

Abstract
Transport of cargoes by kinesin-1 is essential for many cellular processes. Nevertheless, the number of proteins known to recruit kinesin-1 via its cargo binding light chain (KLC) is still quite small. We also know relatively little about the molecular features that define kinesin-1 binding. We now show that a bipartite tryptophan-based kinesin-1 binding motif, originally identified in Calsyntenin is present in A36, a vaccinia integral membrane protein. This bipartite motif in A36 is required for kinesin-1-dependent transport of the virus to the cell periphery. Bioinformatic analysis reveals that related bipartite tryptophan-based motifs are present in over 450 human proteins. Using vaccinia as a surrogate cargo, we show that regions of proteins containing this motif can function to recruit KLC and promote virus transport in the absence of A36. These proteins interact with the kinesin light chain outside the context of infection and have distinct preferences for KLC1 and KLC2. Our observations demonstrate that KLC binding can be conferred by a common set of features that are found in a wide range of proteins associated with diverse cellular functions and human diseases.
Keywords: kinesin-1, KLC, microtubule tranport, motif, vaccinia virus
Introduction
Microtubule-mediated transport of a wide range of cellular cargoes by kinesin family molecular motors is essential for the normal functioning of eukaryotic cells (Hirokawa and Noda, 2008; Verhey and Hammond, 2009). Deficiencies in microtubule transport are associated with a number of diseases, including neurological disorders such as amyotrophic lateral sclerosis, Alzheimer's disease and Huntington's disease (Salinas et al, 2008; Hirokawa et al, 2009). Microtubule-based transport can also have devastating cellular consequences, when it is used inappropriately by viruses to reach their cellular replication sites, as well as providing the means for them to leave their host at the later stages of infection (Dohner and Sodeik, 2005; Greber and Way, 2006; Radtke et al, 2006; Dodding and Way, 2011).
Phylogenetic analysis reveals that the human and mouse genomes encode 45 different kinesin motors that are grouped into 14 families (Hirokawa et al, 2009; Verhey and Hammond, 2009). Each kinesin contains a conserved motor domain, the position of which is generally indicative of directionality: N-terminal and C-terminal motor domain kinesins are plus and minus end directed, respectively. The majority of kinesin motors exist as homodimers due to the presence of extensive coiled-coil regions (Hirokawa et al, 2009; Verhey and Hammond, 2009). In contrast to most of the motors, kinesin-1, which is also known as KIF5, also has two associated light chains (KLC). The motor containing heavy chain is encoded by three distinct genes giving rise to the closely related KIF5A, KIF5B and KIF5C. KIF5B appears to be ubiquitously expressed, while KIF5A and KIF5C are neuronal specific. Each KIF5 heavy chain homodimer associates with two copies of KLC1 or KLC2, which are expressed in most cell types (Hirokawa et al, 2009; Verhey and Hammond, 2009).
Kinesin-1, which was the first plus end directed microtubule motor to be identified, is involved in a wide variety of cell processes by virtue of its ability to bind and transport many different types of cellular cargoes including vaccinia and Herpes viruses (Greber and Way, 2006; Hirokawa and Noda, 2008; Verhey and Hammond, 2009; Dodding and Way, 2011). Cargoes can bind directly to specific sites on the kinesin-1 heavy chain. They also associate with kinesin-1 via the tetratricopeptide repeats (TPRs) in the C-terminal half of the kinesin light chain, which in turn is bound to the heavy chain via its N-terminal heptad repeats. Given the importance of kinesin-1-mediated transport in so many different cellular processes, it is surprising that the number of known interacting proteins is still quite small (Hirokawa and Noda, 2008; Verhey and Hammond, 2009). In addition, relatively little is known about the molecular features that confer the ability of a protein to bind kinesin-1. Understanding how viruses recruit kinesin-1 offers a great opportunity to obtain insights into the molecular basis of motor recruitment.
During the vaccinia virus replication cycle, newly formed intracellular enveloped virus (IEV) particles are transported towards the cell periphery on microtubules by kinesin-1, prior to their fusion with the plasma membrane (Geada et al, 2001; Hollinshead et al, 2001; Rietdorf et al, 2001; Ward and Moss, 2001a, 2001b). The recruitment of kinesin-1 to vaccinia virus is dependent on the integral IEV membrane protein A36 (Rietdorf et al, 2001). In the absence of A36, newly assembled IEV remain largely restricted to their perinuclear site of assembly and are deficient in their cell-to-cell spread (Rietdorf et al, 2001; Ward and Moss, 2001a). The ability of residues 81–111 of A36 to interact directly with the TPRs of the kinesin-1 light chain suggests that it is responsible for the recruitment of kinesin-1 to the virus (Ward and Moss, 2004). An interaction between A36 and the TPRs of KLC2 has also been confirmed in infected cells using fluorescence resonance energy transfer approaches (Jeshtadi et al, 2010). Interestingly, residues 81–111 of A36 contains a short WD motif that is related to the KLC-binding site in Calsyntenin-1 (CSTN1) (Konecna et al, 2006; Araki et al, 2007; Dodding and Way, 2009; Morgan et al, 2010) (Figure 1A). Recent observations have also shown that a closely related WE motif in the cayman ataxia protein, Caytaxin (ATCAY) and γ-BAR also known as Gadkin (AP1AR) can interact with KLC (Aoyama et al, 2009; Schmidt et al, 2009). Curiously, A36 also contains a related WE motif at residues 64–65 of A36 (Morgan et al, 2010) (Figure 1A). In this paper we set out to determine whether these two tryptophan-based motifs in A36 function to recruit kinesin-1 to mediate viral transport to the cell periphery.
Figure 1.

The WD/E motifs in A36 are required for efficient virus transport to the cell periphery. (A) Schematic representation of A36 highlighting its TM domain and positions of the WE and WD motifs at residues 64–65 and 97–98, respectively. (B) Representative immunofluorescence images showing actin tail formation (red) by the indicated A36–YFP WE/WD mutant viruses (green) at 8 h post-infection. Scale bar=10 μm. The graph shows quantification of the number of actin tails per cell for the indicated viruses. A P-value of <0.05 and <0.001 is indicated with * and ***, respectively, while error bars represent standard error of the mean from 50 cells in three independent experiments. (C) Representative immunofluorescence images showing the spread of the indicated recombinant A36–YdF–YFP viruses (green) from their perinuclear site of assembly to the cell periphery at 8 h post-infection compared with the ΔA36R virus. Scale bar=10 μm. The different viruses are co-labelled with anti-A27 (red), which detects both IMV and IEV, the latter of which appear yellow as they contain both A27 and A36–YdF–YFP. The graph shows quantification of viral spread to the cell periphery at 8 h post-infection based upon fluorescence intensity measurements of extracellular B5, an IEV-specific protein, detected with a monoclonal antibody in non-permeabilized cells. A P-value of <0.05 and <0.001 is indicated with * and ***, respectively. Error bars represent standard error of the mean from 50 cells in three independent experiments.
Results
Viral spread is dependent on the bipartite tryptophan motifs in A36
To examine whether the WD and WE motifs in A36 are required to recruit kinesin-1, we generated recombinant viruses in which residues 64 and 65 (WE) or 97 and 98 (WD) of A36 were replaced by alanine in an A36-YFP background (Figure 1A). For simplicity, these recombinant viruses are referred to as WE/AA, WD/AA and WEWD/AAAA (both mutants combined), while the non-mutated A36 at these two positions is designated as wild type (WT). To assess the ability of these recombinant viruses to undergo microtubule-based transport, we initially examined their ability to induce actin tails, as this is indicative of IEV reaching the cell periphery and fusing with the plasma membrane. We found that mutation of the WE motif in A36 resulted in a small but significant reduction in the average number of actin tails per cell at 8 h post-infection (Figure 1B). Mutation of the WD motif had a more dramatic impact, resulting in an ∼50% reduction in the average number of actin tails. Combining the WE and WD mutations into a single recombinant virus resulted in ∼90% reduction in the average number of actin tails formed per cell when compared with the WT (Figure 1B).
While measuring actin tail formation is indicative of microtubule transport, it actually represents an indirect measurement. To circumvent this problem and rule out any possible impact of the WE/WD mutations on the ability of A36 to induce actin tails, we mutated the A36 WD and WE motifs in an A36–YdF–YFP viral background (Rietdorf et al, 2001; Arakawa et al, 2007a). Working in this viral background eliminates any confusion between A36-dependent actin and microtubule transport, as the A36–YdF virus is deficient in actin-based motility (Rietdorf et al, 2001; Ward and Moss, 2001a). Quantification of the amount of B5 exposed on the cell surface as a readout for the number of IEV reaching the cell periphery at 8 h post-infection reveals that mutation of the WE motif in the A36–YdF background has a similar impact on viral spread as seen using actin tail formation as a read out (Figure 1C). In contrast, quantification of the amount of B5 exposed on the infected cell surface reveals that mutation of the WD motif actually results in a far larger reduction in the number of IEV reaching the cell periphery than was inferred from actin tail formation (Figure 1C). By 11 h post-infection, however, IEV did accumulate in the cell periphery (see Figure 3C), suggesting that they were still capable of moving on microtubules albeit less efficiently. When both motifs were mutated in the A36–YdF background, we observed that the WEWD/AAAA virus remained at its perinuclear site of assembly and did not spread to the cell periphery. The inability of the WEWD/AAAA virus to spread from its site of assembly is reminiscent of that observed for a virus that lacks A36 (ΔA36R) (Rietdorf et al, 2001; Ward and Moss, 2001a) (Figure 1C).
To confirm and extend our analysis beyond the single cell level, we examined the consequences of mutating the WD/E motifs on viral spread in confluent monolayers of BSC1 cells during plaque formation (Figure 2A). We found that the size of plaques induced by the different viruses mirrored the results obtained at the single cell level with the WD/AA and WEWD/AAAA viruses forming significantly smaller plaques (Figure 2A and B). Our observations demonstrate that both the WD and WE motifs contribute to efficient IEV movement to the cell periphery and viral spread.
Figure 2.

The A36 WD/E motifs are important for the cell-to-cell spread of the virus. (A) Representative fluorescence images of plaques produced by the WT, WE/AA, WD/AA and WDWE/AAAA viruses in the A36–YdF–YFP background at 48 h post-infection. Scale bar=100 μm. (B) Quantification of plaque sizes produced by the indicated viruses at 48 h post-infection. A P-value of <0.001 is indicated with *** and error bars represent s.e.m. from 30 plaques.
Kinesin-1 recruitment is dependent on the bipartite tryptophan motifs in A36
Our data reveal that the A36 WD motif is the major determinant in promoting viral spread to the cell periphery (Figures 1C and 2B). To obtain insights into the slower spread of the WD/AA virus to the cell periphery compared with the WT virus, we performed live cell imaging of infected cells (Figure 3A). Viruses were unambiguously identified as IEV from the presence of both A36–YdF–YFP and RFP–A3, a viral core protein. Analysis of all three viruses moving in linear trajectories reveals that they move with similar speed distributions that average out at 0.88±0.04, 0.86±0.04, 0.88±0.04 μm/s for WT, WE/AA and WD/AA, respectively (). The WD/AA virus, however, moved with significantly shorter linear runs (2.58±0.14 μm for WD/AA compared with 6.44±0.37 and 6.23±0.38 for WT and WE/AA, respectively) (Figure 3A). We considered that this shorter run length may reflect differences in the ability of the WD/AA virus to recruit and retain kinesin-1. Consistent with this, we found that, in contrast to the WT and WE/AA viruses, the WD/AA virus does not retain KLC1 or KLC2 when it accumulates in the cell periphery at 11 h post-infection (Figure 3B; Supplementary Figure S1). We were also only able to detect low levels of KLC2 on WD/AA virus that had moved away from their perinuclear site of assembly at 8 h post-infection (Supplementary Figure S2). We were unable to detect association of KLC1 or KLC2 with the WEWD/AAAA virus at 8 or 11 h post-infection (Figure 3B; Supplementary Figure S1). In parallel to our immunofluorescence analysis, pull-down assays reveal, that mutation of the WD motif reduced the ability of the cytoplasmic domain of A36 (residues 24–221) to associate with KLC1, KLC2 or the kinesin-1 heavy chain (Figure 3C). Simultaneous mutation of both WE and WD motifs eliminated any detectable binding of A36 to kinesin-1. In contrast, these mutations did not appear to affect the association of A36 with F12 (Figure 3C).
Figure 3.

Both WD/E motifs contribute to KLC1/2 recruitment and viral spread. (A) Histograms and scatter plot showing the distribution of the speeds and run lengths of virus moving towards the cell periphery in cells infected with A36–YdF–YFP (black) as well as WE/AA (green) and WD/AA (red) viruses for 8 h. Inserts show the average values for both parameters and error bars represent s.e.m. for 65 virus particles in four independent experiments. (B) Immunofluorescence images showing RFP–KLC2 is not associated with the WD or WEWD viruses (yellow arrows) after they have fused with the plasma membrane at the cell periphery at 11 h post-infection. Pink arrows highlight association of RFP–KLC2 with DAPI-positive WT and WE viruses. Scale bar=10 μm. (C) Analysis of GST pull-down experiments showing the effect of mutating WD/WE motifs on the ability of the cytoplasmic domain of A36 to interact with HA–KLC1 (left panel), HA–KLC2 (middle panel) or endogenous kinesin-1 detected with anti-Kif5B antibody (right panel). Mutation of the WD motif weakens the association of A36 with KLC1/2 and kinesin-1, while mutation of both motifs abrogates all binding.
Calsyntenin can functionally replace A36 to mediate viral transport
Given the apparent similarity between the WD motif of A36 and those in Calsyntenin, we wondered whether the KLC-binding region of Calsyntenin could substitute for the cytoplasmic domain of A36 to promote microtubule-based transport of vaccinia. We reasoned that if this were the case, then the spread of IEV to the cell periphery would provide a powerful assay to investigate the mechanisms regulating kinesin-1 recruitment. To explore this possibility, we generated a recombinant virus in which residues 879–971 of Calsyntenin, containing both WD motifs were fused to the transmembrane (TM) domain of A36 (A36–TM–CSTN–GFP) (Figure 4A). We found that the TM domain of A36 was sufficient to drive incorporation of the GFP fusion protein into IEV (S3). Moreover, residues 879–971 of Calsyntenin were able to mediate transport of IEV towards the cell periphery in the absence of the cytoplasmic domain of A36 (Figure 4B and C). Immunofluorescence analysis revealed that the A36–TM–CSTN–GFP virus particles were able to recruit KLC1 or KLC2, consistent with their movement to the cell periphery (Figure 4D). The ability of this region of Calsyntenin to mediate IEV transport is dependent on its WD-based motifs (Figure 4B and C). However, in contrast to A36, we found that mutation of either of the WD motifs effectively resulted in loss of viral transport to the cell periphery (Figure 4B and C). Quantification of the size of plaques formed by the A36–TM–CSTN–GFP virus, at 2 days post-infection confirmed that the WD motifs of Calsyntenin can enhance viral spread relative to a virus expressing GFP fused to the TM domain of A36 (A36–TM–GFP) (Figure 4E and F). The ability of calsyntenin to functionally replace A36 opens up the possibility of using spread of IEV to the cell periphery as an assay to assess the ability of a protein to recruit kinesin-1.
Figure 4.

Bipartite tryptophan motifs of Calsyntenin can functionally replace A36. (A) Schematic representation of hybrid protein in which the region following the A36 TM domain (residue 32 onwards) is replaced with residues 879–971 of Calsyntenin. (B) Images showing the spread of the indicated recombinant A36–TM–CSTN1–GFP mutant viruses detected with anti-A27 (virus) from their perinuclear site of assembly to the cell periphery at 11 h post-infection. Scale bar=10 μm. (C) The graph shows quantification of extracellular B5 labelling for the indicated viruses. A P-value of <0.001 is indicated with *** and error bars represent s.e.m. from 50 cells in three independent experiments. (D) Images showing recruitment of RFP-tagged KLC1 and KLC2 to the WT A36–TM–CSTN1–GFP virus (GFP) co-labelled with DAPI to identify the viral genome. Scale bars=2 μm. (E) Representative fluorescence images of plaques produced by the A36–YdF–YFP, A36–TM–GFP and A36–TM–CSTN1–GFP viruses at 48 h post-infection. Scale bar=100 μm. (F) Quantification of plaque sizes produced by the indicated viruses at 48 h post-infection. A P-value of <0.001 is indicated with *** and error bars represent s.e.m. from 30 plaques.
Identification of potential KLC-binding partners in the human genome
We reasoned that it may be possible to use the presence of a bipartite WD/E motif to identify additional potential KLC-binding proteins that could be tested in our IEV transport assay. Examination of Caytaxin (ATCAY) and Gadkin, which both contain a WE kinesin-1-binding motif (Aoyama et al, 2009; Schmidt et al, 2009), reveals the presence of a second WE/Q motif outside of any structured domain (Figure 5A). The WD/E motifs in A36 and Calsyntenin-1 (CSTN1) are also outside of obvious structured domains and the linker sequence contains no additional tryptophan residues. It is also noticeable that the acidic or hydrophilic residues D, E, N or Q preceded the tryptophan residue at the –1 position in these four KLC-binding proteins (or their human homologues and paralogues in other species) (Figure 5A). Likewise, the residue at the +2 position was frequently a D, E, N or Q residue (Konecna et al, 2006; Araki et al, 2007; Aoyama et al, 2009; Dodding and Way, 2009; Schmidt et al, 2009). Using this relatively limited information, we searched the RefSeq database of expressed human proteins for the presence of four combinations of bipartite motifs: W(DENQ)—W(DENQ)(DENQ); W(DENQ)—(DENQ)W(DENQ); W(DENQ) (DENQ)—W(DENQ) and (DENQ)W(DENQ)—W(DENQ) to identify potential kinesin-1 interacting proteins (Figure 5A). We identified >1360 unique genes encoding proteins containing these bipartite motifs within 100 residues of each other and lacking an intervening tryptophan residue. This number was reduced to 516 potential candidates by removing proteins in which the motifs fall within predicted TM or conserved structured domains. In addition, sequences lacking close homologues in the HomoloGene database were also removed (Sayers et al, 2010). The final list of 460 proteins (Supplementary Table SI) contains a number of proteins, which are associated with human diseases including LDLRAP1, RASSF8 and ATF6 (Yu et al, 2008), while others, such as BSDC1 and FAM63B have no known function (Figure 5B and C). In addition to identifying proteins closely related to those used to define the consensus, we also found the known KLC interacting proteins Dynein intermediate Chain-1 (DYNC1I1), Huntingtin-associated protein (HAP1) and SKIP (PLEKHM2) (Ligon et al, 2004; Boucrot et al, 2005; McGuire et al, 2006) (Figure 5B and C). Encouragingly, the bipartite tryptophan motifs fall within their known KLC-binding regions (Boucrot et al, 2005; McGuire et al, 2006; Dumont et al, 2010).
Figure 5.

Potential bipartite tryptophan KLC1/2-binding motifs are found in a wide range of proteins. (A) Sequence alignment showing the WD/E motifs in proteins previously shown to interact with KLC (bold text indicates experimental evidence). W–W indicates the number of residues separating the two tryptophans. A consensus based on these four proteins as well as conserved amino-acid substitutions is shown. A schematic representation of the set of patterns that was used to search the RefSeq database is shown. (B) Sequence alignment of bipartite tryptophan motifs in candidate KLC-binding proteins. SKIP, DYNC1I1 and HAP1 are known KLC-binding proteins. W–W indicates the number of residues between the two tryptophan residues highlighted in red. (C) Schematic representation of the location of bipartite tryptophan motifs in the proteins in (B) relative to other domains (from the Pfam database).
KLC-binding candidates support vaccinia transport and the spread of infection
To begin to verify our bioinformatic analysis and test the utility of our virus transport assay, we examined whether the bipartite tryptophan motifs in SKIP would mediate transport of IEV to the cell periphery in the absence of the cytoplasmic domain of A36. We found that residues 157–264 of SKIP fused to the TM domain of A36 (A36–TM–SKIP–GFP) was targeted to IEV and sufficient to drive their transport to the cell periphery (Figure 6A and B; Supplementary Figure S3). Furthermore, this activity is dependent on the presence of both WD/E motifs, although the WD motif seems to play the more important role (Figure 6A and B). Infection of cells with recombinant viruses encoding A36–TM–ATF6 or A36–TM–BSDC1 reveals that the bipartite tryptophan motifs of ATF6 and BSDC1 were also capable of promoting IEV transport to the cell periphery in the absence of A36 (Figure 6C and D). Quantification of the size of plaques formed by the different recombinant viruses reveals that the bipartite tryptophan motifs of SKIP, ATF6 and BSDC1 were also capable of enhancing viral spread in a cell monolayer when compared with a virus lacking the cytoplasmic domain on A36 (Figure 6E).
Figure 6.

Multiple bipartite tryptophan motifs can rescue vaccinia transport. (A) Images showing the spread of the indicated recombinant A36–TM–SKIP–GFP viruses detected with anti-A27 (virus) from their perinuclear site of assembly to the cell periphery at 11 h post-infection. Scale bar=10 μm. (B) Quantification of viral spread to the cell periphery for the indicated recombinant A36–TM–SKIP–GFP viruses. A P-value of <0.001 is indicated with *** and error bars represent s.e.m. from 50 cells in three independent experiments. (C) Representative immunofluorescence showing the spread of A36–TM–ATF6–GFP and A36–TM–BSDC1–GFP viruses detected with anti-A27 (virus) from their perinuclear site of assembly to the cell periphery at 11 h post-infection. Scale bar=10 μm. (D) Quantification of viral spread to the cell periphery as detected by staining for extracellular B5 in the absence of permeabilization for the indicated recombinant viruses. A P-value of <0.001 is indicated with *** and error bars represent s.e.m. from 50 cells in three independent experiments. (E) Quantification of plaque sizes produced by the indicated recombinant viruses at 48 h post-infection. A P-value of <0.001 is indicated with *** and error bars represent s.e.m. from 30 plaques. (F) Quantification of transient rescue of viral spread to the cell periphery at 10 h post-infection. A P-value of <0.001 relative to the A36–TM control is indicated with *** and error bars represent s.e.m. from 50 cells in three independent experiments.
To extend our analysis further, we examined whether transient expression of the bipartite WD/E motifs from additional candidate KLC interacting proteins could rescue peripheral directed transport of IEV in the absence of A36 when fused to the TM domain of A36. Candidates were chosen to cover a diverse range of cellular functions and/or diseases (Figure 5C). We found that the bipartite WD/E motifs from FAM63B, LDLRAP1, PRKAG3, PARC (CUL9), RASSF8 and RIC3 rescue IEV transport to the cell periphery when expressed in ΔA36R-infected cells (Figure 6F). Our analysis reveals that a variety of subtly different bipartite WD/E motifs are capable of functionally replacing the cytoplasmic domain of A36 to promote virus transport to the cell periphery.
WD/E containing proteins have different preferences for KLC1 and KLC2
A36, as well as the bipartite WD motifs of Calsyntenin, are capable of recruiting both KLC1 and KLC2 to IEV particles (Figures 3C and 4D; Supplementary Figure S1). Immunofluorescence analysis of cells infected with the recombinant virus expressing A36–TM–SKIP reveals that the bipartite WD/E motifs of SKIP are also able to recruit KLC1 and KLC2 (Figure 7A). The A36–TM–BSDC1 virus on the other hand was able to recruit KLC1 but KLC2 was only weakly detected on the virus. In contrast, the A36–TM–ATF6 virus were only able to recruit KLC2 (Figure 7A). To examine this further and confirm that the full-length proteins, and not just their WD/E motifs on vaccinia particles, are capable of interacting with the kinesin-1 light chain, we performed KLC1 and KLC2 pull-down assays in non-infected cells (Figure 7B and C). Consistent with our immunofluorescence analysis of cells infected with the A36–TM–ATF6 virus, we found that full-length ATF6 can interact with KLC2 but not KLC1 (Figure 7B). In contrast, all the other proteins tested were able to interact with KLC1 to varying extents. The ability to bind KLC2, however, was restricted to SKIP, BSDC1 and PRKAG3 (Figure 7C). Our data clearly demonstrate that the full-length candidate proteins we tested are capable of interacting with KLC1 and/or KLC2 outside the context of infection.
Figure 7.

(A) Images showing recruitment of RFP-tagged KLC1 and KLC2 to the different recombinant virus particles. Purple arrows indicate virus particles (IEV) that show good recruitment of the indicated KLC isoform. The white arrows highlight weak recruitment of KLC2 by the A36–TM–BSDC1–GFP virus and yellow arrows indicate A36–TM–ATF6–GFP virus that cannot recruit KLC1. Scale bars=2 μm. (B) Western blot analysis of co-immunoprecipitation with the indicated antibodies reveals that FLAG–ATF6 associates with HA–KLC2 but not HA–KLC1 (left panel). In contrast, GFP–PARC interacts with HA–KLC1 but not HA–KLC2 (right panel). (C) Western blot analysis of co-immunoprecipitation experiments between GFP-tagged SKIP, BSDC1, FAM63B, LDLRAP1, PRKAG3, RASSF8 and RIC3 with either HA–KLC1 (left panel) or HA–KLC2 (right panel) reveals the candidate proteins can bind both isoforms or have distinct binding preferences for KLC1 or KLC2.
Discussion
The A36 WE and WD motifs are needed to recruit kinesin-1 and promote IEV transport
During vaccinia infection, newly assembled IEV enhance their spread to the cell periphery by recruiting kinesin-1 and undergoing microtubule-dependent transport (Geada et al, 2001; Hollinshead et al, 2001; Rietdorf et al, 2001; Ward and Moss, 2001a, 2001b). The recruitment of kinesin-1 to the virus is dependent on the IEV integral membrane protein A36, which interacts with the TPRs of the kinesin-1 light chain (Rietdorf et al, 2001; Ward and Moss, 2004; Jeshtadi et al, 2010). The molecular basis for this interaction had not been clearly defined but was known to involve a direct interaction between the KLC TPR domain and residues 81–111 of A36 (Ward and Moss, 2004). Consistent with this, deletion of this region of A36 results in loss of transport of IEV to the cell periphery (Ward et al, 2003). Our analysis using a panel of recombinant A36 mutant viruses has now demonstrated that two tryptophan-based motifs in A36 are required to recruit kinesin-1 and promote IEV transport to the cell periphery.
Our initial analysis of the ability of the different A36 mutant viruses to undergo microtubule transport was based on their ability to induce actin tails, which only form after IEV fuse with the plasma membrane (Hollinshead et al, 2001; Rietdorf et al, 2001; Ward and Moss, 2001a; Newsome et al, 2004). Mutation of the A36 WE motif results in a small but significant reduction in actin tail formation at 8 h post-infection. However, when the WE mutant was combined with the WD mutant, which by itself results in an ∼50% reduction, we found there was a dramatic ∼90% reduction in actin tail formation. These data suggest that while both motifs participate in IEV transport, the role of the WE motif is only unmasked by the absence of the WD, which plays the dominant role in kinesin-1 recruitment.
Our conclusions differ starkly from a recent study of IEV transport using transient expression of A36 WE and WD mutants in ΔA36 virus-infected cells (Morgan et al, 2010). This study also supported the notion that disruption of the A36 WD motif results in a reduction in actin tail formation, albeit ∼20% rather than the 50% we observed. The same study, however, found that the loss of the WE has no effect on actin tail formation, even when combined with the WD mutant (Morgan et al, 2010). The difference between the two studies may reflect using recombinant viruses as compared with transiently expressing the A36 mutants for extended periods from a synthetic promoter. In addition, we performed our quantification at 8 h when infected cells are less motile rather than 12 h (Sanderson et al, 1998; Valderrama et al, 2006). It is likely that virus-induced cell migration will bring some IEV to the plasma membrane, where they will be able to fuse and induce actin tails in the absence of previous microtubule transport. This notion is consistent with the ability of the WEWD mutant, which cannot recruit kinesin-1 to induce a small number of actin tails. These actin tails are presumably formed by IEV that assemble close to the ventral surface of the cell and fuse with the plasma membrane independently of microtubule-based transport.
To circumvent the potential problems in using actin tail formation as a read out, we examined the impact of the WE and WD mutations on IEV spread in A36–YdF–YFP-infected cells. Using this viral background allows unambiguous assessment of microtubule transport, as the A36–YdF virus is deficient in actin-based motility (Rietdorf et al, 2001; Ward and Moss, 2001a). Quantification of IEV spread in single-infected cells as well as cell monolayers (plaque size) in the absence of actin tail formation confirmed our initial observations that the A36 WD motif is the major determinant in promoting viral transport. Interestingly, the WD but not the WE motif is within the previously described KLC TPR-binding region of A36 that is required for IEV transport to the cell periphery (Ward et al, 2003; Ward and Moss, 2004). Our live cell imaging reveals that the reduction in viral spread in the absence of the A36 WD motif is due to a dramatic reduction in average run length rather than speed of IEV movement. We suggest that this change reflects a reduced affinity for the kinesin-1, as the WD/AA virus is unable to retain KLC1/2 when it accumulates in the cell periphery at longer times post-infection. Consistent with this, it is harder to detect KLC2 on the WD/AA virus at 8 h post-infection (see Supplementary Figure S1) and pull-down assays show that loss of the WD motif leads to reduced KLC binding.
A36 versus F12-mediated kinesin-1 recruitment
The only other viral protein that has been proposed to play a direct role in microtubule-mediated transport of IEV particles is F12 (van Eijl et al, 2002). Loss of F12 leads to an accumulation of IEV at their site of assembly and a dramatic reduction in actin tail formation (van Eijl et al, 2002; Dodding et al, 2009). F12 is also associated with IEV moving on microtubules but is absent from virus-induced actin tails (van Eijl et al, 2002; Dodding et al, 2009). It has now been suggested that F12 is a viral KLC-like mimic that may link kinesin-1 to IEV (Morgan et al, 2010). This hypothesis is based upon the primary sequence of F12, which when combined with structural predictions suggest that the protein contains 14 TPR-like motifs. TPRs, which consist of two small anti-parallel α helices connected by a shorter linker associate with each other to form protein interaction surfaces in a wide range of proteins (D’Andrea and Regan, 2003). If F12 can associate with the kinesin-1 heavy chain, as would be expected of a KLC mimic, then it must do so by a novel mechanism that is fundamentally different to the heptad repeat interaction of KLC1/2. More importantly, there is still no published evidence that F12 can directly associate with the kinesin-1 heavy chain. Interestingly, F12 also contains a single WD motif between its 12th and 13th TPR-like repeats, which is important for its function (Morgan et al, 2010). This may point to it being a possible KLC binding or regulatory protein. Unfortunately, there is no evidence that F12 can bind KLC. F12 has, however, been shown to interact directly with A36 and E2, the latter of which is predicted to contain 15 TPR-like repeats (Johnston and Ward, 2008; Dodding et al, 2009; Morgan et al, 2010). Consistent with them working as a complex, we previously found that loss of F12 or E2 leads to a similar defect in membrane wrapping during IEV assembly prior to their microtubule transport (Dodding et al, 2009). It is possible that F12 together with E2 functions to inhibit A36-mediated kinesin-1 recruitment until IEV formation is complete, as its binding site overlaps with that of KLC on A36 (Ward and Moss, 2004; Johnston and Ward, 2008). Blocking kinesin-1 recruitment during IEV formation would prevent premature transport of incompletely wrapped virions. However, it is not immediately clear why the F12–E2 complex remains associated with IEV undergoing microtubule transport, if its primary function is to inhibit A36-mediated KLC recruitment. F12 is, however, significantly less abundant on IEV particles than A36. This suggests that the F12–E2 complex only associates with a subset of A36 molecules. The task ahead is now to understand whether the F12–E2 complex bound to A36 is inhibiting or modulating either the recruitment or activation of kinesin-1 during IEV transport.
A kinesin-1 light chain binding signature is found in a diverse range of proteins
We have shown that the bipartite WD motifs of calsyntenin can functionally replace A36 to mediate kinesin-1 recruitment and IEV transport. The ability of calsyntenin to rescue IEV transport strongly supports the notion that it is A36 rather than F12 that is principally involved in recruiting kinesin-1 to the virus. During our study, we noticed that there was a second tryptophan-based motif in Caytaxin and Gadkin (γ-BAR), the only two other proteins known to contain a functional KLC-binding tryptophan motif (Aoyama et al, 2009; Schmidt et al, 2009). On the basis that KLC binding might be defined by the presence of bipartite tryptophan motifs, we searched the human proteome for proteins that also contained two related motifs. Our search pattern identified 460 proteins that have the potential to bind KLC1/2. This list will undoubtedly contain false positives and is unlikely to be exhaustive. However, it did identify bipartite tryptophan motifs in the kinesin-1-binding regions of SKIP, HAP1 and the Dynein intermediate chain (Boucrot et al, 2005; McGuire et al, 2006; Dumont et al, 2010). Using our virus transport assay, we confirmed that the bipartite tryptophan-based motifs of SKIP are capable of recruiting kinesin-1 and conveying IEV to the cell periphery. We were also able to show that bipartite tryptophan motifs from ATF6, BSDC1, FAM63B, LDLRAP1, PARC, PRKAG3, RASSF8 and RIC3 could rescue IEV transport to the cell periphery in the absence of A36. In contrast, we found that single clones containing the bipartite tryptophan motifs of DAB2, EMP2AIP, GRP, HYLS1, Nostrin and PCM1 did not rescue transport of the virus to the cell periphery in the absence of A36. However, it is hard to draw an unambiguous conclusion from this negative data, as the ability to recruit kinesin-1 is likely to be highly dependent on how the protein is presented on the surface of the virus, which will vary depending on the position of the fusion to the TM domain of A36. Consistent with this notion, pull-down assays demonstrated that Nostrin can interact with KLC1.
We were also able to confirm that full-length ATF6, BSDC1, FAM63B, LDLRAP1, PARC, PRKAG3, RASSF8 and RIC3 could interact with KLC. An interaction with kinesin-1 fits well with the cellular function of RIC3, which plays an important role in the transport of neurotransmitter receptors (Millar, 2008; Alexander et al, 2010). The connection between kinesin-1 and ATF6 or PARC is more intriguing. ATF6, a sensor of unfolded proteins in the ER, is transported from the ER to the Golgi apparatus where its cytoplasmic domain is liberated to yield a transcription factor that regulates gene expression to minimize ER stress (Kimata and Kohno, 2011). It will be interesting to determine whether any of these transport steps is kinesin-1 dependent, given the ATF6 KLC bipartite binding motifs are in its cytoplasmic domain. PARC binds p53 to maintain its cytoplasmic localization (Nikolaev et al, 2003). It is possible that kinesin-1/PARC interaction plays a role in regulating the nucleo-cytoplasmic localization of p53. It is also curious that 53BP1, which binds p53 and regulates its nuclear import in a Dynein-dependent fashion (Lo et al, 2005) also contains a predicted bipartite tryptophan KLC-binding motif (see Supplementary Table SI). The ability of protein complexes and membrane compartments to simultaneously bind kinesin-1 and Dynein is emerging as a common theme for bidirectional microtubule transport (Welte, 2004; Encalada et al, 2011). Indeed, LDLRAP one of our confirmed KLC-binding proteins is also found in complex with Dynein (Lehtonen et al, 2008). LDLRAP also has a functional AP-2 clathrin adaptor binding sequence in the linker between the bipartite tryptophan motif (Mishra et al, 2005). Curiously, the second WE motif of Gadkin (γ-BAR) has also been shown to be involved in the recruitment of the AP-1 clathrin adaptor complex (Schmidt et al, 2009). This raises the question whether the WD motif in F12 can also interact with a clathrin adaptor, given the defects we see in Golgi organization and wrapping of IEV in the absence of F12 in vaccinia-infected cells (Dodding et al, 2009).
Kinesin light chain binding and specificity
We found that the relative role of each tryptophan motif in promoting virus transport varied depending upon the protein examined. In the case of A36, both tryptophan motifs are capable of promoting virus transport although the second tryptophan motif (WD) appears to be the more effective. In the case of SKIP, it is the first tryptophan motif (WD) that is most effective. In contrast, in calsyntenin both WD motifs were required to rescue IEV transport. Our search of the human proteome for potential KLC interacting proteins was based on the presence of a bipartite tryptophan motif outside of any structured domain. However, our data clearly demonstrate that a single tryptophan motif can be sufficient to recruit kinesin-1 and mediate viral transport. This suggests that in the right context, proteins with a single tryptophan motif may be capable of interacting with kinesin-1. Consistent with this, Smad2 from Xenopus, which can interact with KLC2 (Batut et al, 2007), contains a single DQWDT motif that is conserved in human and found in a unique loop extending from the MH1 domain (Shi et al, 1998). A single conserved motif (TEWNE) is also found in the linker between the RhoGAP and SH2 domains of the p85 subunit of PI3K, which has recently been shown to associate with KLC2 (Amato et al, 2011). There is also a single WD motif in the KLC-binding protein JIP3, which is also known as Sunday Driver in Drosophila (Bowman et al, 2000; Verhey et al, 2001). However, this WD motif is outside the region responsible for binding KLC (Hammond et al, 2008). Curiously, however, the KLC-binding region of JIP3 does contain a closely related sequence, DEWSD.
What determines whether a single tryptophan motif is dominant and sufficient to bind KLC in isolation will depend on the binding affinity of the individual motif as well as additional sequences outside of the motif. It is also highly likely that clustering of KLC-binding proteins will impose steric constraints and/or avidity effects that will influence kinesin-1 recruitment. We assume it is a combination of these factors, including how the protein is presented on the surface of the virus that leads to the variation in ability of our KLC-binding proteins to rescue viral spread in the absence of A36. Another important consideration is the stoichiometric relationship between KLC-binding motifs and individual KLC molecules, which will vary depending on the oligomeric state of the binding protein. Possible scenarios include interaction of both tryptophan motifs with the same KLC or two different KLC molecules, which may be associated with either a single or two different kinesin-1 heavy chain dimers. Clearly, KLC binding is likely to be both protein and context dependent. It is also not immediately obvious what determines the ability of an individual bipartite tryptophan motif to associate with KLC1 and/or KLC2. Understanding this specificity, which is likely to have important regulatory consequences will require analysis of the binding preferences of additional bipartite tryptophan motifs as well as structural determination of KLC1 and KLC2 bound to their binding partners we have defined here.
Summary
We have shown that a bipartite tryptophan-based motif in A36 is required for kinesin-1-dependent transport of the virus to the cell periphery. Moreover, we found that this bipartite kinesin-1-binding motif is not unique to A36 and Calsyntenin, but is found in a diverse range of proteins, a number of which are associated with human diseases. Our list greatly expands the potential number of cellular roles for kinesin-1. It also suggests that kinesin-1 recruitment can occur via many specific cargo-associated proteins in addition to using set of common adaptor or scaffolding proteins. The presence of so many potential KLC-binding proteins also raises issues of how kinesin-1 recruitment to the right cargoes is regulated in space and time. Further biochemical analysis confirming our candidates bind KLC, combined with structural approaches will ultimately provide the molecular basis of kinesin-1 recruitment and facilitate identification of additional KLC-binding proteins.
Materials and methods
Clones and mammalian expression plasmids
I.M.A.G.E. clones of BSDC1 (4812029), CSTN1 (100003893), LDLRAP (5197824), FAM63B (99021679), PRKAG3 (40005883), RASSF8 (5266519) and RIC3 (4792930) were obtained from Source BioScience (Nottingham, UK). Clones of ATF6 (11975) and PARC (20937) were supplied by Addgene (Cambridge, MA, USA). Clone KIAA0842 encoding SKIP (PLEKHM2) was obtained from the Kazusa DNA Research Institute (Japan). ATF6 was expressed in pCMV–3xFLAG-7.1 as supplied by Addgene. The open reading frames of candidate DNA sequences were amplified by PCR and cloned into the CMV promoter driven expression vector CB6 with either N- or C-terminal GFP tags using the following primer pairs.
GFP N-terminal
BSDC1 NotI FOR TATAGCGGCCGCATGGCGGAAGGGGAGGACGTGGG
BSDC1 HindIII REV TATAAAGCTTTCACTCCCAGTCCTCCCACTCTAC
LDLRAP NotI FOR TATAGCGGCCGCATGGACGCGCTCAAGTCGGCGG
LDLRAP EcoRI TATAGAATTCTCAGAAGCTGAAGAGGTCATCCTGC
RASSF8 NotI FOR TATAGCGGCCGCATGGAACTTAAAGTATGGGTGGATG
RASSF8 BamHI REV TATAGGATCCCTAATCTTTACACTCCTGC TTATC
RIC3 NotI FOR TATAGCGGCCGCATGGCGTACTCCACAGTGCAGAG
RIC3 EcoRI REV TATAGAATTCTCACTCTAAACCCTGGGGGTTACG
SKIP NotI FOR TATAGCGGCCGCATGGAGCCGGGGGAGGTGAAGG
SKIP EcoRI REV TATAGAATTCTTAGAGCTCCGTGCAGGCATCCGG CGGG
GFP C-terminal
FAM63B HindIII FOR TATAAAGCTTATGGAGAGCAGCCCCGAGAGCCTG
FAM63B EcoRI REV TATAGAATTCCAAAATAACACAGCTATTTTTTTC
PRKAG3 EcoRI FOR TATAGAATTCATGGAGCCCGGGCTGGAGCACGCAC
PRKAG3 NotI REV TATAGCGGCCGCGGCCCCGAGGGCATCGATGCC
KLC1 and KLC2 expression vectors
The open reading frames of KLC1 and KLC2 were amplified by PCR from mouse KLC1 and KLC2 templates (Rahman et al, 1998), which were provided by Professor Larry Goldstein (University of California, San Diego). The resulting PCR products were cloned into EcoRI–BamHI or NotI–EcoRI sites of pEL–HA or CB6–HA to generate vectors that drive expression of HA–KLC1 and HA–KLC2 in vaccinia-infected cells or non-infected cells, respectively. Alternatively, the PCR products were cloned into the EcoRI–BamHI or NotI–EcoRI sites of pLVX–cherry–puro or pLL3.7–RFP, respectively, to generate lentivirus vectors that drive expression of Cherry–KLC1 and RFP–KLC2. These lentivirus vectors were used to establish stable HeLa cell lines as described previously (Weisswange et al, 2009).
Generation of A36R–YdF–YFP mutant viruses
Recombinant viruses with point mutations in the bipartite tryptophan motifs of A36 were isolated as previously described using a mutated LA–A36R–YdF–YFP–RA targeting vector (Arakawa et al, 2007a). Mutations were introduced into the LA–A36R–YdF–YFP–RA targeting vector using the Stratagene site-directed mutagenesis kit using primer pairs corresponding to the forward primers A36R–W64AE65A CAACTGATAGCGAATCAGACGCAGCGGATCACTGTAGTGCTATG and A36R–W97AD98A CTGGTAGTTTAATAGCAGCCAACGAATCCAATG and their reverse complement sequences. Double mutants were made by sequential rounds of mutagenesis.
Generation of recombinant hybrid viruses
SnaBI and NotI restriction sites were introduced to the 5′ and 3′ ends of the DNA corresponding to residues 2–97 of ATF6; 376–430 of BSDC1; 879–971 of CSTN and 157–264 of SKIP by PCR with the primers.
ATF6 G2 SnaBI FOR/GATCTACGTACTGGGGAGCCGGCTGGGGTTGCCGG
ATF6 S97 NotI REV/TATAGCGGCCGCGGAGGCTGGAGAAAGTGGCT GAG
BSDC1 SnaBI K376 FOR/TATATACGTACTAAGTCTACACCCTCCAACAATGG
BSDC1 NotI E430 REV/TATAGCGGCCGCCTCCCAGTCCTCCCACTCTAC
CSTN1 SnaBI T879 FOR/TATATACGTACTACCATGCGGGATCAGGACACCGGG
CSTN1 NotI Y971 REV/TATAGCGGCCGCGTAGCTGAGGGTGGAGTCATCCCAC
SKIP A157 SnaBI FOR/TATATACGTACTGCCCCTTACCTAGACCTGGCCCCC
SKIP S264 NotI REV/TATAGCGGCCGCGCTCCTGGTGGAGGCCTTGCTGC
The resulting PCR products were cloned into the SnaBI and NotI sites of pEL–A36R–GFP (Rietdorf et al, 2001). The SnaBI site is embedded in the codons for residues 29–31, which immediately follow the TM domain of A36 (Rietdorf et al, 2001). A BglII and BamHI digest was used to release the hybrid clone (A36 TM domain and the candidate sequence) fused to GFP, which was subsequently cloned into the BamHI site of the vector used to generate the WR–ΔA36R–YL126R targeting vector (Dodding and Way, 2009). The fidelity of the targeting vectors was confirmed by sequencing prior to the isolation of recombinant viruses as described previously (Dodding and Way, 2009). The fidelity of the resulting recombinant viruses expressing candidate sequences fused to the first 31 residues of A36 under the control of the endogenous A36R promoter were confirmed by sequencing.
Recombinant hybrid viruses with point mutations in the bipartite tryptophan motifs of CSTN1 and SKIP were isolated as described above using their respective WT templates that had been mutated using the Stratagene site-directed mutagenesis kit using the following primer pairs.
CSTN1-WD893AA FOR GAAGGAGAACGAGATGGACGCAGCAGACTCTGCCCTGACCATC
CSTN1-WD964AA FOR CAGCAGCTGGAGGCAGCTGACTCCACCC
SKIP-WD207AA FOR CACCAACCTGGAGGCAGCAGACAGTGCGATTG
SKIP-WE236AA FOR CTGTACCCAGCACAGACGCAGCAGATGGAGACCTCACAG
Double mutants were made by sequential rounds of mutagenesis. The fidelity of all recombinant hybrid viruses was confirmed by sequencing.
Generation of transient pE/L hybrid expression plasmids
SnaBI and NotI restriction sites were introduced to the 5′ and 3′ ends of the DNA encoding residues 436–566 of FAM63B; 179–308 of LDLRAP; 2410–2517 of PARC; 117–210 of PRKAG3; 158–284 of RASSF8 and 203–311 of RIC3 by PCR using the following primer.
FAM63B. SnaBI Q436 FOR/TATATACGTACTCAGGAAGGAGAACTTTGTGTGTTC
FAM63B NotI Q566 REV/TATAGCGGCCGCTTGTTCCTGTTCCTGATAGTATTG
LDLRAP SnaBI K179 FOR/GATCTACGTACTAAAGAGAAGAGGGACAAAGCCAGCC
LDLRAP1 NotI F308 REV/TATAGCGGCCGCGAAGCTGAAGAGGTCATCCTGC
PARC SnaBI E2410 FOR/GATCTACGTACTGAGCTGCTCCGGCGGATCCAGGAG
PARC NotI D2517 REV/TATAGCGGCCGCGTCATAGGCCTCATCTTCATCCTC
PRKAG3 SnaBI L117 FOR/GATCTACGTACTCTCCCCTCTGACTGTACAGCCTCAGC
PRKAG3 NotI L210 REV/TATAGCGGCCGCCAGCATGGTGTCGAAGATGACTAGC
RASSF8 N158 SnaBI FOR/GATCTACGTACTAACTGCAAAACAACAGCAGATGAG
RASSF8 A284 NotI REV/TATAGCGGCCGCTGCCTCTTGAACTTCCCGTTGC
RIC3 I203 SnaBI FOR/GATCTACGTACTATTGACAGATTTTCTCCAGAG
RIC3 D311 NotI REV/TATAGCGGCCGCATCCTCGTCTTCATGAAAACAGC
The resulting PCR products were subsequently cloned into the SnaBI and NotI sites of pEL–A36R–mCherry (Arakawa et al, 2007b). The fidelity of all pEL hybrid mCherry fusions was confirmed by sequencing.
Infections, antibodies and immunofluorescence analysis
HeLa cells on fibronectin-coated coverslips or dishes were infected with the viruses described above as described previously (Arakawa et al, 2007a). The viral proteins A27 and B5 were detected by indirect immunofluorescence using the monoclonal antibodies C3 (Rodriguez et al, 1985) and 19C2 (Hiller and Weber, 1985), respectively. HA and GFP were detected in western blot analysis using the HA7 (Sigma) and 3E1 (Cancer Research UK) monoclonal antibodies, respectively. Infected cells were fixed and processed for immunofluorescene analysis as previously described (Arakawa et al, 2007b). To visualize extracellular virus, staining using the anti-B5 19C2 antibody was performed prior to permeabilization. Cells infected with the ΔA36R virus were transfected 4 h post-infection with the pEL driven clones described above and fixed as required (Frischknecht et al, 1999). Immunofluorescence images were collected using Apochromat 63/1.40 NA Oil lens or Plan-neofluar × 25 lenses (Carl Zeiss, Germany) on a Zeiss Axioplan 2 controlled by Metamorph (Molecular Devices Corporation). All figures were prepared for publication using the Adobe Photoshop and Illustrator packages (Adobe, CA, USA).
Live cell imaging and virus tracking
Images from live cells grown in 35 mm glass bottom MatTek dishes then infected for 8 h were collected using a CoolSNAP HQ cooled CCD camera (Photometrics, AZ) with 2 × 2 binning on an Axiovert 200 microscope using an Apochromat 63/1.40. NA Oil lens (Carl Zeiss, Germany) under the control of Metamorph (Molecular Devices Corporation). IEV particles were initially identified by the presence of signals from RFP-tagged A3 viral core marker and YFP-tagged A36 as described previously (Dodding et al, 2009). The signal from YFP-tagged A36 alone was subsequently acquired at 10 frames per second. Image sequences and rates of virus movement were determined using Metamorph and supplementary movies assembled and annotated using Adobe After Effects (Adobe).
Quantitative analysis of viral spread and plaque size
Viral transport to the plasma membrane was assessed by measuring the level of peripheral extracellular virus per cell. The integrated intensity levels of extracellular virus detected with anti-B5 antibody and anti-rat-conjugated secondary antibody was measured in unsaturated images taken under identical conditions. The cell periphery intensity was defined by subtracting the integrated intensity from area corresponding to the perinuclear site of viral assembly and nucleus (defined in the other channel containing either A36–YFP or the TM–candidate–GFP/Cherry fusion) from the integrated intensity of total cell area. Mean cellular background from that region was also subtracted from the total. The data are shown in arbitrary units (A.U.) for clarity. Plaque areas were measured at 48 h post-infection by examining the area fluorescence as previously described (Dodding et al, 2009).
Statistical analysis
Data are presented as mean±standard error of the mean and were analysed by ANOVA or Student's t-test using Prism 4.0. (GraphPad Software, CA). A P-value of <0.05 was considered statistically significant.
KLC1 and KLC2 pull downs
For A36, KLC1 or KLC2 pull downs, a 10-cm dish of HeLa cells at 100% confluency was infected with vaccinia lacking A36 and transfected with constructs expressing GST–A36 cytoplasmic domain (residues 24–221), GST and HA–KLC1 or HA–KLC2 under control of the vaccinia early/late promoter. At 16 h post-infection, cells were lysed in 1 ml of buffer consisting of 10 mM Tris pH 7.5, 300 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, 0.5% Triton-X 100, 10 mM sodium fluoride and 20 mM β-glycerol phosphate, 5% glycerol and a protease inhibitor cocktail for 30 min prior to centrifugation at 13 000 g for 10 min at 4 °C. The resulting supernatant was incubated with 20 μl of glutathione beads. The beads were washed 4 × with lysis buffer, boiled in SDS-loading buffer, subjected to SDS–PAGE and analysed by western blot using antibodies against HA, GST and F12 (Dodding et al, 2009). For endogenous kinesin-1 pull-down experiments, a similar protocol was used starting with 4 × 15 cm2 dishes for each sample and lysing in 4 ml of buffer. These samples were analysed by ponceau staining of the membrane and probing with an antibody directed against Kif5B (Kamal et al, 2000).
To analyse the interaction between candiate KLC-binding proteins and KLC, a 10-cm dish of HeLa cells at ∼70% confluency was co-transfected with CB6 expression vectors encoding GFP-tagged candidate kinesin-1-binding proteins and HA-tagged mouse KLC1A or KLC2. After 16 h, transfected cells were lysed in 1 ml of 10 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40 and 5% glycerol containing a protease inhibitor cocktail for 30 min prior to centrifugation at 13 000 g for 10 min at 4 °C. The resulting supernatant was incubated with 10 μl of GFP-Trap beads prewashed in lysis buffer at 4 °C for 2 h. GFP-Trap beads were pelleted by centrifugation at 2000 r.p.m. for 1 min and washed with 1 ml of lysis buffer (without detergent). Beads were washed 4 × , boiled in SDS-loading buffer, subjected to SDS–PAGE and analysed by western blot using antibodies against GFP and HA. In the case of ATF6, 5 μg of polyclonal anti-FLAG antibody (Sigma) was incubated with the cell extract for 2 h at 4 °C prior to the addition of 15 μl of protein A coated agarose beads (Invitrogen). After 1 h at 4 °C, the beads were pelleted and washed as above.
Bioinformatic analysis
Human protein sequences and reports were downloaded from the NCBI Reference Sequence (RefSeq) collection (Release 44) (Pruitt et al, 2005). Reports were scanned for the locations of predicted conserved domains identified by the NCBI Conserved Domains Database (Marchler-Bauer et al, 2009). Protein TM regions were predicted using TMHMM 2.0 from the Center for Biological Sequence Analysis (Sonnhammer et al, 1998). Links to disease information were obtained from the Human Genome Epidemiology (HuGE) encyclopedia (Yu et al, 2008).
Each sequence was searched using four Perl regular expressions representing variations on a consensus kinesin light chain interaction motif.
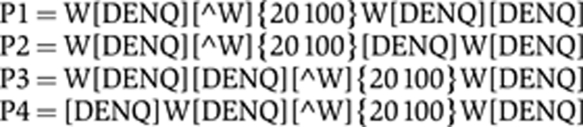 |
Motifs overlapping with a predicted TM domain or a conserved functional domain were flagged as likely false positives. Likewise, Refseq records that did not have associated homologues automatically identified within the NCBI's HomoloGene (Release 64) database (Sayers et al, 2010) were considered to be of lower quality and also flagged.
Supplementary Material
Acknowledgments
We thank Professor Larry Goldstein (University of California, San Diego) for providing mouse KLC1A and KLC2 clones. We thank Dr Jamine Abella (Way laboratory) for providing the pLVX–cherry–puro lentivirus vector. We thank the members of the Way laboratory and Dr Helen Walden for constructive comments on the manuscript.
Author contributions: MPD and MW planned the experiments, analysed the data and wrote the paper. MPD and ACH performed the experiments. RM provided the bioinformatic analysis and database searches.
Footnotes
The authors declare that they have no conflict of interest.
References
- Alexander JK, Sagher D, Krivoshein AV, Criado M, Jefford G, Green WN (2010) Ric-3 promotes alpha7 nicotinic receptor assembly and trafficking through the ER subcompartment of dendrites. J Neurosci 30: 10112–10126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato S, Liu X, Zheng B, Cantley L, Rakic P, Man HY (2011) AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science 332: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Hata S, Nakao T, Tanigawa Y, Oka C, Kawaichi M (2009) Cayman ataxia protein caytaxin is transported by kinesin along neurites through binding to kinesin light chains. J Cell Sci 122: 4177–4185 [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Cordeiro JV, Schleich S, Newsome T, Way M (2007a) The release of vaccinia virus from infected cells requires RhoA-mDia modulation of cortical actin. Cell Host Microbe 1: 227–240 [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Cordeiro JV, Way M (2007b) F11L-mediated inhibition of RhoA-mDia signaling stimulates microtubule dynamics during vaccinia virus infection. Cell Host Microbe 1: 213–226 [DOI] [PubMed] [Google Scholar]
- Araki Y, Kawano T, Taru H, Saito Y, Wada S, Miyamoto K, Kobayashi H, Ishikawa HO, Ohsugi Y, Yamamoto T, Matsuno K, Kinjo M, Suzuki T (2007) The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J 26: 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batut J, Howell M, Hill CS (2007) Kinesin-mediated transport of Smad2 is required for signaling in response to TGF-beta ligands. Dev Cell 12: 261–274 [DOI] [PubMed] [Google Scholar]
- Boucrot E, Henry T, Borg JP, Gorvel JP, Meresse S (2005) The intracellular fate of Salmonella depends on the recruitment of kinesin. Science 308: 1174–1178 [DOI] [PubMed] [Google Scholar]
- Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS (2000) Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell 103: 583–594 [DOI] [PubMed] [Google Scholar]
- D’Andrea LD, Regan L (2003) TPR proteins: the versatile helix. Trends Biochem Sci 28: 655–662 [DOI] [PubMed] [Google Scholar]
- Dodding MP, Newsome TP, Collinson LM, Edwards C, Way M (2009) An E2-F12 complex is required for IEV morphogenesis during vaccinia infection. Cell Microbiol 11: 808–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodding MP, Way M (2009) Nck- and N-WASP-dependent actin-based motility is conserved in divergent vertebrate poxviruses. Cell Host Microbe 6: 536–550 [DOI] [PubMed] [Google Scholar]
- Dodding MP, Way M (2011) Coupling viruses to dynein and kinesin-1. EMBO J 30: 3527–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohner K, Sodeik B (2005) The role of the cytoskeleton during viral infection. Curr Top Microbiol Immunol 285: 67–108 [DOI] [PubMed] [Google Scholar]
- Dumont A, Boucrot E, Drevensek S, Daire V, Gorvel JP, Pous C, Holden DW, Meresse S (2010) SKIP, the host target of the Salmonella virulence factor SifA, promotes kinesin-1-dependent vacuolar membrane exchanges. Traffic 11: 899–911 [DOI] [PubMed] [Google Scholar]
- Encalada SE, Szpankowski L, Xia CH, Goldstein LS (2011) Stable Kinesin and Dynein assemblies drive the axonal transport of Mammalian prion protein vesicles. Cell 144: 551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht F, Moreau V, Röttger S, Gonfloni S, Reckmann I, Superti-Furga G, Way M (1999) Actin based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature 401: 926–929 [DOI] [PubMed] [Google Scholar]
- Geada MM, Galindo I, Lorenzo MM, Perdiguero B, Blasco R (2001) Movements of vaccinia virus intracellular enveloped virions with GFP tagged to the F13L envelope protein. J Gen Virol 82: 2747–2760 [DOI] [PubMed] [Google Scholar]
- Greber UF, Way M (2006) A superhighway to virus infection. Cell 124: 741–754 [DOI] [PubMed] [Google Scholar]
- Hammond JW, Griffin K, Jih GT, Stuckey J, Verhey KJ (2008) Co-operative versus independent transport of different cargoes by Kinesin-1. Traffic 9: 725–741 [DOI] [PubMed] [Google Scholar]
- Hiller G, Weber K (1985) Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J Virol 55: 651–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y (2008) Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol Rev 88: 1089–1118 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S (2009) Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10: 682–696 [DOI] [PubMed] [Google Scholar]
- Hollinshead M, Rodger G, Van Eijl H, Law M, Hollinshead R, Vaux DJ, Smith GL (2001) Vaccinia virus utilizes microtubules for movement to the cell surface. J Cell Biol 154: 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeshtadi A, Burgos P, Stubbs CD, Parker AW, King LA, Skinner MA, Botchway SW (2010) Interaction of poxvirus intracellular mature virion proteins with the TPR domain of kinesin light chain in live infected cells revealed by two-photon-induced fluorescence resonance energy transfer fluorescence lifetime imaging microscopy. J Virol 84: 12886–12894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SC, Ward BM (2008) The vaccinia virus protein F12 associates with IEV through an interaction with A36. J Virol 83: 1708–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28: 449–459 [DOI] [PubMed] [Google Scholar]
- Kimata Y, Kohno K (2011) Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr Opin Cell Biol 23: 135–142 [DOI] [PubMed] [Google Scholar]
- Konecna A, Frischknecht R, Kinter J, Ludwig A, Steuble M, Meskenaite V, Indermuhle M, Engel M, Cen C, Mateos JM, Streit P, Sonderegger P (2006) Calsyntenin-1 docks vesicular cargo to kinesin-1. Mol Biol Cell 17: 3651–3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen S, Shah M, Nielsen R, Iino N, Ryan JJ, Zhou H, Farquhar MG (2008) The endocytic adaptor protein ARH associates with motor and centrosomal proteins and is involved in centrosome assembly and cytokinesis. Mol Biol Cell 19: 2949–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL (2004) A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J Biol Chem 279: 19201–19208 [DOI] [PubMed] [Google Scholar]
- Lo KW, Kan HM, Chan LN, Xu WG, Wang KP, Wu Z, Sheng M, Zhang M (2005) The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J Biol Chem 280: 8172–8179 [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Tasneem A, Thanki N, Yamashita RA, Zhang D, Zhang N, Bryant SH (2009) CDD: specific functional annotation with the conserved domain database. Nucleic Acids Res 37: D205–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ (2006) Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem 281: 3552–3559 [DOI] [PubMed] [Google Scholar]
- Millar NS (2008) RIC-3: a nicotinic acetylcholine receptor chaperone. Br J Pharmacol 153(Suppl 1): S177–S183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SK, Keyel PA, Edeling MA, Dupin AL, Owen DJ, Traub LM (2005) Functional dissection of an AP-2 beta2 appendage-binding sequence within the autosomal recessive hypercholesterolemia protein. J Biol Chem 280: 19270–19280 [DOI] [PubMed] [Google Scholar]
- Morgan GW, Hollinshead M, Ferguson BJ, Murphy BJ, Carpentier DC, Smith GL (2010) Vaccinia protein F12 has structural similarity to kinesin light chain and contains a motor binding motif required for virion export. PLoS Pathog 6: e1000785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, Scaplehorn N, Way M (2004) SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science 306: 124–129 [DOI] [PubMed] [Google Scholar]
- Nikolaev AY, Li M, Puskas N, Qin J, Gu W (2003) Parc: a cytoplasmic anchor for p53. Cell 112: 29–40 [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33: D501–D504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke K, Dohner K, Sodeik B (2006) Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cell Microbiol 8: 387–400 [DOI] [PubMed] [Google Scholar]
- Rahman A, Friedman DS, Goldstein LS (1998) Two kinesin light chain genes in mice. Identification and characterization of the encoded proteins. J Biol Chem 273: 15395–15403 [DOI] [PubMed] [Google Scholar]
- Rietdorf J, Ploubidou A, Reckmann I, Holmström A, Frischknecht F, Zettl M, Zimmermann T, Way M (2001) Kinesin dependent movement on microtubules precedes actin based motility of vaccinia virus. Nat Cell Biol 3: 992–1000 [DOI] [PubMed] [Google Scholar]
- Rodriguez JF, Janeczko R, Esteban M (1985) Isolation and characterization of neutralizing monoclonal antibodies to vaccinia virus. J Virol 56: 482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Bilsland LG, Schiavo G (2008) Molecular landmarks along the axonal route: axonal transport in health and disease. Curr Opin Cell Biol 20: 445–453 [DOI] [PubMed] [Google Scholar]
- Sanderson CM, Way M, Smith GL (1998) Virus-induced cell motility. J Virol 72: 1235–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR et al. (2010) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 38: D5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MR, Maritzen T, Kukhtina V, Higman VA, Doglio L, Barak NN, Strauss H, Oschkinat H, Dotti CG, Haucke V (2009) Regulation of endosomal membrane traffic by a Gadkin/AP-1/kinesin KIF5 complex. Proc Natl Acad Sci USA 106: 15344–15349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP (1998) Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell 94: 585–594 [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 6: 175–182 [PubMed] [Google Scholar]
- Valderrama F, Cordeiro JV, Schleich S, Frischknecht F, Way M (2006) Vaccinia virus-induced cell motility requires F11L-mediated inhibition of RhoA signaling. Science 311: 377–381 [DOI] [PubMed] [Google Scholar]
- van Eijl H, Hollinshead M, Rodger G, Zhang WH, Smith GL (2002) The vaccinia virus F12L protein is associated with intracellular enveloped virus particles and is required for their egress to the cell surface. J Gen Virol 83: 195–207 [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW (2009) Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol 10: 765–777 [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B (2001) Cargo of kinesin identified as jip scaffolding proteins and associated signaling molecules. J Cell Biol 152: 959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Moss B (2001a) Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J Virol 75: 11651–11663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Moss B (2001b) Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J Virol 75: 4802–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Moss B (2004) Vaccinia virus A36R membrane protein provides a direct link between intracellular enveloped virions and the microtubule motor kinesin. J Virol 78: 2486–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM, Weisberg AS, Moss B (2003) Mapping and functional analysis of interaction sites within the cytoplasmic domains of the vaccinia virus A33R and A36R envelope proteins. J Virol 77: 4113–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisswange I, Newsome TP, Schleich S, Way M (2009) The rate of N-WASP exchange limits the extent of ARP2/3-complex-dependent actin-based motility. Nature 458: 87–91 [DOI] [PubMed] [Google Scholar]
- Welte MA (2004) Bidirectional transport along microtubules. Curr Biol 14: R525–R537 [DOI] [PubMed] [Google Scholar]
- Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ (2008) A navigator for human genome epidemiology. Nat Genet 40: 124–125 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.