

Abstract
It has been assumed, based largely on morphologic evidence, that human pluripotent stem cells (hPSCs) contain underdeveloped, bioenergetically inactive mitochondria. In contrast, differentiated cells harbour a branched mitochondrial network with oxidative phosphorylation as the main energy source. A role for mitochondria in hPSC bioenergetics and in cell differentiation therefore remains uncertain. Here, we show that hPSCs have functional respiratory complexes that are able to consume O2 at maximal capacity. Despite this, ATP generation in hPSCs is mainly by glycolysis and ATP is consumed by the F1F0 ATP synthase to partially maintain hPSC mitochondrial membrane potential and cell viability. Uncoupling protein 2 (UCP2) plays a regulating role in hPSC energy metabolism by preventing mitochondrial glucose oxidation and facilitating glycolysis via a substrate shunting mechanism. With early differentiation, hPSC proliferation slows, energy metabolism decreases, and UCP2 is repressed, resulting in decreased glycolysis and maintained or increased mitochondrial glucose oxidation. Ectopic UCP2 expression perturbs this metabolic transition and impairs hPSC differentiation. Overall, hPSCs contain active mitochondria and require UCP2 repression for full differentiation potential.
Keywords: differentiation, metabolism, mitochondria, stem cell
Subject Categories: Development & Differentiation, Metabolism
Short abstract
While studying metabolic fluxes in human pluripotent stem cells, this paper reveals UCP2 as metabolic switch from glycolysis to OXPHOS, facilitating early differentiation events.
Introduction
A distinguishing feature of human pluripotent stem cells (hPSCs) compared with differentiated cells is the capacity for self‐renewal to maintain pluripotency. Self‐renewal is supported by unique chromatin modifications (Meshorer and Misteli, 2006) and a regulatory circuit comprised of OCT4, NANOG, and SOX2 transcription factors (Boyer et al, 2005). hPSCs proliferate relatively fast, with shortened cell cycle times and a higher proportion of cells in S phase compared with most differentiated cell types (Becker et al, 2006). Differences in energy status and biosynthesis from distinct physiologies also distinguish hPSCs from differentiated cells, although the extent and regulation of these differences is unknown. Thus, metabolism could provide a relatively unexplored distinguishing feature between hPSCs and differentiated cells.
Mitochondria are central organelles in carbohydrate, lipid, and amino‐acid metabolism. Studies suggest that there are few, rounded, and non‐fused mitochondria with underdeveloped cristae in human or mouse embryonic stem cells (ESCs) (St John et al, 2005; Cho et al, 2006; Chung et al, 2010; Prigione et al, 2010) Also, hPSCs produce more lactate than differentiated cells (Chung et al, 2010; Prigione et al, 2010), suggesting that glycolysis supercedes oxidative phosphorylation (OXPHOS) and that hPSC mitochondria may be metabolically inactive. However, this important assumption has not been thoroughly tested and it remains unclear how glycolysis and respiration contribute to the hPSC metabolic profile.
Uncoupling proteins (UCPs) are mitochondrial inner membrane proteins that regulate cell metabolism (Nicholls and Rial, 1999; Klingenberg and Echtay, 2001; Brand and Esteves, 2005). UCP1 mediates proton movement from the mitochondrial intermembrane space to the matrix, which uncouples electron transport from ATP synthesis in brown fat to generate heat (Klingenberg and Huang, 1999). In contrast, the function(s) of the widely expressed UCPs, including UCP2 and UCP3, are still controversial (Brand and Esteves, 2005; Bouillaud, 2009). UCP2 and UCP3 show uncoupling activity with in vitro proteoliposome assays (Echtay et al, 2001) or when activated by fatty acids and free radical‐derived alkenals (Considine et al, 2003; Brand et al, 2004a, 2004b). However, UCP2 and UCP3 have not shown physiological uncoupling activity (Cadenas et al, 2002; Couplan et al, 2002). Instead, studies show that UCP2 augments fatty acid or glutamine oxidation and decreases glucose‐derived pyruvate oxidation in mitochondria (Pecqueur et al, 2008, 2009; Bouillaud, 2009; Emre and Nubel, 2010). UCP2 blocking of pyruvate entry into the tricarboxylic acid (TCA) cycle has been postulated but not experimentally validated as a mechanism for enhancing aerobic glycolysis, consistent with UCP2 expression mainly in glycolytic tissues (Pecqueur et al, 2001) and in glycolysis‐switched cancer cells (Pecqueur et al, 2001; Samudio et al, 2008, 2009; Ayyasamy et al, 2011). Therefore, UCP2 could be a gatekeeper for the oxidation of carbon substrates, such as glucose. Notably, no role has been described for UCPs in hPSC bioenergetics to date. Here, we evaluated bioenergetics in human ESCs (hESCs), human‐induced pluripotent stem cells (hIPSCs), and differentiated cells. We report an important role for UCP2 in regulating hPSC energy metabolism and the fate of early differentiating hPSCs.
Results
A conserved mitochondrial mass ratio for hPSCs and differentiated cells
The distribution, abundance, fusion–fission status, and cristae structure of mitochondria regulates O2 consumption, bioenergetics, apoptosis, and autophagy (Frank et al, 2001; Narendra et al, 2008; Chan et al, 2010; Sauvanet et al, 2010). Therefore, the morphology and abundance of hPSC and fibroblast mitochondria were assessed for changes with differentiation or reprogramming. Confocal microscopy with MitoTracker Red CMXRos showed that the mitochondria in hESCs (HSF1 and H1 lines), and to a slightly lesser extent in hIPSCs (HIPS2 and HIPS18 lines), are punctate, or fragmented, compared with the well‐developed filamentous network of normal human dermal fibroblasts (NHDFs, also called fibroblasts) (Figure 1A). hESCs differentiated by bFGF withdrawal also establish a filamentous mitochondrial network within days (Supplementary Figure S1A). Notably, hIPSC lines HIPS2 and HIPS18 were reprogrammed from NHDFs (Lowry et al, 2008), indicating that mitochondrial morphology is reversible with de‐differentiation. Combined, the data show that mitochondrial fusion–fission and network morphology reflect the extent of cell differentiation.
Figure 1.
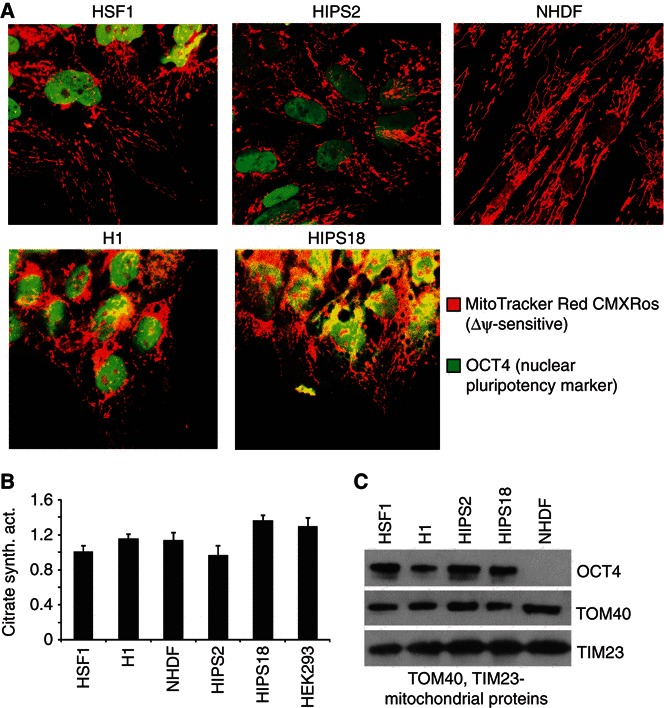
hPSC mitochondrial morphology and abundance. (A) A fragmented hPSC mitochondrial network is shown by confocal microscopy. (B) Ratio of citrate synthase enzyme activity to total cellular protein is plotted, normalized to 1.0 for HSF1. Data are expressed as mean±s.d. (n=3). (C) Representative western blot with 50 μg protein loaded per lane.
The F1F0 ATP synthase appears to play a scaffolding role for the mitochondrial inner membrane cristae structure and impacts OXPHOS activity and cellular ATP levels (Giraud et al, 2002; Paumard et al, 2002; Minauro‐Sanmiguel et al, 2005; Buzhynskyy et al, 2007; Campanella et al, 2008; Strauss et al, 2008). Therefore, the cristae structure of hPSCs was examined as a potential indicator of mitochondrial function. Transmission electron microscopy shows that hPSC cristae are swollen, circular, and disorganized compared with the linear, stacked cristae observed in many differentiated cell types, including fibroblasts (Supplementary Figure S1B). These features are similar to those seen in mouse ESCs and for a different set of hPSCs (Baharvand and Matthaei, 2003; Prigione et al, 2010). Less orderly cristae and a fragmented network suggests that PSC mitochondria are less functional than differentiated cell mitochondria.
Prior studies reported few mitochondria in hESCs (St John et al, 2005; Facucho‐Oliveira et al, 2007). However, the ratio of mitochondrial to total cell protein mass is a more informative comparison between cell types and stages. Citrate synthase marks the mitochondrial matrix (Morgunov and Srere, 1998) and is commonly used as an indicator of mitochondrial function and mass. Citrate synthase enzymatic activity normalized to total protein is similar between hPSCs, hPSCs induced to differentiate by retinoic acid (RA) (Pan et al, 2007), and fibroblasts (Figure 1B; Supplementary Figure S1C). Also, the translocons of the inner (TIM23) and outer (TOM40) mitochondrial membranes accurately reflect mitochondrial mass and both are expressed at similar levels relative to the total protein content in hPSCs, RA‐differentiated hPSCs, and fibroblasts (Figure 1C; Supplementary Figure S1D). Fibroblast cell diameters are larger (∼25–35 μm) and cell volumes (assuming a spherical shape) are much larger (∼16 000 μm3) than hPSCs (∼15–20 μm and ∼3000 μm3, respectively) (Supplementary Figure S1E). Fibroblasts also have 1.5‐ to 2‐fold more protein than hPSCs, suggesting on a per cell basis there will be more mitochondrial mass in fibroblasts than in hPSCs, as reported (St John et al, 2005; Facucho‐Oliveira et al, 2007). However, a similar mitochondrial mass per unit protein for hESCs, hIPSCs, and fibroblasts indicates that hPSCs and differentiated cells are equally committed to generating or maintaining mitochondria.
hPSC and differentiated cell mitochondria show similar O2 consumption rates
hPSCs were examined to determine how much O2 they consume. O2 consumption rate (OCR) studies showed that intact hPSCs consume O2 in room air at ∼5–8 nmol O2/min/5 × 106 cells (or ∼1.0–1.6 fmol/min/cell) (Figure 2A). By contrast, fibroblasts consume O2 at ∼13–15 nmol O2/min/5 × 106 cells (or ∼2.6–3.0 fmol/min/cell) in room air, an ∼2‐fold higher rate than hPSCs. However, because there is ∼1.5‐ to 2‐fold more mitochondrial mass per fibroblast than hPSC, the OCR normalized to mitochondrial mass is ∼equivalent between fibroblasts and hPSCs.
Figure 2.
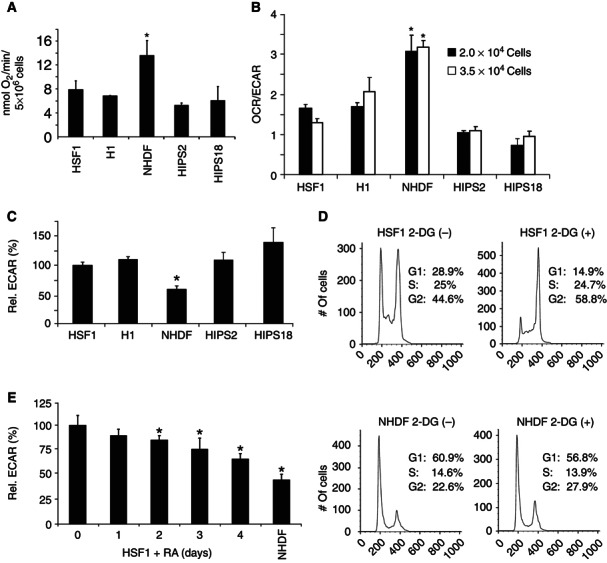
hPSCs consume O2 and are programmed for glycolysis. (A) OCR in room air measured with a Clark‐type O2 electrode in intact cells. Data are expressed as mean±s.d. (n=3), * P<0.05. (B) OCR to ECAR ratios measured by the XF24 extracellular flux analyser is shown. Data are expressed as mean±s.d. (n=3), * P<0.005. (C) ECAR normalized to protein concentration is shown. Data are expressed as mean±s.d. (n=3), * P<0.05. (D) Cells were incubated with 150 mM 2‐DG for 45 min, followed by flow cytometry and cell‐cycle profile analyses. (E) HSF1 cells were induced to differentiate by 10 μM RA for 4 days and ECAR determined. Data are expressed as mean±s.d. (n=3), * P<0.05.
Electron transport chain (ETC) complexes I–IV transfer electrons from substrates to O2 and the activity of each complex can vary between cell types. Substrate feeding into each ETC complex was performed with digitonin‐treated HSF1 and HEK293 cells to determine relative complex activities. HEK293 cells were chosen because of a similar mitochondrial content (Figure 1B) and size (Supplementary Figure S1E) relative to hPSCs and for ease of substrate feeding compared with fibroblasts. Results show that ETC complex I–IV activities are similar between HSF1 and HEK293 cells (Supplementary Figure S2A) and, overall, the data indicate that hPSCs have a functional respiratory chain and OCR similar to fibroblasts when normalized to mitochondrial mass.
hPSCs are programmed for glycolysis
Different cell types in distinct microenvironments rely upon different bioenergetic processes for ATP production (Jezek et al, 2010). hPSCs and fibroblasts were therefore analysed for respiration and glycolysis in the same environment. To do this, OCR and the extracellular acidification rate (ECAR), which can approximate glycolysis from lactate production (Xie et al, 2009), were determined using a XF24 Extracellular Flux Analyser (Wu et al, 2007). The XF24 measures the rates of change in pmol O2 and mpH simultaneously for cultured cells, providing a comparison for a defined cell population. For accurate analyses, hPSCs were plated with ROCK inhibitor (Watanabe et al, 2007) to form monolayers. Control or ROCK inhibitor‐treated fibroblasts showed no statistical difference in the OCR/ECAR ratio (Supplementary Figure S2B). The number of cells seeded per well was adjusted for the linear range of measurements (Supplementary Figure S2C and D).
The OCR/ECAR ratios at two cell densities for hESCs and hIPSCs are ∼50–65% lower than for fetal‐derived NHDFs (Figure 2B) and ECAR values for hPSCs are ∼1.5‐ to 2‐fold higher than for fibroblasts (Figure 2C). Analysis of adult‐origin fibroblasts (NHDFs) showed results that were statistically similar to those obtained with fetal‐derived fibroblasts (Supplementary Figure S2E). These data suggest that hPSC bioenergetics favours glycolysis over OXPHOS, in contrast to fibroblasts. Addition of the competitive glycolysis inhibitor, 2‐deoxyglucose (2‐DG), causes hPSCs to G2 arrest (Figure 2D) and to minimally increase apoptosis (Supplementary Figure S2F) rather than shifting to respiration in room air, suggesting an immutable energetic preference. To determine a glycolytic transition with differentiation, HSF1 cells were exposed to RA and ECAR measured over time. Results showed a gradual and significant decrease in glycolysis with differentiation, with decreased ECAR (Figure 2E) and lactate in the culture media (Supplementary Figure S2G), indicating a progressive metabolic reprogramming during differentiation. Notably, hPSC OCRs and ECARs are mainly unaffected by 5–25 mM glucose (Supplementary Figure S2H) or by 0–17.5 mM insulin (Supplementary Figure S2I) incubations, indicating that the glucose supplied in these studies is not limiting. Furthermore, glucose transporters GLUT1 (widely expressed) and GLUT4 (insulin‐regulated) are expressed at ∼equivalent high or barely detectable levels in hPSCs and fibroblasts, respectively (Supplementary Figure S2J and K), indicating an equivalent ability to bind and uptake glucose. Combined, the data strongly suggest that differences in OCRs and ECARs between hPSCs and differentiated cells are not from differences in glucose uptake but rather from unique intracellular metabolic programmes.
hPSCs are more sensitive to glycolysis inhibitors than are differentiated cells
OXPHOS was inhibited with oligomycin, a F1F0 ATP synthase inhibitor. Fibroblasts showed an ∼80% reduction in O2 consumption within 10 min (Figure 3A). By contrast, hPSCs showed an ∼50% decrease in OCR, suggesting that hPSC are less dependent on OXPHOS than fibroblasts. Notably, there is a greater compensatory increase in ECAR for differentiated cells than for hPSCs with oligomycin exposure (Supplementary Figure S3A). Supplementary glucose (from 2.5 mM to 25 mM) maximized ECAR, with fibroblasts increasing ∼55% in 20 min, whereas hPSC ECAR increased by a more modest ∼20–30% (Figure 3B), indicating that hPSCs are closer to their maximal glycolytic capacity compared with fibroblasts. Following additions of 2‐DG, which competes for uptake with glucose, hPSC glycolysis was repressed ∼65% from the ECAR maximum within 34 min, whereas fibroblast glycolysis did not change for 6 min and then fell by only ∼45% from the ECAR maximum after 34 min (Figure 3B), indicating that the hPSC glycolysis level is greater than in fibroblasts. Combined, the data indicate that hPSCs are less sensitive to OXPHOS inhibition, more sensitive to glycolysis disruption, and function closer to their maximum glycolytic capacity than do fibroblasts.
Figure 3.
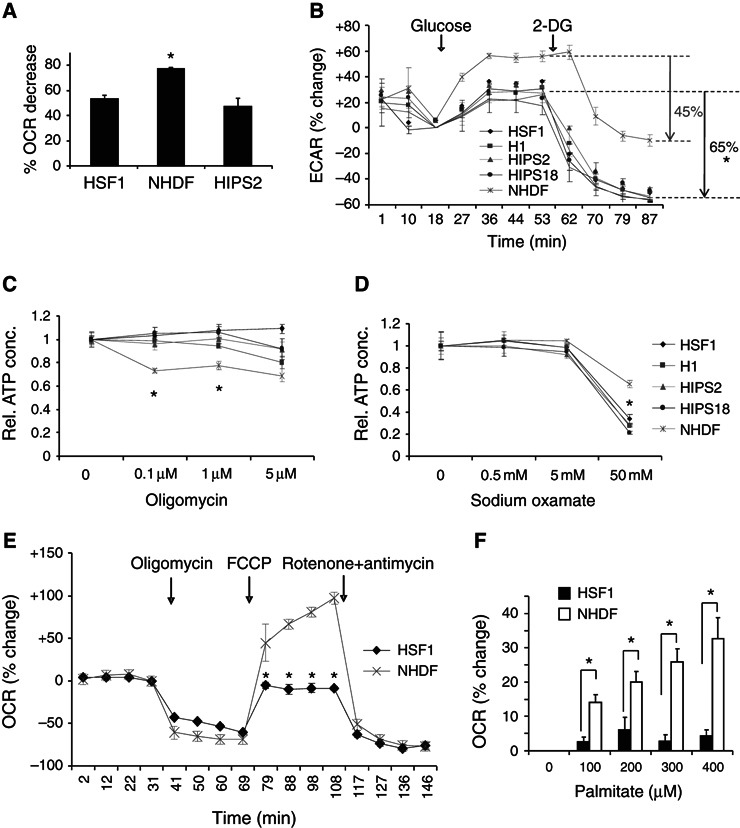
hPSCs require glycolysis for ATP production despite maximal O2 consumption. (A) OCR change with 1 μM oligomycin for 10 min. Data are expressed as mean±s.d. (n=3), * P<0.05. (B) Cells were cultured in assay medium with 2.5 mM glucose. ECAR change with glucose (25 mM final) and 2‐DG (150 mM final) additions is shown. Arrows indicate percentage reduction from maximal ECAR (n=3), * P<0.05. (C) Change in cellular ATP with increasing doses of oligomycin at 45 min (n=3), * P<0.05. (D) Change in cellular ATP with increasing doses of sodium oxamate at 45 min (n=3), * P<0.05. (E) OCR changes in response to F1F0 ATP synthase inhibitor (1 μM oligomycin), uncoupler (0.3 μM FCCP), and ETC blockade (1 μM rotenone plus 1 μM antimycin) (n=3), * P<0.05. OCR due to non‐mitochondrial sources is the distance between the post‐rotenone+antimycin OCR (time points 127–146 min) and the abscissa. (F) OCR change in response to increasing doses of palmitate (n=3), * P<0.05.
ATP in hPSCs is mainly produced by glycolysis
The steady‐state ATP level is ∼2‐fold higher per cell in fibroblasts than in hPSCs, reflecting ATP production, consumption, and the size difference between these two cell types (Supplementary Figure S3B). OXPHOS inhibition with 0.1 μM oligomycin caused an ∼30% ATP drop in fibroblasts, whereas the ATP drop in hPSCs was <5% (Figure 3C), suggesting that hPSCs produce minimal ATP by OXPHOS. By contrast, sodium oxamate, a lactate dehydrogenase inhibitor that blocks ATP production by glycolysis, caused the ATP level in hPSCs to drop >60%, whereas fibroblasts showed <40% ATP reduction (Figure 3D), indicating that hPSCs depend more on glycolysis for ATP production than do fibroblasts. ATP reductions with short‐term inhibitor exposures are not due to a significant increase in cell death (Supplementary Figure S3C). Combined, the data indicate that the ATP level in hPSCs is sensitive to glycolysis inhibition.
hPSCs consume O2 at maximal capacity
Although hPSC and fibroblast mitochondria consume O2 at an ∼equivalent rate, it is unclear what proportion of their maximal electron transport capacity is being utilized. FCCP is a mitochondrial uncoupler (protonophore) that dissipates the mitochondrial membrane potential (Δψ) to stimulate maximal electron transport and O2 consumption (Heytler, 1979). hPSCs failed to increase OCR, whereas fibroblasts showed an ∼90–100% increase in OCR at 0.1–0.4 μM FCCP exposures (Supplementary Figure S3D). To establish a baseline at which coupled respiration is inhibited, HSF1 cells and fibroblasts were exposed to 1 μM oligomycin to prevent proton movement through the F1F0 ATP synthase (Figure 3E). No significant changes in cell viability were detected during the course of oligomycin exposure (Supplementary Figure S3C). Addition of 0.3 μM FCCP re‐establishes proton movement and results in maximal O2 consumption. Then, incubation with rotenone and antimycin (complex I and III inhibitors, respectively) completely blocked mitochondrial respiration. The maximal mitochondrial respiration capacity is the difference between blocked and FCCP‐induced OCR. Maximal hPSC respiration is equivalent to its pre‐oligomycin respiration, which contrasts with the up to ∼100% increase in OCR measured for fibroblasts (Figure 3E). OCR does not increase further for hPSCs with increased sodium pyruvate supplemented media and FCCP exposure, excluding limited substrate as a cause for an hPSC ceiling (Supplementary Figure S3E). Because oligomycin inhibits and FCCP stimulates OCR in coupled mitochondria, the smaller difference between FCCP‐stimulated and oligomycin‐inhibited OCR also suggests that hPSC mitochondria could be less coupled for ATP synthesis than fibroblast mitochondria (Supplementary Figure S3E). Finally, free fatty acid palmitate, an OXPHOS substrate, was added to cells incubated with l‐carnitine, which transports fatty acids from the cytosol into the mitochondria. In contrast to fibroblasts that showed a dose‐dependent OCR increase of ∼35%, HSF1 cells showed only a dose‐independent ∼5% OCR increase (Figure 3F). The lack of a robust free fatty acid response in HSF1 cells could be from ETC saturation with glucose or internally produced fatty acids. Notably, the essential enzymes for fatty acid oxidation are equally expressed in HSF1 cells and fibroblasts by QPCR analysis. Combined, these data show that hPSCs consume O2 at maximal capacity.
Glycolytic ATP hydrolysis helps to maintain Δψ in hPSCs
A reduction in Δψ has been linked to mitochondrial dysfunction and possible mitophagy (Narendra et al, 2008). The presence of PARKIN protein expression in HSF1 cells that increases with RA‐induced differentiation is consistent with the need to maintain Δψ in hPSCs (Supplementary Figure S4A), although this suggestion requires further investigation (Narendra et al, 2008, 2010). In differentiated cells, Δψ is maintained by a proton gradient formed during the passage of electrons through the ETC. Alternatively, Δψ is maintained by hydrolysis of glycolytic ATP in the F1F0 ATP synthase when ETC function is reduced (Nicholls and Lindberg, 1972; Hatefi, 1985). For mouse ESCs, the relative level of Δψ has a role in regulating differentiation (Schieke et al, 2008). Therefore, the mechanism(s) for hPSC Δψ maintenance was examined. Immunofluorescence microscopy showed an intact Δψ for all HSF1 mitochondria imaged that was dissipated with 100 μM FCCP (Supplementary Figure S4B). To determine what portion of Δψ is provided by electron transport versus hydrolysis of glycolytic ATP, antimycin was used to dissipate the ETC‐generated component of Δψ and sodium oxamate was used to dissipate the glycolysis‐generated component of Δψ, followed by quantification using Δψ‐sensitive TMRM staining and flow cytometry. Antimycin and FCCP equivalently and robustly dissipate Δψ in fibroblasts, whereas antimycin only partially dissipates Δψ in hPSCs (Figure 4A; Supplementary Figure S4C, with JC‐1 staining). Sodium oxamate partially dissipates Δψ in hPSCs but had no effect on Δψ in fibroblasts (Figure 4B). Combined, the data indicate that Δψ in fibroblasts is maintained by the ETC, whereas Δψ in hPSCs is partially maintained by glycolysis.
Figure 4.
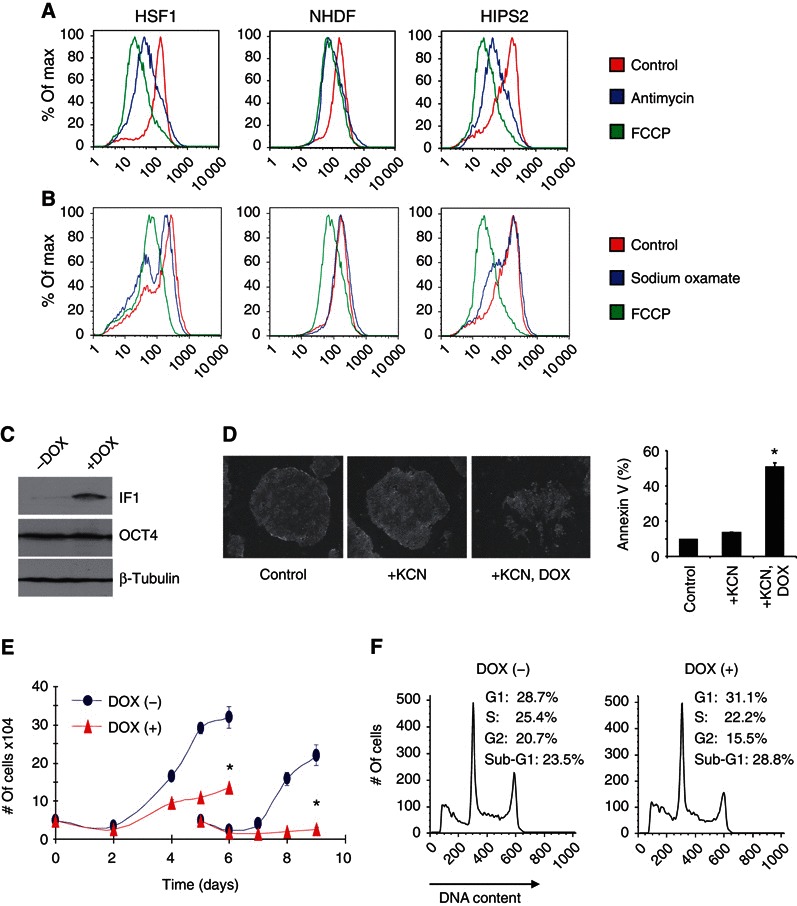
ATP hydrolysis maintains Δψ, viability, and proliferation of hPSCs. (A) Dissipation of Δψ with antimycin (10 μM, 10 min) or FCCP (0.3 μM, 45 min) determined by Δψ‐sensitive TMRM dye staining and flow cytometry (n=2). (B) Dissipation of Δψ with sodium oxamate (50 mM, 45 min) or FCCP analysed as described for (A). (C) Representative western blot of H1 hESCs transduced with a DOX‐inducible IF1 lentiviral expression construct, with or without 2 μg/ml DOX for 3 days. (D) Representative colony morphology and percent Annexin V‐positive cells for DOX‐inducible IF1‐containing H1 hESCs, with or without DOX and 1 mM KCN at 3 days. Data are expressed as mean±s.d. (n=3), * P<0.05. (E) Cell counts of DOX‐inducible IF1‐containing H1 cells, with or without DOX. In all, 5 × 104 cells were plated after 3 days of DOX. Live cells were counted every other day. Cells were split at 4 days, re‐plated at 5 × 104 cells/well, and counted until 9 days. Data are expressed as mean±s.d. (n=3), * P<0.05. (F) Representative cell‐cycle analysis of DOX‐inducible IF1‐containing H1 hESCs, with or without DOX at 3 days (n=3).
In differentiated cells with an impaired ETC, ATP hydrolysis maintains Δψ, proliferation, and viability (Buchet and Godinot, 1998). Conversion of F1F0 ATP synthase to hydrolase activity is blocked by inhibitory factor‐1 (IF1) (Campanella et al, 2008). Notably, the ratio of IF1 to the F1F0 ATP synthase β‐subunit protein, which is the target of IF1 (Ichikawa et al, 2005), is significantly lower in hPSCs than in differentiated cells, which is suggestive of a potential ATP hydrolase function for the F1F0 ATP synthase in hPSCs (Supplementary Figure S4D). To investigate this possibility, a doxycycline‐inducible IF1 lentiviral expression construct was transduced into H1 hESCs (Figure 4C), followed by ETC impairment using KCN, a complex IV inhibitor. hPSCs with ectopic IF1 expression showed abundant cell death in KCN (Figure 4D). Notably, ectopic IF1‐expressing H1 cells with intact ETC activity in room air also showed a significant growth defect in extended culture (Figure 4E) due to increases in G0/G1 phase of the cell cycle and apoptosis (Figure 4D and F). Combined, the data suggest that the ATP hydrolase activity of the F1F0 ATP synthase is essential for maintaining the Δψ, proliferation, and viability of hPSCs.
UCP2 is expressed in hPSCs and repressed during differentiation
To identify a mechanism(s) controlling the unique mitochondrial metabolism of hPSCs, we re‐analysed three expression profiling studies to search for metabolism‐regulating genes that were differentially expressed between hPSCs and differentiated cells. Two studies compared gene expression between hPSCs and fibroblasts (Lowry et al, 2008; Maherali et al, 2008) and a third study evaluated gene expression with hESC differentiation to embryoid bodies (EBs) and cardiomyocytes (Cao et al, 2008). Notably, UCP2 expression was 5‐ to 10‐fold higher in hPSCs than in human fibroblasts (Figure 5A; Supplementary Figure S5A) and ∼30–40% lower in EBs and cardiomyocytes than in hESCs (Supplementary Figure S5B). Validating these in‐silico data, UCP2 gene and protein expression was significantly higher in hPSCs than in differentiated cells (Figure 5B–D; Supplementary Figure S5C). HSF1 and HIPS2 cells were stimulated to differentiate by medium replacement (20% KSR → 20% FBS) (Figure 5D) or HSF1 cells were induced to differentiate by replacing bFGF with 10 μM RA (Figure 5E). UCP2 was repressed in both settings. In contrast to UCP2, UCP1 and UCP3 expression was very low or absent in hPSCs (Figure 5F). UCP2 and UCP3 typically account for 0.01–0.1% of mitochondrial inner membrane proteins, whereas UCP1, whose expression is confined to brown adipose tissue cells, accounts for up to 10% of the inner membrane protein mass (Brand and Esteves, 2005). Comparatively abundant UCP2 expression suggests potential functional significance in hPSCs.
Figure 5.
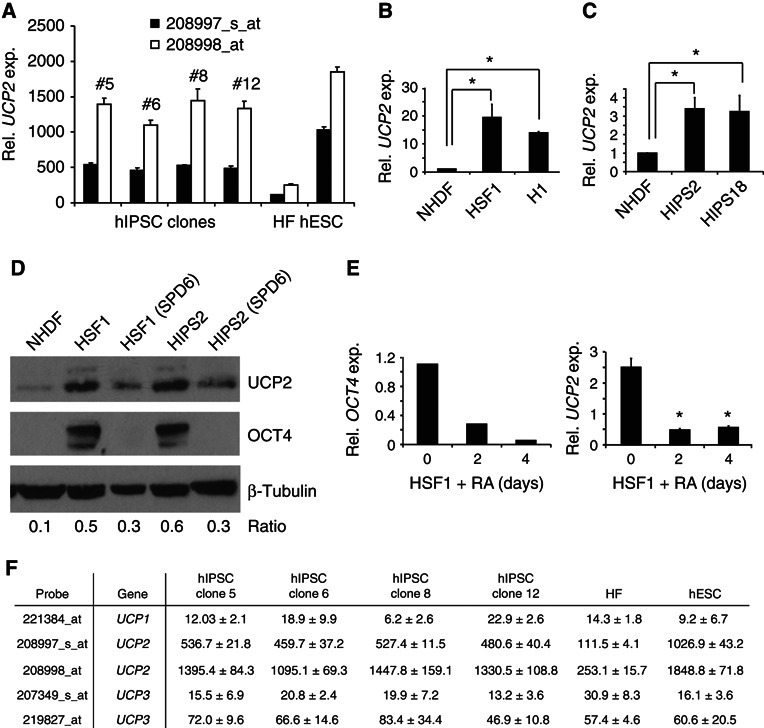
UCP2 is highly expressed in hPSCs and is repressed during differentiation. (A) In‐silico analyses of UCP2 expression from expression profiling studies (Maherali et al, 2008). Two probes detecting UCP2 RNA are shown. HF, human fibroblasts. (B, C) UCP2 expression by QPCR (n=3), * P<0.05. (D) Western blots under native conditions and conditions that promote hPSC differentiation. SPD6: media change‐induced differentiation by replacing 20% KSR with 20% FBS for 6 days. (E) OCT4 and UCP2 expression by QPCR with 10 μM RA‐induced differentiation for 4 days (n=3), * P<0.05. (F) In‐silico analysis of UCP1, UCP2, and UCP3 expression from expression profiling studies in the same lines as in (A) (Maherali et al, 2008).
UCP2 is a regulator of hPSC energy metabolism
UCP2 stimulates aerobic glycolysis in leukaemia cells (Samudio et al, 2008, 2009; Bouillaud, 2009) and one hypothesized, but experimentally untested mechanism for UCP2 activity based on UCP1 studies is to shunt pyruvate out of mitochondria, thereby reducing glucose oxidation in mitochondria and increasing glycolytic flux (Bouillaud, 2009; Samudio et al, 2009). UCP2 expression in hPSCs suggests that it could regulate hPSC glucose metabolism. To test this hypothesis, shUCP2 HSF1 cells were generated. shUCP2 cells retained OCT4 (Figure 6A) and SSEA4 (Supplementary Figure S6A) pluripotency markers with an ∼33% decrease in ECAR (Figure 6B) and an ∼40% decrease in cellular ATP (Figure 6C), indicating reduced metabolic activity compared with control cells without affecting the expression of OCT4. A similar decrease in ECAR occurred with a second shUCP2 construct in HSF1 cells (Supplementary Figure S6B). shUCP2 also reduced ECAR in highly proliferative HEK293 cells that express UCP2 at a level similar to hPSCs, suggesting a conserved metabolic regulatory mechanism for UCP2‐expressing cells (Supplementary Figure S6C and D).
Figure 6.
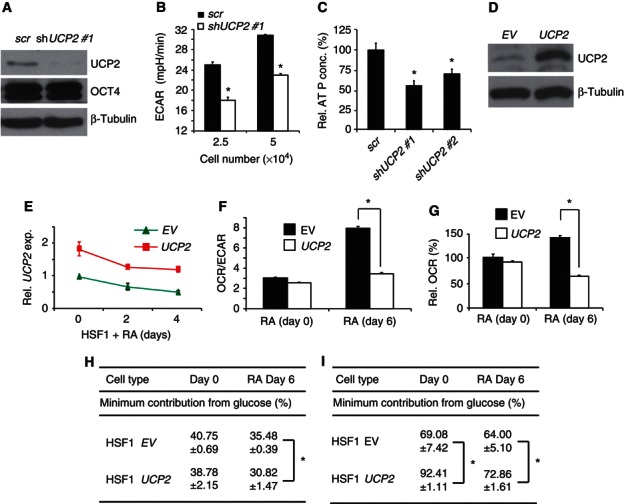
UCP2 regulates energy metabolism in hPSCs and during differentiation. (A) Western blots in HSF1 cells transduced with shUCP2 #1 and a scramble (scr) control. (B) ECAR for shUCP2 #1 and scr HSF1 cells (n=3), * P<0.05. (C) ATP normalized for HSF1 cell number with shUCP2 knockdown (n=3), * P<0.05. (D) UCP2 immunoblot in HSF1 cells transduced with empty vector and UCP2‐expressing lentiviruses. (E) UCP2 expression by QPCR in EV and UCP2 transduced HSF1 cells with 10 μM RA‐induced differentiation for up to 4 days (n=3). (F, G) OCR/ECAR and OCR for EV or UCP2 transduced HSF1 cells exposed to 10 μM RA for up to 6 days (n=3–7), * P<0.005. (H) Stable isotope tracing analysis. hPSCs were grown in StemPro SFM medium (17.5 mM glucose) supplemented with 7.5 mM [U13C6]‐labelled glucose for 48 h. ∑mn (average 13C/molecule) was calculated from the mass isotopomer distribution in glutamate. Minimum contribution from glucose was calculated through dividing the observed ∑mn glutamate by the theoretical ∑mn glutamate (7.5/25 × 4), when all glucose carbons become ∑mn carbons in the C2–C5 fragment of glutamate. Data are expressed as mean±s.d. (n=3), * P<0.02. (I) Stable isotope tracing analysis with cells grown as in (H), ∑mn was calculated from the mass isotopomer distribution in ribose. The minimum contribution from glucose was calculated through dividing the observed ∑mn ribose by the theoretical ∑mn ribose (7.5/25 × 5). Data are expressed as mean±s.d. (n=3), * P<0.05.
UCP2 has been postulated to block glucose‐derived pyruvate oxidation in mitochondria (Jezek and Garlid, 1990; Jezek and Borecky, 1998; Bouillaud, 2009). Therefore, UCP2 repression could maintain or enhance mitochondrial oxidation of glucose in differentiated cells. To test this idea, UCP2 was ectopically expressed during early hPSC differentiation (Figure 6D and E). HSF1 cells exposed to RA for 6 days showed a >2‐fold increase in the OCR/ECAR ratio, indicating a transition from glycolysis to mitochondrial glucose oxidation, whereas ectopic UCP2 expression blocked this transition as shown by an almost unchanged OCR/ECAR ratio before and after differentiation (Figure 6F). Interestingly, UCP2 expression repressed OCR in differentiated cells, consistent with UCP2 blocking mitochondrial glucose oxidation (Figure 6G). In order to further test this hypothesized mechanism for UCP2 regulation of metabolism, stable isotope tracing studies were performed to determine whether UCP2 directly prevents pyruvate from entering into mitochondrial oxidation. Cells were incubated in media supplemented with [U13C6]‐labelled glucose, during which the expression of UCP2 and pluripotent transcription factors were unaltered (Supplementary Figure S6E). Labelled glucose is converted to pyruvate, which then enters mitochondria and is further oxidized into different mass isotopomers of TCA cycle intermediates, such as α‐ketoglutarate (Lee et al, 2010). Since α‐ketoglutarate is in equilibrium with glutamate, the isotopomers of glutamate were used to measure the amount of labelled glucose contributing to mitochondrial oxidation (Boren et al, 2003). Ectopic UCP2 expression had no significant effect on glucose oxidation in undifferentiated hPSCs (Figure 6H), possibly because of already high UCP2 expression (Figure 5). By contrast, a significant ∼13% decrease in 13C incorporation into glutamate (Σmn) was detected in RA‐differentiated hPSCs (Figure 6H), indicating UCP2 impairs glucose oxidation. Notably, RA‐induced differentiation also caused an ∼13% decrease in glucose oxidation, likely from reduced proliferation and resultant lowered overall metabolism with differentiation (see Supplementary Figure S7A). Ectopic UCP2 expression also increased the amount of ribose generated from glucose by ∼30% in undifferentiated hPSCs and by ∼12% with hPSC differentiation (Figure 6I), strongly supporting a role for UCP2 expression in shunting pyruvate away from oxidation in mitochondria and towards the pentose phosphate pathway via an increased glycolytic flux. Combined, the data show that UCP2 repression during differentiation is required to transition from glycolysis to glucose oxidation in mitochondria.
To determine whether UCP2‐mediated uncoupling contributes to this metabolic transition, the effect of oligomycin and FCCP was examined in UCP2 knockdown and ectopic‐expression HSF1 cells. Neither manipulation altered responses to oligomycin nor FCCP compared with control hPSCs (Supplementary Figure S6F). Ectopic UCP2 expression also did not increase OCR (Figure 6G) as uncouplers do. The data suggest that UCP2 does not function like UCP1 with its established uncoupling activity, but rather UCP2 helps to regulate hPSC bioenergetics by controlling substrate access to oxidation in mitochondria.
UCP2 impacts ROS and differentiation potential of hPSCs
Reactive oxygen species (ROS) are respiration by‐products that cause oxidative damage to proteins, lipids, and DNA. ROS also increases with hPSC differentiation (Cho et al, 2006; Saretzki et al, 2008). In immune cells, UCP2 attenuates ROS accumulation (Echtay et al, 2001). To evaluate UCP2 in hPSC ROS regulation, ROS was measured in control and shUCP2 HSF1 cells. The data show that ROS was substantially increased with shUCP2 (Figure 7A) and resulted in elevated hPSC apoptosis (Figure 7B). Ectopic UCP2‐expressing HSF1 cells showed decreased ROS compared with control cells (Figure 7C, panels i, iv, and vii). By contrast, HSF1 cells with or without ectopic UCP2 expression induced to differentiate for 2 days by media‐replacement (20% KSR → 20% FBS, or spontaneous differentiation day 2 (SPD2)) showed increased ROS accumulation (Figure 7C, panels iii and vi). However, ectopic UCP2‐expressing HSF1‐differentiated cells show less ROS than differentiated control cells (Figure 7C, panel viii), indicating that UCP2 repression with differentiation contributes to ROS accumulation.
Figure 7.
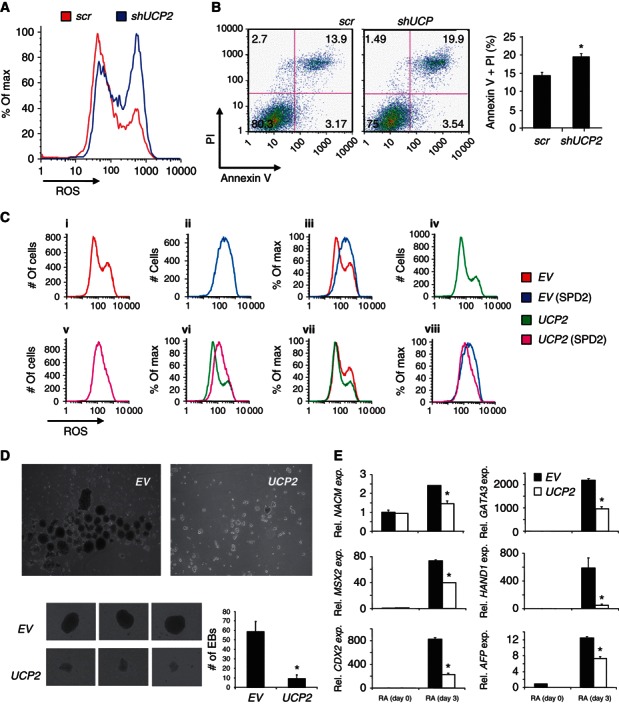
UCP2 regulates ROS, hPSC viability, and differentiation. (A) Representative ROS determination in transduced HSF1 cells by 2‐dihydroethidium staining and flow cytometry (n=3). (B) Apoptosis staining and flow cytometry. Data are plotted in the right panel. Data are expressed as mean±s.d. (n=3), * P<0.05. (C) ROS determination in transduced HSF1 cells with or without differentiation induction by media replacement for 2 days (SPD2). Panel (iii) is an overlay of (i, ii); panel (vi) is an overlay of (iv, v); panel (vii) is an overlay of (i, iv); and panel (viii) is an overlay of (ii, v). (D) EB formation assay with transduced HSF1 cells. Data are expressed as mean±s.d. (n=3), * P<0.05. (E) mRNA expression of developmental genes in transduced HSF1 cells. Data are expressed as mean±s.d. (n=3), * P<0.05.
The data thus far show that UCP2 helps to regulate metabolic reprogramming during hPSC differentiation. Whether this regulation contributes to hPSC differentiation is unknown. To address this critical question, EB formation, as a measure of differentiation, was determined with control and ectopic UCP2‐expressing HSF1 cells. Strikingly, ectopic UCP2 expression caused significant defects in EB quantity and quality (Figure 7D). Ectopic UCP2 expression did not alter the expression of developmental genes representing the three‐germ layers of the definitive embryo or the trophoblast lineage in hPSCs (Figure 7E) as they are all lowly expressed before differentiation. By contrast, UCP2 significantly impaired the induction of these developmental genes in hPSCs induced to differentiate with RA (Figure 7E) without affecting cell proliferation or viability (Supplementary Figure S7A). Notably, addition of ROS scavengers such as N‐acetyl‐cysteine or glutathione did not change the repression of differentiation from ectopic UCP2 expression (Supplementary Figure S7B and C), strongly suggesting that ROS is not the main factor in UCP2 regulation of hPSC differentiation. Instead, the data show that UCP2 is a metabolic regulator of hPSC differentiation and its repression facilitates a metabolic transition favouring oxidative metabolism, which is required for the proper early differentiation of hPSCs.
Discussion
Little is known and much is assumed about hPSC energy production and utilization. Here, we show that hPSCs are ‘hardwired’ for metabolic activity that is heavily skewed towards glycolysis. ATP from glycolysis is used to meet energy and biosynthetic demands and to maintain Δψ in support of cell proliferation and viability. Despite their underdeveloped appearance, hPSC mitochondria contain functional ETC complexes and consume O2 maximally, although they rely mainly on glycolytic ATP compared with differentiated cells, such as fibroblasts. There is a shift from glycolysis to OXPHOS that becomes more pronounced as hPSCs progressively differentiate. UCP2 expression is repressed with hPSC differentiation, and this repression is necessary for a proper shift from glycolysis to mitochondrial glucose oxidation. Ectopic UCP2 expression during early differentiation impairs this metabolic transition, retards ROS accumulation, and inhibits EB formation and differentiated gene induction. These unique metabolic features are conserved in multiple hESC and hIPSC lines, suggesting that hPSC energy metabolism is an underappreciated pre‐set programme and a core component of ‘stemness’, similar to the epigenetic and genetic programmes that maintain hPSC self‐renewal (Boyer et al, 2005; Meshorer and Misteli, 2006).
Support for an hPSC metabolic programme comes from several lines of evidence. First, glycolysis is the main ATP production mechanism, regardless of O2 availability, with hPSCs grown in ambient O2 utilizing glycolysis in preference to OXPHOS outside of the hypoxic blastocyst inner cell mass (Fischer and Bavister, 1993). Second, hPSC mitochondria hydrolyze glycolytic ATP to partially maintain Δψ, which is required for cell proliferation and survival. By contrast, differentiated cell mitochondria do not hydrolyze appreciable glycolytic ATP unless their ETC becomes compromised (Buchet and Godinot, 1998; Campanella et al, 2008). hESCs in room air with intact ETC functions show reduced proliferation and viability with IF1‐induced F1F0 ATP hydrolase inhibition, rather than adapting to OXPHOS to maintain Δψ. Third, hPSCs proliferate rapidly compared with most differentiated cell types (Supplementary Figure S7D) (Becker et al, 2006), and they may require a high glycolytic flux to support anabolic metabolism. Glycolysis provides intermediate metabolites such as ribose and NADPH from the pentose phosphate pathway for nucleotide and lipid biosynthesis (DeBerardinis et al, 2008). In addition to a gradual decrease in glycolysis with differentiation (Figure 2E), QPCR showed that most key enzymes in the pentose phosphate pathway and lipid biosynthesis pathways are repressed during media‐replacement or RA‐induced differentiation (Supplementary Figure S7E and F). Finally, hESCs typically die or differentiate with oxidative stress (Saretzki et al, 2008) or the DNA damage that results from oxidative stress (Lin et al, 2005). Mouse ESCs and hESCs also show elevated ROS with differentiation, and ROS is a known DNA mutagen (Sauer et al, 2000; Cho et al, 2006). The impact of UCP2 on hPSC metabolism provides a potential mechanism by which hPSCs reduce ROS to maintain genomic integrity and cell viability.
Distinct bioenergetic metabolism in hPSCs and during early differentiation or reprogramming suggests a coordinated molecular switching mechanism of unknown origin. Here, we show that UCP2 is a molecular regulator of hPSC energy metabolism, although it is most likely one of the several thus far unidentified regulators of hPSC metabolism. UCP2 knockdown shifts hPSC bioenergetics away from glycolysis, whereas ectopic UCP2 expression blocks glucose oxidation. In Supplementary Figure S7G, we provide a simple model for our study results and the mechanism for UPC2 regulation of hPSC metabolism based upon the study results. In hPSCs, UCP2 shunts substrates such as pyruvate from glucose oxidation, favouring glycolysis and nucleotide biosynthesis through the pentose phosphate shunt pathway. Glycolysis provides ATP to partially maintain Δψ in hPSC mitochondria. In early hPSC differentiation, overall cell metabolism drops with reduced proliferation, glycolysis, and glucose oxidation in parallel with UCP2 repression. Physiologic UCP2 repression sustains glucose oxidation, ATP production, and provides TCA cycle intermediates which could be important co‐factors for hPSC differentiation, such as α‐ketoglutarate, which is required for the activity of epigenetic enzymes that regulate gene expression (Figueroa et al, 2010; Xu et al, 2011). Disruption of UCP2 repression interrupts this metabolic transition and blocks early hPSC differentiation. Lowered UCP2 expression facilitates ROS accumulation, which could stimulate hPSC differentiation to certain lineages, such as cardiomyocytes. Since hIPSCs show a mitochondrial network structure, metabolic profile, and UCP2 expression pattern similar to hESCs, the hPSC metabolism programme appears reversible with de‐differentiation.
These results may have significant implications for utilizing hPSCs in emerging cell‐based therapies in the clinic. When hPSCs are differentiated for tissue transplantation, residual undifferentiated cells, even if rare, could retain metabolic characteristics seen in human cancers, such as a high glycolytic flux, perhaps making them more prone to tumour formation. The characterization of mitochondria‐related energy metabolism before and after hPSC differentiation, and the discovery of one molecular mechanism regulating a metabolic transition to oxidation during differentiation, will potentially aid in identifying hPSCs with reduced tumourigenic potential or the discovery of drugs targeting metabolic pathways that increase the safety of hPSC‐based therapies in the future.
Materials and methods
Cell lines
hESCs and hIPSCs (Lowry et al, 2008) were grown on feeder‐free Matrigel (BD Biosciences) in StemPro SFM medium (Invitrogen) for all experiments, as described (Khvorostov et al, 2008; Zhang et al, 2008a, 2008b). Fibroblast and HEK293 cells were cultured in DMEM (Cellgro) with 10% FBS and supplements.
Fluorescence microscopy
Cells were fixed in 3.7% formaldehyde for 10 min, blocked in 0.2% BSA for 5 min, and incubated with OCT4 Ab (1:200; Cell Signaling) in 0.1% Triton‐X100 and 1 × PBS, pH 7.4 overnight at 4°C, followed by staining with FITC‐conjugated rabbit anti‐IgG Ab (1:500; Jackson ImmunoResearch) for 1 h. For mitochondrial staining, 100 nM of either Δψ‐dependent MitoTracker Red CMXRos or independent MitoTracker Green FM was added to the culture medium for 10 min at 37°C. A LSM 5 Pa laser scanning microscope (Zeiss) was used to visualize mitochondrial morphology.
Transmission electron microscopy
hPSCs were processed for TEM as described previously (Sun et al, 2007). Briefly, cells were fixed in a solution containing 3.0% formaldehyde, 1.5% glutaraldehyde in 100 mM cacodylate containing 2.5% sucrose, pH 7.4 and subsequently osmicated in Palade's OsO4 (1 h at 4°C). The pellet was washed three times in 100 mM cacodylate, pH 7.4, treated with 1% tannic acid for 30 min at RT, washed three times in double‐distilled water, and incubated overnight in Kellenberger's uranyl acetate. The pellet was then dehydrated through a graded series of ethanol and embedded in EMBED‐812. Sections were cut on a Leica Ultracut UCT ultramicrotome, collected onto 400‐mesh thin‐bar nickel grids, post‐stained with uranyl acetate and lead citrate, and observed in a Tecnai 12 at 100 kV. Images were collected with an Olympus SIS Megaview III CCD. The figures in Supplementary Figure S1B were assembled in Adobe Photoshop with only linear adjustments in brightness and contrast.
Citrate synthase assay
Whole‐cell protein (8 μg/sample) was mixed with acetyl‐CoA, 5,5′‐dithiobis‐2‐nitrobenzoic acid (DTNB), and oxaloacetic acid in a 96‐well plate and 412 nm absorption was recorded using a Citrate Synthase Assay kit (Sigma).
Immunoblotting
Cells were lysed in lysis buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton‐X100) containing 1 × protease inhibitor cocktail (Sigma). Protein (50 μg/lane) was resolved by 8–15% SDS–PAGE, transferred to nitrocellulose membranes, and incubated for 1 h with 5% milk TBS‐T and overnight with primary Abs in 5% BSA. Abs included OCT4, PARKIN (Santa Cruz), TOM40, TIM23, IF1, β‐TUBULIN (Abcam), and UCP2 (BioLegend). ECL Plus (GE Healthcare) was used for chemiluminescent detection.
O2 consumption
A Clark‐type O2 electrode was used in a magnetically stirred, thermostatically controlled 0.3 ml chamber at 25°C (Oxytherm; Hansatech). Cells (5 × 106) in 300 μl buffer (0.137 M NaCl, 5 mM KCl, 0.7 mM NaH2PO4, 25 mM Tris–HCl, pH 7.4) were assessed for O2 consumption. For determination of individual ETC complex activities, 0.03% digitonin was used to permeabilize cells. Complex activities were measured by feeding complex‐specific substrates while inhibiting the prior complexes in the chain. OCR for complexes I–IV was measured by adding 8 mM pyruvate+2.5 mM malate; OCR for complexes II–IV was measured by first inhibiting complex I with 3.3 μM rotenone followed by the addition of 10 mM succinate; OCR for complexes III–IV was measured by first inhibiting complex II with 10 mM malonate followed by the addition of 20 mM glycerophosphate; and OCR for complex IV was measured by first inhibiting complex III with 33 mM antimycin followed by the addition of 10 mM ascorbate plus 0.2 mM TMPD (tetramethyl‐p‐phenylenediamine).
Cell diameters
Measurements were made using the Countess® Automated Cell Counter (Invitrogen) according to the manufacturer's instructions.
XF24 extracellular flux analyser
hPSCs were seeded onto an XF24 Cell Culture Microplate (Seahorse Bioscience) at 2–7.5 × 104 cells/well with the ROCK inhibitor dihydrochloride (Sigma) (Watanabe et al, 2007) and incubated at 37°C overnight. Cell metabolic rates were measured using an XF24 Extracellular Flux Analyser (Seahorse Bioscience) in unbuffered DMEM assay medium supplemented with 1 mM pyruvate and 25 mM glucose after 45 to 60 min equilibration. For studies with palmitate addition, KHB assay medium (Seahorse Bioscience) supplemented with 500 μM l‐carnitine was used. OCR and ECAR were normalized to protein concentration using the Protein Assay reagent (Bio‐Rad) in all experiments.
Lactate quantification
Culture media (50 μl) was harvested per sample and lactate content was measured using a Lactate Assay kit (Biovision) according to the manufacturer's instructions.
ATP quantification
Cells (104/well) were incubated with oligomycin or sodium oxamate (Sigma) for 45 min at 37°C. The CellTiter‐Glo Luminescent Cell Viability Assay kit (Promega) was used to assay for ATP. Luminescence intensity from each well was measured using a GloMax 96 Luminometer (Promega).
Mitochondrial membrane potential determination
Δψ was determined using a MitoPT TMRM Assay kit (ImmunoChemistry Technologies) or BD MitoScreen kit (BD Biosciences Pharmingen) according to the manufacturers' instructions.
Lentiviral hPSC infection
The cDNA for ATPIF1 (Gene ID 93974), encoding IF1, was cloned into the lentiviral vector pLTET1 (Vieyra and Goodell, 2007), which contains a tetracycline controlled promoter and dsRed expression cassette. Two UCP2 (TRCN0000060143 and TRCN0000060144) or scramble shRNAs in the lentiviral vector of pLKO were bought from Sigma. The cDNA for UCP2 (Gene ID 7351) was cloned into a modified lentiviral vector pCCL with CMV promoter replaced by EF1α promoter. Lentivirus was made by transfecting 2 × 106 HEK293T cells with 10 μg lentiviral vector, 6.5 μg pCMV‐ΔR8.2 packaging vector, and 3.5 μg pCMV‐VSV‐G envelope vector using Fugene (Roche). Two days later, medium was collected, filtered, and virus concentrated using the Lenti‐X concentrator (Clontech). Concentrated virus was added to HSF1 hESCs or H1 hESCs stably expressing the tetracycline transactivator, rtTA2SM2 (Vieyra and Goodell, 2007), followed by mixing for 2 h at 37°C in the presence of 8 μg/μl polybrene, plating onto Matrigel plates in StemPro SFM medium.
Measurement of ROS
HSF1 cells (106) were trypsinized, collected, and washed by PBS. In all, 5 μM 2‐dihydroethidium (Sigma) was added to cells followed by incubation at 37°C for 10 min. Stained cells were washed by PBS and applied to a BD FACSCalibur flow cytometer.
Reverse transcription and real‐time PCR (QPCR)
Total RNA was extracted from hESCs, hIPSCs, fibroblasts, and HEK293 cells using Trizol reagent (Invitrogen). cDNA was synthesized using Superscript III first‐strand cDNA synthesis kit (Invitrogen). Real‐time PCR was performed using SYBR green real‐time PCR kit (Diagenode) with denaturation 94°C; 15 s, annealing 60°C; 30 s, extension 72°C; 45 s for 40 cycles. Primer sequences are available upon request.
Cell‐cycle analysis
Cells were stained in a hypotonic buffer containing 0.1 g/l propidium iodide, 1 g/l sodium citrate, 0.3% Triton‐X100, and 0.02 g/l ribonuclease A for 5 min, and analysed on a BD FACSCalibur flow cytometer.
Embryoid body formation
EB formation was assessed using AggreWell400 plates (Stemcell Technologies) according to the manufacturer's instructions. In brief, 106 cells were plated and the next day EBs were harvested and further cultured in IMDM medium supplemented with 20% FBS for 10 days. An inverted light microscope was used to assess and count EB colonies.
Stable isotope tracing
Cells were cultured in StemPro SFM medium (17.5 mM unlabelled glucose) supplemented with 7.5 mM [U13C6]‐glucose (Cambridge Isotope Laboratories) for 48 h. Three independent replicates of 2 × 106 cells for each line were collected, and the cell pellets were suspended in 0.5 ml of water and lysed by sonication. Cell debris was separated by centrifugation and proteins precipitated by treating the clear supernatant with 1 ml of cold acetone. The final supernatant was air‐dried and the free glutamic acid was converted to its trifluoroacetamide butyl ester for GC/MS analysis (Lee et al, 1996). Glutamate mass isotopomers were determined from the m/z 198 cluster representing the C2–C5 fragment of glutamate. ∑mn, which is given by m0+m1+2 × m2+3 × m3+…, is the average 13C per C2–C5 of the glutamate molecule and the equivalent of specific activity in radioisotope studies. The observed ∑mn divided by the theoretical ∑mn (calculated from ∑mn of glucose adjusted for the difference in the number of carbons per molecule) provides an estimate of the contribution of glucose to the TCA cycle. For ribose, RNA was extracted with TriReagent (Sigma, Cat#T9424). Ribose was isolated after RNA acid hydrolysis with 2 N HCl and derivatization into its aldonitrile acetate derivative for GC/MS analysis (Lee et al, 1998). The observed ∑mn divided by the theoretical ∑mn provides an estimate of the contribution of glucose to the pentose phosphate shunt.
Statistical analysis
Data are presented as mean±s.d. A two‐tailed t‐test was used for most comparisons, with P<0.05 considered statistically significant.
Supplementary data
Supplementary data are available at The EMBO Journal Online (http://www.embojournal.org).
Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Supplementary Data
Review Process File
Acknowledgements
We thank William Lowry and Jinghua Tang for hPSC lines; and Stephen Smale, Heather Christofk, Amander Clark, and Angara Zambrano for helpful discussions. This study was supported by CIRM Grants RS1‐00313 (MAT and CMK), RB1‐01397 (MAT and CMK), and TG2‐01169 (JZ), the Eli & Edythe Broad Center of Regenerative Medicine & Stem Cell Research at UCLA training grant (JZ), the Biomedical Mass Spectrometry Lab of the GCRC (M01‐RR00425; WNPL), and NIH Grants DK58132 (IJK), GM061721 (CMK), GM073981 (CMK and MAT), PNEY018228 (MAT), P01GM081621 (MAT), CA156674 (MAT), and CA90571 (MAT). CMK is an Established Investigator of the American Heart Association and MAT is a Scholar of the Leukemia and Lymphoma Society. This project was supported by Award Number S10RR026744 from the National Center for Research Resources.
Author contributions: JZ, CMK, and MAT designed the experiments. JZ, IK, JSH, YO, LV, EN, PNW, KS, GW, AD, and HJJ carried out the experiments. JMM performed the EM experiments. WNPL analysed the MS data. IJK and KR gave detailed comments of the paper. JZ and MAT wrote the manuscript.
There is a Have you seen? (December 2011) associated with this Article.
References
- Ayyasamy V, Owens KM, Desouki MM, Liang P, Bakin A, Thangaraj K, Buchsbaum DJ, Lobuglio AF, Singh KK (2011) Cellular model of warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS One 6: e24792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baharvand H, Matthaei KI (2003) The ultrastructure of mouse embryonic stem cells. Reprod Biomed Online 7: 330–335 [DOI] [PubMed] [Google Scholar]
- Becker KA, Ghule PN, Therrien JA, Lian JB, Stein JL, van Wijnen AJ, Stein GS (2006) Self‐renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J Cell Physiol 209: 883–893 [DOI] [PubMed] [Google Scholar]
- Boren J, Lee WN, Bassilian S, Centelles JJ, Lim S, Ahmed S, Boros LG, Cascante M (2003) The stable isotope‐based dynamic metabolic profile of butyrate‐induced HT29 cell differentiation. J Biol Chem 278: 28395–28402 [DOI] [PubMed] [Google Scholar]
- Bouillaud F (2009) UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim Biophys Acta 1787: 377–383 [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122: 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S, Pakay JL, Parker N (2004a) Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37: 755–767 [DOI] [PubMed] [Google Scholar]
- Brand MD, Buckingham JA, Esteves TC, Green K, Lambert AJ, Miwa S, Murphy MP, Pakay JL, Talbot DA, Echtay KS (2004b) Mitochondrial superoxide and aging: uncoupling‐protein activity and superoxide production. Biochem Soc Symp 71: 203–213 [DOI] [PubMed] [Google Scholar]
- Brand MD, Esteves TC (2005) Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab 2: 85–93 [DOI] [PubMed] [Google Scholar]
- Buchet K, Godinot C (1998) Functional F1‐ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA‐depleted rho degrees cells. J Biol Chem 273: 22983–22989 [DOI] [PubMed] [Google Scholar]
- Buzhynskyy N, Sens P, Prima V, Sturgis J, Scheuring S (2007) Rows of ATP synthase dimers in native mitochondrial inner membranes. Biophys J 93: 2870–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas S, Echtay KS, Harper JA, Jekabsons MB, Buckingham JA, Grau E, Abuin A, Chapman H, Clapham JC, Brand MD (2002) The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein‐3. J Biol Chem 277: 2773–2778 [DOI] [PubMed] [Google Scholar]
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR (2008) Regulation of mitochondrial structure and function by the F1Fo‐ATPase inhibitor protein, IF1. Cell Metab 8: 13–25 [DOI] [PubMed] [Google Scholar]
- Cao F, Wagner RA, Wilson KD, Xie X, Fu JD, Drukker M, Lee A, Li RA, Gambhir SS, Weissman IL, Robbins RC, Wu JC (2008) Transcriptional and functional profiling of human embryonic stem cell‐derived cardiomyocytes. PLoS One 3: e3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC, Chen HC, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park do J, Park KS, Lee HK (2006) Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem Biophys Res Commun 348: 1472–1478 [DOI] [PubMed] [Google Scholar]
- Chung S, Arrell DK, Faustino RS, Terzic A, Dzeja PP (2010) Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J Mol Cell Cardiol 48: 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Considine MJ, Goodman M, Echtay KS, Laloi M, Whelan J, Brand MD, Sweetlove LJ (2003) Superoxide stimulates a proton leak in potato mitochondria that is related to the activity of uncoupling protein. J Biol Chem 278: 22298–22302 [DOI] [PubMed] [Google Scholar]
- Couplan E, del Mar Gonzalez‐Barroso M, Alves‐Guerra MC, Ricquier D, Goubern M, Bouillaud F (2002) No evidence for a basal, retinoic, or superoxide‐induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J Biol Chem 277: 26268–26275 [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20 [DOI] [PubMed] [Google Scholar]
- Echtay KS, Winkler E, Frischmuth K, Klingenberg M (2001) Uncoupling proteins 2 and 3 are highly active H(+) transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc Natl Acad Sci USA 98: 1416–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre Y, Nubel T (2010) Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS Lett 584: 1437–1442 [DOI] [PubMed] [Google Scholar]
- Facucho‐Oliveira JM, Alderson J, Spikings EC, Egginton S, St John JC (2007) Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci 120: 4025–4034 [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel‐Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18: 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Bavister BD (1993) Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil 99: 673–679 [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann‐Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin‐related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1: 515–525 [DOI] [PubMed] [Google Scholar]
- Giraud M, Paumard P, Soubannier V, Vaillier J, Arselin G, Salin B, Schaeffer J, Brèthes D, di Rago J, Velours J (2002) Is there a relationship between the supramolecular organization of the mitochondrial ATP synthase and the formation of cristae? Biochim Biophys Acta 1555: 174–180 [DOI] [PubMed] [Google Scholar]
- Hatefi Y (1985) The mitochondrial electron transport and oxidative phosphorylation system. Annu Rev Biochem 54: 1015–1069 [DOI] [PubMed] [Google Scholar]
- Heytler PG (1979) Uncouplers of oxidative phosphorylation. Methods Enzymol 55: 462–472 [DOI] [PubMed] [Google Scholar]
- Ichikawa N, Chisuwa N, Tanase M, Nakamura M (2005) Mitochondrial ATP synthase residue betaarginine‐408, which interacts with the inhibitory site of regulatory protein IF1, is essential for the function of the enzyme. J Biochem 138: 201–207 [DOI] [PubMed] [Google Scholar]
- Jezek P, Borecky J (1998) Mitochondrial uncoupling protein may participate in futile cycling of pyruvate and other monocarboxylates. Am J Physiol 275: C496–C504 [DOI] [PubMed] [Google Scholar]
- Jezek P, Garlid KD (1990) New substrates and competitive inhibitors of the Cl‐ translocating pathway of the uncoupling protein of brown adipose tissue mitochondria. J Biol Chem 265: 19303–19311 [PubMed] [Google Scholar]
- Jezek P, Plecita‐Hlavata L, Smolkova K, Rossignol R (2010) Distinctions and similarities of cell bioenergetics and the role of mitochondria in hypoxia, cancer, and embryonic development. Int J Biochem Cell Biol 42: 604–622 [DOI] [PubMed] [Google Scholar]
- Khvorostov I, Zhang J, Teitell M (2008) From MEFs to Matrigel 2: splitting hESCs from MEFs onto Matrigel. J Vis Exp pii: 831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M, Echtay KS (2001) Uncoupling proteins: the issues from a biochemist point of view. Biochim Biophys Acta 1504: 128–143 [DOI] [PubMed] [Google Scholar]
- Klingenberg M, Huang SG (1999) Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta 1415: 271–296 [DOI] [PubMed] [Google Scholar]
- Lee WN, Boros LG, Puigjaner J, Bassilian S, Lim S, Cascante M (1998) Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2‐13C2]glucose. Am J Physiol 274: E843–E851 [DOI] [PubMed] [Google Scholar]
- Lee WN, Edmond J, Bassilian S, Morrow JW (1996) Mass isotopomer study of glutamine oxidation and synthesis in primary culture of astrocytes. Dev Neurosci 18: 469–477 [DOI] [PubMed] [Google Scholar]
- Lee WN, Wahjudi PN, Xu J, Go VL (2010) Tracer‐based metabolomics: concepts and practices. Clin Biochem 43: 1269–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, Xu Y (2005) p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 7: 165–171 [DOI] [PubMed] [Google Scholar]
- Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, Clark AT, Plath K (2008) Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci USA 105: 2883–2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K (2008) A high‐efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell 3: 340–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E, Misteli T (2006) Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol 7: 540–546 [DOI] [PubMed] [Google Scholar]
- Minauro‐Sanmiguel F, Wilkens S, García JJ (2005) Structure of dimeric mitochondrial ATP synthase: novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc Natl Acad Sci USA 102: 12356–12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgunov I, Srere PA (1998) Interaction between citrate synthase and malate dehydrogenase. Substrate of oxaloacetate. J Biol Chem 273: 29540–29544 [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. Plos Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Lindberg O (1972) Inhibited respiration and ATPase activity of rat liver mitochondria under conditions of matrix condensation. FEBS Lett 25: 61–64 [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Rial E (1999) A history of the first uncoupling protein, UCP1. J Bioenerg Biomembr 31: 399–406 [DOI] [PubMed] [Google Scholar]
- Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA (2007) Whole‐genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1: 299–312 [DOI] [PubMed] [Google Scholar]
- Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller D, Brèthes D, di Rago J, Velours J (2002) The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J 21: 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecqueur C, Alves‐Guerra C, Ricquier D, Bouillaud F (2009) UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life 61: 762–767 [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Alves‐Guerra MC, Gelly C, Levi‐Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, Miroux B (2001) Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem 276: 8705–8712 [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Bui T, Gelly C, Hauchard J, Barbot C, Bouillaud F, Ricquier D, Miroux B, Thompson CB (2008) Uncoupling protein‐2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis‐derived pyruvate utilization. FASEB J 22: 9–18 [DOI] [PubMed] [Google Scholar]
- Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J (2010) The senescence‐related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 28: 721–733 [DOI] [PubMed] [Google Scholar]
- Samudio I, Fiegl M, Andreeff M (2009) Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res 69: 2163–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudio I, Fiegl M, McQueen T, Clise‐Dwyer K, Andreeff M (2008) The warburg effect in leukemia‐stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res 68: 5198–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saretzki G, Walter T, Atkinson S, Passos JF, Bareth B, Keith WN, Stewart R, Hoare S, Stojkovic M, Armstrong L, von Zglinicki T, Lako M (2008) Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cells 26: 455–464 [DOI] [PubMed] [Google Scholar]
- Sauer H, Rahimi G, Hescheler J, Wartenberg M (2000) Role of reactive oxygen species and phosphatidylinositol 3‐kinase in cardiomyocyte differentiation of embryonic stem cells. FEBS Lett 476: 218–223 [DOI] [PubMed] [Google Scholar]
- Sauvanet C, Duvezin‐Caubet S, di Rago JP, Rojo M (2010) Energetic requirements and bioenergetic modulation of mitochondrial morphology and dynamics. Semin Cell Dev Biol 21: 558–565 [DOI] [PubMed] [Google Scholar]
- Schieke S, Ma M, Cao L, McCoy JJ, Liu C, Hensel N, Barrett A, Boehm M, Finkel T (2008) Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J Biol Chem 283: 28506–28512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John JC, Ramalho‐Santos J, Gray HL, Petrosko P, Rawe VY, Navara CS, Simerly CR, Schatten GP (2005) The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells. Clon Stem Cell 7: 141–153 [DOI] [PubMed] [Google Scholar]
- Strauss M, Hofhaus G, Schroder RR, Kuhlbrandt W (2008) Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J 27: 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MG, Williams J, Munoz‐Pinedo C, Perkins GA, Brown JM, Ellisman MH, Green DR, Frey TG (2007) Correlated three‐dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol 9: 1057–1065 [DOI] [PubMed] [Google Scholar]
- Vieyra DS, Goodell MA (2007) Pluripotentiality and conditional transgene regulation in human embryonic stem cells expressing insulated tetracycline‐ON transactivator. Stem Cells 25: 2559–2566 [DOI] [PubMed] [Google Scholar]
- Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, Sasai Y (2007) A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25: 681–686 [DOI] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 292: C125–C136 [DOI] [PubMed] [Google Scholar]
- Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, Sukhatme VP, Seth P (2009) LDH‐A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther 8: 626–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM et al (2011) Oncometabolite 2‐hydroxyglutarate is a competitive inhibitor of alpha‐ketoglutarate‐dependent dioxygenases. Cancer Cell 19: 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Khvorostov I, Teitell M (2008a) From MEFs to Matrigel 3: passaging hESCs from Matrigel onto Matrigel. J Vis Exp pii: 832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Khvorostov I, Teitell M (2008b) From MEFs to matrigel I: passaging hESCs in the presence of MEFs. J Vis Exp pii: 722 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data
Review Process File