

Abstract
Missorting of Tau from axons to the somatodendritic compartment of neurons is a hallmark of Alzheimer's disease, but the mechanisms underlying normal sorting and pathological failure are poorly understood. Here, we used several Tau constructs labelled with photoconvertible Dendra2 to analyse its mobility in polarized neurons. This revealed a novel mechanism of sorting—a retrograde barrier in the axon initial segment (AIS) operating as cellular rectifier. It allows anterograde flow of axonal Tau but prevents retrograde flow back into soma and dendrites. The barrier requires binding of Tau to microtubules but does not require F-actin and thus is distinct from the sorting of membrane-associated proteins at the AIS. The barrier breaks down when Tau is phosphorylated in its repeat domain and detached from microtubules, for example, by the kinase MARK/Par1. These observations link the pathological hallmarks of Tau missorting and hyperphosphorylation in neurodegenerative diseases.
Keywords: axon initial segment, microtubules, neuronal polarization, Tau protein
Introduction
An early sign of neurodegeneration in Alzheimer's disease is the missorting of Tau protein. In normal mature neurons, it has a mostly axonal distribution (Binder et al, 1985; Kosik and Finch, 1987; Migheli et al, 1988; Mandell and Banker, 1995), but in disease it appears in the somatodendritic compartment. This occurs before Tau aggregation and correlates with the incipient loss of synapses (Coleman and Yao, 2003; Ballatore et al, 2007; Haass and Selkoe, 2007; Pimplikar et al, 2010). There has been a debate on the causes of the polarized distribution of microtubule-associated proteins (MAPs) in mature neurons, notably axonal Tau and somatodendritic MAP2. Suggested contributing factors include the preferential routing of Tau or MAP2 mRNAs towards their appropriate compartment (Garner et al, 1988; Aronov et al, 2002), selective protection of Tau or MAP2 protein against degradation, and directional cues based on the interplay of microtubules, motors, and cargo (Hirokawa et al, 1996; Nakata and Hirokawa, 2003; Konishi and Setou, 2009). The sorting of Tau can break down in abnormal situations, for example, following excitotoxicity, cell stress, or exposure to amyloid-β peptide (Mattson, 2004; Roberson et al, 2007; Zempel et al, 2010). This causes the accumulation of Tau in cell bodies and dendrites and heralds the gradual pathological aggregation. Considering Tau's role in the disease process it would be important to gain a detailed understanding of the sorting pathway: How does it function in normal neurons? What causes it to break down in disease? Where does the missorted dendritic Tau come from, what is its fate?
The establishment of polarity in a neuron is essential for its function. It involves cytoskeletal fibres, notably microtubules and motor proteins of the kinesin superfamily (KIFs) for selective long-haul delivery of cargoes to somatodendritic and axonal compartments (e.g., dendritic neurotransmitter receptors or axonal synaptic vesicles; Burack et al, 2000; Lazarov et al, 2007; Hirokawa et al, 2010; Scott et al, 2011). A further element in maintaining polarity is the buildup of selective barriers (Ledesma and Dotti, 2003; Witte and Bradke, 2008). The best-known example for neurons is the axon initial segment (AIS), located just beyond the axon hillock. It has been characterized as an F-actin-based diffusion barrier for membrane-associated proteins and lipids, and for large (>70 kDa) cytosolic particles (Winckler et al, 1999; Nakada et al, 2003; Kole et al, 2008; Song et al, 2009).
Because of the key role of Tau in AD, we and others studied tau transport, diffusion, and MT binding in the axon by GFP-tagged proteins and FRAP (Samsonov et al, 2004; Konzack et al, 2007; Weissmann et al, 2009). Rapid diffusion appears to be a major mechanism for axonal Tau distribution in the short-to-intermediate range, while microtubule-dependent transport of Tau dominates over long distances. Here, we describe a new approach, photoconversion of Tau tagged with Dendra2, to directly visualize and compare the movement of different Tau variants over long distances in differentiated cortical neurons. This revealed a novel barrier mechanism that allows Tau to enter the axon but prevents it from flowing back to the somatodendritic compartment. We show that this barrier is uniquely localized in the same initial segment as the actin-based barrier of membrane components, but it depends on microtubules rather than F-actin and is regulated by Tau phosphorylation.
Results
Tau diffuses rapidly in neurons, but is restrained by a retrograde barrier in the initial axon
One of the enigmas of Tau in AD is its early redistribution from axons into the somatodendritic compartments. This begs two questions: how is Tau sorted into axons but not into dendrites, and why does it stay in axons in spite of its ability to diffuse rapidly? To study the redistribution of Tau, a common approach is to transfect neurons with GFP-tagged Tau into cells and then observe its redistribution after photobleaching (Samsonov et al, 2004; Konzack et al, 2007). In this approach, a bleached zone must be observed against a bright background, which limits the signal/noise. To overcome these limitations, we have employed new photoconvertible variants of GFP that enable one to observe the overall distribution at any time point, and then, after photoconversion in a defined region, reveal the spreading with high contrast, in spite of an apparent steady state. We chose Dendra2 which is monomeric (230 residues), highly photostable, matures rapidly at 37°C and displays bright fluorescence before and after photoconversion (Gurskaya et al, 2006; Gauthier and Brandt, 2010). An overview figure depicting the raw data and ratiometric analysis before and after photoconversion is shown in Supplementary Figure S1, illustrating a photoconversion efficiency of >60% in our experiments. When Dendra2 is transfected by lipofectamine into mature rat cortical neurons (7–11 DIV), the protein becomes visible in the cell body by its intrinsic green fluorescence (Figure 1A). In Figure 1B–D, Dendra2 is photoconverted from green to red fluorescence by irradiation at 405 nm in the cell body, and its spreading can be traced in real time (Figure 1C and D). Within seconds, Dendra2 diffuses into the proximal cell processes, and within minutes it appears along the length of dendrites and axons that can be distinguished by their length, diameter, and brightness (Figure 1D; Supplementary Movie S1). The rate of spreading is roughly consistent with the free diffusion of monomeric Dendra2 in the cytosol (D∼25 μm2/s, for details see Konzack et al, 2007).
Figure 1.

Photoconversion of TauDendra2 in transfected neurons. Top: Bar diagram of human tau40 with domains, phosphorylation sites, and antibody epitopes. The C-terminal half contains the repeat domain (repeats R1–R4, red, R2 alternatively spliced) plus flanking regions (green), responsible for microtubule binding. The N-terminal half (‘projection domain’) does not bind to microtubules and contains alternatively spliced inserts I1 and I2 (blue). Major target sites of kinases MARK (KXGS motifs with S262, S293, S324, and S356) and PKA (S214) that control binding of Tau to MT are labelled in red. Seventeen major target sites of proline-directed kinases (in the flanking regions of the repeats and projection domain) and some of the responsible kinases are labelled in green. Epitopes of some phosphorylation-dependent antibodies (12E8, AT8, and PHF1) are indicated. Dendra2 is linked to the N-terminus of Tau as indicated. Bottom: (A) Cortical neuron transfected with Dendra2 for 2 days, showing the green fluorescence before photoconversion. Arrow indicates the axon and arrowhead indicates the somatodendritic compartment. (B) Photoconversion of Dendra2 by UV illumination of the soma (ROI1, boxed in B–D). Images were taken in the red fluorescent channel (B–D) at the indicated time points (0–8 min). (C, D) Time-lapse images trace the rapid redistribution of photoconverted Dendra2 from the soma (ROI1) into the dendrites (arrowhead in D) and the axon (arrow in D). (E) Distribution of TauD2 in a cortical neuron transfected for 2 days before photoconversion. TauD2 appears both in the somatodendritic compartment (arrowheads, top) and in the axon (arrow). (F) Photoconversion of TauD2 by UV illumination of the soma (ROI1, boxed in F–H). (G, H) Time-lapse images in red fluorescent channel trace the redistribution of photoconverted TauD2 from the soma into the dendrites (arrowhead in H) and the axon (arrow in H). Compared with Dendra2 (B–D), the spreading of TauD2 is much slower (images at 0–95 min). The asterisk indicates the non-photoconverted neuron. (I) Intensity versus time in ROI2 in axon (∼150 μm from the soma, red circle in D or H). The signal of Dendra2 reaches a half-maximal level at ∼300 s, whereas TauD2 takes much longer (∼5300 s). Data shown here are representations of at least three independent experiments.
Figure 1E–H shows the same type of experiment with full-length human Tau (441 residues, ‘2N4R’), N-terminally fused to Dendra2 (688 aa residues, including a 17 aa linker, construct termed TauD2; Figure 1, top). As expected from the higher mass, TauD2 spreads more slowly than Dendra2, but also fills dendrites and axons within ∼48 h, as seen either by the green fluorescence of Dendra2 or by immunostaining. This behaviour is unexpected since mature neurons possess a sorting machinery that routes Tau into axons but not into dendrites (Binder et al, 1985; Mandell and Banker, 1995), and indeed endogenous rat Tau is sorted axonally if expressed alone (see below). On the other hand, overexpression or microinjection of Tau is known to overwhelm the sorting machinery so that Tau becomes ubiquitous in neurons (Hirokawa et al, 1996), as is the case here (Figure 1E). When photoconverting the exogenous TauD2 in the cell body, the protein spreads gradually throughout the neuron, but again the rate is much slower than that of Dendra2 alone (∼20-fold), and there is a preferential filling of axons first (Figure 1F–H). The arrival of photoactivated TauD2 was monitored in selected regions of interest, for example, ∼150 μm from the soma (Figure 1H, ROI2). In this case, it took ∼70 min for TauD2 to redistribute from the soma to the examined area (Figure 1I; Supplementary Movie S2). Thus, this method provides a direct visualization of the dynamics of Tau in neurons.
Since the above observations suggested that the sorting machinery of neurons was still functional, in spite of a background of unsorted Tau, we began a systematic investigation of the transfer of TauD2 and Dendra2 (as control) between different regions of mature neurons. Figure 2A1 illustrates part of an axon ∼60 μm from the soma of a Dendra2-transfected neuron (ROI1). Photoconverted Dendra2 redistributed rapidly into the distal axon and somatodendritic compartment (Figure 2A3). In these neurons, a homogeneous distribution of fluorescence was reached across the AIS (Figure 2A2 and A4, intensity profiles at t=1 and 5 min; Supplementary Movie S3).
Figure 2.

A barrier mechanism in the initial axon prevents axonal TauD2, but not Dendra2 alone, from entering the cell body. (A1–A4) Time-lapse red fluorescent images of Dendra2 alone (without Tau, A1) after photoconversion in the axon in ROI1 (inset in A1 shows the even overall distribution of Dendra2). The intensity profile (A2) corresponds to the fluorescent intensity in ROI2 in the initial axon (between 0 and 50 μm distance from the cell body) at 1 min. Only a low level of Dendra2 has diffused into ROI2, but fluorescence is already visible in the cell body (dashed circle). (A3, A4) At 5 min, the level of Dendra2 is increased by diffusion in ROI2, but even more pronounced in the cell body. Only a shallow intensity gradient is observed along ROI2, indicating that there is no barrier for diffusion for Dendra2. (B1–B4) A similar experiment was performed by photoconversion in the axon in ROI1of a neuron transfected with TauD2 (B1). The intensity profile of ROI2 at 5 min (B2) shows the pronounced gradient of TauD2 in the initial axon, with intensity decaying towards the soma. (B3, B4) The gradient is maintained throughout the experimental period (up to 30 min and more) and the intensity in the cell body (dashed circle) remains low, indicating that a barrier in the initial axons (ROI2) prevents the axonal TauD2 from missorting into the cell body. (C1–C4) Photoconversion of a dendrite (C1) in a neuron transfected with TauD2 (ROI1). The intensity profile in ROI2 at 5 min (C2) in the axon emanating from the cell body shows the smooth intensity flow from the somatodendritic compartment into the axon without indication of a barrier. Even at later time points, there is no appearance of a gradient of TauD2 (30 min, C3, C4). Data represent at least three independent experiments.
When performing this experiment with TauD2, a very different picture emerged: the photoconverted TauD2 travelled without hindrance to the distal axon; but was prevented from redistributing towards the somatodendritic compartment (Figure 2B1 and B3). An intensity gradient of photoconverted TauD2 was observed in the AIS, decreasing towards the soma and maintained throughout the experimental period (Figure 2B2 and B4, intensity profiles at t=5 and 30 min; Supplementary Movie S4). This means that once TauD2 is localized in the axon, it cannot return retrogradely to the soma and dendrites. This indicates the presence of a barrier in the AIS, which functions akin to a ‘diode’ to allow directional entry of Tau from the soma to the axon but not reverse. In fact, photoconversion directly at the AIS also resulted in an anterograde flow of TauD2 towards the distal axon and not towards the soma (Supplementary Figure S2). We also tested the effect of photoconversion of TauD2 in the distal axons, and even after repeated photoconversion the barrier was maintained throughout the experimental period (Supplementary Figure S4).
We asked whether a similar barrier exists between the soma and the dendrites and performed photoconversion in dendrites of a TauD2-expressing neuron (11 DIV) (Figure 2C1, ROI1). In contrast to axonal Tau, the dendritic TauD2 readily re-entered the somatodendritic compartment and the axon (Figure 2C3). The intensity profiles (Figure 2C2 and C4) indicate that there was no barrier within the somatodendritic compartment (see Supplementary Movie S5). The same effects were observed after photoconversion at the distal ends of dendrites (Supplementary Figure S3). The data demonstrate the unique axonal localization of the barrier mechanism in the AIS to trap Tau once it has entered the axon.
The retrograde barrier depends on microtubules
What cell components are responsible for the retrograde barrier? As Tau is a microtubule-binding protein, we suspected that intact microtubules are one of the requirements. This is indeed the case: once they are depolymerized by nocodazol, the barrier breaks down and allows retrograde axonal flow of TauD2 into the soma (Figure 3A–D; Supplementary Movie S6). This property of a microtubule-based barrier is distinct from the AIS barrier described previously for membrane-associated proteins, characterized by the accumulation of AnkyrinG, βIV-spectrin, ion channels and cell adhesion molecules and F-actin (Winckler et al, 1999; Ogawa and Rasband, 2008; Grubb and Burrone, 2010; Rasband, 2010). The AIS components are indeed stable against nocodazol, and therefore cannot account for the retrograde barrier of Tau (Figure 4). Likewise, the barrier of Tau is distinct from the porous filter described by Song et al (2009), which blocks axonal access for diffusible molecules greater than ∼70 kDa. Their filter is based on F-actin and operates in the anterograde direction, not retrogradely. Our results were confirmed by destroying the actin-based diffusion filter of Song et al (2009) in the AIS by latrunculin A (Supplementary Figure S5), yet the barrier for Tau remained intact (Figure 4G–I). The F-actin staining was significantly reduced after treatment with latrunculin A, including in the AIS (Supplementary Figure S5).
Figure 3.

The diffusion barrier depends on intact microtubules. (A) Green fluorescent image of an 11 DIV neuron transfected with TauD2 for 2 days before photoconversion. (B) Red fluorescent image of the same neuron immediately after illumination of its axon near the soma (ROI1) with UV light. (C) Time-lapse images up to 30 min show the anterograde diffusion of axonal Tau but retrograde blockage at the initial axon (arrowhead) so that TauD2 does not enter the cell body (dashed circle). At 30 min, cells were treated with 5 μM nocodazol to disrupt microtubules. (D) After addition of nocodazol, there is diffusion of TauD2 across the barrier in the initial axon (arrowhead) into the somatodendritic compartment (dashed circle). Data represent at least three independent experiments.
Figure 4.

The axonal diffusion barrier for Tau coincides with the axon initial segment (AIS) but depends on a different mechanism. (A) Cortical neuron (10 DIV) transfected with TauD2. Note the even distribution in axon (arrow), soma, and dendrites. (B) Same neuron, fixed and stained for AnkyrinG, a marker of the AIS, (C) merged images. (D) Cortical neuron (10 DIV) transfected with TauD2 and then treated with 10 μM nocodazol. This treatment destroys microtubules but not the localization of AnkyrinG in the AIS (E, F). (G) Green fluorescent image of a neuron (10 DIV) expressing TauD2 and incubated in 2.5 μM latrunculin A for 1 h to disrupt the actin cytoskeleton before photoconversion. (H) Red fluorescent image of the same neuron in the presence of latrunculin A where part of its axon was photoconverted (ROI1) (circles indicate the soma in H and I). (I) Time-lapse image after 75 min showing the blockage at the AIS (arrowhead) for TauD2 in the presence of latrunculin A, indicating that the barrier for Tau does not depend on the actin cytoskeleton.
In order to test the spatial relationship of the Tau diffusion barrier with the AIS, we stained the AIS with AnkyrinG after photoconversion in the axon. The data indicate that the classical AIS, as defined by AnkyrinG, and the Tau diffusion barrier overlap significantly. The Tau diffusion barrier is localized within the broad decreasing gradient of the AnkyrinG staining beginning from the axon hillock to the more distal parts of the axon (Supplementary Figure S6).
The retrograde barrier depends on Tau's microtubule affinity and state of phosphorylation
Tau occurs in several isoforms and phospho-variants, and therefore we asked whether all forms of Tau are restrained by the retrograde barrier in the same way, and whether the affinity for microtubules plays a role. This was tested by varying the phosphorylation state of Tau (which in turn alters the affinity for microtubules), or by changing the balance of kinases and phosphatases. The latter is achieved by the phosphatase inhibitor okadaic acid (OA), resulting in a gradual hyperphosphorylation of Tau at multiple sites similar to AD Tau, and loss of MT binding (Lichtenberg-Kraag et al, 1992). Figure 5A–C illustrates a neuron with a stable barrier, but after exposure to 0.5 μM OA at 55 min the barrier breaks down quickly (Figure 5D). The blots (Figure 5E) illustrate that Tau becomes highly phosphorylated at the phospho-epitopes AT8, PHF1, 12E8, and others upon exposure to OA.
Figure 5.

Hyperphosphorylation by okadaic acid (OA) treatment allows TauD2 to pass the barrier. (A) Green fluorescent image of a 9 DIV neuron expressing TauD2 before photoconversion. (B) Red fluorescent image of the same neuron where part of its axon was photoconverted (ROI1). Time-lapse image after 20 min showing the anterograde spreading of TauD2 but blockage at the AIS near the cell body (arrowheads in B–D indicate the AIS). (C) The same protocol of photoconversion as in (B) was repeated in the same axonal region (ROI1) 55 min after the first illumination; at this time, 500 nM OA was added. The diffusion barrier at the AIS is still visible (arrowhead). (D) Redistribution of axonal Tau to the somatodendritic compartment after OA treatment. Increasing fluorescence intensity in the soma is observed shortly after addition of OA, and the gradient in distribution of axonal Tau in the AIS disappears. Dashed circles indicate the soma in (B–D). (E) Western blot of extracts of cortical neurons (11 DIV) treated with OA (500 nM, 1 h) using phosphorylation-dependent Tau antibodies AT8, PHF1, and 12E8. The signals of hyperphosphorylated Tau increase strongly after phosphatases are inhibited by OA treatment (right lanes), especially in the case of 12E8 (pS262/pS356 in the repeats, right panel); whereas in the untreated cells the signals of phosphorylated Tau at these epitopes remain low (left lanes). Note the clear upward shift of the bands of phosphorylated Tau in the OA-treated cells. A blot with anti-actin antibody was used as loading control.
In the OA experiment, the phosphorylation of Tau is heterogeneous and includes sites of minor importance for MT binding (e.g., some of the SP or TP motifs). In order to demonstrate a direct and specific relationship between the barrier and the Tau-MT affinity, we transfected TauD2 modified at the four KXGS motifs in the repeat: the ‘4KXGE’ mutant mimics constitutive phosphorylation, whereas the ‘4KXGA’ is non-phosphorylatable at these motifs. The 4KXGE mutant binds to MT only weakly (Biernat and Mandelkow, 1999), and indeed there is no retrograde barrier for this Tau mutant (Figure 6A and B). By contrast, the 4KXGA mutant retains high affinity to MT, and the barrier remains intact (Figure 6C and D). This illustrates that the barrier depends not on the overall degree of phosphorylation, but on specific phosphorylation sites that detach Tau from microtubules.
Figure 6.
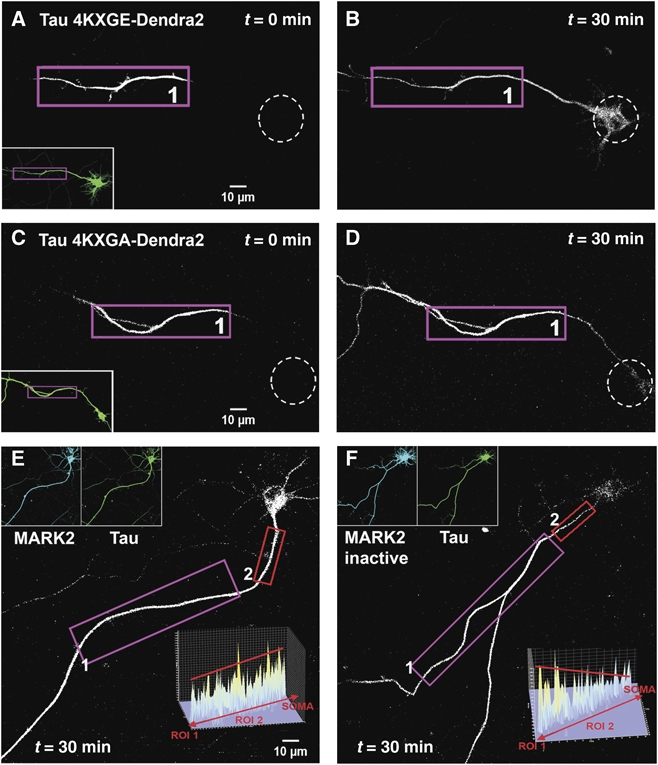
Effect of pseudo-phosphorylation of TauD2 and MARK activity on axonal Tau redistribution and diffusion barrier. (A, B) Cortical neuron transfected with pseudo-phosphorylated TauD2-4KXGE (at all four KXGS → E motifs in the repeat domain, thus weakening the binding to microtubules). After photoconversion in the axon at time t=0 (ROI1 in A) the soma (dashed circles) lights up in a time-dependent manner, indicating that loss of microtubule-binding enables TauD2-4KXGE to pass the retrograde diffusion barrier between the axon and the cell body. The inset in (A) shows the green fluorescent image of the neuron before photoconversion. (C, D) Similar experiment to (A, B) performed in neurons transfected with TauD2-4KXGA (non-phosphorylatable in the repeat domain and therefore tightly binding to microtubules). ROI1 in (C) indicates the axonal region for UV illumination. There is no retrograde flow of TauD2-4KXGA into the soma of the neuron (D, 30 min). (E) Time-lapse images taken in the red fluorescent channel at 1 h after photoconversion in the axon (ROI1) of a neuron co-transfected with CFP-MARK2 and TauD2. Note that the soma shows strong fluorescence since the phosphorylated TauD2, not bound to microtubules, can pass the barrier. The intensity profile of the AIS (ROI2 in E) (inset) shows no gradient of Tau distribution in this region, indicating that TauD2 can pass through the barrier after phosphorylation by MARK2. (F) Same experimental procedure as in (E), performed in a neuron co-transfected with the inactive mutant CFP-MARK2T208A+S212A and TauD2. ROI1 indicates the axonal region for UV illumination. The fluorescent intensity in the somatodendritic compartment remains low compared with (E), indicating that the kinase-dead mutant of MARK could not phosphorylate TauD2 at the KXGS motifs in the repeat domain, and therefore TauD2 still binds strongly to microtubules and cannot pass the barrier. The intensity profile in the AIS (ROI2 in F) (inset) shows the gradient of TauD2 typical of an intact barrier. Data represent at least three independent experiments.
The KXGS motifs of Tau, notably S262, are phosphorylated early in neurodegeneration by kinases of the MARK/Par-1 family which play a role in establishing cell polarity (Matenia and Mandelkow, 2009). We, therefore, transfected TauD2 together with MARK2CFP. The kinase became distributed in the cell body, axon, and dendrites (Figure 6E, inset), Tau became strongly phosphorylated at the KXGS motifs (Thies and Mandelkow, 2007), and the barrier indeed broke down, allowing axonal TauD2 to flow into the cell body (Figure 6E). When the same experiment was done with inactive MARK2CFP (with mutations T208A+S212A; Timm et al, 2008), Tau remained in a low state of phosphorylation, and the barrier remained intact (Figure 6F). Together, the data show that the retrograde barrier restrains Tau only when its affinity to microtubules is high, but disappears when microtubules break down or when Tau becomes detached by phosphorylation.
In the above experiments, we focused on factors that determine whether a retrograde barrier is present or not. However, the question arises whether there are intrinsic differences in the mobilities of axonal Tau, independent or superimposed on the properties of the barrier. For this purpose, we tested the spreading of photoactivated TauD2 variants in the anterograde axonal direction. In the initial steady state (∼2 days after transfection), the cells are homogeneously filled with green fluorescent Tau (Figure 7A). Photoactivation of area ROI1 (∼50–70 μm from the cell body) generates red fluorescence that travels retrogradely until it is stopped by the barrier, but travels anterogradely without interruption (Figure 7B). The rate of migration was measured by the red intensity at ROI2 further downstream (Figure 7C). During the initial 10–20 min the intensity shows a hyperbolic approach to an apparent steady state, consistent with diffusion of TauD2 into ROI2 and beyond whose half time reflects the effective diffusion rate. Comparing half times for different protein species we observed roughly four categories: Dendra2 alone is ‘very fast’ (t1/2 ∼7 s), corresponding to a protein that does not interact with microtubules and diffuses freely. Among the Tau species, those binding weakly to MT are ‘fast’, with t1/2 ∼50 s (red in Figure 7D). This includes the phospho-mimicking full-length Tau mutants 4KXGE, mutant 214E (mimicking phosphorylation at S214) and 17EP (17 SP/TP motifs turned into EP; Figure 1, top). The ‘slow’ variants, with t1/2 around 120–150 s, included wild-type Tau and the 4KXGA mutant, both of which bind well to MT. Finally, there are the ‘ultra-slow’ full-length Tau mutants 17AP (17 SP/TP motifs turned into AP and thus non-phosphorylatable), and 8-repeat-Tau (with duplicated repeat domain), both of which bind tightly to MT (Gustke et al, 1994). The data are explained by the diffusion of Tau while it is not bound to MT, which is related to the rates of association and dissociation (kon, koff; Konzack et al, 2007). Here, the important point is that the retrograde barrier is observed only with ‘slow’ or ‘ultra-slow’ variants of Tau with tight MT binding, suggesting that the barrier is dominated by kinetic parameters.
Figure 7.

Anterograde axonal diffusion of Tau depends on phosphorylation and binding to microtubules. (A) Green fluorescent image of a cortical neuron expressing TauD2, before photoconversion. A region of 55 × 35 μm2 containing a 65 μm stretch of the axon (ROI1 in B) and lying at a 50–70 μm distance from the soma is chosen for photoconversion. (B) Red fluorescent image of the same neuron at 15 min after photoconversion. ROI2 is selected for intensity integration (30 μm segment of the axon, at 10 μm distance to ROI 1). Note that TauD2 diffuses rapidly in the anterograde direction, but is restrained towards the cell body (dashed circle). (C) Intensity in ROI2 versus time (0=time of photoconversion), rising with a half time of t1/2∼150 s which reflects the effective rate of Tau diffusion in the anterograde direction. (D) Half times of spreading of different Tau variants tagged with Dendra2 (n⩾3 cells each; error bars indicate s.e.). Note that Dendra2 alone diffuses rapidly (t1/2∼7 s). Tau wild type and tightly binding non-phosphorylatable mutants 4KXGA and 17AP diffuse slowly (t1/2∼130 to 220 s), whereas phospho-mimicking mutants binding weakly to microtubules (4KXGE, 214E, and 17EP) and diffuse more rapidly (t1/2∼70 s). The strongly binding mutant with duplicated repeats (8R-Tau) diffuses very slowly (t1/2 ∼260 s). **P<0.01 and *P<0.05.
When refining this analysis, the comparison of half times measured for Tau spreading proximally or distally from the region of photoconversion (ROI) reveals some directional bias which becomes significant for Tau variants with high MT affinities (4KXGA; 17AP and 8-repeat-Tau). It shows somewhat enhanced spreading to the distal direction (Supplementary Figure S7). The likely explanation is that Tau diffusion is superimposed on (slow) anterograde transport of Tau which is known to depend on binding to microtubules or tubulin oligomers (Baas and Buster, 2004).
Breakdown versus maintenance of the barrier for endogenous and exogenous Tau
One of the enigmas in Tau's behaviour is the apparent contradiction between three key features: (1) At the start of experiments (∼7–9 DIV) the neurons are differentiated, and intrinsic Tau is already sorted into axons, suggesting preferential shuttling to the axon and an intact retrograde barrier (consistent with the literature, e.g., Binder et al, 1985; Mandell and Banker, 1995). (2) Transfection with exogenous Tau variants, for example, TauD2, leads to unpolarized distribution into axons, dendrites and cell bodies (e.g., Figures 1E, 3A, and 5A), suggesting that the sorting machinery might no longer function properly. (3) However, testing axonal Tau directly by photoactivation shows that the retrograde barrier is functional, as described above (e.g., Figure 2).
To clarify this contradiction, we asked if endogenous Tau changes its polarized distribution after transfection with exogenous Tau. In order to distinguish the two types of Tau, we transfected cells with the juvenile isoform (hTau23GFP) lacking the N-terminal inserts and repeat R2 (Tau ‘0N3R’; Figure 8A). Endogenous rat Tau is a mixture of different isoforms so that some of them can be recognized by antibodies against the first insert (e.g., SA4473, Figure 8B and D). Unexpectedly, the exclusive axonal sorting of endogenous Tau breaks down after transfection of exogenous hTau23GFP so that endogenous Tau now appears in all compartments (Figure 8B). This means that exogenous Tau can induce the missorting of endogenous Tau, while the retrograde barrier is still functional in axons. The effect can be explained by a limited capacity of the axonal sorting machinery, combined with the low degree of phosphorylation of axonal Tau that prevents re-entry into the cell body and dendrites. To minimize possible artefacts of overexpression, we expressed Tau at very low levels by reducing the transfecting virus and/or exposure times up to four-fold. In these conditions, transfected TauD2 could no longer be detected by its fluorescence, but even after antibody amplification with an anti-Dendra2 antibody we could not find physiologically sorted cells with Tau in axons only. At the earliest time point when transfected Tau becomes detectable, it is predominantly present in the cell body and dendrites (Supplementary Figure S8A), but with time it propagates into the axons. We quantified the axonal spreading on the basis of the fluorescent front (following Utton et al, 2002) and fitted it with a square root function as described (Konzack et al. 2007), consistent with the assumption that the spreading in our regime of time and distance is dominated by diffusion (Supplementary Figure S8B). However, even at very low expression levels of TauD2 the endogenous Tau became missorted into the somatodendritic compartment, suggesting that the capacity of sorting is readily overwhelmed.
Figure 8.

Transfection of neurons with TauGFP causes missorting of both exogenous and endogenous Tau. (A) Cortical neuron (7 DIV) transfected with hTau23GFP lacking the two N-terminal inserts (see Figure 1, top). The exogenous Tau distributes into all compartments (axon indicated with arrows in A–C, dendrites indicated with arrowheads in A–C, soma), similar to the longest isoform used above. (B) Cells were stained with antibody SA4473 against the first insert, present only in endogenous Tau but not in the transfected Tau. The antibody stains not only axons but all compartments, indicating that the transfection of exogenous Tau leads to the missorting of endogenous and exogenous Tau. (C) Same field stained with MAP2 as a marker of soma and dendrites, illustrating that MAP2 maintains its normal sorting. (D) Blot showing that exogenous hTau40D2 is recognized by antibody SA4473 against the first insert, but hTau23GFP is not recognized, confirming the specificity of this antibody. Endogenous Tau is recognized in both cases. Actin is shown as a loading control.
It is noteworthy that the missorting of Tau at elevated levels does not affect the sorting of the somatodendritic MAP2 (Figure 8C), in spite of its Tau-like microtubule-binding domain. This argues that the two proteins are constrained by different mechanisms (e.g., MAP2 would be too large to pass through the porous anterograde filter; Nakata and Hirokawa, 2003; Song et al, 2009).
Discussion
This study was motivated by the observation that Tau is sorted into axons of mature healthy neurons, whereas it acquires a somatodendritic distribution in Alzheimer's disease and other tauopathies (Binder et al, 1985). This raises the questions of how Tau becomes axonal in the first place, how the neuron makes the transition from the immature state (with ubiquitous Tau distribution) to the mature state, and why the sorting mechanism breaks down during neurodegeneration.
Several authors have addressed the problem of sorting of axonal Tau, dendritic MAP2, and other MAPs during neuronal polarization (Binder et al, 1985; Kosik and Finch, 1987; Hirokawa et al, 1996; Aronov et al, 2001). These studies established that the bulk of Tau is indeed restricted to axons in mature neurons, even though minor amounts might occur in other compartments, for example, in dendrites (Papasozomenos and Binder, 1987; Ittner et al, 2010), the nucleus (Sultan et al, 2011), or in other cell types (notably oligodendrocytes; LoPresti et al, 1995; Klein et al, 2002). The emerging picture for axonal Tau sorting is that there are several overlapping mechanisms, rather than a single one. On the level of mRNA, the 3′UTR contains a zip code that directs it into the axon for local translation (Aronov et al, 2001). The 5′ UTR contains a terminal oligopyrimidine tract which enhances translation of Tau in axons under control of mTOR (Morita and Sobue, 2009). On the level of Tau protein, sorting could be achieved by a combination of selective transport, barriers, phosphorylation, and degradation. For example, Tau can be transported into the axon while MAP2 is excluded from it, a given MAP is more readily degraded if it appears in the ‘wrong’ compartment, and selective transport can occur along microtubules through special combinations of motors, adaptors, and tubulin modifications (Hirokawa et al, 1996; Nakata and Hirokawa, 2003; Hirokawa and Takemura, 2005; Konishi and Setou, 2009). The long-haul transport mechanisms tend to be microtubule based because they form extended tracks for vesicles, organelles, and nucleoprotein complexes (Stokin and Goldstein, 2006; Hirokawa et al, 2010), whereas known barriers such as the AIS are based on F-actin filaments because they can form meshworks, for example, for membrane-associated components (Ogawa and Rasband, 2008; Song et al, 2009).
In the case of Tau, we are faced with several seemingly contradictory observations:
Tau is associated with microtubules, yet it diffuses fairly rapidly (Samsonov et al, 2004; Konzack et al, 2007). This can be explained by the short dwell time of Tau on microtubules. However, by this argument one cannot explain why Tau is mostly restricted to axons.
In immature axons, Tau is ubiquitous in the cytoplasm. During differentiation, the Tau level rises several fold (Drubin and Kirschner, 1986), yet Tau becomes axonally restricted. This could be explained because the high initial state of phosphorylation (∼4 Pi per molecule) drops to the normal low adult levels (∼2 Pi; Kopke et al, 1993), and the isoforms shift from small (fetal, 3R-Tau) to a mix of larger 4-repeat isoforms (Goedert et al, 1989; Kosik et al, 1989). These features argue for a tighter binding of adult Tau to microtubules, coincident with increased microtubule stabilization.
Nevertheless, the axonal sorting of Tau during differentiation is in stark contrast to the missorting after microinjection or transfection of Tau (which causes even missorting of endogenous Tau, Figure 8, and missorting is already present at extremely low expression levels detectable only via antibody amplification of transfected Tau; Supplementary Figure S8A). A plausible interpretation is that the Tau sorting machinery is readily overwhelmed by elevated Tau, such that even very low levels of additional Tau suffice for impairing the tight and precise sorting machinery of Tau.
How can we reconcile the observations on Tau sorting with the mechanisms described in the literature so far? Three major modes of selection have been described: (a) There is a diffusion barrier for membrane-associated components in the AIS, based on F-actin but not on microtubules (Rasband, 2010). (b) There is an anterograde diffusion barrier for cytoplasmic components greater than ∼70 kDa (Song et al, 2009), also dependent on F-actin but not on microtubules. (c) There is active, microtubule, and motor-based transport of selected cargoes that can overcome the F-actin-dependent barriers. How does this apply to Tau? Since the bulk of Tau is not membrane-associated, option (a) does not apply. Since Tau is smaller than 70 kDa, the cytoplasmic pore filter does not apply either, consistent with the observation that Tau can readily diffuse into axons (Konzack et al, 2007). Third, Tau and other axonal MAPs (e.g., MAP1b) can be transported into axons by long-haul microtubule-based transport, piggy backing on microtubular structures (Baas and Buster, 2004). This system is apparently able to carry almost all endogenous Tau into the axon, leaving only trace amounts in dendrites.
How is this polarized distribution maintained, vis-a-vis the high mobility of Tau by diffusion? Our experiments argue that there is a novel type of retrograde filter, also located in the AIS but dependent on microtubules, not on F-actin. Thus, Tau can move into the axon, but not out. What conditions are necessary for the filter to work? First, Tau must be able to bind to microtubules, because Tau phosphorylated at critical sites, which disrupt microtubule binding can bypass the barrier. Second, microtubules must be intact, because disassembly by nocodazol destroys the retrograde Tau barrier (but not the F-actin-based AIS barrier). Significantly, excess Tau (by transfection) can overwhelm the anterograde sorting machinery, but does not destroy the retrograde barrier.
The results suggest answers to an important question in Tau research: where does the missorted dendritic Tau come from in neurodegeneration? One possibility is that Tau synthesized in the cell body is not axonally sorted because the sorting machinery has broken down, so that the dendritic Tau would come straight from the cell body. However, our experiments show that even axonal Tau can transit directly past the barrier into dendrites, once it is hyperphosphorylated. This places a new importance on Tau phosphorylation which causes detachment from microtubules: a perturbed balance of kinase/phosphatase activities in the axon can make the retrograde barrier leaky and promote accumulation of Tau in dendrites, where it can aggregate into PHFs.
What is the molecular basis of the retrograde barrier? One could think of several possibilities:
A (passive) size exclusion filter (analogous to the filter described by Song et al, 2009)—this is unlikely because it would not discriminate between different states of phosphorylation. Conversely, the data imply that the mechanism involves some preferential interaction of Tau regulated by phosphorylation.
A barrier imposed by preferential binding to the F-actin meshwork in the AIS—also unlikely because the barrier remains active even when F-actin is depolymerized (Figure 4).
Preferential interactions with microtubules in the AIS—this would be partly consistent with the observations that only Tau with high microtubule affinity is restrained by the barrier. However, this model requires further assumptions. One is that microtubules in the AIS must be different from the remainder of the axon, which is indeed the case. The microtubules in the AIS are selectively modified and differ in their dynamics (Nakata and Hirokawa, 2003; Konishi and Setou, 2009; Sanchez-Ponce et al, 2011). A problem with this model is that trapping of Tau on microtubules of the AIS would lead to a local accumulation of Tau, contrary to the observations. One way to remove this dilemma is to postulate that trapped Tau is continuously swept forward into the axon by slow anterograde transport of Tau–tubulin complexes (Baas and Buster, 2004). This phenomenon could explain the observation that Tau constructs with high affinity for microtubules have a preference for anterograde over retrograde propagation (Supplementary Figure S7).
The neurofilament meshwork could provide a further preferential trap for Tau. Although neurofilaments are also mobile and move down axons in a stop-and-go fashion (Wang et al, 2000), the meshwork in the AIS of mature neurons is particularly static and dense due to their extended sidearms (Hirokawa, 1982; Yuan et al, 2009). Neurofilaments interact with microtubules, which would be consistent with the dependence of the retrograde barrier on intact microtubules (Figure 3). We have carried out computational modelling (not shown) to confirm that a combination of these factors would suffice to explain the ‘diode’ effect of Tau distribution. In this case, the barrier exerts its function through differential binding affinities for several cytoskeleton components.
Materials and methods
Plasmids construction
pDendra2-C vector was purchased from Evrogen and cDNA of human full-length Tau was subcloned into the C-terminal of Dendra2. The cDNA of Dendra2–Tau (termed TauD2) was then subcloned into the pShuttle vector under control of the human cytomegalovirus (CMV) promotor. The SfiI–NheI fragments containing parts of the cDNAs of Tau-S214E, pseudo-phosphorylated Tau-4KXGE (Ser to Glu at all four KXGS motifs in the repeat domain), non-phosphorylatable Tau-4KXGA (Ser to Ala at all four KXGS motifs in the repeat domain), Tau-17EP (Ser/Thr to Glu at all SP/TP sites in the flanking region), Tau-17AP (Ser/Thr to Ala at all SP/TP sites in the flanking region) and Tau-8-repeat (Tau without N-terminal inserts and including a duplicated insertion of the four repeats; Gustke et al, 1994) were subcloned into the respective restriction sites of the pShuttle-CMV vector containing Dendra2–Tau. The cloning of CFP-MARK2 and its inactive mutant CFP-MARK2-T208A/S212A was reported previously (Timm et al, 2008). All plasmids were verified by restriction analysis and DNA sequencing.
Cell culture and transfection
Dissociated cortical neurons were prepared from embryonic day 18 (E18) Sprague Dawley rats according to Banker and Goslin (1988) and plated on poly-D-lysine (50 μg/ml)-coated cover glass at a density of 1 × 105 cells/cm2. After 4 days, 0.5 μM cytosine arabinoside (Sigma, Munich, Germany) was added to the cultures to reduce glial growth. Cultures were maintained in NeuroBasal medium with B27 (Invitrogen) at 37°C, 5% CO2, and 100% relative humidity for 7–9 days. Transfection with the plasmids was carried out with Lipofectamine 2000 (Invitrogen). Expression was allowed for 48–72 h.
Immunostaining
Primary cortical neurons were fixed at indicated experimental time with 3.7% paraformaldehyde (Sigma) for 15 min at room temperature and washed with PBS. Cells were then permeabilized with 80% methanol at −20°C for 5 min. This was followed by blocking with 10% goat serum for 30 min at 37°C. After incubation with primary antibodies for 1 h at 37°C, cells were washed with PBS and labelled with secondary antibodies for 1 h at 37°C. The following antibodies were used: monoclonal anti-MAP2 (2a+2b) (AP20) (Sigma) (1:300), monoclonal anti-AnkyrinG (Invitrogen) (1:300), polyclonal SA4473 against tau N-terminal inserts (1:100), polyclonal anti-Dendra2 (Evrogen) (1:500), fluorescently labelled secondary antibodies FITC or TRITC against mouse (1:200), and Cy5 against rabbit (1:200) were from Dianova (Hamburg, Germany).
Imaging, photoconversion, and analysis
A Fluoview1000 confocal microscope (Olympus, Hamburg, Germany) equipped with a SIM scanner (which allows for simultaneous stimulation and imaging) and a × 60 objective live-cell imaging chamber and ZDC system for Z-drift compensation was used for photoconversion experiments and image acquisition. For time-lapse live-cell imaging, cells were plated on glass bottom dishes and were kept in the imaging chamber (37°C, 65% humidity and supplied with 5% CO2). Images were taken every 10 s for the calculation of axonal Tau distribution rate; every 20 s for other applications. Before photoconversion, green fluorescence of Dendra2 was acquired by excitation at 488nm and detection over the range of 500--550nm. Photoconversion was performed during the imaging interval by rapidly illuminating the selected area with a 405-nm laser scanner (3–4 exposures of 0.3 s each). Thereafter, red fluorescence was acquired by excitation at 561nm and detection over the range 570--670nm. Fluorescence intensity profiles and values according to the indicated time points and area were determined by the microscopy software. Microscopic settings as well as the selection of the regions of interest were kept similar in order to allow comparison between different experiments, for example, region for photoconversion of TauD2 ROI1=55 × 35 μm2 containing a 65-μm stretch of the axon and lying at a 50–70-μm distance from the soma, region ROI2 for observing translocated TauD2=30 μm segment of the axon ∼10 μm away from ROI1 (see e.g., Figure 7). Ratiometric analysis was carried out by taking the ratio of red/green channel after photoconversion in the region of interest (ROI), with the aid of Metamorph Software. Identification, imaging, and analysis of photoconverted cells after AnkyrinG staining, and Tau propagation analysis after different transfection periods was done using a Leica DMI 4000 B Metamorph-based microscope equipped with an HQCoolSnap2 camera.
Western blotting
Proteins were separated onto SDS gels and electrotransferred onto polyvinylidene difluoride membranes by semi-dry blotting (1 mA/cm2, 1 h). The membranes were blocked with 5% non-fat dry milk in 1 × Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h at room temperature and then treated with the selected primary antibody (12E8 1:1000, Elan Pharmaceuticals; PHF1 1:500, gift from Peter Davies; AT8 1:500, Thermo Scientific; β-actin 1:1000, Sigma) in 1 × TBST at 37°C for 1 h. The membranes were washed three times with 1 × TBST. The corresponding secondary antibody in 1 × TBST was added, and the membranes were incubated at 37°C for 45 min followed by washing with 1 × TBST (three times). The substrate reaction was carried out with ECL detection reagents (GE Healthcare) and visualized using the LAS 3000 system (Raytest).
Supplementary Material
Acknowledgments
We thank Annika Eikhof and Ilka Lindner for excellent technical assistance and Alexander Marx for help with statistical analysis. The project was supported by grants from EU (FP7-Memosad), BMBF (KNDD), DFG (FOR629), Breuer Foundation and Metlife Foundation.
Author contributions: XL and EMM designed the study; XL performed experiments; JB supplied Tau plasmids; XL, EMM, and EM analysed the data; XL, EMM and EM wrote the paper. HZ and YK conducted experiments for the revisions of the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aronov S, Aranda G, Behar L, Ginzburg I (2001) Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J Neurosci 21: 6577–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronov S, Aranda G, Behar L, Ginzburg I (2002) Visualization of translated tau protein in the axons of neuronal P19 cells and characterization of tau RNP granules. J Cell Sci 115: 3817–3827 [DOI] [PubMed] [Google Scholar]
- Baas PW, Buster DW (2004) Slow axonal transport and the genesis of neuronal morphology. J Neurobiol 58: 3–17 [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer′s disease and related disorders. Nat Rev Neurosci 8: 663–672 [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K (1988) Developments in neuronal cell culture. Nature 336: 185–186 [DOI] [PubMed] [Google Scholar]
- Biernat J, Mandelkow EM (1999) The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol Biol Cell 10: 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI (1985) The distribution of tau in the mammalian central nervous system. J Cell Biol 101: 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burack MA, Silverman MA, Banker G (2000) The role of selective transport in neuronal protein sorting. Neuron 26: 465–472 [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ (2003) Synaptic slaughter in Alzheimer's disease. Neurobiol Aging 24: 1023–1027 [DOI] [PubMed] [Google Scholar]
- Drubin D, Kirschner M (1986) Purification of tau protein from brain. Methods Enzymol 134: 156–160 [DOI] [PubMed] [Google Scholar]
- Garner CC, Brugg B, Matus A (1988) A 70-kilodalton microtubule-associated protein (MAP2c), related to MAP2. J Neurochem 50: 609–615 [DOI] [PubMed] [Google Scholar]
- Gauthier A, Brandt R (2010) Live cell imaging of cytoskeletal dynamics in neurons using fluorescence photoactivation. Biol Chem 391: 639–643 [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8: 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J (2010) Building and maintaining the axon initial segment. Curr Opin Neurobiol 20: 481–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurskaya NG, Verkhusha VV, Shcheglov AS, Staroverov DB, Chepurnykh TV, Fradkov AF, Lukyanov S, Lukyanov KA (2006) Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol 24: 461–465 [DOI] [PubMed] [Google Scholar]
- Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E (1994) Domains of tau protein and interactions with microtubules. Biochemistry 33: 9511–9522 [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer′s amyloid beta-peptide. Nat Rev Mol Cell Biol 8: 101–112 [DOI] [PubMed] [Google Scholar]
- Hirokawa N (1982) Cross-linker system between neurofilaments, microtubules, and membranous organelles in frog axons revealed by the quick-freeze, deep-etching method. J Cell Biol 94: 129–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Funakoshi T, Sato-Harada R, Kanai Y (1996) Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J Cell Biol 132: 667–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68: 610–638 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R (2005) Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6: 201–214 [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J (2010) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer′s disease mouse models. Cell 142: 387–397 [DOI] [PubMed] [Google Scholar]
- Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J (2002) Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci 22: 698–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ (2008) Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci 11: 178–186 [DOI] [PubMed] [Google Scholar]
- Konishi Y, Setou M (2009) Tubulin tyrosination navigates the kinesin-1 motor domain to axons. Nat Neurosci 12: 559–567 [DOI] [PubMed] [Google Scholar]
- Konzack S, Thies E, Marx A, Mandelkow EM, Mandelkow E (2007) Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J Neurosci 27: 9916–9927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 268: 24374–24384 [PubMed] [Google Scholar]
- Kosik KS, Finch EA (1987) MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J Neurosci 7: 3142–3153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Bakalis S, Neve RL (1989) Developmentally regulated expression of specific tau sequences. Neuron 2: 1389–1397 [DOI] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J, Ho H, Brady ST, Sisodia SS (2007) Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer′s disease-linked mutant presenilin 1. J Neurosci 27: 7011–7020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma MD, Dotti CG (2003) Membrane and cytoskeleton dynamics during axonal elongation and stabilization. Int Rev Cytol 227: 183–219 [DOI] [PubMed] [Google Scholar]
- Lichtenberg-Kraag B, Mandelkow EM, Biernat J, Steiner B, Schroter C, Gustke N, Meyer HE, Mandelkow E (1992) Phosphorylation-dependent epitopes of neurofilament antibodies on tau protein and relationship with Alzheimer tau. Proc Natl Acad Sci USA 89: 5384–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI (1995) Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci USA 92: 10369–10373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell JW, Banker GA (1995) The microtubule cytoskeleton and the development of neuronal polarity. Neurobiol Aging 16: 229–237; discussion 238 [DOI] [PubMed] [Google Scholar]
- Matenia D, Mandelkow EM (2009) The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci 34: 332–342 [DOI] [PubMed] [Google Scholar]
- Mattson MP (2004) Pathways towards and away from Alzheimer′s disease. Nature 430: 631–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migheli A, Butler M, Brown K, Shelanski ML (1988) Light and electron microscope localization of the microtubule-associated tau protein in rat brain. J Neurosci 8: 1846–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T, Sobue K (2009) Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J Biol Chem 284: 27734–27745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada C, Ritchie K, Oba Y, Nakamura M, Hotta Y, Iino R, Kasai RS, Yamaguchi K, Fujiwara T, Kusumi A (2003) Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol 5: 626–632 [DOI] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N (2003) Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J Cell Biol 162: 1045–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Rasband MN (2008) The functional organization and assembly of the axon initial segment. Curr Opin Neurobiol 18: 307–313 [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC, Binder LI (1987) Phosphorylation determines two distinct species of Tau in the central nervous system. Cell Motil Cytoskeleton 8: 210–226 [DOI] [PubMed] [Google Scholar]
- Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH (2010) Amyloid-independent mechanisms in Alzheimer′s disease pathogenesis. J Neurosci 30: 14946–14954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN (2010) The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci 11: 552–562 [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316: 750–754 [DOI] [PubMed] [Google Scholar]
- Samsonov A, Yu JZ, Rasenick M, Popov SV (2004) Tau interaction with microtubules in vivo. J Cell Sci 117: 6129–6141 [DOI] [PubMed] [Google Scholar]
- Sanchez-Ponce D, Munoz A, Garrido JJ (2011) Casein kinase 2 and microtubules control axon initial segment formation. Mol Cell Neurosci 46: 222–234 [DOI] [PubMed] [Google Scholar]
- Scott DA, Das U, Tang Y, Roy S (2011) Mechanistic logic underlying the axonal transport of cytosolic proteins. Neuron 70: 441–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song AH, Wang D, Chen G, Li Y, Luo J, Duan S, Poo MM (2009) A selective filter for cytoplasmic transport at the axon initial segment. Cell 136: 1148–1160 [DOI] [PubMed] [Google Scholar]
- Stokin GB, Goldstein LS (2006) Axonal transport and Alzheimer's disease. Annu Rev Biochem 75: 607–627 [DOI] [PubMed] [Google Scholar]
- Sultan A, Nesslany F, Violet M, Begard S, Loyens A, Talahari S, Mansuroglu Z, Marzin D, Sergeant N, Humez S, Colin M, Bonnefoy E, Buee L, Galas MC (2011) Nuclear tau, a key player in neuronal DNA protection. J Biol Chem 286: 4566–4575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies E, Mandelkow EM (2007) Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci 27: 2896–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm T, Balusamy K, Li X, Biernat J, Mandelkow E, Mandelkow EM (2008) Glycogen synthase kinase (GSK) 3beta directly phosphorylates Serine 212 in the regulatory loop and inhibits microtubule affinity-regulating kinase (MARK) 2. J Biol Chem 283: 18873–18882 [DOI] [PubMed] [Google Scholar]
- Utton MA, Connell J, Asuni AA, van Slegtenhorst M, Hutton M, de Silva R, Miller CC, Anderton BH (2002) The slow axonal transport of the microtubule-associated protein tau and the transport rates of different isoforms and mutants in cultured neurons. J Neurosci 22: 6394–6400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ho CL, Sun D, Liem RK, Brown A (2000) Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol 2: 137–141 [DOI] [PubMed] [Google Scholar]
- Weissmann C, Reyher HJ, Gauthier A, Steinhoff HJ, Junge W, Brandt R (2009) Microtubule binding and trapping at the tip of neurites regulate tau motion in living neurons. Traffic 10: 1655–1668 [DOI] [PubMed] [Google Scholar]
- Winckler B, Forscher P, Mellman I (1999) A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature 397: 698–701 [DOI] [PubMed] [Google Scholar]
- Witte H, Bradke F (2008) The role of the cytoskeleton during neuronal polarization. Curr Opin Neurobiol 18: 479–487 [DOI] [PubMed] [Google Scholar]
- Yuan A, Sasaki T, Rao MV, Kumar A, Kanumuri V, Dunlop DS, Liem RK, Nixon RA (2009) Neurofilaments form a highly stable stationary cytoskeleton after reaching a critical level in axons. J Neurosci 29: 11316–11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM (2010) Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci 30: 11938–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.