

Abstract
Multiple K+ conductances are targets for many peripheral and central signals involved in the control of energy homeostasis. Potential K+ channel targets are the KCNQ subunits that form the channels underlying the M-current, a subthreshold, non-inactivating K+ current that is a common target for G-protein-coupled receptors. Whole-cell recordings were made from GFP (Renilla)-tagged neuropeptide Y (NPY) neurons from the arcuate nucleus of the hypothalamus using protocols to isolate and characterize the M-current in these orexigenic neurons. We recorded robust K+ currents in the voltage range of the M-current, which were inhibited by the selective KCNQ channel blocker 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride (XE991) (40 μm), in both intact males and ovariectomized, 17β-estradiol (E2)-treated females. Since NPY neurons are orexigenic and are active during fasting, the M-current was measured in fed and fasted male mice. Fasting attenuated the XE991-sensitive current by threefold, which correlated with decreased expression of the KCNQ2 and KCNQ3 subunits as measured with quantitative real-time PCR. Furthermore, E2 treatment augmented the XE991-sensitive M-current by threefold in ovariectomized (vs oil-treated) female mice. E2 treatment increased the expression of the KCNQ5 subunit in females but not KCNQ2 or KCNQ3 subunits. Fasting in females abrogated the effects of E2 on M-current activity, at least in part, by decreasing KCNQ2 and KCNQ3 expression. In summary, these data suggest that the M-current plays a pivotal role in the modulation of NPY neuronal excitability and may be an important cellular target for neurotransmitter and hormonal signals in the control of energy homeostasis in both males and females.
Introduction
In the hypothalamic arcuate nucleus, two distinct, neuronal cell types differentially modulate energy homeostasis—the proopiomelanocortin (POMC) and neuropeptide Y (NPY) neurons. NPY neurons are orexigenic and respond to many peripheral and central hormonal signals that control energy homeostasis (Elmquist, 2001; Takahashi and Cone, 2005; Gao and Horvath, 2007; Kohno et al., 2007; van den Pol et al., 2009). NPY neurons coexpress agouti-related peptide (AgRP), the endogenous antagonist to the melanocortin MC4 receptors, thus blocking the anorectic effects of the POMC peptide, α-MSH (Haskell-Luevano et al., 2001; Lu et al., 2001). Ablation of NPY neurons in adults causes a rapid starvation (Gropp et al., 2005; Luquet et al., 2005) while intracerebroventricular injection of NPY into the paraventricular nucleus stimulates food intake (Stanley and Leibowitz, 1985). The anorectic, ovarian steroid 17β-estradiol (E2) also suppresses NPY expression in the arcuate nucleus (Crowley et al., 1985; Pelletier et al., 2007) and may exert its effects on NPY neurons by modulating neuronal excitability.
NPY neurons are more excitable after fasting in a leptin-dependent manner (Takahashi and Cone, 2005). The activity of NPY neurons is modulated by peripheral signals (leptin, ghrelin, gastrin-releasing peptide) via targeting potassium and calcium channels (Wang et al., 2008; van den Pol et al., 2009; Yang et al., 2010). These K+ channels hyperpolarize the membrane potential and inhibit neuronal excitability. Another K+ channel that modulates neuronal excitability, action potential kinetics, and burst firing are the KCNQ channels (Robbins, 2001; Delmas and Brown, 2005). KCNQ channels constitute the M-current, a subthreshold, non-inactivating, voltage-dependent, outward K+ current. The M-current, ubiquitously expressed in the brain and in neurons, is generated by expression of the KCNQ subunits, KCNQ2, -3, -4, and -5. Coexpression of KCNQ2/3 or KCNQ3/5 heteromultimers produces currents with similar kinetics to the native M-current (Robbins, 2001). Moreover, the M-current is a target of numerous neurotransmitters and neuropeptides that modulate neuronal excitability through G-protein-coupled receptors including acetylcholine, serotonin, substance P, and gonadotropin-releasing hormone (Delmas and Brown, 2005; Xu et al., 2008). The role of the M-current in feeding behavior has, to our knowledge, not been reported.
Previously, we have shown that E2 (24 h and long-term) increases the expression of KCNQ5 in guinea pig arcuate nucleus (Roepke et al., 2007, 2008). Due to the heterogeneous nature of the arcuate nucleus, the exact identity of the neurons involved in the increase in KCNQ5 expression was unknown. However, since E2 reduces feeding and NPY expression (for review, see Roepke, 2009) and the M-current dampens neuronal excitability, we hypothesized that an increase in M-current activity specifically in NPY neurons is a cellular mechanism used by E2 to reduce food intake. Furthermore, the effects of fasting, which are known to increase NPY excitability, may attenuate the expression and activity of the M-current. Therefore, in this study, we examined the effects of fasting and E2 treatment on the expression and activity of the M-current using whole-cell patch-clamp recordings, single-cell harvesting, and quantitative real-time PCR.
Materials and Methods
Animal care and experimental treatments.
All animal treatments are in accordance with institutional guidelines based on National Institutes of Health standards, and were performed with Institutional Animal Care and Use Committee approval at the Oregon Health & Science University. Female and male GFP-NPY transgenic mice (provided by Dr. Brad Lowell, Harvard University, Cambridge, MA) (van den Pol et al., 2009) were selectively bred in-house and maintained under controlled temperature (25°C) and photoperiod conditions (12 h light/dark cycle) with food and water ad libitum. Male mice were divided into three groups: fed, 24 h fasted, and 24 h refed. The refed treatment entailed a 24 h fast followed by a 24 h refeeding before experimentation. A 24 h fast has previously been reported to alter the neuronal excitability of the NPY neurons (Takahashi and Cone, 2005). NPY females were ovariectomized under isoflurane anesthesia, implanted with sterile capsules containing sesame oil or 35 μl of 180 μg/ml E2 in sesame oil and allowed to recover for 1 week. This E2 treatment produces low, diestrus levels of E2 (∼30 pg/ml). After 1 week, the females were injected subcutaneously with either oil or 1 μg of 17β-estradiol benzoate (E2B) 24 h before experimentation. The estrogen replacement paradigm has been previously used to mimic the endogenous levels of E2 during proestrous [average serum E2 levels, 77.6 ± 10.7 pg/ml (n = 9) in our animals] (Bronson and Vom Saal, 1979; Zhang et al., 2009). A subset of E2-treated females was also subjected to a 24 h fast starting at the time of injection. Average weight loss after a 24 h fast was 2.9 ± 0.4 g (n = 10) for females and 3.5 ± 0.2 g (n = 21) for males. Refed males recovered on average 90% of their prefast weight. Fasting had no effect on animal activity, fur condition, and response to handling.
Drugs.
E2 and E2B were purchased from Steraloids and dissolved in ethanol before oil dissolution. 10,10-bis(4-Pyridinylmethyl)-9(10H)-anthracenone dihydrochloride (XE991), (2R)-amino-5-phosphonovaleric acid (d-AP5), and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) were all purchased from Tocris. (−)-Bicuculline methiodide (bicuculline) was purchased from Sigma-Aldrich. Tetrodotoxin (TTX) was purchased from Alomone Labs and dissolved in H2O.
Preparation of basal hypothalamic slices.
Slices were prepared as described previously (Ibrahim et al., 2003; Qiu et al., 2003, 2010). Transgenic GFP-NPY mice were killed quickly by decapitation at 10:00–11:00 A.M. The brain was rapidly removed from the skull and a block containing the basal hypothalamus (BH) was immediately dissected. The BH block was submerged in cold (4°C) oxygenated (95% O2, 5% CO2) high-sucrose CSF (in mmol: 208 sucrose, 2 KCl, 26 NaHCO3, 10 glucose, 1.25 NaH2PO4, 2 MgSO4, 1 MgCl2, 10 HEPES, pH 7.3, 300 mOsm). Coronal slices (250 μm) from the BH were cut on a vibratome during which time (10 min) the slices were bathed in high-sucrose CSF at 4°C. The slices were then transferred to an auxiliary chamber in which they were kept at room temperature (25°C) in artificial CSF (aCSF) consisting of the following (in mm): 124 NaCl, 5 KCl, 2.6 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 10 glucose, pH 7.3, 310 mOsm until recording (recovery for 2 h). A single slice was transferred to the recording chamber mounted on an Olympus BX51W1 upright microscope equipped with video-enhanced, infrared-differential interference contrast (IR-DIC) and Exfo X-Cite 120 Series fluorescence light source. The slice was continually perfused with warm (35°C), oxygenated aCSF at 1.5 ml/min. Targeted neurons were viewed using both IR-DIC and blue excitation light with an Olympus 40× water-immersion lens.
Visualized whole-cell patch recording.
Normal aCSF and pipette solutions were used in electrophysiological recording (Ibrahim et al., 2003; Qiu et al., 2003, 2010). Standard whole-cell patch recording procedures and pharmacological testing were as previously described (Xu et al., 2008; Qiu et al., 2010). Whole-cell voltage- and current-clamp recordings were performed using pipettes made of borosilicate glass and pulled using a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument). Pipettes were filled with normal internal solution (in mm: 10 NaCl, 128 K-gluconate, 1 MgCl2, 10 HEPES, 1 ATP, 1.1 EGTA, 0.25 GTP, pH 7.3, 300 mOsm) with a 3–5 MΩ resistance. An Axopatch 200A amplifier, Digidata 1322A Data Acquisition System and pCLAMP software (version 9.2; Molecular Devices) were used for data acquisition and analysis. Input resistance, series resistance, and membrane capacitance were monitored throughout the experiments. Only cells with stable series resistance (<30 MΩ; <20% change) and an input resistance >500 MΩ were used for analysis. The access resistance was 80% compensated and the calculated liquid junction potential (∼10 mV) was corrected. To display reversal potential and rectification characteristics of the ligand-activated currents, I–V plots constructed by voltage steps from −50 to −140 mV at 10 mV increments applied at 1 s intervals from a holding potential of −60 mV. From this protocol, the input resistance was determined from the slope of the I–V plot in the range between −60 and −80 mV. To calculate the rheobase and elicit an action potential in current clamp, the resting membrane potential was held to −80 mV for all neurons with a small hyperpolarizing current, followed by injection of incremental current steps (∼6 pA/step) for 500 ms before and after bathing the slice in XE991 (40 μm). In voltage clamp, two protocols were used to measure the deactivation and steady-state currents. The deactivation protocol measured the currents elicited during 500 ms voltage steps from −25 to −75 mV after a 300 ms prepulse to −20 mV, which included the membrane potential at which the maximal M-current could be obtained (Schweitzer, 2000; Xu et al., 2008). The amplitude of M-current relaxation or deactivation was measured as the difference between the initial (<10 ms) and sustained current (>475 ms) of the current trace in the control conditions (TTX only; 1 μm; 5 min) minus the difference in the XE991 (40 μm; +TTX; 10 min) conditions. To examine the steady-state current, a series of command voltage steps from −80 to +10 mV in 10 mV increments (1 s) were applied from a holding potential of −90 mV (500 ms) and returning to −60 mV holding potential (Leão et al., 2009). The XE991-sensitive current was determined by subtracting the steady-state (SS) current (the last 50 ms of the voltage steps) of the whole K+ currents in the XE991-treated traces from steady-state current in the control traces. Both voltage-clamp protocols were repeated twice for each bath solution and averaged for analysis.
Cell harvesting of dispersed mouse NPY neurons, single-cell RT-PCR, and quantitative real-time PCR.
These procedures were conducted as described previously (Ibrahim et al., 2003; Xu et al., 2008; Zhang et al., 2008, 2009). Briefly, four 250 μm basal hypothalamic slices were cut on a vibratome and placed in an auxiliary chamber containing oxygenated aCSF. The slices were allowed to recover for 1–2 h in the auxiliary chamber. Thereafter, the arcuate nucleus and median eminence was microdissected and incubated in 10 ml of aCSF, pH 7.3, 300 mOsm, containing 1 mg/ml protease for ∼15 min at 37°C. The tissue was then washed four times in low-calcium CSF (1 mm CaCl2) and two times in aCSF. The cells were isolated by trituration with flame-polished Pasteur pipettes, dispersed onto a 60 mm glass-bottomed Petri dish, and perfused continuously with aCSF at a rate of 2.0 ml/min. The dispersed, bipolar or unipolar fluorescent cells were visualized using a Leitz inverted fluorescent microscope, patched, and then harvested with gentle suction to the pipette using the Xenoworks manipulator system (Sutter Instrument) and expelled into siliconized microcentrifuge tubes. Each harvested cell or pools of cells were heat denatured and reverse transcribed as described previously (Zhang et al., 2008, 2009). For quantitative PCR, six to eight pools of 5 cells (KCNQ2 and -3) or 10 cells (KCNQ5) each were harvested from individual ovariectomized, oil- and E2-treated (both fed and fasted) females or fed and fasted males (age 10–14 weeks). Perfused aCSF was regularly collected along with the harvested cells and processed together with the cells and control samples. Single-cell and tissue RNA used as negative controls were processed similarly but without reverse transcriptase (RT).
The single-cell RT-PCR primers for KCNQ2 and KCNQ4 were as previously published (Xu et al., 2008). The single-cell RT-PCR primers for KCNQ3, KCNQ5, and NPY were as follows: KCNQ3 (94 bp product; accession no. NM_152923.1; forward primer, GCTGCTGGAAACCTTTGC; reverse primer, ACGCCAGCCTTTGTATCG; 474–567 nt; annealing temperature, 60°C), KCNQ5 (143 bp product; accession no. NM_023872.2; forward primer, GATGGCAAGGAAGCCTGAG; reverse primer, TAGGAACCGGAGACTTCTG; 572–714 nt; annealing temperature, 57°C), and NPY (182 bp product; accession no. NM_023456; forward primer, ACTGACCCTCGCTCTATCTC; reverse primer, TCTCAGGGCTGGATCTCTTG; 106–287 nt; annealing temperature, 64°C). Each reaction was amplified for 50 cycles using a Bio-Rad C1000 Thermal Cycler (Bio-Rad). All single-cell RT-PCR products were confirmed by sequencing. Negative (cell and tissue samples without RT and harvested CSF) and positive tissue controls were used for each PCR run. KCNQ subunit colocalization patterns were determined by counting some cells more than once (e.g., each cell that had all three subunits also were included with those that coexpressed only KCNQ2 plus -3, KCNQ3 plus -5, or KCNQ2 plus -5).
To determine the changes in KCNQ gene expression between fed and fasted males, and oil- and E2-treated females, pools of dispersed GFP-NPY neurons were harvested, reverse transcribed, and analyzed using quantitative real-time PCR. Due to the technical limitations of single-cell expression analysis of low-abundant genes, we could not measure KCNQ subunit transcripts in individual neurons reliably with quantitative real-time PCR. Therefore, pooling of neurons was required especially when targeting low-expression genes such as cation channel subunits. This has allowed us to do careful quantitative analysis of mRNA expression that often correlates with the whole-cell current activity (Zhang et al., 2009) as in the current study. The procedures for quantitative real-time PCR were previously described (Zhang et al., 2009). For KCNQ2 and KCNQ3, pools of 5 NPY neurons were sufficient, while pools of 10 NPY neurons were needed for the analysis of KCNQ5 gene expression, which was most likely due to lower expression of the KCNQ5 subunit in NPY neurons or the fewer number of neurons that expressed this subunit. Negative (cell and tissue samples without RT and harvested CSF) and positive tissue controls were used for each single-cell and quantitative PCR run. The primers for quantitative real-time PCR for KCNQ3 were previously described above. For KCNQ2 and KCNQ5, the primers were as follows: KCNQ2 (171 bp product; accession no. NM_133322; forward primer, GGTGCTGATTGCCTCCATTG; reverse, TCCTTGCTGTGAGCGTAGAC; 644–814 nt; annealing temperature, 60°C) and KCNQ5 (99 bp product; accession no. NM_023872.2; forward primer, GGGCACAATCACACTGACAAC; reverse primer, GAAATGCCAAGGAGTGCGAAG; 915–1013 nt; annealing temperature, 58°C). The relative standard curve model was used to determine amplification efficiency using serial dilutions of basal hypothalamic cDNA. The calculated efficiencies for all four primers were >94%. The small difference between the efficiencies (<5%) does not affect the analysis of relative gene expression (Livak and Schmittgen, 2001; Pfaffl, 2001; Schmittgen and Livak, 2008). Relative KCNQ5 expression was also analyzed using quantitative real-time PCR in ovariectomized, oil- and E2-treated wild-type (C57BL/6) and αERKO mice (C57BL/6 from Prof. Pierre Chambon, CNRS/Inserm, Illkirch Cedex, France).
To determine which estrogen receptor (ER) was expressed in NPY neurons, we also analyzed the expression of ERα and ERβ in NPY pools (five cells) from ovariectomized, female mice. The primers for ERs were as follows: ERα (107 bp product; accession no. NM_007956; forward primer, GCGCAAGTGTTACGAAGTG; reverse primer, TTCGGCCTTCCAAGTCATC; 919–1025 nt; annealing temperature, 60°C) and ERβ (113 bp product; accession no. NM_010157; forward primer, AATGTCCACCCGCTAGGCATTC; reverse primer, CTCCATGTCTTGCGTAGGTCTC; 298–410 nt; annealing temperature, 60°C). The reference gene for all quantitative real-time PCR was β-actin as previously published (Zhang et al., 2009). All quantitative real-time PCR was conducted using the Power SYBR Green master mix on an ABI 7500 Fast Real-Time PCR System (Invitrogen).
Colocalization of NPY and ERα using immunohistochemistry.
We performed immunohistochemistry to document ERα expression in mouse NPY neurons. Four ovariectomized female GFP-NPY mice were sedated by ketamine injection and decapitated 7–10 d after ovariectomy. The brains were quickly extirpated and cooled in aCSF (4°C) for 1 min. One millimeter blocks of the basal hypothalamus were sliced, fixed in 4% paraformaldehyde for 6 h with gentle shaking on ice, and then cryopreserved in 20% sucrose overnight in the refrigerator. Tissue blocks were frozen at −55°C, coronally sectioned on a cryostat at 20 μm, and thaw-mounted on Superfrost Plus glass slides (Thermo Fisher Scientific). Sections were rinsed in PBS (0.1 m phosphate buffer, pH 7.4, and 0.15 m NaCl; all rinses were in PBS for 30 min unless noted), and then incubated for 24 h at 4°C in antiserum against ERα (1:10,000; C1355; Millipore). After rinsing, sections were incubated for 1 h at room temperature in biotinylated goat anti-rabbit IgG (1:1000; Vector Laboratories), washed, and then incubated in Cy3-conjugated streptavidin (1:1000; Jackson ImmunoResearch). Following a final rinse for 3 h, slides were coverslipped using a glycerol–glycine buffer (2:1), pH 8.6, containing 5% n-propyl gallate to reduce photobleaching (Giloh and Sedat, 1982). Both the primary and secondary antisera and tertiary marker were diluted in Tris-(hydroxymethyl)aminomethane (0.5%; Sigma-Aldrich) in PBS containing 0.7% seaweed gelatin (Sigma-Aldrich), 0.4% Triton X-100 (Sigma-Aldrich), and 3% BSA (Sigma-Aldrich) adjusted to pH 7.6. Slides were analyzed and photographed using Nikon E800 microscope equipped with epifluorescence and a DS digital camera. A total of 10–12 arcuate sections from each female were examined. The number of GFP-NPY cells colocalizing ERα was counted per section.
Data analysis.
Comparisons of the I–V plots between different feeding states and steroid treatments were performed using a two-way ANOVA analysis with the Bonferroni-Dunn post hoc test. Rheobase and quantitative real-time PCR results were analyzed by unpaired Student's t test. Differences were considered statistically significant if the probability of error was <5%. All data are presented as mean ± SEM.
Results
Feeding states modulate the activity of the M-current in males
We initially determined the basal activity of the NPY neurons and the role of the M-current in modulating neuronal excitability in fed male mice. In current clamp, action potentials were evoked by the injection of incremental current steps (∼6 pA/step) before and after the application of XE991 (40 μm), a selective blocker of KCNQ channels (Fig. 1A). Under control conditions, no action potentials were evoked after the first current injection of 6 pA (gray trace); however, the first current injection did produce an action potential after treatment with XE991. The first action potential evoked under control conditions (black trace) was after ∼30 pA of current, which produced two action potentials (or more in other cells) in the XE991 conditions. Therefore, the average rheobase was significantly reduced by XE991 from a mean of 27.4 ± 1.2 to 18 ± 0.9 pA (n = 15; p < 0.0001) (Fig. 1B). During a 10 min perfusion of XE991, the resting membrane potential (RMP) of each neuron was continuously recorded in current clamp. The average RMP of NPY neurons from the fed males was −73.9 ± 0.9 mV, and after a 10 min XE991 perfusion the membrane depolarized to −57.3 ± 1.1 mV (p < 0.0001). XE991 application also increased the input resistance in the fed males from 0.75 ± 0.05 to 1.43 ± 0.27 GΩ (p < 0.01), indicating a reduction in membrane conductance. The majority of the neurons (10 of 15) were quiescent before XE991 application (Fig. 1D) yet became active after 8–9 min of XE991 perfusion. The remaining neurons were active but depolarized (and increased firing) after XE991. To determine whether action potential generation during XE991 perfusion was dependent on presynaptic inputs (glutaminergic, GABAergic), we repeated these experiments in the presence of glutamate receptor (CNQX, AP5) and GABAA receptor (bicuculline) blockers (Fig. 1D). Blockade of the M-current by XE991 in NPY neurons did not increase firing, although the neurons were significantly depolarized from baseline to a similar level of depolarization [14.9 ± 3.2 mV (n = 3) with blockers compared with 16.8 ± 1.3 mV (n = 15) without blockers]. The level of depolarization did not reach the action potential threshold for hypothalamic arcuate neurons (Kelly et al., 1990; Loose et al., 1990) to fire independently of excitatory inputs. However, the neurons treated with the synaptic blockers did fire when sufficient depolarizing current was applied, and the response was greater in the XE-treated neurons (Fig. 1E). Therefore, the data suggest that the M-current in NPY neurons contributes to the control of neuronal excitability and therefore is a potential target for modulation during different physiological states.
Figure 1.

Blocking the M-current increases firing activity in NPY neurons from fed male mice. A, In current clamp, the rheobase was determined in both control and XE991 (40 μm) conditions. To elicit action potentials, the RMP was held at −80 mV, followed by injection of small amounts of current (∼6 pA/step; 500 ms). The black trace is the first current injection to evoke an AP in the control conditions and multiple spikes in the XE991 conditions. The gray trace is the first current injection that evokes a spike in XE991 conditions but not the control conditions. B, In fed males, the average rheobase in control conditions was significantly higher than after XE991 (40 μm; 10 min) treatment (n = 15; ****p < 0.0001). C, The firing activity of NPY neurons in fed males was continuously recorded in control and XE991 (40 μm) conditions. A typical example of an NPY neuron at rest (RMP, −75 mV) and the effects of XE991 to depolarize the cell (to −58.5 mV) and induce firing. D, The firing activity of NPY neurons in fed males was continuously recorded in control and XE991 (40 μm) conditions but in the presence of glutamate receptor blockers [CNQX (10 μm); AP5 (50 μm)] and a GABAA receptor blocker [bicuculline (20 μm)]. A typical example of an NPY neuron at rest (RMP, −74 mV) and the effects of XE991 to depolarize the cell (to −57.5 mV) without inducing firing are shown. The average depolarization with and without blockers was 14.9 ± 3.2 mV (n = 3, at 20–22 min) and 16.8 ± 1.3 mV (n = 15, at 12–14 min), respectively. E, Action potentials were evoked by current injection before (control) and after XE991 perfusion from the neuron recorded in D. The black trace is the first current injection to evoke a response in the control conditions, and the gray trace is the first current injection during XE991 application. Error bars indicate SEM.
To measure the M-current in voltage clamp, we measured the deactivation or relaxation of the whole-cell K+ currents elicited by an established protocol (Schweitzer, 2000; Xu et al., 2008) before and after application of XE991 (40 μm) in the presence of TTX (1 μm). The XE991-sensitive current (M-current) was calculated by subtracting the current relaxation [the difference between the instantaneous and steady state (arrows) (Fig. 2A)] in XE991 from the control conditions. Because the M-current has been shown to decrease or “run down” after cell dialysis in whole-cell recordings in other neuronal cell types (Shen et al., 2005), the whole-cell K+ currents evoked by the deactivation protocol were monitored over 30 min to determine the change in the relaxation currents (Fig. 2B). In NPY neurons from fed males, the outward K+ currents evoked by the protocol did not significantly decrease over the 30 min, which is beyond the experimental time needed for each recording, yet the KCNQ channel blocker XE991 was fully efficacious after this time period.
Figure 2.

Fasting reduces the activity of the M-current in NPY neurons. A, The slow deactivation of the M-current (IM) in NPY neurons was recorded from fed, male mice using whole-cell patch-clamp recording. From a holding potential of −60 mV, a voltage jump to −20 mV (300 ms) was followed by steps from −25 to −75 mV in 5 mV increments (500 ms). Currents were recorded under control conditions and after 10 min incubation with XE991 (40 μm), a selective KCNQ blocker in the presence of TTX (1 μm). The XE991-sensitive current was calculated by subtracting the current relaxation [the difference between the instantaneous and steady state (arrows)] in XE991-treated traces from the control traces. This XE991-sensitive current is graphed as an I–V plot (D) (Xu et al., 2008). B, The K+ currents evoked by the deactivation protocol do not run down over the 30 min recording period. T = 0 is 3 min after whole-cell access. XE991 (40 μm) was perfused after 30 min to demonstrate the amount of M-current still active. The figure is from one cell, but rundown was examined in two other cells with the same results. C, To determine the most effective dose to measure the M-current across treatments, a sequential concentration response of XE991 (1, 10, 40 μm) was applied to same neuron (n = 5). I–V plots were analyzed by a two-way ANOVA (p < 0.05, F = 4.67, df = 3) followed by Bonferroni–Dunn comparison. ap < 0.05; bp < 0.01; cp < 0.001 compared with control. D, In NPY neurons, the I–V plot of A from fed males (n = 11), fasted males (n = 17), and 24 h refed males (n = 10). I–V plots were analyzed by a two-way ANOVA (p < 0.01, F = 7.3, df = 2) followed by Bonferroni–Dunn comparison. ap < 0.05; cp < 0.001, compared with fasted. Error bars indicate SEM.
To determine the appropriate concentration to use in the bath perfusion, a sequential concentration–response was performed using 1, 10, and 40 μm XE991 in five NPY neurons from fed males (Fig. 2C). The lowest concentration of XE991 did not significantly inhibit the K+ current evoked by the deactivation protocol, while 10 μm did significantly attenuate the current at the highest voltages (−35 and −30 mV). At 40 μm, XE991 significantly attenuated the current at voltages more depolarized than −45 mV. The effects of XE991 at this concentration were not fully reversible within the experimental time period (>30 min) (data not shown). However, this concentration ensured that all possible homomultimers and heteromultimers of KCNQ channel subunits (KCNQ2, -3, and -5), which have different sensitivity to XE991 (Robbins, 2001), were inhibited. For example, in cell expression systems the IC50 of XE991 for KCNQ5 homomultimers is ∼65 μm (Schroeder et al., 2000), while the IC50 for KCNQ2/3 heteromultimers is 0.6 μm (Wang et al., 1998). Higher concentrations of XE991 (>100 μm) can block ERG (ether-a-go-go-related gene) channels (Elmedyb et al., 2007) and may affect Kv4.x (A-type) channels (Wang et al., 1998); although similar to other investigators (van den Pol et al., 2009), we did not measure a robust A-current in NPY neurons (data not shown). Therefore, the 40 μm concentration of XE991 was used for all subsequent experiments to ensure the maximum amount of KCNQ blockade without alteration of other K+ currents.
Because NPY neurons play a pivotal role in the hypothalamic control of feeding behaviors, we determined the effects of fasting and refeeding on the M-current in NPY neurons from adult male mice in the presence of 1 μm TTX to block Na+-spike-dependent synaptic inputs. The deactivation protocol examines the voltage range (−30 to −75 mV) in which the M-current has its most profound effect on excitability. In males, the XE991-sensitive current (the M-current) was significantly attenuated by a 24 h fasting (p < 0.01) (Fig. 2D). A 24 h refeeding restored a significant portion of the M-current in the NPY neurons. A 2 h refeeding had no effect on the M-current in fasted males (data not shown). The maximum XE-sensitive current was recorded at −35 mV [fed males, 43.6 ± 5.4 pA (n = 11); fasted, 14.8 ± 3.3 pA (n = 17); refed, 35.7 ± 2.6 pA (n = 10)] (Fig. 2D). In addition, the RMP between fed and fasted males was significantly different (fed, −71.5 ± 1.6 mV, vs fasted, −64.8 ± 1.6 mV; p < 0.01), while the capacitance was not significantly different (fed, 17.9 ± 2.0 pF; fasted, 16.4 ± 0.7 pF). XE991 application significantly reduced the RMP in both states but only significantly increased the input resistance in fed males. The RMP in refed males was not significantly different from either of the other treatments, but XE991 significantly increased the input resistance (Table 1), which correlates with the increased conductance due to the M-current in the fed and refed states compared with the fasted state (Fig. 2D).
Table 1.
The membrane potential and input resistance for each feeding state and steroid treatment
| Treatment | Control |
XE991 |
||
|---|---|---|---|---|
| Membrane potential (mV) | Input resistance (GΩ) | Membrane potential (mV) | Input resistance (GΩ) | |
| Males | ||||
| Fed | −71.5 ± 1.6** | 0.64 ± 0.12 | −62.5 ± 2.9a | 1.02 ± 0.18a |
| Fasted | −64.8 ± 1.6 | 0.65 ± 0.12 | −57.1 ± 1.9b | 0.81 ± 0.09 |
| Refed | −68.5 ± 0.9 | 0.63 ± 0.07 | −58.3 ± 1.5d | 0.97 ± 0.11b |
| Females | ||||
| Oil | −68.1 ± 1.8* | 0.65 ± 0.09 | −62.7 ± 1.3a | 1.19 ± 0.28 |
| E2 | −72.4 ± 1.1 | 0.68 ± 0.08 | −62.4 ± 2.0c | 1.0 ± 0.12b |
| Fasted E2 | −65.6 ± 1.5*** | 0.63 ± 0.06 | −58.8 ± 2.0a | 0.79 ± 0.12 |
ap < 0.05, bp < 0.01, cp < 0.001, and dp < 0.0001 denote control to XE991 comparisons.
*p < 0.05, **p < 0.01, and ***p < 0.001 denote fed versus fasted comparisons in males, and oil and fasted E2 compared with E2 in females. For males: fed n = 11; fasted n = 17; refed n = 10. For females: oil n = 14; E2 n = 15; fasted E2 n = 11.
To determine the steady-state conductance and kinetics of the M-current in NPY neurons, we used an established activation protocol (Leão et al., 2009) in the presence of 1 μm TTX (Fig. 3A). The steady-state currents at the higher voltages were significantly higher in the fed and refed males compared with the fasted males (p < 0.001). The XE-sensitive current at +10 mV was 484 ± 37.6 pA (n = 11) for fed males, 259 ± 63.3 pA (n = 15) for fasted males, and 480 ± 53.6 pA (n = 10) for refed males (Fig. 3B). The percentage of steady-state current at +10 mV that was XE991 sensitive was 51.6 ± 5.2% in the fed state, 29.3 ± 3.7% in the fasted state, and 38.7 ± 3.3% in the refed state (fed vs fasted, p < 0.01). The M-current is active at subthreshold potentials and increases in amplitude around −50 to −40 mV (Robbins, 2001). The amplitude of the currents evoked by the activation protocol at these voltages was similar to the amplitude of the currents evoked by the deactivation protocol for all treatments (greater than −50 mV). A Boltzmann equation fit to the G/Gmax plot (Gmax at +10 mV) yielded a V1/2 for activation of the M-current of −22.9 ± 2.4 mV in the fasted state. This V1/2 is well within the range of the V1/2 for M-currents expressed in heterologous cell systems (Robbins, 2001). Since the XE-sensitive current did not plateau at the maximum test potential of +10 mV, a Boltzmann fit was not done for the fed or refed states.
Figure 3.

Fasting reduces the activity of the M-current but not XE991-insensitive K+ currents in NPY neurons. A, The activation of the M-current (IM) in NPY neurons was recorded from fed, fasted, or refed male mice using the following protocol: in the present of TTX (1 μm) and from a holding potential of −60 mV, the voltage was jumped to −90 mV (500 ms) followed by steps from −80 to +10 mV in 10 mV increments (1 s) and returned to −60 mV. Currents were recorded in control conditions and after 10 min incubation with XE991 (40 μm). The XE991-sensitive current was calculated by subtracting the SS current (the last 50 ms of the voltage steps) in the XE991-treated traces from the control traces. This difference is graphed in the I–V plots as the amplitude of the XE991-sensitive current (Leão et al., 2009). B, The I–V curve of A is shown for fed males (n = 11), fasted males (n = 15), and 24 h refed males (n = 10). The currents at +10 mV for the three feeding states were 484 ± 37.6 pA for fed males, 259 ± 63.3 pA for fasted males, and 480 ± 53.6 pA for refed males. I–V plots were analyzed by a two-way ANOVA (ns) followed by Bonferroni–Dunn comparison. ap < 0.05; bp < 0.01; cp < 0.001, compared with fasted. C, A plot of the conductances at 0 mV between each feeding state comparing total, XE991-sensitive and -insensitive currents. D, Summary of the ratio [(Inst-SS)/SS] of the time-dependent (Inst-SS) and SS currents from control and XE991-treated conditions in response to a +10 mV step. **p < 0.01. Error bars indicate SEM.
To determine whether changes in the K+ conductance were due mainly to the M-current and not other K+ currents, the total conductances and the XE991-sensitive and XE991-insensitive currents were compared between each feeding state (Fig. 3C). The differences in the total conductance were directly correlated to the decrease in XE991-sensitive conductance in the fasted state. The ratio of time-dependent current (Inst-SS) to the SS current at +10 mV was calculated to determine the relative effect of XE991 on outward currents between the feeding states (Fig. 3D). The ratio at the +10 mV step was significantly higher after XE991-treated compared with control in the fasted state (p < 0.001), indicating that the reduction in the control currents was mostly due to the decrease in the steady-state M-current. Conversely, in the fed and refed males, blocking the M-current with XE991 did not significantly affect the ratio.
E2 increases activity of the M-current in ovariectomized females
Because E2 suppresses food intake and NPY neurons are orexigenic, we hypothesized that E2 may suppress feeding behavior by increasing the KCNQ5 expression and activity of the M-current in NPY neurons. To initially test this hypothesis, we used the deactivation protocol described in Figure 2 in the presence of 1 μm TTX. In oil-treated, ovariectomized females (n = 14), the M-current (XE991 sensitive) was significantly less than in E2-treated, ovariectomized females (n = 15; p < 0.01) (Fig. 4). The relaxation current at −35 mV was 15.2 ± 2.5 and 42.5 ± 3.9 pA for oil-treated and E2-treated females, respectively (Fig. 4) (p < 0.001). The RMP between oil- and E2-treated females was significantly different (−68.1 ± 1.8 vs −72.4 ± 1.1 mV; p < 0.05), while the capacitance between the treatments was not significantly different (oil treated, 18.5 ± 1.3 pF; E2 treated, 16.1 ± 0.9 pF). XE991 application significantly reduced the RMP in both treatments but only significantly increased input resistance in E2-treated females (Table 1), again indicating that the M-current contributed a significant portion of the resting conductance in the E2-treated NPY neurons. Interestingly, fasting (24 h) abrogated the E2-induced potentiation of the M-current and significantly reduced the relaxation current to 14.7 ± 2.6 pA (n = 11; p < 0.001) (Fig. 4). The RMP in NPY neurons from fasted, E2-treated females was significantly lower (−65.6 ± 1.5 mV) than in fed, E2-treated females. XE991 depolarized NPY neurons but did not significantly increase the input resistance in the fasted, E2-treated females (Table 1).
Figure 4.

17β-Estradiol increases the activity of the M-current in NPY neurons. Using the same protocol described in Figure 2, the I–V curves in the E2-treated females (n = 15), the oil-treated females (n = 14), and 24 h fasted E2-treated females (n = 11) are shown. I–V plots were analyzed by a two-way ANOVA (p < 0.01, F = 6.0, df = 2) followed by Bonferroni–Dunn comparison. ap < 0.05; bp < 0.01; cp < 0.001, compared with oil-treated and fasted E2-treated. Error bars indicate SEM.
To determine whether E2 alters the steady-state conductances and kinetics of the M-current in NPY neurons, we used the activation protocol described in Figure 3. In the presence of 1 μm TTX, the steady-state XE991-sensitive currents were significantly greater in fed, E2-treated females (n = 12) compared with both oil-treated (n = 14) and fasted, E2-treated females (n = 11; p < 0.001) (Fig. 5A). The current at +10 mV for the three treatments was 231 ± 24 pA for oil-treated females; 513 ± 82 pA for fed, E2-treated females; and 250 ± 31 pA for fasted, E2-treated females (p < 0.001, oil and fasted E2 compared with E2). The current at +10 mV for the fed, E2-treated, ovariectomized females was not significantly different from that of the fed, intact males. The percentage of steady-state current at +10 mV that was XE991 sensitive was 28.9 ± 2.8% in oil-treated females, 44.1 ± 3.1% in fed, E2-treated females, and 23.1 ± 1.9% in fasted, E2-treated females (oil vs fed E2, p < 0.01; fed E2 vs fasted E2, p < 0.001). As in the males, the M-current is active at subthreshold potentials and increases significantly in amplitude at −50 to −40 mV. Using G/Gmax and a Boltzmann equation fit, the V1/2 of the M-current in the oil-treated females was −22.2 ± 1.6 mV. Since the XE-sensitive current did not plateau at the maximum test potential of +10 mV, a Boltzmann fit was not done for the fed or fasted, E2-treated females.
Figure 5.
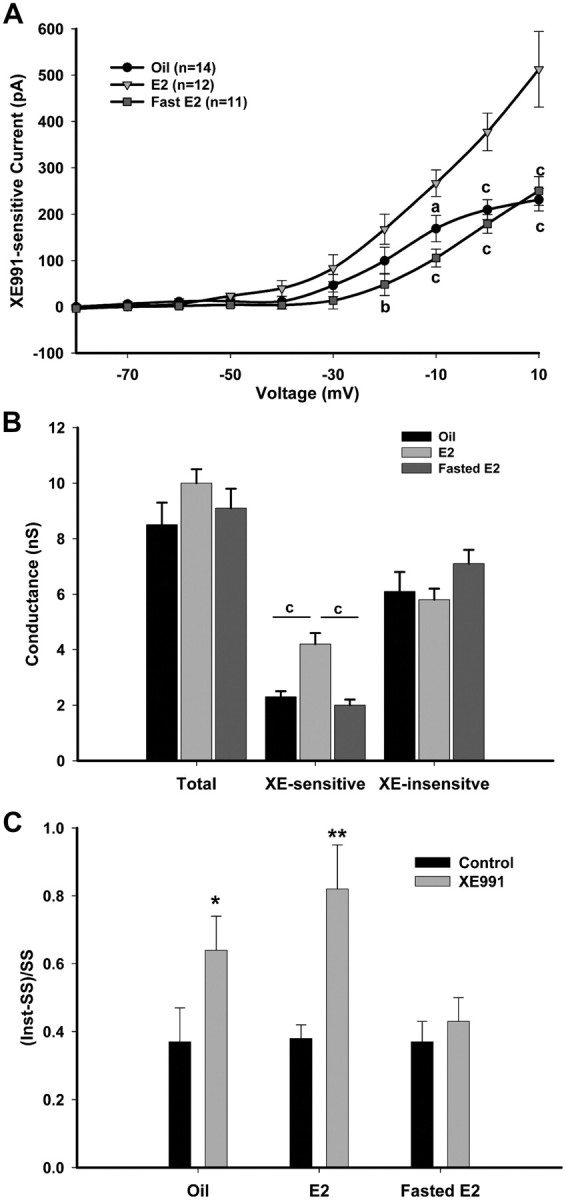
E2 increases M-current activity but not XE991-insensitive K+ currents in NPY neurons. A, The activation of the IM in NPY neurons was recorded from ovariectomized, oil-treated (n = 14), E2-treated (n = 12), and fasted, E2-treated (n = 11) female mice using the protocol described in Figure 3. The current at +10 mV for the three treatments was 231 ± 24, 513 ± 82, and 250 ± 31 pA, respectively. I–V plots were analyzed by a two-way ANOVA (p < 0.001, F = 11.0, df = 2) followed by Bonferroni–Dunn comparison. ap < 0.05; bp < 0.01; cp < 0.001, compared with E2 treated. B, A plot of the conductances at 0 mV between each steroid treatment comparing control, XE991-sensitive and -insensitive currents. C, Summary of the ratio [(Inst-SS)/SS] of the time-dependent (Inst-SS) and SS currents from control and XE991-treated conditions in response to a +10 mV step. *p < 0.05; **p < 0.01. Error bars indicate SEM.
As in the males, the total conductance and the XE991-sensitive and XE991-insensitive current were compared between steroid treatments and feeding states to determine whether the changes in total conductance were due solely to the M-current (Fig. 5B). The E2-treated, XE991-sensitive conductances were significantly greater than the oil-treated or fasted, E2-treated (p < 0.001). However, unlike the males, the total conductances were not significantly different, which suggests that the contribution of the M-current to the total conductance is smaller in female NPY neurons compared with male NPY neurons. The ratio of time-dependent current (Inst-SS) to the SS current at +10 mV was significantly different between control and XE991-treated conditions in both oil-treated and fed, E2-treated females (p < 0.001) (Fig. 5C).
KCNQ expression in mouse NPY neurons
Previously, we have shown that KCNQ2, -3, and -5 are all expressed in guinea pig NPY neurons (Roepke et al., 2007). Based on the present electrophysiological findings, we hypothesized that all three subunits would also be expressed in mouse NPY neurons. Using single-cell RT-PCR collected from eight ovariectomized females, the average percentage expression was 72 ± 2, 65 ± 2, and 48 ± 3% for KCNQ2, KCNQ3, and KCNQ5, respectively. A representative coexpression pattern is illustrated in Figure 6A (n = 8 animals; average, 15 cells/animal; 110 total cells). Only GFP cells positive for NPY mRNA were used in the analysis and 99% of all GFP-labeled arcuate neurons were NPY neurons. The coexpression pattern of the three KCNQ subunits showed that a majority (∼65%) of NPY neurons expressed more than one subunit. Among the harvested NPY neurons, 37% coexpressed KCNQ2 and -3, 33% coexpressed KCNQ2 and -5, and 25% coexpressed KCNQ3 and -5 (Fig. 6B). Among the 110 NPY neurons, 15% coexpressed all three subunits, 50% expressed two transcripts, 32% expressed only one, and the remaining 3% did not express any KCNQ transcripts. The majority of the cells expressing only one subunit expressed KCNQ2 or KCNQ3. One or more KCNQ subunits were detected in 97% of NPY neurons by single-cell RT-PCR, and the whole-cell recording of the M-current revealed that the channels are expressed in the vast majority of cells. All three KCNQ subunits may be expressed in the majority of cells but at levels beyond the sensitivity of the single-cell RT-PCR and therefore cannot be coupled directly to the electrophysiological analysis. We and others have previously discussed this technical limitation with other channels expressed in CNS neurons (Tkatch et al., 2000; Zhang et al., 2007).
Figure 6.
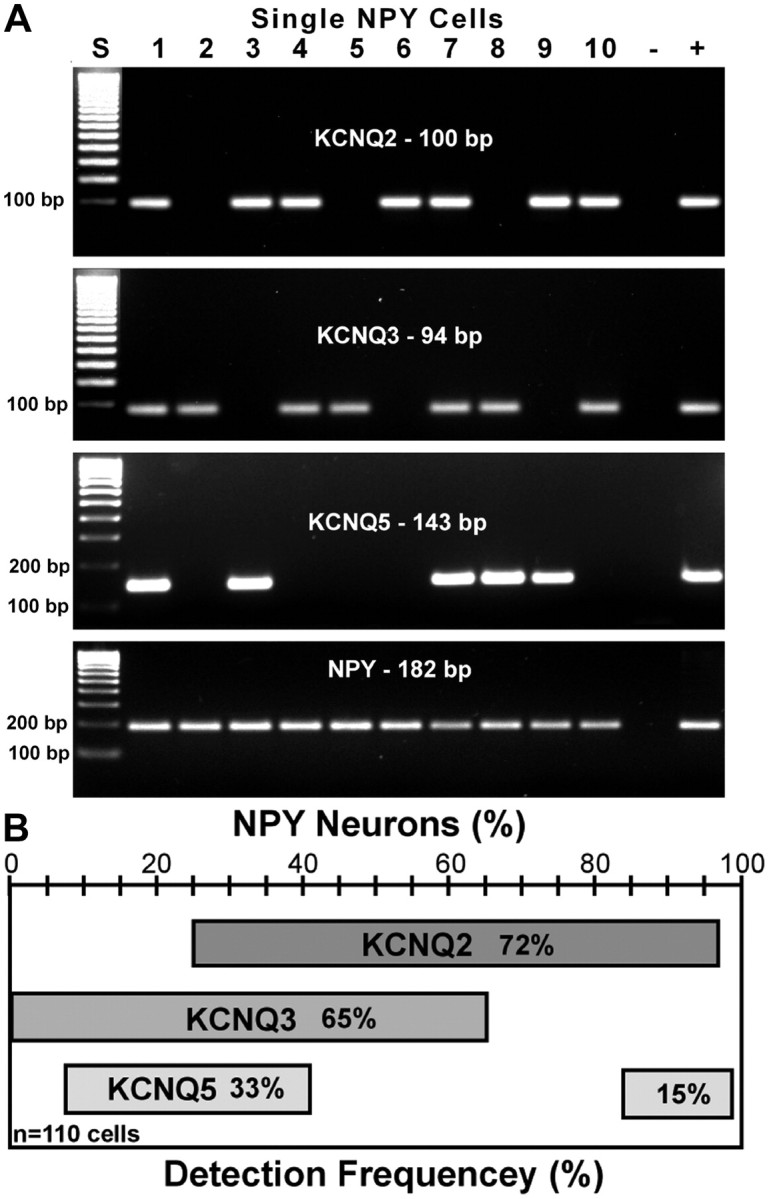
KCNQ subunits are highly expressed in NPY neurons. A, B, A representative gel (A) and a frequency distribution plot (B) illustrating the average number of NPY neurons expressing KCNQ2, KCNQ3, and KCNQ5 subunits from ovariectomized females. The total number of cells analyzed was 110 with an average of 15 cells per female. KCNQ4 is not expressed in NPY neurons. The ladder for the KCNQ2 and -3 gels has been overexposed to visualize the lower molecular (100 bp) weight bands.
Differential regulation of KCNQ channel subunits by fasting and E2
To determine whether regulation of KCNQ subunit gene expression is, at least, partially responsible for the reduction of the M-current, we examined the relative mRNA expression of KCNQ2, KCNQ3, and KCNQ5 (Fig. 7A–C, respectively) in pooled NPY neurons from fed and fasted males using quantitative real-time PCR. The expression of KCNQ2 and KCNQ3 in fasted males was significantly reduced compared with fed males (Fig. 7D) (n = 5; p < 0.01) without any effect on KCNQ5 expression (Fig. 7D). The regulation of KCNQ channel mRNA expression and whole-cell M-current by fasting indicates that the M-current is an important neuronal current in the control of energy homeostasis.
Figure 7.

Fasting reduces KCNQ2 and KCNQ3 expression and 17β-estradiol increases KCNQ5 expression in NPY neurons. A–C, Quantitative real-time PCR assays for KCNQ2 (A), KCNQ3 (B), and KCNQ5 (C) transcripts using the SYBR Green method. Standard curves were prepared with basal hypothalamic cDNA serial dilutions as follows: 1:50, 1:100, 1:500, 1:1000, 1:5000, 1:7500, and 1:10,000. Cycle number was plotted against the normalized fluorescence intensity (Delta Rn) to visualize the PCR amplification. The cycle threshold (CT, arrow) is the point in the amplification at which the sample values were calculated. The BH cDNA serial dilutions and one representative NPY pool (black squares) are illustrated in A–C. The insets in A–C show standard curve regression lines, slopes from serial dilution data, and the dissociation (melting) curves for all three transcripts. The amplification efficiencies calculated from the slopes were 96% for KCNQ2, 100% for KCNQ3, and 94% for KCNQ5. These efficiencies allowed us to use the ΔΔCT method for quantification. The melting curves depict single-product melting at 83, 78, and 80°C for KCNQ2, -3, and -5, respectively. Single peaks illustrate that only one product was formed in NPY pools. β-Actin was used as control and its primer pair was 100% efficient (data not shown). Negative (CSF, -RT cell, and tissue samples) and positive tissue controls were analyzed on each plate along with the experimental samples. D, Using cell harvesting coupled with quantitative real-time PCR, fasting significantly reduced the expression of KCNQ2 and KCNQ3 in NPY neurons [3–4 pools of 5 (KCNQ2/3) or 10 (KCNQ5) cells each from 5 animals; n = 5] from male mice. **p < 0.01. E, E2 treatment significantly increased the expression of KCNQ5 while fasting significantly reduced the expression of KCNQ2 and KCNQ3 in NPY neurons [3–4 pools of 5 (KCNQ2/3) or 10 (KCNQ5) cells each from 5 animals; n = 5] from female mice. *p < 0.05; **p < 0.01, compared with oil-treated. KCNQ2 and KCNQ3 expression in fed, E2-treated, and fasted, E2-treated were significantly different (p < 0.05). Error bars indicate SEM.
Previously, we have shown that estrogen replacement in ovariectomized female guinea pigs increased the expression of KCNQ5 in the arcuate nucleus after both 24 h and long-term treatment (Roepke et al., 2007, 2008). To determine whether regulation of KCNQ subunit gene expression is, at least, partially responsible for the increase in M-current activity in female arcuate NPY neurons, we also examined the relative mRNA expression of KCNQ subunits in pooled NPY neurons from oil- and E2-treated females using quantitative real-time PCR. The expression of KCNQ2 and KCNQ3 was not affected by E2 treatment, while KCNQ5 was significantly increased by E2 treatment (Fig. 7E) (n = 5; p < 0.05). As in the males (Fig. 7D), fasting significantly reduced the expression of KCNQ2 and KCNQ3 in NPY neurons from E2-treated females compared with fed, E2-treated females (Fig. 7E) (n = 5; p < 0.05) but did not affect the elevated KCNQ5 expression observed in fed, E2-treated females (Fig. 7E) (n = 5; p < 0.05 compared with oil-treated). The increase in M-current activity after E2 replacement indicates that the M-current has a role in mediating the effects of E2 on NPY neuronal activity, which may be, in part, due to the upregulation of KCNQ5 expression.
ERα expression and colocalization with KCNQ5 in NPY neurons
Because E2 increased the expression of KCNQ5 in NPY neurons, we wanted to determine whether this was dependent on ERα. Tissue RNA was extracted from the microdissected arcuate nucleus obtained from WT and ERα KO oil- and E2-treated mice. As found in NPY neurons, E2 replacement significantly increased the mRNA expression of KCNQ5 in RNA extracted from the arcuate nucleus dissected from wild-type females but failed to alter KCNQ5 expression in ERα KO females (Fig. 8A). Because E2 regulates KCNQ5 and NPY gene expression in the arcuate (Roepke et al., 2007; Roepke, 2009) but fails to increase KCNQ5 expression in NPY neurons in ERα KO mice, we determined the expression of ERα and ERβ in single and pooled NPY neurons collected from ovariectomized females. In pools of NPY neurons (n = 12; 5 cells per pool), ERα mRNA was detected in every pool using quantitative real-time PCR with an average CT value of 33.6 ± 0.19 and a single product at 80.5°C. ERβ was not detected in any NPY pools (data not shown). Using single-cell RT-PCR, we found that ERα mRNA was expressed in 19% [23 of 122; average, 18 cells/animal (n = 7)] of NPY neurons, while ERβ was not expressed in any of these neurons (Fig. 8B). Interestingly, 50% of ERα-positive NPY neurons coexpressed KCNQ5 mRNA, but only 18% of KCNQ5-positive NPY neurons coexpressed ERα. In contrast to mRNA expression, immunocytochemistry detected only a small percentage (<4%) of NPY neurons that colocalized ERα (Fig. 8C), which is similar to a previous report (Simonian et al., 1999).
Figure 8.

17β-Estradiol regulation of KCNQ5 expression is ERα dependent, and a small population of NPY neurons express ERα. A, E2 treatment in ovariectomized, wild-type females increased the mRNA expression of KCNQ5 in the arcuate nucleus but failed to regulate KCNQ5 expression in ERα knock-out mice. *p < 0.05. The number in the column equals the number of animals per treatment. B, A representative gel illustrating the expression of ERα in NPY neurons harvested from ovariectomized females and the coexpression of KCNQ5. C, Immunocytochemistry showed a small population of GFP-NPY neurons colocalizing ERα: NPY-GFP neurons to left and ERα-immunoreactive neurons in middle with an overlay illustrating colocalization (indicated by arrows) to right. Ninety-nine percent of GFP-labeled neurons express NPY mRNA. 3V demarks the third ventricle. The white bar in the overlay represents 25 μm. Error bars indicate SEM.
Discussion
For the first time, we show that energy states (fasting) regulate the expression and activity of the M-current in hypothalamic NPY neurons. In fasted males, the M-current was suppressed by approximately threefold in NPY neurons compared with fed males, in part, due to a reduction in KCNQ2/3 channel expression. This study is also the first to characterize the effects of E2 on M-current activity in a native neuronal cell type. E2 replacement in ovariectomized females potentiated the M-current compared with oil-treated females, in part, by increasing the mRNA expression of the KCNQ5 subunit. Fasting abrogated the effects of E2 on the M-current, indicating that the activity of the M-current is critical for the control of food intake via NPY neurons.
The M-current controls resting membrane potential and is suppressed during fasting in NPY neurons
Arcuate NPY neurons are involved in the control of food intake and are responsive to multiple central and peripheral signals that are responsive to satiety and fasting (for review, see Saper et al., 2002; Gao and Horvath, 2007; Woods, 2009). In fed animals, NPY neurons are in a less active state with reduced firing activity (Takahashi and Cone, 2005) and NPY expression, but NPY release is increased during periods of food restriction or deprivation (Brady et al., 1990; Yoshihara et al., 1996). Indeed, in our studies, fed NPY neurons were relatively quiescent and were more hyperpolarized compared with the fasted state. The depolarized state reported in fasted males may be due to presynaptic inputs, peripheral hormonal signals such as leptin, or the modulation of endogenous cation conductances (e.g., the M-current).
Most arcuate NPY neurons express multiple KCNQ subunits and exhibit a robust outward K+ current in fed males. One of the primary functions of the M-current is to control action potential firing, spike-frequency adaptation, depolarizing afterpotentials, and afterhyperpolarization (AHP) currents (Yue and Yaari, 2004; Gu et al., 2005; Peters et al., 2005; Tzingounis and Nicoll, 2008). Therefore, we examined the effects of XE991, a selective blocker of KCNQ channels, on the excitability of NPY neurons from fed males. We found that, by blocking the M-current, quiescent NPY neurons became depolarized and started firing continuously with a concomitant reduction in the rheobase. Although action potential firing did not occur spontaneously in the presence of glutaminergic and GABAergic blockers, the membrane potential was similarly depolarized. Therefore, the M-current plays a role in action potential generation in arcuate NPY neurons as in other neuronal cell types (Hu et al., 2007; Leão et al., 2009).
Interestingly, under our examination, NPY-GFP neurons in the fed state were more hyperpolarized compared with the initial description of this transgenic strain (van den Pol et al., 2009). In the study by van den Pol et al. (2009), NPY neurons exhibited a mean RMP of −56.6 mV and were highly active, exhibiting regular, irregular, and burst firing patterns. However, the neurons in the study by van den Pol et al. (2009) were mostly from juvenile mice (2–6 weeks of age) of unreported sex examined with different external and internal solutions. Therefore, the differences in membrane properties and excitability between the two studies may be due to differences in experimental conditions and/or changes in feeding patterns between weanling-juvenile mice used in the latter study and the mature, adult mice used in the present experiments, as suggested in previous studies (Leibowitz et al., 2005).
Fasting significantly reduces the current while increasing the neuronal excitability of the NPY neurons. This effect on M-current activity is partially restored by a 24 h refeeding but not by a shorter 2 h refeeding. The suppression of the M-current due to fasting appears to be an NPY-specific effect as a similar effect is not found in POMC neurons between fed and fasted male mice (n = 4, 5 respectively) (our unpublished observations). Moreover, the suppression of the M-current in NPY neurons by fasting may involve peripheral signals such as serum leptin concentrations, which are higher during states of satiety, driving KCNQ channel expression. However, it is not known, at this time, whether leptin affects KCNQ2/3 or M-current expression or that another hormone, peptide, or neurotransmitter is responsible for altered expression in the different feeding states in these hypothalamic neurons.
The role of the M-current in the control of food intake by 17β-estradiol
E2 is known to reduce NPY/AgRP expression in ovariectomized females (Crowley et al., 1985; Bonavera et al., 1994; Pelletier et al., 2007) and during proestrous when serum E2 levels are elevated (Olofsson et al., 2009). Short-term (3–48 h) E2 replacement reduces NPY mRNA expression and protein immunoreactivity in the arcuate nucleus (Crowley et al., 1985; Pelletier et al., 2007), and long-term (18 d) E2 treatment reduces the in vitro release of NPY in the paraventricular nucleus (Bonavera et al., 1994). The suppression of NPY/AgRP mRNA expression in intact females is associated with a reduction in food intake and body weight, and E2 attenuates food intake during refeeding (2 h) after fasting, indicating that the interplay between fasting and E2 is complex (Olofsson et al., 2009). E2 replacement also attenuates the increase in food intake following NPY infusion into the lateral ventricle (Santollo and Eckel, 2008). These data suggest that the anorectic effects of E2 involve the suppression of NPY expression and neuronal function, which may involve the potentiation of the M-current.
NPY neurons are significantly hyperpolarized in the E2- versus oil-treated, ovariectomized females, in part, because of the potentiation of the M-current. This potentiation could be due, in part, to the KCNQ5 subunit expression. Furthermore, the KCNQ5 subunit contributes to the medium and slow AHP current (Tzingounis et al., 2010) in hippocampal CA3 neurons, which may also be altered in the E2-treated NPY neurons to reduce firing frequency. Ultimately, a reduction in NPY activity would lead to less NPY/AgRP release and subsequently an attenuation in feeding activity.
Fasting abrogated the effects of E2 treatment on M-current activity. This fasting effect was due, in part, to reduction of KCNQ2/3 expression in the native neurons, resulting in a significant depolarization of the membrane potential, similar to the fasted males. The effects of fasting overwhelmed the effects of E2 (elevated KCNQ5 expression) at the neuronal level via the reduction of the other KCNQ subunits, especially KCNQ3. Coexpression of KCNQ3 with KCNQ5 in heterologous cell systems increases current amplitudes twofold to threefold over KCNQ5 homomultimers and also produces currents with similar amplitudes to the native M-current in hippocampal neurons and KCNQ2/3 heteromultimers in the heterologous cells (Lerche et al., 2000; Tzingounis et al., 2010). Thus, the more robust M-current measured in fed, E2-treated NPY neurons may be due to a greater association of KCNQ3/5, while the effects of fasting reduces this association via a reduction in KCNQ3.
We have previously shown that E2 will increase KCNQ5 expression in the guinea pig arcuate nucleus both after 24 h and long-term treatment using unbiased microarray analysis followed by quantitative real-time PCR (Roepke et al., 2007, 2008). In the mouse, E2 replacement also increased the expression of KCNQ5 both in the whole arcuate and specifically in NPY neurons. The gene regulation is lost in ERα KO transgenic mice, suggesting that ERα is involved in KCNQ5 gene regulation. However, ERα mRNA and ERα protein are detected in only 19% and in <4%, respectively, of native NPY neurons and colocalizes with only ∼20% of KCNQ5-expressing NPY neurons. Therefore, these data suggest that cellular localization of ERα in NPY neurons is below detectability or that ERα-mediated gene regulation is not the sole mechanism. Similarly, previous immunohistochemical studies have reported low levels or absence of colocalization between NPY neurons and ERα (Simonian et al., 1999; Olofsson et al., 2009). The estrogenic regulation of KCNQ5 may be via ER–protein associations (Sp-1, Ap-1, etc.) because a putative ERE (estrogen response element) has not been identified within the KCNQ5 gene using an online promoter sequence search tool (Roepke et al., 2007). Consequently, the low level of ERα colocalization suggests that the effects of E2 on the M-current occur via multiple mechanisms including nuclear receptor-mediated gene regulation, presynaptic inputs from other arcuate neurons, and/or potentially via a membrane-mediated (G-protein-coupled receptor) mechanism (Kelly and Rønnekleiv, 2008) that will be examined in future experiments.
In conclusion, NPY neurons are orexigenic and active during states of negative energy balance. Suppression of the M-current by gene regulation, and potentially other pathways, is a novel mechanism for the increase in NPY neuronal activity during fasting. Furthermore, E2 treatment augments the M-current, in part, via KCNQ5 gene regulation and is abrogated during fasting. The opposing actions of E2 and fasting on the M-current suggests that the M-current plays a pivotal role in NPY neuronal excitability and may be an important cellular target for neurotransmitter and hormonal signals to control energy homeostasis in both males and females.
Footnotes
This work was supported by NIH Grants DK 68098, NS 43330 and NS 38809, and K99 DK 83457 (T.A.R.). We thank Martha A. Bosch for her expert technical assistance with single-cell harvesting and quantitative real-time PCR. We thank Dr. Brad Lowell (Harvard University, Cambridge, MA) for providing the transgenic GFP-NPY mice.
References
- Bonavera JJ, Dube MG, Kalra PS, Kalra SP. Anorectic effects of estrogen may be mediated by decreased neuropeptide-Y release in the hypothalamic paraventricular nucleus. Endocrinology. 1994;134:2367–2370. doi: 10.1210/endo.134.6.8194462. [DOI] [PubMed] [Google Scholar]
- Brady LS, Smith MA, Gold PW, Herkenham M. Altered expression of hypothalamic neuropeptide mRNA in food-restricted and food-deprived rats. Neuroendocrinology. 1990;52:441–447. doi: 10.1159/000125626. [DOI] [PubMed] [Google Scholar]
- Bronson FH, Vom Saal FS. Control of the preovulatory release of luteinizing hormone by steroids in the mouse. Endocrinology. 1979;104:1247–1255. doi: 10.1210/endo-104-5-1247. [DOI] [PubMed] [Google Scholar]
- Crowley WR, Tessel RE, O'Donohue TL, Adler BA, Kalra SP. Effects of ovarian hormones on the concentrations of immunoreactive neuropeptide Y in discrete brain regions of the female rat: correlation with serum luteinizing hormone (LH) and median eminence LH-releasing hormone. Endocrinology. 1985;117:1151–1155. doi: 10.1210/endo-117-3-1151. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Elmedyb P, Calloe K, Schmitt N, Hansen RS, Grunnet M, Olesen SP. Modulation of ERG channels by XE991. Basic Clin Pharmacol Toxicol. 2007;100:316–322. doi: 10.1111/j.1742-7843.2007.00048.x. [DOI] [PubMed] [Google Scholar]
- Elmquist JK. Hypothalamic pathways underlying the endocrine, autonomic, and behavioral effects of leptin. Physiol Behav. 2001;74:703–708. doi: 10.1016/s0031-9384(01)00613-8. [DOI] [PubMed] [Google Scholar]
- Gao Q, Horvath TL. Neurobiology of feeding and energy expenditure. Annu Rev Neurosci. 2007;30:367–398. doi: 10.1146/annurev.neuro.30.051606.094324. [DOI] [PubMed] [Google Scholar]
- Giloh H, Sedat JW. Fluorescence microscopy: reduced photobleaching of rhodamine and fluorescein protein conjugates by n-propyl gallate. Science. 1982;217:1252–1255. doi: 10.1126/science.7112126. [DOI] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Brüning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not Kca2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskell-Luevano C, Cone RD, Monck EK, Wan YP. Structure activity studies of the Melanocortin-4 receptor by in vitro mutagenesis: identification of agouti-related protein (AGRP), melanocortin agonist and synthetic peptide antagonist interaction determinants. Biochemistry. 2001;40:6164–6179. doi: 10.1021/bi010025q. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Rønnekleiv OK, Low MJ, Kelly MJ. Hypothalamic proopiomelanocortin neurons are glucose-responsive and express K-ATP channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Rønnekleiv OK. Membrane-initiated estrogen signaling in hypothalamic neurons. Mol Cell Endocrinol. 2008;290:14–23. doi: 10.1016/j.mce.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Loose MD, Rønnekleiv OK. Opioids hyperpolarize β-endorphin neurons via μ-receptor activation of a potassium conductance. Neuroendocrinology. 1990;52:268–275. doi: 10.1159/000125597. [DOI] [PubMed] [Google Scholar]
- Kohno D, Nakata M, Maekawa F, Fujiwara K, Maejima Y, Kuramochi M, Shimazaki T, Okano H, Onaka T, Yada T. Leptin suppresses ghrelin-induced activation of neuropeptide Y neurons in the arcuate nucleus via phosphatidylinositol 3-kinase- and phosphodiesterase 3-mediated pathway. Endocrinology. 2007;148:2251–2263. doi: 10.1210/en.2006-1240. [DOI] [PubMed] [Google Scholar]
- Leão RN, Tan HM, Fisahn A. Kv7/KCNQ channels control action potential phasing of pyramidal neurons during hippocampal gamma oscillations in vitro. J Neurosci. 2009;29:13353–13364. doi: 10.1523/JNEUROSCI.1463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz SF, Sepiashvili K, Akabayashi A, Karatayev O, Davydova Z, Alexander JT, Wang J, Chang GQ. Function of neuropeptide Y and agouti-related protein at weaning: relation to corticosterone, dietary carbohydrate and body weight. Brain Res. 2005;1036:180–191. doi: 10.1016/j.brainres.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loose MD, Rønnekleiv OK, Kelly MJ. Membrane properties and response to opioids of identified dopamine neurons in the guinea pig hypothalamus. J Neurosci. 1990;10:3627–3634. doi: 10.1523/JNEUROSCI.10-11-03627.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XY, Nicholson JR, Akil H, Watson SJ. Time course of short-term and long-term orexigenic effects of agouti-related protein (86–132) Neuroreport. 2001;12:1281–1284. doi: 10.1097/00001756-200105080-00045. [DOI] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- Olofsson LE, Pierce AA, Xu AW. Functional requirement of AgRP and NPY neurons in ovarian cycle-dependent regulation of food intake. Proc Natl Acad Sci U S A. 2009;106:15932–15937. doi: 10.1073/pnas.0904747106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier G, Li S, Luu-The V, Labrie F. Oestrogenic regulation of pro-opiomelanocortin, neuropeptide Y and corticotrophin-releasing hormone mRNAs in mouse hypothalamus. J Neuroendocrinol. 2007;19:426–431. doi: 10.1111/j.1365-2826.2007.01548.x. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Rønnekleiv OK, Kelly MJ. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–9540. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Fang Y, Rønnekleiv OK, Kelly MJ. Leptin excites proopiomelanocortin neurons via activation of TRPC channels. J Neurosci. 2010;30:1560–1565. doi: 10.1523/JNEUROSCI.4816-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Roepke TA. Oestrogen modulates hypothalamic control of energy homeostasis through multiple mechanisms. J Neuroendocrinol. 2009;21:141–150. doi: 10.1111/j.1365-2826.2008.01814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepke TA, Malyala A, Bosch MA, Kelly MJ, Rønnekleiv OK. Estrogen regulation of genes important for K+ channel signaling in the arcuate nucleus. Endocrinology. 2007;148:4937–4951. doi: 10.1210/en.2007-0605. [DOI] [PubMed] [Google Scholar]
- Roepke TA, Xue C, Bosch MA, Scanlan TS, Kelly MJ, Rønnekleiv OK. Genes associated with membrane-initiated signaling of estrogen and energy homeostasis. Endocrinology. 2008;149:6113–6124. doi: 10.1210/en.2008-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santollo J, Eckel LA. Estradiol decreases the orexigenic effect of neuropeptide Y, but not agouti-related protein, in ovariectomized rats. Behav Brain Res. 2008;191:173–177. doi: 10.1016/j.bbr.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Schweitzer P. Cannabinoids decrease the K+ M-current in hippocampal CA1 neurons. J Neurosci. 2000;20:51–58. doi: 10.1523/JNEUROSCI.20-01-00051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonian SX, Spratt DP, Herbison AE. Identification and characterization of estrogen receptor α-containing neurons projecting to the vicinity of the gonadotropin-releasing hormone perikarya in the rostral preoptic area of the rat. J Comp Neurol. 1999;411:346–358. doi: 10.1002/(sici)1096-9861(19990823)411:2<346::aid-cne13>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Stanley BG, Leibowitz SF. Neuropeptide Y injected in the paraventricular hypothalamus: a powerful stimulant of feeding behavior. Proc Natl Acad Sci U S A. 1985;82:3940–3943. doi: 10.1073/pnas.82.11.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi KA, Cone RD. Fasting induces a large, leptin-dependent increase in the intrinsic action potential frequency of orexigenic arcuate neurons neuropeptide Y/agouti-related protein neurons. Endocrinology. 2005;146:1043–1047. doi: 10.1210/en.2004-1397. [DOI] [PubMed] [Google Scholar]
- Tkatch T, Baranauskas G, Surmeier DJ. Kv4.2 mRNA abundance and A-type K+ current amplitude are linearly related in basal ganglia and basal forebrain neurons. J Neurosci. 2000;20:579–588. doi: 10.1523/JNEUROSCI.20-02-00579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci U S A. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, Jentsch TJ. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci U S A. 2010;107:10232–10237. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Yao Y, Fu LY, Foo K, Huang H, Coppari R, Lowell BB, Broberger C. Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing stong Renilla green fluorescent protein in NPY neurons. J Neurosci. 2009;29:4622–4639. doi: 10.1523/JNEUROSCI.3249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wang JH, Wang F, Yang MJ, Yu DF, Wu WN, Liu J, Ma LQ, Cai F, Chen JG. Leptin regulated calcium channels of NPY and POMC neurons by activation of different signal pathways. Neuroscience. 2008;156:89–98. doi: 10.1016/j.neuroscience.2008.04.079. [DOI] [PubMed] [Google Scholar]
- Woods SC. The control of food intake: behavioral versus molecular perspectives. Cell Metab. 2009;9:489–498. doi: 10.1016/j.cmet.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Roepke TA, Zhang C, Rønnekleiv OK, Kelly MJ. GnRH activates the M-current in GnRH neurons: an autoregulatory negative feedback mechanism? Endocrinology. 2008;149:2459–2466. doi: 10.1210/en.2007-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang MJ, Wang F, Wang JH, Wu WN, Hu ZL, Cheng J, Yu DF, Long LH, Fu H, Xie N, Chen JG. PI3K integrates the effects of insulin and leptin on large-conductance Ca2+-activated K+ channels in neuropeptide Y neurons of the hypothalamic arcuate nucleus. Am J Physiol Endocrinol Metab. 2010;298:E193–E201. doi: 10.1152/ajpendo.00155.2009. [DOI] [PubMed] [Google Scholar]
- Yoshihara T, Honma S, Honma K. Effects of restricted daily feeding on neuropeptide Y release in the rat paraventricular nucleus. Am J Physiol. 1996;270:E589–E595. doi: 10.1152/ajpendo.1996.270.4.E589. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Levine JE, Rønnekleiv OK, Kelly MJ. Gonadotropin-releasing hormone neurons express KATP channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. J Neurosci. 2007;27:10153–10164. doi: 10.1523/JNEUROSCI.1657-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Roepke TA, Kelly MJ, Rønnekleiv OK. Kisspeptin depolarizes GnRH neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28:4423–4434. doi: 10.1523/JNEUROSCI.5352-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Rick EA, Kelly MJ, Rønnekleiv OK. 17β-Estradiol regulation of T-type calcium channels in gonadotropin-releasing hormone neurons. J Neurosci. 2009;29:10552–10562. doi: 10.1523/JNEUROSCI.2962-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]