

Abstract
Phylogenetic inference is fundamental to our understanding of most aspects of the origin and evolution of life, and in recent years, there has been a concentration of interest in statistical approaches such as Bayesian inference and maximum likelihood estimation. Yet, for large data sets and realistic or interesting models of evolution, these approaches remain computationally demanding. High-throughput sequencing can yield data for thousands of taxa, but scaling to such problems using serial computing often necessitates the use of nonstatistical or approximate approaches. The recent emergence of graphics processing units (GPUs) provides an opportunity to leverage their excellent floating-point computational performance to accelerate statistical phylogenetic inference. A specialized library for phylogenetic calculation would allow existing software packages to make more effective use of available computer hardware, including GPUs. Adoption of a common library would also make it easier for other emerging computing architectures, such as field programmable gate arrays, to be used in the future. We present BEAGLE, an application programming interface (API) and library for high-performance statistical phylogenetic inference. The API provides a uniform interface for performing phylogenetic likelihood calculations on a variety of compute hardware platforms. The library includes a set of efficient implementations and can currently exploit hardware including GPUs using NVIDIA CUDA, central processing units (CPUs) with Streaming SIMD Extensions and related processor supplementary instruction sets, and multicore CPUs via OpenMP. To demonstrate the advantages of a common API, we have incorporated the library into several popular phylogenetic software packages. The BEAGLE library is free open source software licensed under the Lesser GPL and available from http://beagle-lib.googlecode.com. An example client program is available as public domain software.
Keywords: Bayesian phylogenetics, GPU, maximum likelihood, parallel computing
Most modern approaches to statistical phylogenetic inference involve computing the probability of observed character data for a set of taxa given a phylogenetic model—often a tree and continuous-time Markov chain model of character state evolution. Felsenstein (1981) demonstrated an efficient algorithm to calculate this probability, which is often referred to as the likelihood of the model. His algorithm recursively computes partial likelihoods via simple sums and products. These partial likelihoods track the probability of the observed data descended from an internal node conditional on a particular state at that internal node. A library that implements the calculations required by Felsenstein's algorithm is appealing because this procedure accounts for the majority of computing time in most likelihood-based phylogenetic operations. Furthermore, the algorithm offers opportunities for parallelization.
In typical phylogenetic models, likelihood calculation operations assume independence at several levels. These independencies provide the opportunity to perform operations in parallel. For example, models often assume that sites in a sequence alignment evolve independently, so that one can compute the likelihood for each site separately. The product of site likelihoods yields the likelihood for the alignment. In models that include among-site rate variation via a finite mixture, it is often possible to calculate conditional likelihoods given each rate category in parallel. Several other opportunities for parallelism exist at a finer scale.
We have developed the software library BEAGLE: Broad-platform Evolutionary Analysis General Likelihood Evaluator. BEAGLE provides a uniform interface for calculating phylogenetic likelihoods under a variety of different phylogenetic models. The library implements parallelism in the likelihood calculation on important emerging computer hardware technology, including graphics processing units (GPUs) and multicore central processing units (CPUs). We intend for users to install the library as a shared resource to be used by any phylogenetic software that supports the library. This approach allows developers of phylogenetic software to share any optimizations of the core calculations and any package that uses BEAGLE will automatically benefit from the improvements to the library. For researchers, this centralization provides a single installation to take advantage of new hardware and parallelization techniques. We now describe the interface to the library and some details regarding its implementation.
APPLICATION PROGRAMMING INTERFACE
Key Concepts
The key to BEAGLE performance lies in delivering fine-scale parallelization while minimizing data transfer and memory copy overhead. To accomplish this, the library lacks the concept or data structure for a tree, in spite of the intended use for phylogenetic analysis. Instead, BEAGLE acts directly on flexibly indexed data storage (called buffers) for observed character states and partial likelihoods. The client program can set the input buffers to reflect the data and can calculate the likelihood of a particular phylogeny by invoking likelihood calculations on the appropriate input and output buffers in the correct order. Because of this design simplicity, the library can support many different tree inference algorithms and likelihood calculation on a variety of models. Arbitrary numbers of states can be used, as can nonreversible substitution matrices via complex eigen decompositions, and mixture models with multiple rate categories and/or multiple eigen decompositions. Finally, BEAGLE application programming interface (API) calls can be asynchronous, allowing the calling program to implement other coarse-scale parallelization schemes such as evaluating independent genes or running concurrent Markov chains.
Usage
To use the library, a client program first creates an instance of BEAGLE by calling beagleCreateInstance (further API method names can be found in the documentation distributed with the library); multiple instances per client are possible and encouraged. All additional functions are called with a reference to this instance. The client program can optionally request that an instance run on certain hardware (e.g., a GPU) or have particular features (e.g., double-precision math). Next, the client program must specify the data dimensions and specify key aspects of the phylogenetic model. Character state data are then loaded and can be in the form of discrete observed states or partial likelihoods for ambiguous characters. The observed data are usually unchanging and loaded only once at the start to minimize memory copy overhead. The character data can be compressed into unique “site patterns” and associated weights for each. The parameters of the substitution process can then be specified, including the equilibrium state frequencies, the rates for one or more substitution rate categories and their weights, and finally, the eigen decomposition for the substitution process.
In order to calculate the likelihood of a particular tree, the client program then specifies a series of integration operations that correspond to steps in Felsenstein's algorithm. Finite-time transition probabilities for each edge are loaded directly if considering a nondiagonalizable model or calculated in parallel from the eigen decomposition and edge lengths specified. This is performed within BEAGLE's memory space to minimize data transfers. A single function call will then request one or more integration operations to calculate partial likelihoods over some or all nodes. The operations are performed in the order they are provided, typically dictated by a postorder traversal of the tree topology. The client needs only specify nodes for which the partial likelihoods need updating, but it is up to the calling software to keep track of these dependencies. The final step in evaluating the phylogenetic model is done using an API call that yields a single log likelihood for the model given the data.
Aspects of the BEAGLE API design support both maximum likelihood (ML) and Bayesian phylogenetic tree inference. For ML inference, API calls can calculate first and second derivatives of the likelihood with respect to the lengths of edges (branches). In both cases, BEAGLE provides the ability to cache and reuse previously computed partial likelihood results, which can yield a tremendous speedup over recomputing the entire likelihood every time a new phylogenetic model is evaluated.
MATERIALS AND METHODS
The core BEAGLE library is implemented in C++ with C and Java JNI interfaces. BEAGLE uses a runtime module loading system to load hardware-specific plugins (shared libraries) when suitable hardware is available. Current plugins implement BEAGLE on GPUs using CUDA and OpenCL (in development), CPUs with vector instructions using Streaming SIMD Extensions (SSE), and multicore systems via OpenMP. BEAGLE is available for Linux, Mac, and Windows operating systems and is packaged with conventional installer methods for each.
GPU Implementation
The GPU implementation of BEAGLE supports both single- and double-precision arithmetic. Single precision requires more frequent use of a rescaling scheme to avoid underflow but allows BEAGLE to run on a greater variety of graphics processors since initial generations of such hardware did not include support for double-precision math. The GPU does fine-scale parallelization of the likelihood calculation, primarily by parallelizing across alignment sites, rate categories, and state values. Models such as amino acid (20 states) or codon models (64 states), therefore, permit a greater degree of parallelization than nucleotide models (4 states) and also yield the most notable speedups on GPU hardware (Suchard and Rambaut 2009). The CUDA kernels load using the CUDA driver API, which enables them to be compiled at runtime and utilize features specific to the particular hardware and CUDA version installed. Multiple GPUs can be seamlessly utilized simultaneously via multiple BEAGLE instances.
CPU-based Implementations
In addition to a standard serial CPU implementation, BEAGLE includes two other CPU-based implementations that exploit parallelism in different ways. An SSE implementation in double precision uses vector processing extensions present in many CPUs to parallelize computation across character state values. Single-precision SSE vectorization has not been a BEAGLE priority as other phylogenetic tools already provide this feature (Ronquist and Huelsenbeck 2003, Swofford 2003) and, so, is not yet available in BEAGLE. The OpenMP implementation uses multiple threads to parallelize computation across rate categories. Although finer-scale parallelization, equivalent to that achieved for GPU devices, could be attempted, it is unlikely to yield significant speedups due to the thread synchronization overhead in the OpenMP model.
EXAMPLE
Program Speedups
Currently, three popular phylogenetic software packages interface with BEAGLE: MrBayes (Ronquist and Huelsenbeck 2003) and BEAST (Drummond and Rambaut 2007), which use Bayesian inference, and GARLI (Zwickl 2006), which uses an ML approach. We benchmarked each of these programs to compare the speed of their native likelihood calculators to the BEAGLE implementations. In order to better exploit the parallelism offered by the GPU implementation, we used a data set with a large number of alignment sites and ran it under both nucleotide and codon models. More specifically, the data set used had 15 taxa and 18,792 nucleotide columns, 8558 of which were unique; for the codon model, 6080 of the 6264 site patterns were unique. This data set was a subset of a larger arthropod data set (Regier et al. 2010). We performed these benchmarks on a standard desktop PC with a 2.9 GHz Intel Core i7-930 CPU and 6 GB of 1.6 GHz DDR3 RAM. The PC was equipped with an NVIDIA GTX 580 GPU, with 1.5 GB of RAM and 512 processing cores running at 1.5 GHz. Figure 1 shows runtime speedups for each program when using BEAGLE CPU, SSE, and GPU implementations under nucleotide and codon models. For the GPU implementation, we also benchmarked in single-precision mode. Reported speedups are relative to the runtime when using the native sequential CPU implementation of each program. We note that the GARLI interface with BEAGLE is not fully optimized. Although we expect that further integration work will produce positive results, in our tests, only the GPU implementation achieved effective speedups. We have thus omitted the results from the CPU-based implementations.
FIGURE 1.
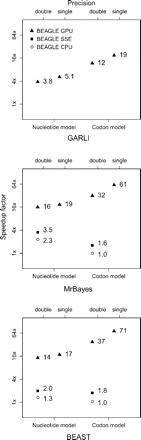
Performance using the BEAGLE library relative to the native sequential CPU implementations of phylogenetic analysis programs GARLI, MrBayes, and BEAST. Speedup factors are on a log scale.
For the BEAGLE GPU implementation, we observe significant speedups across all programs. The speedups are largest under the codon models, as they allow for better utilization of the GPU cores. We also observe the higher performance cost of double-precision calculation on the GPU relative to single precision. Overall, the highest speedup is 71-fold, for the BEAGLE GPU single-precision implementation when compared with the BEAST native implementation, under the codon model.
We note that not every analysis run on a GPU will achieve the same speedups we report, and, in some circumstances, using the BEAGLE GPU implementation may result in a slower overall runtime than using a CPU implementation. Several factors affect the relative performance. Beyond state-space size and numerical precision, the number of unique alignment columns and the hardware specifications of the GPU, especially numbers of cores and memory bandwidth, are important factors. We recommend that users first assess the relative performance of the GPU implementation with their setup by performing short comparative runs, which specify a smaller chain length or fewer generations.
CONCLUSION
BEAGLE is an API and library for high-performance evaluation of phylogenetic likelihoods. The API provides a uniform interface for performing calculations on an expanding variety of computer hardware platforms including GPUs, multicore CPUs, and SSE vectorization. On GPUs, the library provides novel algorithms and methods for evaluating likelihoods under arbitrary molecular evolutionary models, harnessing the large number of processing cores to efficiently parallelize calculations. Current results show speedups of up to 71-fold on a single GPU over CPU-based likelihood calculators. BEAGLE is currently integrated with three state-of-the-art phylogenetic software packages: MrBayes, BEAST, and GARLI, and compatible with many more. Forthcoming extensions include OpenCL support, single-precision SSE vectorization, improved performance for highly partitioned data sets, and additional high-level language wrappers, such as Python.
BEAGLE is freely available from http://beagle-lib.googlecode.com under the GNU Lesser General Public License and new collaborators are welcome.
FUNDING
This work was supported by the National Science Foundation [grant numbers DBI-0755048, DEB-0732920, DEB-1036448, DMS-0931642, EF-0331495, EF-0905606, EF-0949453]; the National Institutes of Health [grant numbers R01-HG006139, R01-GM037841, R01-GM078985, R01-GM086887, R01-NS063897]; the Biotechnology and Biological Sciences Research Council [grant number BB/H011285/1]; the Wellcome Trust [grant number WT092807MA]; and Google Summer of Code.
Acknowledgments
Some of the development of BEAGLE occurred at meetings at the University of Maryland, the National Evolutionary Synthesis Center through a working group (Software for Bayesian Evolutionary Analysis), the Mathematical Biosciences Institute at The Ohio State University, and several instances of the Workshop on Molecular Evolution (Europe and North America). We thank Jerome Regier for providing the data set used in evaluating the programs.
References
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Regier J, Shultz J, Zwick A, Hussey A, Ball B, Wetzer R, Martin J, Cunningham C. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463:1079–1083. doi: 10.1038/nature08742. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. Mr Bayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Suchard MA, Rambaut A. Many-core algorithms for statistical phylogenetics. Bioinformatics. 2009;25:1370–1376. doi: 10.1093/bioinformatics/btp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP*: phylogenetic analysis using parsimony (* and other methods) 2003 Version 4. Sunderland (MA): Sinauer Associates. [Google Scholar]
- Zwickl DJ. Austin (TX): University of Texas; 2006. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion [PhD dissertation] pp. 1–115. [Google Scholar]