros I., Peterson J. D., Hauser C. R., Sundaram J. P., Nelson W. C., Madupu R., Brinkac L. M., Dodson R. J., Rosovitz M. J., Sullivan S. A., Daugherty S. C., Haft D. H., Selengut J., Gwinn M. L., Zhou L., Zafar N., Khouri H., Radune D., Dimitrov G., Watkins K., O'Connor K. J., Smith S., Utterback T. R., White O., Rubens C. E., Grandi G., Madoff L. C., Kasper D. L., Telford J. L., Wessels M. R., Rappuoli R., Fraser C. M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Song J. H., Ko K. S., Lee J. Y., Baek J. Y., Oh W. S., Yoon H. S., Jeong J. Y., Chun J. (2005) Mol. Cells 19, 365–374 [PubMed] [Google Scholar]
- 90. Ulijasz A. T., Falk S. P., Weisblum B. (2009) Mol. Microbiol. 71, 382–390 [DOI] [PubMed] [Google Scholar]
- 91. Johnson D. S., Bai L., Smith B. Y., Patel S. S., Wang M. D. (2007) Cell 129, 1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jankowsky E. (2011) Trends Biochem. Sci. 36, 19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ochem A. E., Rechreche H., Skopac D., Falaschi A. (2008) Arch. Biochem. Biophys. 470, 1–7 [DOI] [PubMed] [Google Scholar]
- 94. Yang L., Yang J., Huang Y., Liu Z. R. (2004) Biochem. Biophys. Res. Commun. 314, 622–630 [DOI] [PubMed] [Google Scholar]
- 95. Banu L. D., Conrads G., Rehrauer H., Hussain H., Allan E., van der Ploeg J. R. (2010) Infect. Immun. 78, 2209–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kang C. M., Abbott D. W., Park S. T., Dascher C. C., Cantley L. C., Husson R. N. (2005) Genes Dev. 19, 1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sureka K., Hossain T., Mukherjee P., Chatterjee P., Datta P., Kundu M., Basu J. (2010) PLoS ONE 5, e8590. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98. Rogers J. P., Beuscher A. E., 4th, Flajolet M., McAvoy T., Nairn A. C., Olson A. J., Greengard P. (2006) J. Med. Chem. 49, 1658–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.