

Background: We previously identified a synthetic glycolipid (named CCL-34) that activates Toll-like receptor 4 (TLR4).
Results: CCL-34 induces cancer cell death via TLR4-dependent activation of immune cells, which requires its sugar moiety.
Conclusion: CCL-34 exhibits anticancer immunity via TLR4, and its sugar moiety plays an essential role.
Significance: A new TLR4 agonist with anticancer activity and a broadening molecular basis of TLR4-activating glycolipids is revealed.
Keywords: Anticancer Drug, Cancer Therapy, Chemical Biology, Drug Discovery, Glycolipids, Molecular Pharmacology, Toll-like Receptors (TLR), Structure-and-Activity Relationship, TLR4 Agonist, Anticancer Immunity
Abstract
Activation of Toll-like receptor 4 (TLR4) triggers the innate immune response and leads to the induction of adaptive immunity. TLR4 agonists are known to function as immunostimulants and exhibit promising therapeutic potential for cancer immunotherapy. We have previously developed a synthetic serine-based glycolipid (designated as CCL-34) that can activate TLR4-dependent signaling pathways. In this study, the anticancer immunity of CCL-34 was further demonstrated. CCL-34-activated macrophages induced cancer cell death via the apoptotic pathway, and this cytotoxicity was significantly inhibited by NG-monomethyl-l-arginine (an inducible NOS inhibitor). Notably, conditioned medium collected from CCL-34-treated splenocytes also induced cytotoxicity toward cancer cells. Furthermore, CCL-34 treatment suppressed tumor growth and increased the survival rate in TLR4-functional C3H/HeN mice but not in TLR4-defective C3H/HeJ mice. Increased apoptosis, the induction of cytokines (IFN-γ and IL-12) and chemokines (CXCL9 and CXCL10), and the elevation of leukocyte markers (CD11b, CD11c, CD4, and CD8) were detected at tumor sites in C3H/HeN mice but not in C3H/HeJ mice. Structure-and-activity relationship analysis of CCL-34 and its structural analogs revealed that a sugar moiety is essential for its activity. However, the substitution of the galactose in CCL-34 with glucose or fucose did not reduce its activity. Altogether, this study reveals the anticancer activity of a new synthetic TLR4 agonist and broadens the molecular basis of TLR4-activating glycolipids.
Introduction
Toll-like receptor 4 (TLR4)3 plays a central role in the initiation of innate immunity and the induction of adaptive immunity (1). TLR4 is commonly expressed in innate immune cells (such as neutrophils, macrophages, and dendritic cells) but can also be detected in nonimmune cells (1). Diverse exogenous and endogenous ligands, such as lipopolysaccharide (LPS), heparin sulfate, and high mobility group box 1, have been identified to trigger TLR4-mediated responses (1). An extracellular TLR4-associated protein, MD-2, is required for ligand recognition and activation of TLR4 (2, 3). Stimulation of TLR4 can trigger both myeloid differentiation factor 88 (MyD88)-dependent and -independent signaling pathways. MyD88-dependent signaling subsequently leads to activation of nuclear factor-κB (NF-κB) and MAPK (ERK, p38, and JNK). Alternatively, TIR domain-containing adaptor protein-inducing IFN-β (TRIF), and TRIF-related adaptor molecule can induce a MyD88-independent pathway, which subsequently mediates the activation of NF-κB and interferon regulatory factor 3 (IRF-3). In both pathways, activated NF-κB induces the expression of cytokines such as TNF-α, IL-6, and IL-12. Moreover, the activation of the IRF-3 stimulates the expression of type I interferon (IFN-α/β) (4).
TLR4 agonists have been shown as potent immunostimulants for vaccine adjuvant and cancer treatment. Two major types of TLR4 agonists have been developed. Lipid A derivatives and mimics, such as monophosphoryl lipid A and OM-174, have been demonstrated to exhibit adjuvant activity and anticancer activity (5, 6). Furthermore, bacterially derived components, such as Bacillus Calmette-Guerin, OK-432 (a streptococcal agent), and attenuated Salmonella choleraesuis, are found to exhibit anticancer effects via TLR4 (7–9). Immunogenic cancer cell death via TLR4-signaling pathways, induced by chemotherapy and radiotherapy, has been recently demonstrated, supporting the involvement of TLR4 in anticancer immunity (5, 10). Together, these observations indicate that TLR4 is an emerging target for cancer immunotherapy.
The molecular basis for the anticancer activity of TLR4 agonists involves both innate and adaptive immunity. Macrophages stimulated by TLR4 agonists produce significant amounts of anticancer molecules like TNF-α, IFN-γ, and nitric oxide (NO) and can induce cytotoxicity of cancer cells (11). Functional activation of dendritic cells by TLR4 agonists, which subsequently activate other immunocompetent cells (such as helper and cytotoxic T cells, NK cells, and macrophages), is a critical step in the induction of the host antitumor response (7). Furthermore, various cytokines and chemokines induced by TLR4 agonists increase the number of tumor-infiltrating immune cells and cause apoptosis within the tumors (9, 12).
We have previously identified a synthetic serine-based glycolipid (CCL-34) that induced macrophage activation through the TLR4-signaling pathway (13). CCL-34 comprises a galactose α-linked to a hydrophobic lipid moiety that contains both acyl and lipid amine chains. Compared with other known TLR4 agonists, CCL-34 has the advantage of a defined structure, easier synthesis, and less contamination with bioactive microbial components. To further explore the therapeutic potential of CCL-34, we determined its anticancer activity in vitro and in vivo. Moreover, to facilitate the development of effective CCL-34 derivatives, the structure-and-activity relationship analysis was performed to investigate the role of sugar moiety in CCL-34 activity.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
CCL-34 and its derivatives were synthesized as described previously (13, 14). OK-432 was purchased from Chugai Pharmaceutical (Tokyo, Japan).
Animals
C3H/HeN and C3H/HeJ mice were maintained at the Animal Center of Yang-Ming University according to the Guidelines for the Care and Use of Laboratory Animals (National Institutes of Health). All experiments related to the animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Yang-Ming University.
Cells and Culture Medium
The murine macrophage cell line RAW264.7, the human monocyte cell line THP1, and the human kidney cell line HEK293 were obtained from the Food Industry Research and Development Institute (Hsinchu, Taiwan). The 293-hTLR4/MD2/CD14 cells were purchased from Invivogen (San Diego). The RAW264.7, 293-hTLR4/MD2/CD14, and THP1 cells were cultured as described previously (13, 15). Further differentiated THP-1 cells were generated by treatment with 5 ng/ml phorbol 12-myristate 13-acetate for 48 h in the same growth medium. The mouse lung carcinoma cell line LLC1 (ATCC CRL-1642) (16) and the mouse bladder cancer cell line MBT-2 from Dr. Lih-Hwa Hwang at National Yang-Ming University (17) were cultured in DMEM containing 10% heat-inactivated FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 μm l-glutamine (16, 17). Primary peritoneal macrophages were isolated and cultured as described previously (18). For all experiments, early passage cells were used.
Macrophage-mediated Cytotoxicity Assay
Macrophage-mediated tumor cell cytotoxicity was measured using a LIVE/DEAD cell-mediated cytotoxicity kit (Invitrogen). The cancer cells (2 × 105/well) were labeled with DiOC18 and incubated with drug-treated macrophages (6-h treatment followed by PBS washing) for 24 h. The ratio of cancer cells to macrophages was 1:10. Next, all cancer cells/macrophages were stained with propidium iodide (PI; 20 μg/ml) and analyzed using a FACSCalibur (BD Biosciences). The dead cancer cells were identified by their concomitant green and red fluorescence. Alternatively, the drug-treated macrophages were first transferred into culture plate inserts (pore size 0.4 μm, Millipore, MA) and co-incubated with cancer cells for 24 h, followed by PI staining and flow cytometry.
Microarray Analysis
The quality of RNA for microarray analysis was determined using Spectra Max Plus (Molecular Devices) and had an A260/A280 ratio from 1.9 to 2.1. The mRNA was purified using the Oligotex mRNA kit (Qiagen, Valencia, CA). The cDNA was prepared from mRNA using the GeneChip® expression 3′-amplification reagents two-cycle cDNA synthesis kit (Affymetrix, P/N 900432) with the exception that the primer used for the reverse transcription reaction was a modified T7 primer with 24 thymidines. Labeled cRNA was synthesized from the cDNA using the Bioarray High Yield RNA transcript labeling kit (Affymetrix, P/N 900182) according to the manufacturer's instructions. Fragmentation of biotinylated cRNA (15 μg) was performed by employing the GeneChip Sample Cleanup Module (Affymetrix, P/N 900371). Additional protocols and reagents for hybridization, washing, and staining are described in Affymetrix instructions.
Analysis of Microarray Data
Following a quantitative scan of the chip, a grid was aligned to the image using the dimension of the microarray chips and the corner control regions as markers. The images were transformed into text files containing the intensity information using GeneChip® operating software developed by Affymetrix. The microarray data were analyzed using the GeneSpring® 7.0 software (Silicon Genetics, Redwood City, CA). Sequences on the Affymetrix chips were generally represented by an average of 16 perfect match probe sets and corresponding mismatch sequences serving as false-positive controls for nonspecific hybridization. The absolute intensity statistics were based on the feature intensities and corresponding standard deviations provided in the microarray chip-generated CEL files. In addition, normalization between different arrays was performed for all regions, and each gene expression signal was normalized to the median value of all genes in each chip.
TUNEL Assay
Apoptotic cancer cells after co-cultivation with drug-treated macrophages were detected using the in situ cell death detection kit (Roche Applied Science) followed by flow cytometry. For detecting apoptosis in tumor tissues, tumor sections were analyzed using the same kit and counterstained with DAPI (Invitrogen). Apoptotic cells were detected under a fluorescence microscope (Olympus BX61, Tokyo, Japan).
Caspase-3 Activity Assay
Caspase-3 activity was measured using a caspase-3 assay kit (BD Biosciences). In brief, cell lysates were incubated with the caspase-3 substrate Ac-DEVD-7-amino-4-methylcoumarin at 37 °C for 1 h, and the fluorescent 7-amino-4-methylcoumarin was measured using AutoLumat LB953 (Berthold Technologies, Bad Wildbad, Germany).
Splenocyte Isolation and SCM-mediated Cytotoxicity Assay
Splenocytes from male C3H/HeN mice (8–9 weeks old) were isolated as described previously (19). The splenocytes (1 × 107 cells/ml) were cultured in growth medium supplemented with H2O, vehicle (0.1% DMSO), CCL-34, or OK-432, respectively. After 24 or 48 h, the supernatants were collected as SCM. Fresh growth medium supplemented with H2O, vehicle, or the indicated drugs was prepared as control groups for subsequent experiments.
To measure SCM-mediated cytotoxicity, LLC1 cancer cells (2 × 105 in 6-well plates) were treated with different SCM or corresponding control medium for 24 h. Then the viability of cancer cells were determined by PI staining and flow cytometry. The percentages of dead cells between the SCM-treated and control groups were compared.
In Vivo Tumorigenesis Assay and H&E Stain
A syngeneic tumor model for evaluating cancer immunotherapeutic potential was performed as described previously (20). In brief, MBT-2 cells (5 × 105) were injected subcutaneously into the dorsum of CH3/HeN or CH3/HeJ mice (8–9-week-old mice). When tumors reached 80 mm3 in volume (day 10), the mice were randomly divided into several groups (5–7 mice each group) and injected intraperitoneally with 100 μl of vehicle (DMSO, 5.5 μl/mouse), CCL-34 (100 μg/mouse), or OK-432 (100 μg/mouse) on days 10, 13, 17, 20, 24, and 27. Tumor volumes were measured twice a week from day 10 (1/2 ab2; a and b represent the largest and smallest diameter, respectively). The body weights and survival rates were also recorded. To analyze tumor-infiltrating immune cells, tumors were excised and fixed with 4% formalin, followed by H&E staining. Expression of leukocyte markers (CD11b, CD11c, CD4, and CD8) were examined using quantitative (q) RT-PCR as described below.
Measurement of Cytokine Production
To measure cytokine production induced by CCL-34 in vitro, the 293-hTLR4/MD2/CD14 cells (2 × 105) or phorbol 12-myristate 13-acetate-pretreated THP-1 (2 × 105 cells) were incubated with vehicle or drugs for various time periods, and the culture medium was then harvested for cytokine measurement using enzyme-linked immunosorbent assay (ELISA).
To measure cytokine expression in tumors, the tumors were lysed with cell extraction buffers (10 mm Tris-HCl (pH 7.8), 0.5% Nonidet P-40, 0.05 m NaCl, 0.5 mm EGTA, 5% sucrose, 5 mm HEPES (pH 7.6), 1 mm MgCl2, 25 mm NaCl, 2.5 mm EGTA, 0.05% Triton X-100) containing protease inhibitors. The protein lysate (500 μg) from each sample was analyzed for the concentrations of IFN-γ, IL-12, and TNF-α using ELISA kits (R&D Systems, Minneapolis, MN).
qRT-PCR
Total RNA was isolated using TRIzol reagent according to the manufacturer's instructions (Invitrogen). Total RNA (5 μg) from each sample was subjected to reverse transcription using the ThermoScript RT-PCR system (Invitrogen). The cDNA product from each sample was analyzed by qPCR, using reagents from ABgene (Epsom, UK). The primer sequences used in this study are summarized in supplemental Table 1. The level of GAPDH mRNA was measured as an internal control for normalization of the data. All assays were performed in triplicate and repeated three times using an Applied Biosystems model 7000 instrument (Applied Biosystems, CA).
Transient Transfection and Luciferase Assays
The 293-hTLR4/MD2/CD14 cells and HEK293 cells were transfected with the indicated plasmids using FuGENE 6 reagent (Roche Applied Science). For the 293-hTLR4/MD2/CD14 cells (2 × 106), 3 μg of reporter plasmid (pELAM1-Luc or IFN-β-Luc) was delivered. The pELAM1-Luc plasmid was described previously (13). The IFN-β-Luc (−125) plasmid (provided by Dr. Nien-Jung Chen, National Yang-Ming University) contains the IFN-β promoter followed by firefly luciferase. For HEK293 cells (2 × 105 in 24-well plates), the pELAM1-Luc plasmid (100 ng) was introduced together with various combinations of plasmids expressing TLR4 (50 ng), MD-2 (125 ng), or CD14 (125 ng). The luciferase assays were carried out as described previously (13).
Statistical Analysis
Continuous data were compared using the Mann-Whitney U test, and categorical data were compared using Fisher's exact test. All of the statistical analyses were performed with SAS/STAT 8e (SAS Institute, Cary, NC). All of the p values were two-tailed. The results are shown as the means ± S.E. from three independent experiments. A p < 0.05 was considered statistically significant for all experiments.
RESULTS
CCL-34 Induces Macrophage-mediated Cancer Cell Death via an iNOS-dependent Pathway
To examine whether CCL-34 exhibits tumor cytotoxicity, murine macrophage RAW264.7 cells were treated with vehicle, LPS (positive control), or CCL-34 for 6 h, washed with fresh medium, and co-cultured with lung cancer LLC1 cells. As shown in Fig. 1A, the percentage of dead LLC1 cells was significantly increased in the CCL-34-treated group but not in the vehicle-treated group. Notably, direct treatment of LLC1 with CCL-34 (30 μm, 24 h) did not cause any cytotoxicity (supplemental Fig. 1). To investigate whether direct contact between macrophages and cancer cells is required for this macrophage-mediated cytotoxicity, we used plate inserts to separate the macrophages and cancer cells during co-cultivation. The results showed that macrophage-mediated cytotoxicity was still significantly induced by CCL-34 (Fig. 1B). Such macrophage-mediated cytotoxicity was also observed in the MBT-2 bladder cancer cells (Fig. 1B). Furthermore, the CCL-34-treated primary peritoneal macrophages also induced significant cytotoxicity on LLC1 cells (Fig. 1C). The tumor-associated macrophages can be classified as anti-tumor (M1) or pro-tumor (M2) macrophages, which respectively express distinct chemokines, cytokines, and other immune-modulating molecules (21–23). We therefore performed microarray analyses of vehicle- and CCL-34-treated RAW264.7 cells and cross-referenced our data with the gene list described by previous publications (21, 23). Interestingly, 12 M1-associated genes (il6, nos, tnf, nf-kb1, ccl4, ccl2, ccl5, mmp9, cxcl11, ccl3, tlr2, and cxcl10) were up-regulated in CCL-34-treated RAW264.7 cells, but only three M2-associated genes (msr1, cd36, and il10) were induced (Fig. 1D). In addition, a similar gene expression profile was observed in primary peritoneal macrophages treated with CCL-34 (supplemental Fig. 2). These data suggest that CCL-34-treated macrophages exhibit M1-associated features, which supports our observations that CCL-34-activated macrophages can induce cancer cell cytotoxicity.
FIGURE 1.

CCL-34 induces the macrophage-mediated cytotoxicity of cancer cells. A, DiOC18-labeled LLC1 cells were co-cultured with drug-treated RAW264.7 cells, followed by PI staining and flow cytometry analysis. B, RAW264.7 cells were treated with vehicle or drugs for 6 h, washed, transferred into culture plate inserts, and co-cultured with LLC1 or MBT-2 cells for 24 h. The percentage of dead LLC1/MBT-2 cells was measured using PI staining and flow cytometry. C, peritoneal macrophages were isolated and treated according to the same protocol described in B to measure their cytotoxicity to LLC1 cells. *, p < 0.05 versus control groups. D, RAW264.7 cells were treated with vehicle (0.1% DMSO) and CCL-34 (30 μm) for 6 h. The gene expression profile was analyzed by microarray analysis using the protocol described under “Experimental Procedure.” The differentially expressed genes were cross-referenced with M1 and M2 microphage-associated genes described previously (21, 23).
We next investigated the molecular mechanisms underlying macrophage-mediated cytotoxicity. Macrophages activated by CCL-34 could produce anti-cancer-associated molecules such as NO and TNF-α (supplemental Fig. 3) (13). Notably, macrophage-mediated cytotoxicity was significantly repressed by l-NMMA (an inhibitor for iNOS synthase) (Fig. 2A) but was not affected by the addition of Enbrel (a TNF-α inhibitor) (supplemental Fig. 4). Taken together, these findings suggest that CCL-34-activated macrophages induce cancer cell death via iNOS-dependent but TNF-α-independent pathways.
FIGURE 2.

CCL-34-treated macrophages induce apoptosis of LLC1 via an iNOS-dependent pathway. A, RAW264.7 cells, in the absence or presence of l-NMMA, were treated with vehicle or CCL-34 for 6 h and washed and then their cytotoxicity to LLC1 cells was measured using the protocol described in Fig. 1B. B, LLC1 cells were treated according to the protocol described in A and analyzed using TUNEL assays. C, LLC1 cells were treated with the protocol described in A and analyzed for their caspase 3 activities. D, LLC1 cells, in the absence or presence of z-VAD-FMK for 1 h, were co-cultured with vehicle- or CCL-34-treated RAW264.7 cells following the protocol described in Fig. 1B. z, benzyloxycarbonyl; FMK, fluoromethyl ketone. The percentage of dead LLC1 cells was measured using PI staining and flow cytometry. *, p < 0.05 versus vehicle-treated group; †, p < 0.05 versus CCL-34-treated group.
CCL-34-activated Macrophages Induced Apoptosis of Cancer Cells
TUNEL assays were carried out to determine whether such cytotoxicity on LLC1 cells occurred through apoptosis, and the results indicated that CCL-34-treated macrophages induced more apoptosis than vehicle-treated macrophages (Fig. 2B). Consistent with this observation, the caspase-3 activity in LLC1 cells co-incubated with CCL34-activated macrophages was higher than in those co-cultured with vehicle-treated macrophages (Fig. 2C). Moreover, both the apoptotic population and the induction of caspase-3 activity induced by CCL-34 were significantly diminished upon l-NMMA treatment (Fig. 2, B and C). Notably, the CCL-34 induced macrophage-mediated cytotoxicity was significantly reduced by benzyloxycarbonyl-VAD-fluoromethyl ketone, a pan-caspase inhibitor (Fig. 2D and supplemental Fig. 5). These findings demonstrated that CCL-34 triggers the macrophage-mediated cytotoxicity of LLC1 cells through induction of the apoptotic pathway.
We also determined whether CCL-34-treated primary splenocytes induce cytotoxicity on LLC1 cells. SCM from primary splenocytes after various treatments was collected and incubated with the LLC1 cells. The results showed that SCM from CCL-34-treated splenocytes had significant cytotoxic effect on LLC1 cells (Fig. 3). Collectively, the above data demonstrated that CCL-34 could elicit anticancer immunity in vitro.
FIGURE 3.
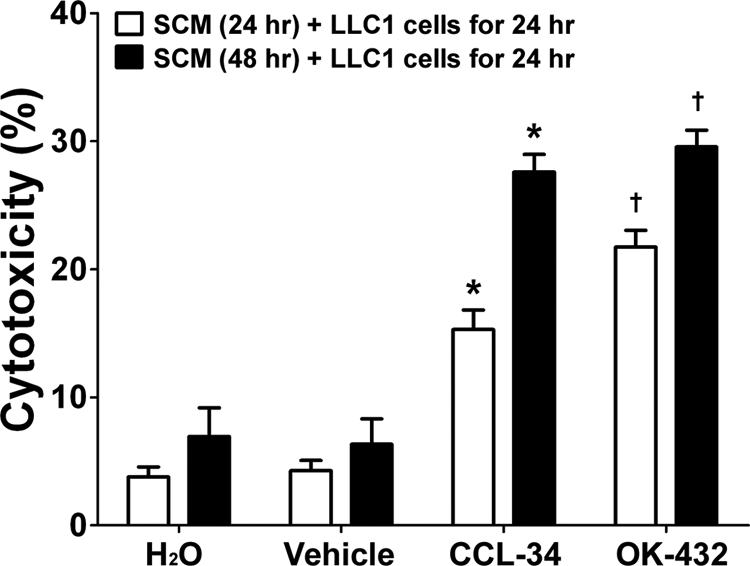
CCL-34 increases SCM-mediated cytotoxicity of LLC1 cells. Conditioned medium from splenocytes under various treatments for 24 or 48 h (SCM) was incubated with LLC1 cells using the protocol described above. The viability of the LLC1 cells was measured using PI staining and flow cytometry. *, p < 0.05 versus vehicle-treated group. †, p < 0.05 versus H2O-treated group.
CCL-34 Exhibits Anticancer Activity via TLR4-dependent Pathways in Vivo
In vivo anticancer activities of CCL-34 were next examined in wild-type (C3H/HeN) or TLR4-defective (C3H/HeJ) mice inoculated with bladder tumor cells (MBT-2). Twenty four days after inoculation, the mean tumor volumes in C3H/HeN mice that received intraperitoneal injections of CCL-34 and OK-432 (positive control) were significantly smaller than in the control groups. However, there were no differences among the C3H/HeJ mice receiving different treatments (Fig. 4A). Moreover, the survival rates of the C3H/HeN mice treated with CCL-34 were higher than the groups treated with vehicle, but there was no significant difference among the survival rates of C3H/HeJ mice receiving various treatments (data not shown). CCL-34 treatment had no influence on body weight (data not shown). Consistent with the above observation, the numbers of apoptotic cells were significantly increased in the tumors in CCL-34-treated C3H/HeN mice but not in C3H/HeN mice receiving vehicle or C3H/HeJ mice from the different treatment groups (Fig. 4B; supplemental Fig. 6). Overall, our data indicate that CCL-34 elicits anticancer activity in vivo in a TLR4-dependent manner.
FIGURE 4.

CCL-34 delays tumor growth, increases survival rate, and induces the expression of cytokines and chemokines in a TLR4-dependent manner. A, top panel, the experimental scheme for measuring the anticancer activity of CCL-34. Left panel, the mean tumor volumes of tumor-bearing C3H/HeN mice after drug treatments (n = 7). Right panel, the mean tumor volumes of tumor-bearing C3H/HeJ mice (n = 5). B, apoptotic cell death in tumors was detected by TUNEL assays on day 28. C, expression of IFN-γ, IL-12, and TNF-α in the tumors of mice under different treatments was measured by ELISA. * indicates p < 0.05. D, expression of chemokines (CXCL10 and CXCL9) in the tumors of mice under different treatments was measured by quantitative RT-PCR. * indicates p < 0.05.
CCL-34 Induces the Production of Cytokines and Chemokines as Well as Accumulation of Immune Cells at Tumor Sites
To reveal the molecules involved in CCL-34-mediated tumor suppression, the levels of Th1 cytokines associated with anticancer activity, including IFN-γ, IL-12, and TNF-α, were measured in tumor-bearing mice under different treatments. As shown in Fig. 4C, IFN-γ and IL-12 levels within the tumors of CCL-34-treated C3H/HeN mice were significantly higher than in tumors of mice that received vehicle. However, TNF-α levels were not increased at the tumor sites. Moreover, these treatments did not cause cytokine induction in C3H/HeJ mice. These data suggest that IFN-γ and IL-12 may contribute to the induction of anticancer activity mediated by CCL-34.
We next determined whether CCL-34 could stimulate the expression of IFN-γ downstream chemokines (CXCL9 and CXCL10) and enhance the recruitment of tumor-infiltrating immune cells. As shown in Fig. 4D, the expression of CXCL9 and CXCL10 was increased in tumors from C3H/HeN mice treated with CCL-34 but not in tumors from mice treated with saline or vehicle, and the expression remained unchanged in tumors from C3H/HeJ mice regardless of the treatment. Notably, tumor-infiltrating cells were found in C3H/HeN but not in C3H/HeJ mice (Fig. 5A). Furthermore, qRT-PCR showed that CD11b (a monocyte marker), CD11c (a dendritic cell marker), CD4 and CD8 (markers for T cells) were elevated in tumors of CCL-34-treated C3H/HeN mice but not in vehicle-treated C3H/HeN mice or in C3H/HeJ receiving various treatments (Fig. 5B). Together, these data demonstrated CCL-34 could induce the recruitment of immune cells to tumor sites in TLR4 functional mice.
FIGURE 5.

Recruitment of immune cells into tumor sites of CCL-34-treated tumor-bearing mice. The tumor-bearing C3H/HeN or C3H/HeJ mice were treated according to the experimental scheme shown in Fig. 4A. On day 28, the tumors were harvested and analyzed for detection of tumor-infiltrating leukocytes by H&E staining (A) and for expression of leukocyte markers by qRT-PCR (B). The arrows indicate infiltrating leukocytes. * represents p < 0.05.
Effect of CCL-34 Analogs on Activation of TLR4-downstream Molecules
To investigate whether the sugar moiety in CCL-34 is crucial for its activity, a series of saccharide-modified analogs of CCL-34 were synthesized (Fig. 6A). A HEK293 cell line (293-hTLR4/MD2/CD14) constitutively expressing human TLR4, MD-2, and CD14 was employed to examine the effects of the CCL-34 analogs on molecules downstream of TLR4 (NF-κB and IL-8). As shown in Fig. 6B, CCL-34-S2, CCL-34-S3, CCL-34-S13, and CCL-34-S14 induced comparable NF-κB activity to CCL-34, whereas β-CCL-34 did not induce NF-κB activation. Notably, these four compounds did not activate NF-κB in HEK293 cells transfected with the same reporter plasmid (data not shown). In addition, these CCL-34 analogs also could induce IL-8 production (Fig. 6B). In summary, the substitution of galactose in CCL-34 with glucose (CCL-34-S3 and CCL-34-S14) or fucose (CCL-34-S13) did not significantly alter its activity. However, removing the galactose (CCL-34-S19) or changing the orientation of the anomeric glycosidic bond (β-CCL-34) in CCL-34 completely abolished its activity.
FIGURE 6.

Structures and activities of the glycoside analogs of CCL-34. A, structures of the CCL-34 analogs tested in this study. B, effects of CCL-34 and its analogs on the induction of NF-κB activity and IL-8 production. HEK-293/TLR4/CD14/MD2 cells were transfected with the pELAM-1 plasmid for 24 h before treatment with vehicle or CCL-34 analogs. The NF-κB activity (at 6 h) and IL-8 secretion (at 24 h) were measured as described previously (13).
Although no analogs exhibited higher bioactivity than CCL-34 in these assays, CCL-34-S3 and CCL-34-S14, in which the anomeric glycosidic bond is connected to the α- or β-position of glucose, respectively, retain compatible activities to CCL-34. Particularly, the β-glycosidic bond of CCL-34-S14 is easier to be synthesized than the α-glycosidic bond, and thus it can be produced with a much higher yield than CCL-34. We further characterized CCL-34-S14 and CCL-34-S3 in the THP-1 human monocytes and found both could activate proinflammatory cytokines downstream of TLR4, including TNF-α and IL-8 (Fig. 7A). The induction of TNF-α in THP-1 cells by these compounds was significantly abrogated by the TLR4-neutralizing antibody (Fig. 7B). Moreover, induction of IFN-β expression by CCL-34-S14 and CCL-34-S3 was also observed (Fig. 7C). Together, these results demonstrate that both CCL-34-S14 and CCL-34-S3 can stimulate TLR4 activation in a mechanism similar to CCL-34.
FIGURE 7.

TLR4-activating ability and anticancer activity of selected CCL-34 analogs. A, cytokine induction by selected CCL-34 analogs. After THP-1 cells were treated with vehicle (0.1% DMSO) or drug (10 μm) for 6 h, the culture medium was collected to measure cytokine production using ELISA for TNF-α (left panel) and IL-8 (right panel). * means p < 0.05 versus vehicle-treated groups. B, TNF-α secretion by THP-1 cells treated with various drugs (10 μm), in the absence or presence of the TLR4-neutralizing antibody, was determined by ELISA. *, p < 0.05. C, left panel, the HEK-293/TLR4/CD14/MD-2 cells were transfected with a reporter plasmid containing the IFN-β promoter (−125) for 24 h, treated with various drugs (poly(I:C) 3 μg/ml; CCL-34-related compounds 30 μm) for 6 h, and analyzed using luciferase assays. *, p < 0.05 versus control groups. Right panel, the THP-1 cells were treated with vehicle, CCL-34, or CCL-34-S14 for 6 h. The expression of IFN-β was measured by qRT-PCR, as described above. * means p < 0.05 versus vehicle-treated group. D, macrophage-mediated cytotoxicity of LLC1 cells induced by the indicated compounds (LPS, 100 ng/ml; CCL-34-related compounds, 30 μm) was investigated as described in Fig. 1B. *, p < 0.05 versus control groups. E, tumor-bearing C3H/HeN or C3H/HeJ mice were treated with CCL-34 or CCL-34-S14 using the same protocol described in Fig. 4A, and the tumor volumes in the treated mice were measured accordingly (n = 5 in each group). *, p < 0.05 versus vehicle-treated groups.
Anticancer Activity of CCL-34 Analogs
The anticancer activity of the selected CCL-34 analogs was further investigated using macrophage-mediated cancer cell cytotoxicity assays. As shown in Fig. 7D, macrophages treated with CCL-34-S14 or CCL-34-S3 had a significant cytotoxic effect in LLC1 cells. Notably, several analogs incapable of activating TLR4-dependent signaling (Fig. 6B), such as β-CCL-34, CCL-34-S7, and CCL-34-S9, also failed to induce cancer cell death in the cytotoxicity assays. Therefore, the TLR4-activating ability of these compounds correlates well with their anticancer effects in vitro.
The in vivo anticancer effect of CCL-34-S14 was further investigated. As shown in Fig. 7E, the mean tumor volumes of C3H/HeN mice receiving CCL-34-S14 were smaller than those in mice receiving vehicle. However, the in vivo anticancer activity of CCL-34-S14 was lower than that of CCL-34. Consistent with this observation, CCL-34-S14 did not significantly increase the survival rates of tumor-bearing C3H/HeN and C3H/HeJ mice (data not shown).
DISCUSSION
A major goal for cancer immunotherapy is to induce effective immunity against malignant cancer phenotypes. In this study, we demonstrate that a synthetic glycolipid, CCL-34, can induce anticancer immunity in a TLR4-dependent manner, revealing a novel compound with therapeutic potential for cancer immunotherapy.
CCL-34 induces macrophage-mediated and SCM-mediated cytotoxicity on cancer cells (Figs. 1 and 3). Because the induction of macrophage-mediated cytotoxicity was independent of physical contact (Fig. 1B), we believe that both types of cytotoxicity mainly resulted from secreted molecules released by CCL-34-treated cells. Indeed, macrophages treated with CCL-34 could produce cytotoxic molecules, such as TNF-α and NO (supplemental Fig. 3) (13). The inhibition of iNOS significantly, but not completely, suppressed CCL-34-induced macrophage-mediated cytotoxicity, indicating that NO plays an important role in such cytotoxicity, and other molecules might also be involved (Fig. 2A). Nevertheless, the involvement of TNF-α has been excluded (supplemental Fig. 4). We further demonstrated such cancer cell death was via induction of NO-dependent apoptotic pathways (Fig. 2, B–D). It has been shown that NO derived from TLR4-mediated macrophage activation can induce apoptosis of cancer cells, and the mechanisms of NO-mediated apoptosis involves accumulation of tumor suppressor p53, decreased expression of Bcl-2, and activation of caspases (5, 24). Interestingly, MBT-2 cancer cells co-cultured with CCL-34-treated macrophage had reduced Bcl-2 expression (supplemental Fig. 7). Thus, we speculate that NO released from CCL-34-activated macrophages may lead to the inhibition of Bcl-2 expression in cancer cells, resulting in apoptosis of these cells.
Anticancer activity of CCL-34 was also demonstrated in vivo (Fig. 4). The tumor-suppressive events induced by CCL-34 only occurred in C3H/HeN but not in C3H/HeJ mice, suggesting that the anticancer activity of this compound is TLR4-dependent and is not via direct cytotoxic effects on cancer cells (Fig. 4, A and B). The induction of cytokines (IFN-γ and IL-12) and chemokines (CXCL9 and CXCL10) (Fig. 4, C and D) and the recruitment of tumor-infiltrating leukocytes (Fig. 5) in tumors of CCL-34-treated C3H/HeN mice further suggest that the anticancer activity of CCL-34 is highly associated with the enhancement of the host immune response. Furthermore, our observation that the reduction of tumor growth continues for a few weeks after the last drug treatment also suggests the activation of adaptive immunity by CCL-34 treatment. IFN-γ is a hallmark Th1 cytokine and a key mediator of the anticancer immunity initiated by TLR4 agonists (7, 9, 25). CXCL9 and CXCL10 are chemokines known to be downstream of IFN-γ and can induce the infiltration of immunocompetent cells into tumors, leading to tumor cell apoptosis (26, 27). Based on our data, the elevation of IFN-γ in tumors is also a crucial step underlying the anticancer effect of CCL-34. Presumably, the elevation of IFN-γ upon CCL-34 treatment could increase the expression of CXCL10 and CXCL9, which subsequently attracts CD11b+ cells, CD11c+ dendritic cells, and CD8+ T cells into tumors, eventually causing tumor regression. In addition, the activation of dendritic cells by TLR4 agonists has been demonstrated to be a critical step required for induction of the host anti-tumor response (7, 9, 12). The induction of IL-12 and the elevation of CD11c expression in tumors indicate the involvement of dendritic cells in the anticancer activity of CCL-34 in vivo (Figs. 4C and 5B). Indeed, we recently found that CCL-34 could trigger dendritic cell maturation in a TLR4-dependent manner (data not shown). Collectively, our data indicate that CCL-34 elicits an anticancer effect by stimulating TLR-4-dependent immune responses.
Our previous study demonstrated that the lengths of the lipid chains in CCL-34 were crucial for its TLR4-activating effect and that the optimal number of carbons for the lipid amine and acyl chains is 10 to 11 and 12, respectively (13, 14). In this study, the role of the monosaccharide in CCL-34 was further investigated. Removal of the sugar moiety in CCL-34 completely abolished its activity (CCL-34-S19), but changing galactose to other sugars either insignificantly or moderately affected its activity (Fig. 6B). The functions of the sugar moiety in TLR4-activating glycolipids have never been investigated previously. Our observation, for the first time, provides evidence that the presence of the sugar moiety is essential for the activity of a TLR4 glycolipid agonist, which presumably may occur in other TLR4 glycolipid agonists. Such a finding expands the molecular basis of TLR4-activating glycolipids and provides structural information for future development of therapeutic TLR4 agonists.
It is known that MD-2 mediates ligand recognition and TLR4 activation by forming an MD-2-TLR4-ligand complex (2). Recent structural analysis of the MD-2-TLR4-LPS complex further demonstrates that most of the lipid chains in LPS directly interact with hydrophobic pockets of MD-2 for efficient TLR4 activation (3, 28). The involvement of MD-2 in TLR4 activation by lipid A and its analogs has been demonstrated, and the acyl lengths in lipid A appear to be crucial for effective TLR4 activation (29). Interestingly, we found that CCL-34-mediated TLR4 activation also required MD2 (supplemental Fig. 8). Based on current data, we speculate that the lipid moiety in CCL-34 may interact with MD-2 and that the presence of monosaccharide might be crucial for maintaining the spatial arrangement of lipid chains for such an interaction to occur. Notably, the phosphate structure in LPS has been suggested to contribute to receptor multimerization by forming an ionic interaction with positive charges in MD-2 and TLR4 protein (3, 28). How the absence of a phosphate group in CCL-34 affects its interactions with TLR4 and MD-2 merits further investigation.
The structure-and-activity relationship analysis identified CCL-34-S3 and CCL-34-S14, which substitute galactose with glucose but contain the lipid linkage in α- or β-orientation respectively, have comparable TLR4-activating and anticancer activity to CCL-34 in vitro (Fig. 7, A–D). However, CCL-34-S14 had a weaker tumor-suppressive effect in vivo (Fig. 7E), which might result from the easier hydrolysis and metabolism of the glucose in CCL-34-S14 (30). Alternatively, the β-linkage in CCL-34-S14 might affect its activity or stability in vivo.
Several TLR4 activators, such as Bacillus Calmette-Guerin, OK-432, monophosphoryl lipid A, and OM-174, have been reported to exhibit strong adjuvant and/or anticancer activity in animal models (12, 25, 31–33). However, most of them have clinical limitations. OK-432 and Bacillus Calmette-Guerin have been occasionally used as cancer treatments, but both are bacterial derivatives with multiple effects. Monophosphoryl lipid A, derived from lipid A by chemical purification (34), is a clinically effective vaccine adjuvant, but its anticancer activity has not been fully demonstrated. OM-174, a water-soluble diphosphorylated triacylated lipid A, is also purified from Escherichia coli. As compared with the above agonists, the well defined and simple structure of CCL-34 as well as its easy synthesis without microbial contamination make CCL-34 an appealing TLR4 activator. This study demonstrates the anticancer immunity induced by CCL-34 in vivo is compatible to that by OK-432 (Figs. 4 and 5). Notably, CCL-34 treatments under such experimental conditions did not significantly decrease body weights of treated mice (data not shown). We also found CCL-34 treatment (30 μm, 24 h) did not induce cytotoxicity of primary cells such as bone marrow cells and mouse embryo fibroblasts, indicating the low toxicity of this compound. Collectively, our study reveals the immunotherapeutic potential of CCL-34 for cancer treatment. Because TLR4 activation has recently been shown to enhance the effectiveness of chemotherapy and radiotherapy (5, 10, 33), the potential clinical application of CCL-34 for use in combination treatment warrants further investigation.
Supplementary Material
Acknowledgments
We are grateful for the technical support provided by Dr. Ting-Fen Tsai, Dr. Chun-Ming Chen, and the Microarray and Gene Expression Analysis Core Facility of the National Yang-Ming University VGH Genome Research Center. We also thank Dr. Joseph Lipsick for suggestions on the manuscript.
This work was supported in part by National Science Council Grants NSC 99-2320-B-010-009-MY3 and 98-2323-B-007-001 and Ministry of Education, Aim for the Top University Plan, Grant 95A-C-D01-PPG-03.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–8 and Table 1.
- TLR4
- Toll-like receptor 4
- iNOS
- inducible nitric-oxide synthase
- SCM
- splenocyte-conditioned medium
- PI
- propidium iodide
- qRT-PCR
- quantitative RT-PCR
- l-NMMA
- NG-monomethyl-l-arginine.
REFERENCES
- 1. Kanzler H., Barrat F. J., Hessel E. M., Coffman R. L. (2007) Nat. Med. 13, 552–559 [DOI] [PubMed] [Google Scholar]
- 2. Miyake K. (2004) Trends Microbiol. 12, 186–192 [DOI] [PubMed] [Google Scholar]
- 3. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., Lee J. O. (2009) Nature 458, 1191–1195 [DOI] [PubMed] [Google Scholar]
- 4. Akira S., Uematsu S., Takeuchi O. (2006) Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 5. Garay R. P., Viens P., Bauer J., Normier G., Bardou M., Jeannin J. F., Chiavaroli C. (2007) Eur. J. Pharmacol. 563, 1–17 [DOI] [PubMed] [Google Scholar]
- 6. Cluff C. W. (2010) Adv. Exp. Med. Biol. 667, 111–123 [DOI] [PubMed] [Google Scholar]
- 7. Okamoto M., Oshikawa T., Tano T., Ohe G., Furuichi S., Nishikawa H., Ahmed S. U., Akashi S., Miyake K., Takeuchi O., Akira S., Moriya Y., Matsubara S., Ryoma Y., Saito M., Sato M. (2003) J. Natl. Cancer Inst. 95, 316–326 [DOI] [PubMed] [Google Scholar]
- 8. Akazawa T., Masuda H., Saeki Y., Matsumoto M., Takeda K., Tsujimura K., Kuzushima K., Takahashi T., Azuma I., Akira S., Toyoshima K., Seya T. (2004) Cancer Res. 64, 757–764 [DOI] [PubMed] [Google Scholar]
- 9. Lee C. H., Wu C. L., Shiau A. L. (2008) Clin. Cancer Res. 14, 1905–1912 [DOI] [PubMed] [Google Scholar]
- 10. Apetoh L., Ghiringhelli F., Tesniere A., Obeid M., Ortiz C., Criollo A., Mignot G., Maiuri M. C., Ullrich E., Saulnier P., Yang H., Amigorena S., Ryffel B., Barrat F. J., Saftig P., Levi F., Lidereau R., Nogues C., Mira J. P., Chompret A., Joulin V., Clavel-Chapelon F., Bourhis J., André F., Delaloge S., Tursz T., Kroemer G., Zitvogel L. (2007) Nat. Med. 13, 1050–1059 [DOI] [PubMed] [Google Scholar]
- 11. Pfannes S. D., Müller B., Körner S., Bessler W. G., Hoffmann P. (2001) J. Leukocyte Biol. 69, 590–597 [PubMed] [Google Scholar]
- 12. D'Agostini C., Pica F., Febbraro G., Grelli S., Chiavaroli C., Garaci E. (2005) Int. Immunopharmacol. 5, 1205–1212 [DOI] [PubMed] [Google Scholar]
- 13. Hung L. C., Lin C. C., Hung S. K., Wu B. C., Jan M. D., Liou S. H., Fu S. L. (2007) Biochem. Pharmacol. 73, 1957–1970 [DOI] [PubMed] [Google Scholar]
- 14. Huang L. D., Lin H. J., Huang P. H., Hsiao W. C., Reddy L. V., Fu S. L., Lin C. C. (2011) Org. Biomol. Chem. 9, 2492–2504 [DOI] [PubMed] [Google Scholar]
- 15. Takashiba S., Van Dyke T. E., Amar S., Murayama Y., Soskolne A. W., Shapira L. (1999) Infect. Immun. 67, 5573–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ho M. Y., Tang S. J., Ng W. V., Yang W., Leu S. J., Lin Y. C., Feng C. K., Sung J. S., Sun K. H. (2010) Cancer Sci. 101, 2411–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen S. A., Tsai M. H., Wu F. T., Hsiang A., Chen Y. L., Lei H. Y., Tzai T. S., Leung H. W., Jin Y. T., Hsieh C. L., Hwang L. H., Lai M. D. (2000) Clin. Cancer Res. 6, 4381–4388 [PubMed] [Google Scholar]
- 18. Zhao W., Wang L., Zhang L., Yuan C., Kuo P. C., Gao C. (2010) J. Biol. Chem. 285, 20452–20461 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Liao H. F., Chen Y. J., Yang Y. C. (2005) Life Sci. 77, 400–413 [DOI] [PubMed] [Google Scholar]
- 20. Kamat A. M., Tharakan S. T., Sung B., Aggarwal B. B. (2009) Cancer Res. 69, 8958–8966 [DOI] [PubMed] [Google Scholar]
- 21. Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., Locati M. (2004) Trends Immunol. 25, 677–686 [DOI] [PubMed] [Google Scholar]
- 22. Martinez F. O., Gordon S., Locati M., Mantovani A. (2006) J. Immunol. 177, 7303–7311 [DOI] [PubMed] [Google Scholar]
- 23. Bingle L., Brown N. J., Lewis C. E. (2002) J. Pathol. 196, 254–265 [DOI] [PubMed] [Google Scholar]
- 24. Umansky V., Schirrmacher V. (2001) Adv. Cancer Res. 82, 107–131 [DOI] [PubMed] [Google Scholar]
- 25. Lee C. F., Chang S. Y., Hsieh D. S., Yu D. S. (2004) Cancer Gene Ther. 11, 194–207 [DOI] [PubMed] [Google Scholar]
- 26. Mei K., Wang L., Tian L., Yu J., Zhang Z., Wei Y. (2008) J. Exp. Clin. Cancer Res. 27, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harlin H., Meng Y., Peterson A. C., Zha Y., Tretiakova M., Slingluff C., McKee M., Gajewski T. F. (2009) Cancer Res. 69, 3077–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim H. M., Park B. S., Kim J. I., Kim S. E., Lee J., Oh S. C., Enkhbayar P., Matsushima N., Lee H., Yoo O. J., Lee J. O. (2007) Cell 130, 906–917 [DOI] [PubMed] [Google Scholar]
- 29. Stöver A. G., Da Silva Correia J., Evans J. T., Cluff C. W., Elliott M. W., Jeffery E. W., Johnson D. A., Lacy M. J., Baldridge J. R., Probst P., Ulevitch R. J., Persing D. H., Hershberg R. M. (2004) J. Biol. Chem. 279, 4440–4449 [DOI] [PubMed] [Google Scholar]
- 30. Spadiut O., Nguyen T. T., Haltrich D. (2010) J. Agric. Food Chem. 58, 3465–3471 [DOI] [PubMed] [Google Scholar]
- 31. Okamoto M., Furuichi S., Nishioka Y., Oshikawa T., Tano T., Ahmed S. U., Takeda K., Akira S., Ryoma Y., Moriya Y., Saito M., Sone S., Sato M. (2004) Cancer Res. 64, 5461–5470 [DOI] [PubMed] [Google Scholar]
- 32. Persing D. H., Coler R. N., Lacy M. J., Johnson D. A., Baldridge J. R., Hershberg R. M., Reed S. G. (2002) Trends Microbiol. 10, S32–37 [DOI] [PubMed] [Google Scholar]
- 33. Udagawa M., Kudo-Saito C., Hasegawa G., Yano K., Yamamoto A., Yaguchi M., Toda M., Azuma I., Iwai T., Kawakami Y. (2006) Clin. Cancer Res. 12, 7465–7475 [DOI] [PubMed] [Google Scholar]
- 34. Thompson B. S., Chilton P. M., Ward J. R., Evans J. T., Mitchell T. C. (2005) J. Leukocyte Biol. 78, 1273–1280 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.