

Abstract
The IL-6/STAT3 and TNFα/NFκB pathways are emerging as critical mediators of inflammation-associated colon cancer. TNFR2 expression is increased in inflammatory bowel diseases, the azoxymethane/dextran sodium sulfate (AOM/DSS) model of colitis-associated cancer, and by combined IL-6 and TNFα. The molecular mechanisms that regulate TNFR2 remain undefined. This study used colon cancer cell lines to test the hypothesis that IL-6 and TNFα induce TNFR2 via STAT3 and/or NFκB. Basal and IL-6 + TNFα-induced TNFR2 were decreased by pharmacological STAT3 inhibition. NFκB inhibition had little effect on IL-6 + TNFα-induced TNFR2, but did inhibit induction of endogenous IL-6 and TNFR2 in cells treated with TNFα alone. Chromatin immunoprecipitation (ChIP) revealed cooperative effects of IL-6 + TNFα to induce STAT3 binding to a -1578 STAT response element in the TNFR2 promoter, but no effect on NFκB binding to consensus sites. Constitutively active STAT3 was sufficient to induce TNFR2 expression. Over-expression of SOCS3, a cytokine-inducible STAT3 inhibitor, which reduces tumorigenesis in preclinical models of colitis-associated cancer, decreased cytokine-induced TNFR2 expression and STAT3 binding to the -1578 STAT response element. SOCS3 over-expression also decreased proliferation of colon cancer cells and dramatically decreased anchorage-independent growth of colon cancer cells, even cells over-expressing TNFR2. Collectively, these studies demonstrate that IL-6 and TNFα-induced TNFR2 expression in colon cancer cells is mediated primarily by STAT3, and provide evidence that TNFR2 may contribute to the tumor-promoting roles of STAT3.
Keywords: TNFR2, STAT3, Colon Cancer, Inflammation, SOCS3
Introduction
Patients with inflammatory bowel diseases (IBD) such as Crohn's disease and ulcerative colitis have an increased lifetime risk of developing inflammation-associated colorectal cancer (CRC) (1-3). Chronic increases in proliferation of intestinal epithelial cells (IEC) driven by pro-inflammatory factors have been shown to promote tumorigenesis. The IL-6/STAT3 (4-9) and TNFα/NFκB (10-12) pathways are both major mediators of inflammation-associated CRC and recent studies show that TNFα blockade decreases intestinal tumor formation in mice (13, 14).
TNFα signals through two receptors: TNFR1 and TNFR2. TNFR1 exerts pro-apoptotic functions due to its intracellular death domain (15). TNFR2, which lacks a death domain, has been linked to increased proliferation of IEC in animal models of colitis and colon cancer cells (16, 17). This supports a concept that TNFR2 may mediate pro-tumorigenic effects of TNFα. The role of TNFR2 in inflammation-associated cancer is a topic of increasing interest, as recent studies indicate that TNFR2 is up-regulated in IBD and in the azoxymethane/dextran sodium sulfate (AOM/DSS) model of inflammation-associated cancer (14, 16, 18). In vitro studies have shown that TNFR2 is induced in colon cancer cells by TNFα and IL-6 combined, but neither cytokine alone (16). Other studies have demonstrated TNFR2 induction by IFNγ (19). These findings suggest that the STAT pathways activated by IL-6 or IFNγ and/or NFκB pathways typically activated by TNFα may interact to induce TNFR2 expression. In support of this possibility, the human TNFR2 promoter contains two consensus STAT binding sites as well as two consensus NFκB binding sites (20). The present study tested the hypothesis that IL-6 and TNFα interact to induce TNFR2 expression by activation of STAT3, NFκB or both STAT3 and NFκB.
Suppressors of cytokine signaling (SOCS) proteins are negative feedback inhibitors of the JAK-STAT pathway (21). IEC-specific SOCS3 gene deletion increased tumor load in the AOM/DSS model of colitis-associated CRC (22). This effect was associated with enhanced activation of both STAT3 and NFκB (22). In vitro, SOCS3 over-expression reduced proliferation of colon cancer cell lines and inhibited both IL-6-induced STAT3 activation and TNFα-induced NFκB activation (22). Furthermore, SOCS3 genes are silenced by promoter hyper-methylation in various human cancers, including CRC (23-26). Together these data provide strong evidence that SOCS3 normally acts as a suppressor of inflammation-associated colorectal cancer. The current study tested the hypothesis that SOCS3 over-expression limits the cytokine induction of TNFR2 and/or growth promoting effects of TNFR2 in colon cancer cells.
Our studies reveal a novel pathway where IL-6 and TNFα cooperatively induce TNFR2 in colon cancer cells by interactions at multiple levels. TNFα, acting through NFκB, induces endogenous IL-6, but combined effects of IL-6 and TNFα to induce TNFR2 gene expression depend on STAT3 activation. We also provide direct evidence that TNFR2 promotes growth of colon cancer cells. Over-expression of the known STAT3 inhibitor SOCS3 decreases TNFR2 expression, decreases STAT3 binding to the TNFR2 promoter, and potently inhibits proliferation and anchorage-independent growth of colon cancer cells.
Materials and Methods
Cell culture and cytokine treatments
The two colon cancer cell lines primarily utilized in this study were SW480 and COLO205 cells, which both express low levels of endogenous SOCS3. SW480 cells were used for the majority of studies because our prior studies demonstrated that these cells are responsive to both IL-6 and TNFα, which robustly activate STAT3 and NFκB, respectively (22). COLO205 cells were used as an independent cell line to confirm cytokine-induction of TNFR2. Since COLO205 cells grow well in soft agar they were also used to address effects of TNFR2 and SOCS3 on anchorage-independent growth. SW480, COLO205, and Caco2 cells were obtained from the American Type Culture Collection (ATCC). The human intestinal epithelial cell line (HIECs) were generously provided by Dr. Jean-Francois Beaulieu (University of Sherbrooke, Quebec, Canada). Cells were grown in RPMI 1640 media (Gibco) supplemented with 10% heat-inactivated fetal bovine serum, 50U/mL penicillin, and 50mg/mL streptomycin. Given prior findings that both IL-6 and TNFα were required to induce TNFR2 (16), cells were treated with recombinant human IL-6 and/or TNFα (Peprotech) at 50ng/mL in serum-free medium. Cells were harvested at various times after cytokine treatment for evaluation of TNFR2 mRNA and protein, or STAT3 and NFκB binding to consensus regions in the TNFR2 promoter.
Semi-quantitative real time PCR analyses
Total RNA was extracted from cell lines using the RNeasy Mini Kit (Qiagen) according to manufacturer's instructions. Reverse-transcription was performed using AMV-RT (Promega). PCR and analyses were completed on the Rotorgene 2000 (Qiagen) using Invitrogen Platinum qPCR Supermix-UDG and the following Taqman primer-probe sets (Applied Biosystems): human TNFR2 Hs00961755_m1, human TNFR1 Hs00533568_g1, human IL-6 (Hs00985639_m1), human ICAM-1 (positive control as NFκB-induced gene). Hydroxymethylbilane synthase (human HMBS) Hs00609297_m1 was used as an invariant control. Non-reverse transcribed samples were used as negative controls. Gene expression was calculated using the R = 2-[delta][delta]Ct method, where changes in Ct values for the genes of interest were normalized to HMBS. In all cases, gene expression for particular treatment groups was expressed as fold change versus mean values for no treatment control. Real time PCR reactions were performed in triplicate and replicated in at least three independent experiments.
ELISA for TNFR2
Soluble TNFR2 levels in cell supernatants were measured using Quantikine ELISA system (R&D System) according to manufacturer's instructions. Samples were normalized to total protein as measured by BCA protein assay (Pierce).
Flow cytometric analysis of surface TNFR2 expression
Cell surface expression of TNFR2 was assessed using flow cytometry as previous described (16). SW480 cells were trypsinized, washed with serum-free RPMI 1640 media (Gibco) and incubated medium alone or medium plus IL-6 and TNFα (50ng/mL) for 10 hours at 37°C with rotation in 15mL conical tubes. Cells were then resuspended in wash buffer (phosphate-buffered saline supplemented with 1% bovine serum albumin and 1mg/mL DNase (Roche)), and Fc-blocked with 1μg human IgG (R&D Systems) for 15 minutes. Cells were incubated with fluorescein-conjugated anti-TNFR2 (R&D Systems) or isotype control (BD Pharmingen) for 45 minutes at 4°C. Following antibody incubation, cells were washed and resuspended in 2% paraformaldehyde. Flow cytometric analysis of surface TNFR2 expression was then performed using a CyAn flow cytometer (Beckman-Coulter-Dako). Effect of cytokine treatment on TNFR2 surface expression was measured based on fluorescein intensity.
STAT3 and NFκB inhibition
To test if cytokine-induced TNFR2 mRNA induction requires STAT3 and/or NFκB activation, SW480 cells were treated with the STAT3 inhibitor Cucurbitacin I (Tocris) at 20μM, or IκB kinase inhibitor Bay 11-7082 at 5μM. Bay 11-7082 was kindly provided by Dr. Albert Baldwin (University of North Carolina, Chapel Hill, NC). Inhibitor doses were based on maximum effective doses used in pilot and published studies (27-29). SW480 cells were seeded in serum-free media, grown for 24 hours, and treated with inhibitors and/or IL-6, TNFα, or both cytokines at 50ng/mL for 10 hours prior to mRNA extraction. No treatment controls were treated with vehicle (dimethyl sulfoxide (DMSO)). ICAM-1, which is known to be up-regulated by cytokine-induction of NFκB (30), was measured by Taqman qRT-PCR to confirm effectiveness of Bay 11-7082.
Chromatin immunoprecipitation (ChIP) to assess STAT3 and NFκB binding to the TNFR2 promoter
For ChIP, SW480 cells were serum-deprived overnight followed by treatment with IL-6, TNFα, or both cytokines (50ng/mL) for 5-60 minutes. After treatment, cells were cross-linked with 1% formaldehyde for 10 minutes. Subsequent steps were performed as specified in the ChIP-IT Express (Active Motif) user manual. Briefly, cross-linked cells were lysed and sonicated, followed by overnight immunoprecipitation with anti-STAT3 (SC-483×) and anti-p65 NFκB (SC-372) (Santa Cruz, Biotechnology), or Negative Control IgG antibody (Active Motif). Eluted, reverse cross-linked protein:DNA complexes were digested with proteinase K for 1 hour. Digested samples were then purified using QIAquick columns according to manufacturer's instructions (QIAquick PCR Purification Kit, Qiagen). PCR amplification was subsequently performed using primers specific to STAT3 and NFκB-binding elements within the TNFR2 promoter. Oligomers used to amplify these binding sites were purchased from Sigma and sequences are shown in Table 1. Densitometry was performed to quantify the PCR amplified transcription factor binding sites.
TABLE 1. Oligomers Used in Chromatin Immunoprecipitation Assays.
| Site Within TNFR2 Promoter | Oligomers | Product Size (bp) |
|---|---|---|
| STAT (-1578) | F-5′-CTGCAGTGAGCTATGGGTGA-3′ | 223 |
| R-5′-GAGGGTGTGGCTGGTATGAC-3′ | ||
|
| ||
| STAT (-364) | F-5′-CTGCAGTGAGCTATGGGTGA-3′ | 172 |
| R-5′-GGGTGAGGCACTAATTTGGA-3′ | ||
|
| ||
| NFκB (-1890) | F-5′-TTGAATTCGTTCCCAGGATG-3′ | 171 |
| R-5′-CTAGTTGTCCCCCACACACC-3′ | ||
|
| ||
| NFκB (-1517) | F-5′-AAGGCTCTGTGGGTCATACCAG-3′ | 228 |
| R-5′-GGCTGCCTGAAGAGGTACAG-3′ | ||
Western blot for activated STAT3 in nuclear extracts
Western blots were performed on nuclear extracts from SW480 cells to determine whether IL-6 combined with TNFα enhanced levels of activated or total nuclear STAT3. Nuclear extracts were prepared as previously described (31). Briefly, SW480 cells were grown to confluence, serum-deprived overnight, and treated with IL-6 and TNFα (50 ng/mL) for 30 minutes. Cells were then pelleted in lysis buffer containing 10mM Hepes pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothretol (DTT), 2 ìg/mL aprotonin and 1 mM phenylmethylsulfonyl fluoride (PMSF). Nuclei were obtained by adding 10% NP40 and centrifuging for 5 minutes at 15,000 × g. Pellets were then resuspended in buffer containing 20 mM Hepes pH 7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, and 1 mM PMSF to release nuclear proteins. The nuclear suspension was centrifuged at 15,000 × g for 5 minutes, and supernatants containing nuclear extracts were then subjected to immunoprecipitation and immunoblot with the following antibodies: anti-phospho-tyrosine STAT3: rabbit polyclonal pTyr705 (#9131, Cell Signaling); anti-STAT3 (total): rabbit polyclonal sc-7179 (Santa Cruz Biotechnologies). Coomasie-stained protein gels verified equivalent amounts of nuclear protein in samples used for immunoprecipitation.
Constitutive activation of STAT3
Constitutively-active STAT3 (CA-STAT3) adenovirus was kindly provided by Dr. Christian Jobin. This vector is constitutively activated due to C661A and C663N mutations and has been functionally characterized in prior studies (32, 33). SW480 cells were treated with CA-STAT3 at 50 multiplicity of infection (MOI) or with IL-6 and TNFα for 10 hours prior to mRNA extraction for evaluation of TNFR2 expression.
TNFR2 and SOCS3 expression constructs
Cells treated with TNFR2 or SOCS3 expression constructs were used to directly evaluate the proliferative and growth-promoting effects of TNFR2 over-expression and test if SOCS3 could inhibit TNFR2 expression or its growth promoting-effects. Retroviral expression vector pQCXIP was obtained from BD Biosciences Clontech. pQCXIP containing c-myc-tagged human TNFR2 was kindly provided by Dr. Daniella Männel, Regensburg, Germany. HEK293 cells were co-transfected with retroviral vectors and packaging vector as previously described (34) using jetPei (Polyplus Transfection) according to manufacturers instructions. Media containing TNFR2 retrovirus was collected from transfected HEK293 cells and used to treat SW480 or COLO205 cells for 24-48 hours. TNFR2 over-expression was confirmed by western immunoblot (data not shown). Plasmid pBIG2i expressing human SOCS3 and empty vector were kindly provided by Drs. Richard Furlanetto and Peter Nissley. These plasmids were used to generate adenovirus expressing human SOCS3 or empty vector control as previously described (22). Adenoviruses were used at a MOI of 100 unless otherwise noted. Cells were treated with adenovirus for 24-48 hours in complete media and switched to serum-free media overnight prior to cytokine stimulation. Adenovirus-mediated over-expression of SOCS3 was confirmed by Northern blot and qRT-PCR (data not shown).
TNFR2 or SOCS3 knockdown
SW480 cells were grown to approximately 50-70% confluence in RPMI 1640 medium (Gibco, with 10% FBS, plus antibiotics). Cells were trypsinized and counted. 5 × 104 cells per condition were transfected using nucleofector technology according to manufacturers instructions (Kit V, Lonza) or by using Lipofectamine 2000 (Invitrogen) in Optimem (Gibco) with scramble control, TNFR1 or TNFR2 siRNAs (Applied Biosystems) according to manufacturer instructions. Additional TNFR2 knockdown experiments were performed using the Santa Cruz plasmid sc-36689-SH. Knockdown was confirmed using qRT-PCR. Caco2 cells, which are known to express SOCS3 and show cytokine-induced SOCS3 expression (30), were used to test the effects of siRNA-mediated SOCS3 knockdown using siRNAs sense 5′-CCAAGAACCUGCGCAUCCAdTdT-3′, antisense, 5′-UGGAUGCGCAGGUUCUUGGdTdT-3′, Dharmacon) and methods previously described for prostate cancer cells (35).
Analysis of cell proliferation and anchorage-independent growth
Assays of [3H]-thymidine incorporation into DNA were used to assess S-phase of the cell cycle as one measure or proliferation and were performed as previously described (36). Briefly, SW480 cells were plated in 24-well plates at a density of 1 × 104 cells per well and treated with TNFR2 and/or SOCS3 expression constructs for 24 hours. Medium was then supplemented with 2 μCi/mL [3H]-thymidine overnight, and thymidine incorporation into DNA was measured using scintillation counting. Values are expressed as fold change compared to empty vector control. As an independent measure of effects of TNFR2 knockdown on cell growth, WST-1 assays (Roche), which measure numbers of viable cells, were performed according to manufacturer's instructions. Briefly, cells were transfected with control or TNFR2 siRNAs in 96-well plates, then switched to media containing IL-6 and TNFα (50 ng/mL) for 48 hours followed by WST-1 assay.
COLO205 cells, which show robust colony formation in soft agar, were used to test the effects of over-expression of TNFR2 and/or SOCS3 on anchorage-independent growth. Cells were treated with empty vector, TNFR2, SOCS3, or both TNFR2 and SOCS3 expression constructs. COLO205 cells were trypsinized and suspended in complete culture media supplemented with 0.3% agar followed by plating in 6-well culture dishes coated with 5% agar. Cells were treated with expression constructs at Days 1, 7, and 14. At day 21, viable cells were stained with 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) for 4 hours and colonies were quantified using NIH ImageJ (37).
Evaluation of TNFR2 in colon of azoxymethane/dextran sodium sulfate treated mice with intestinal epithelial cell (IEC)-specific disruption of SOCS3 genes
C57BL6 mice homozygous for pLox-modified SOCS3 alleles were crossed with mice hemizygous for the villin-Cre transgene (Jackson Laboratories) as previously described (22). Study mice are denoted as VC-SOCS3Δ/Δ and WT-SOCS3fl/fl controls with LoxP-modified, but intact SOCS3 alleles. Genotyping primers are listed in Table 2. All procedures were performed with Institutional Animal Care and Use Committee approval and in accordance with the National Institutes of Health guidelines for use of live animals
TABLE 2. Genotyping Oligomers for IEC-SOCS3Δ/Δ Mice.
| Target | Oligomers | Product Size (bp) |
|---|---|---|
| villin-Cre Transgene | F-5′-CTGCCACGACCAAGTGACAGC-3′ | 324 |
| R-5′-CTTCTGTAGACCTGCGGTGCT-3′ | ||
|
| ||
| Wild-type1 or pLox-modified2 SOCS3 alleles | F-5′-GAGTTTTCTCTGGGCGTCCTCCTA-3′ | 4021 or 5342 |
| R-5′-TGGTACTCGCTTTTGGAGCTGAA-3 | ||
VC-SOCS3Δ/Δ and WT-SOCS3fl/fl were treated azoxymethane/dextran sodium sulfate (AOM/DSS) as previously described (22). Briefly, 8-10 week-old mice were given an intraperitoneal injection of 10 mg/kg AOM. After seven days, mice were given three cycles of 2.5% DSS in the drinking water for 5 days, allowing 14 days of recovery between each DSS treatment. Animals were euthanized 80 days after initial AOM injection. Colon samples were collected and fixed in 10% zinc-buffered formalin for immunohistochemical staining.
Immunostaining for TNFR2 was performed on paraffin-embedded sections of distal colon tumor and non-tumor tissues. Only VC-SOCS3Δ/Δ had histologically detectable tumors in these experiments. After deparaffinizing and hydrating, sections were pretreated with 0.05M Tris Triton epitope retrieval solution at room temperature for 30 minutes. Endogenous peroxidase activity was blocked with 3% H2O2 for 10 minutes, followed by blocking in 5% normal goat serum (NGS)/TT buffer for 1 hour. The sections were incubated with TNFR2 antibody (NBP1-03130, Novus Biologicals) at a dilution of 1:250 in 5% NGS/TT buffer overnight at 4°C in a humidity chamber. After washing 3 times in 0.05M Tris buffer, the sections were incubated with biotinylated goat anti-rabbit IgG (11-065-114, Jackson Immunoresearch) for 1 hr at room temperature. ABC (Vector Labs) was applied for 1 hour, followed by adding DAB (Zymed) for 10-14 minutes and the reaction was stopped in dH2O. Counter-staining was performed with hematoxylin for 25 seconds and cover slips were mounted in DPX mount (VWR International Ltd.). Pictures were taken by Imager A2 microscope (Zeiss).
Statistics
Values are expressed as mean ± standard error (S.E.). Comparisons between cell treatments were analyzed using student's t-test for comparisons between two treatments or one-way analysis of variance for comparisons of multiple treatments followed by post-hoc, pair-wise comparisons using Fisher's PLSD. A p-value of <0.05 was considered statistically significant for all experiments.
Results
IL-6 and TNFα induce TNFR2 in COLO205 and SW480 cells
Prior studies in colon cancer cells indicate that TNFR2 expression is increased by combined IL-6 or TNFα treatment, with only modest effects due to either cytokine alone (16). Fig. 1A and C confirm elevation of TNFR2 mRNA levels by combined IL-6 and TNFα in SW480 and COLO205 cells. TNFR1 mRNA levels were not significantly altered with cytokine treatment. Evaluation of TNFR2 protein by ELISA and flow cytometry (Fig. 1C, D, and E) verified these data. In SW480 cells, treatment with TNFα alone induced TNFR2 to levels similar to those observed with combined IL-6 and TNFα. Since TNFα has been reported to induce IL-6 in cancer cell lines from other organs (38-42), we tested whether TNFα up-regulated endogenous IL-6 mRNA levels. As shown in Fig. 1F, TNFα treatment, and TNFα combined with IL-6, significantly increased endogenous IL-6 mRNA. This result suggests that autocrine or paracrine actions of TNFα-induced IL-6 may contribute to the ability of TNFα to induce TNFR2.
FIGURE 1.
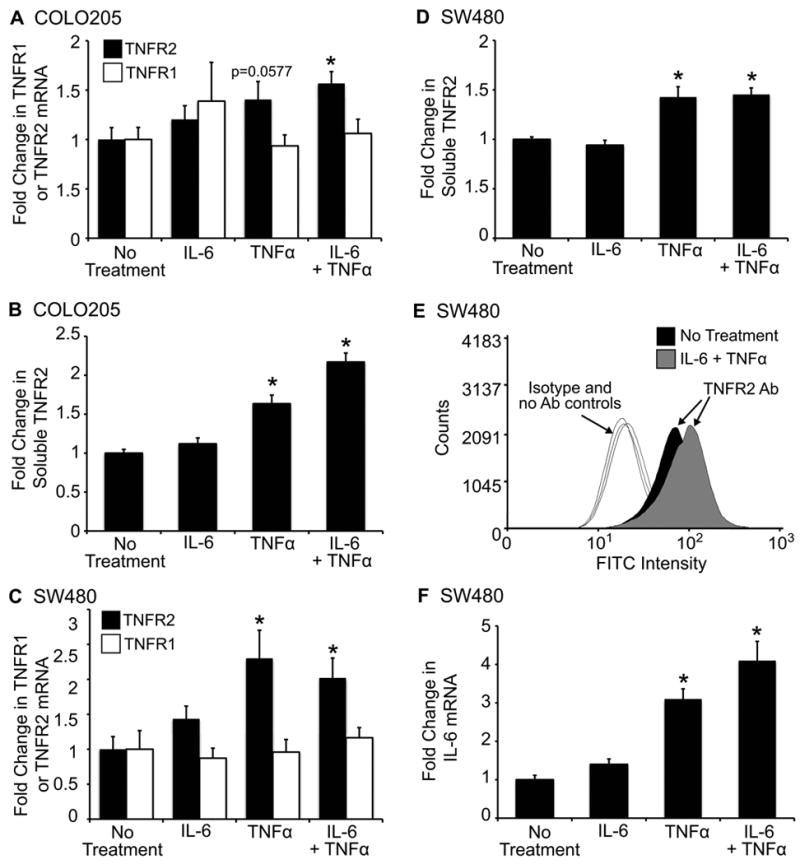
Increased TNFR2 mRNA and protein levels in colon cancer cell lines treated with IL-6 and TNFα. Histograms A –D show levels of TNFR2 mRNA or soluble TNFR2 in SW480 and COLO205 cells treated with 50ng/mL IL-6 plus 50ng/mL TNFα for 10 hours. TNFR2 mRNA was normalized to HMBS. sTNFR2 was measured by ELISA on cell supernatants and values were normalized to total protein. All values are expressed as fold change (mean ±SE) versus mean levels in untreated controls. (* = p ≤ 0.05 compared to no treatment controls). TNFR2 mRNA was significantly increased by IL-6 and TNFα treatment in both cell lines (A and C). Consistent with findings for TNFR2 mRNA, protein levels of soluble TNFR2 were significantly increased with IL-6 and TNFα treatment (B and D). (E) Representative histogram of surface TNFR2 expression as measured by flow cytometry. Cells treated with IL-6 and TNFα exhibit an increase in surface TNFR2 expression. Negative controls include isotype and no antibody controls for each condition. (F) Histograms show IL-6 mRNA levels in SW480 cells. Note that TNFα alone or TNFα + IL-6 elicit similar, significant increases IL-6 mRNA (*p ≤ 0.05 compared to no treatment). For all experiments n ≥ 3 independent experiments performed in duplicate.
A predominant role of STAT3 in mediating basal and cytokine-induced TNFR2 expression
To assess the functional role of STAT3 or NFκB in regulating TNFR2 expression, we examined the effects of a STAT3 (cucurbitacin) or NFκB (Bay 11-7082) inhibitor on basal and IL-6 + TNFα -induced TNFR2 mRNA (Fig. 2A). Based on published and pilot studies, we used maximally effective doses of 20μM cucurbitacin or 5μM Bay 11-7082. Cucurbitacin significantly inhibited basal TNFR2 mRNA and TNFR2 mRNA induction by combined IL-6 and TNFα. In contrast Bay 11-7082 had no significant effect on basal or IL-6 +TNFα-induced TNFR2 mRNA. Furthermore, effects of combined inhibition of STAT3 and NFκB on basal or IL-6 + TNFα-induced TNFR2 was not significantly different than the inhibitory effect of cucurbitacin alone. These date suggest a predominant role of STAT3 compared with NFκB pathways in induction of TNFR2 by combined IL-6 and TNFα. Confirmatory studies were performed in cells treated with IL-6 or TNFα alone plus STAT or NFκB inhibitors. IL-6 alone did not induce TNFR2 mRNA above basal levels, but cucurbitacin and not Bay 11-7082 reduced TNFR2 mRNA in IL-6 treated cells.
FIGURE 2.
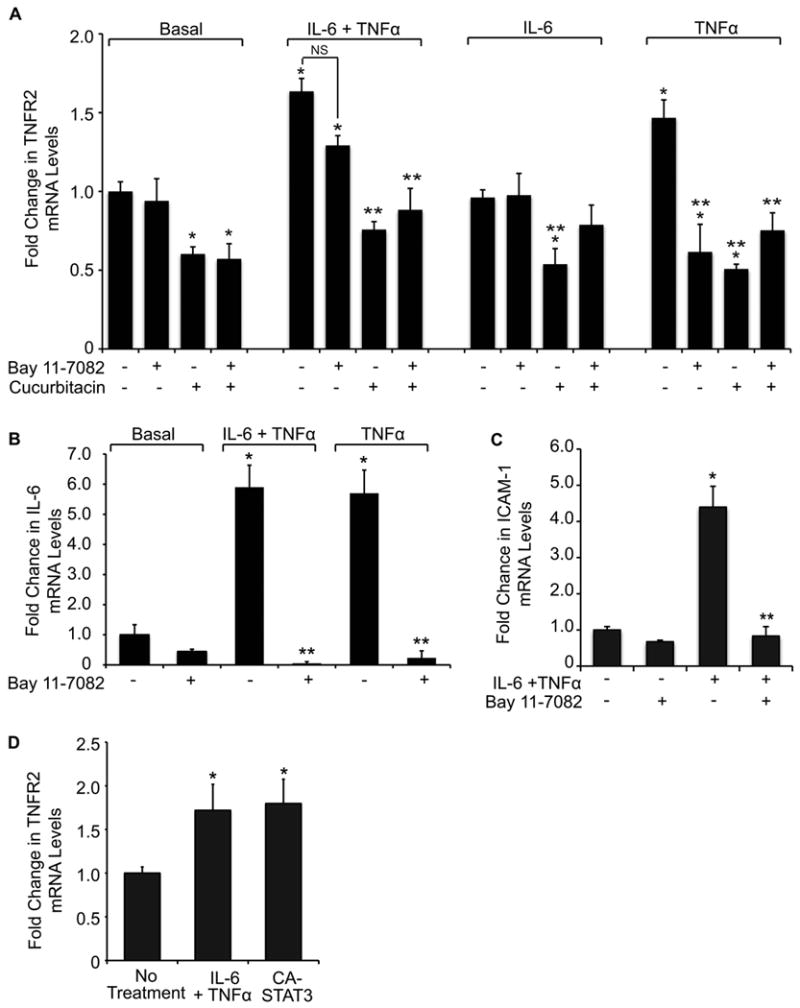
Effects of STAT3 or NFκB inhibitors and constitutively activated STAT3 on TNFR2, and effects of NFκB inhibitor on IL-6 and ICAM. (A) Histograms show levels of TNFR2 mRNA in SW480 cells treated with vehicle, IL-6 + TNFα, IL-6 alone, or TNFα alone, for 10 hours in the absence (-) or presence (+) of the STAT3 inhibitor cucurbitacin (20μM) or the NFκB inhibitor Bay 11-7082 (5μM). (Values are expressed as mean fold change versus basal values in the absence of inhibitor; *p ≤ 0.05 compared to basal; ** p ≤0.05 for effect of STAT or NFκB inhibitor; NS= not significant). Note the dramatic inhibitory effect of cucurbitacin on both basal and IL-6 + TNFα-induced TNFR2 compared with Bay 11-7082. Note that IL-6 alone did not induce TNFR2, but cucurbitacin reduced TNFR2 mRNA in IL-6 treated cells. TNFα alone induced TNFR2, and this was inhibited by Bay 11-7082 or cucurbitacin, with no greater inhibition when both STAT3 and NFκB inhibitors were combined. (B) IL-6 mRNA levels were measured in SW480 cells treated with TNFα or IL-6 + TNFα in the absence (-) or presence (+) of Bay 11-7082 (5μM). Note that Bay 11-7082 completely abolished TNFα or IL-6 + TNFα-mediated increases in IL-6 mRNA levels. (*p ≤ 0.05 compared to no treatment; **p ≤ 0.05 for effect of NFκB inhibitor). For all experiments n ≥ 3 independent experiments were performed in duplicate. (C) ICAM mRNA induction in SW480 cells treated with IL-6 + TNFα in the absence (-) or presence (+) of Bay 11-7082 (5μM). Bay 11-7082 completely abolished IL-6 + TNFα-mediated increases in ICAM mRNA levels. (*p ≤ 0.05 compared to no treatment; **p ≤ 0.05 for effect of NFκB inhibitor). (D) SW480 cells were treated with adenovirus to over-express constitutively-active STAT3 (CA-STAT3) for 10 hours followed by mRNA collection. Expression of CA-STAT3 significantly increased TNFR2 mRNA levels to the same level as that found with IL-6 and TNFα treatment. (*p ≤ 0.05 compared to empty vector).
Analyses of the effects of cucurbitacin and Bay 11-7082 on TNFR2 mRNA in cells treated with TNFα alone revealed that either the NFκB inhibitor or the STAT3 inhibitor significantly attenuated TNFα-induction of TNFR2, but combined inhibitors did not have a more dramatic effect than either inhibitor alone. These results are consistent with our findings that TNFα induces IL-6, and suggest that NFκB likely mediates TNFα induction of IL-6 in SW480 cells. To directly test this, we measured IL-6 mRNA levels in cells treated with TNFα or TNFα + IL-6 in the presence of Bay 11-7082 or DMSO control. Treatment with Bay 11-7082 dramatically decreased IL-6 mRNA in TNFα or TNFα + IL-6 treated cells, confirming that TNFα-induction of IL-6 in SW480 cells is mediated through NFκB (Fig. 2B). Since it has been established that cytokines induce ICAM-1 through NFκB-dependent mechanisms, we also verified the efficacy of Bay 11-7082 by showing that IL-6 and TNFα induced a 4.4 ± 0.6-fold increase in ICAM-1 mRNA, and treatment with Bay 11-7082 potently inhibited this effect (Fig. 2C). Thus, the modest effects of NFκB inhibitor versus STAT3 inhibitor on TNFR2 expression in cells treated with IL-6 and TNFα were not due to a lack of effective Bay 11-7082 dosing. Together these findings suggest a predominant role of STAT3 in mediating cytokine-induced TNFR2 expression.
To confirm a role for STAT3 in TNFR2 expression, we treated SW480 cells with a constitutively-active STAT3 (CA-STAT3) adenovirus. Expression of CA-STAT3 in the absence of cytokine treatment induced TNFR2 mRNA levels to the same degree as observed with combined IL-6 and TNFα treatment (Fig. 2D). Together, the data with STAT3 inhibitor and constitutively activated STAT3 suggest that STAT3 is necessary and sufficient for basal and IL-6/TNFα-induced TNFR2 expression.
IL-6 and TNFα induce STAT3, but not NFκB binding to the TNFR2 promoter
We verified a predominant role of STAT3 in the control of TNFR2 expression by ChIP assays that allowed us to directly assess the effect of cytokines on STAT3 and NFκB binding to putative binding elements in the TNFR2 promoter (Fig. 3A). Combined IL-6 and TNFα treatment induced binding of STAT3 to both of the putative STAT binding sites, with maximal binding at 30 and 60 minutes for -364 and -1578 sites, respectively (Fig. 3B). Densitometry revealed that IL-6 and TNFα induced 2.0 ± 0.4-fold and 3.8 ± 1.6-fold and increases in STAT3 binding to the -364 and -1578 sites respectively. There was a small degree of basal NFκB binding, but this was not enhanced by IL-6 and TNFα treatment.
FIGURE 3.
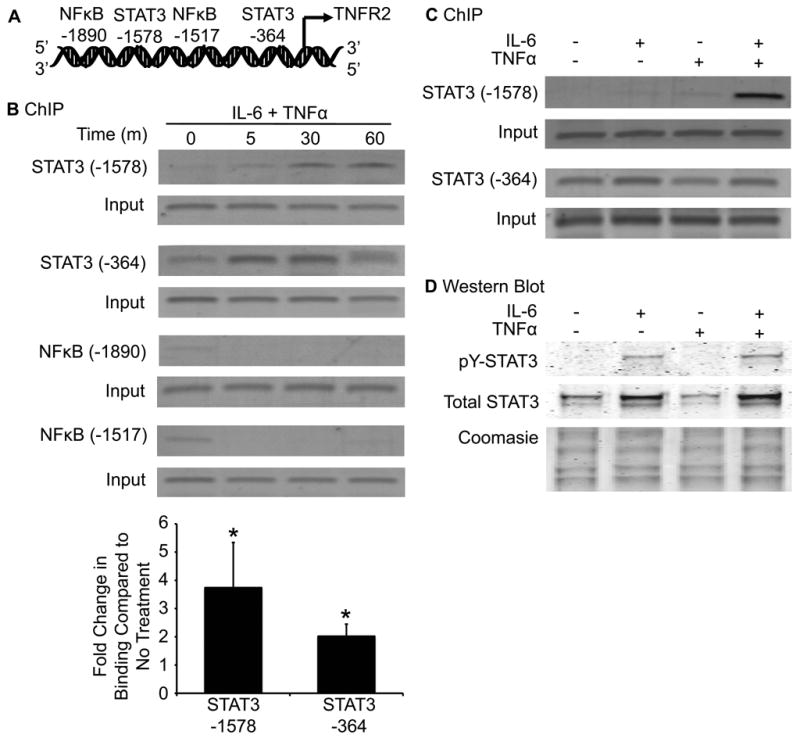
IL-6 and TNFα induce STAT3 binding to the TNFR2 promoter. (A) Schematic shows the locations of two putative STAT binding elements and two putative NFκB binding elements in the human TNFR2 promoter. (B) PCR products from ChIP assays of STAT3 or NFκB binding to consensus STAT3 or NFκB sites in untreated cells or cells treated with IL-6 and TNFα for the indicated times. IL-6 and TNFα treatment lead to a time-dependent increase in STAT3 binding at both consensus sites. Cytokine treatment has no effect on NFκB binding. Densitometric analysis revealed a 3.8 ± 1.6-fold and 2.0 ± 0.4-fold increase in overall STAT3 binding at the -1578 and -364 site, respectively. (*p ≤ 0.05 compared to no treatment). (n =3). (C) PCR products from ChIP assays in SW480 cells treated with IL-6 or TNFα alone, or in combination (30 minutes). Note the dramatically enhanced effect of combined IL-6 and TNFα on STAT3 binding to the -1578 site, but not for the -364 site. (n =3). (D) Western immunoblots on nuclear extracts from SW480 cells treated with IL-6, TNFα, or both cytokines for 30 minutes. Upper panels show immunoblots for tyrosine-phosphorylated (pY) and total STAT3. IL-6 treatment induced pY-STAT3 and increased total nuclear STAT3, while TNFα alone had no detectable effect, and IL-6 and TNFα combined gave a similar effect as IL-6 alone. Lower panel confirms equal protein loading by Coomasie-stain of proteins in nuclear extracts. Blot is representative of 3 total experiments.
To delineate the individual contribution of IL-6 and TNFα on STAT3 binding to the TNFR2 promoter, we treated cells with either cytokine alone or in combination and performed ChIP for STAT3. Treatment with IL-6 and TNFα alone for 30 minutes modestly induced STAT3 binding to the -1578 STAT binding site, but both cytokines combined induced dramatic increases in STAT3 binding to the -1578 binding site (Fig. 3C). Putative STAT3 binding site -364 differed in that IL-6 alone, but not TNFα, induced STAT3 binding and combined cytokines had similar effects as IL-6 alone. Thus, the cooperative effects of IL-6 and TNFα to activate STAT3 appear selective for the -1578 STAT3 binding site.
Because combined IL-6 and TNFα led to a dramatic increase in STAT3 binding to the -1578 STAT3 binding site in the TNFR2 promoter, we assessed whether the two cytokines in combination enhanced tyrosine phosphorylation of STAT3 relative to IL-6 or TNFα alone. Western blot on nuclear extracts revealed that IL-6 increased phosphorylated and total STAT3 in the nucleus, whereas TNFα alone had no effect (Fig. 3D). Both cytokines together did not dramatically augment tyrosine phosphorylation of STAT3 or total nuclear STAT3 as compared to IL-6 alone. Thus, enhanced tyrosine phosphorylation of STAT3 does not appear to account for the combinatorial effects of IL-6 and TNFα on STAT3 binding to the -1578 STAT3 binding site. It is important to note that the treatment time points used to examine effects of TNFα alone on STAT3 binding by ChIP or STAT3 tyrosine phosphorylation are much shorter than the times (10 hours) needed for TNFα to induce IL-6. Thus, the western immunoblot and ChIP data together indicate that IL-6 alone enhances STAT3 activation and nuclear localization, but interaction with TNFα is required to promote IL-6-induced binding of STAT3 to the TNFR2 promoter.
Effects of TNFR2 over-expression or knockdown on proliferation and cell number
Prior in vivo studies suggest that TNFR2 null mice show reduced crypt proliferation during intestinal inflammation (16). To directly test the effects of TNFR2 on colon cancer cell proliferation, we over-expressed TNFR2 in SW480 and COLO205 cells and measured [3H]-thymidine incorporation into DNA. TNFR2 over-expression significantly enhanced [3H]-thymidine incorporation in both cell lines (Fig. 4A). In complementary experiments we knocked down TNFR1 or TNFR2 using siRNA (Fig. 4B) and measured [3H]-thymidine incorporation over 24 hours. The maximal knockdown achieved after testing multiple TNFR2-targeted siRNAs was a 60% reduction in TNFR2 mRNA levels (Fig. 4B). However, this was a specific effect since expression of TNFR1 mRNA was unaffected by the TNFR2 siRNA. Similarly, TNFR1 siRNA had no effect on TNFR2 mRNA, but inhibited TNFR1 expression by 80%. TNFR2 knockdown resulted in a modest, but significant decrease (14± 2.5%) in [3H]-thymidine incorporation in SW480 cells (Fig. 4C). WST-1 assays, which measure number of viable cells rather than just S-phase, revealed that knockdown of TNFR1 significantly increased cell number, while knockdown of TNFR2 reduced cell number up to 40% compared to control siRNA, and up to 70% compared to cells with knockdown of TNFR1 (in which TNFR2 is the predominant TNFR) (Fig. 4D). We have observed similar effects of TNFR2 knockdown in COLO205, Caco2, and HIECs (Supp. Tables 1-3). The more dramatic effect of TNFR2 knockdown on cell number than [3H]-thymidine incorporation suggests that TNFR2 knockdown likely impacts cell survival and/or other phases of cell cycle than S-phase.
FIGURE 4.
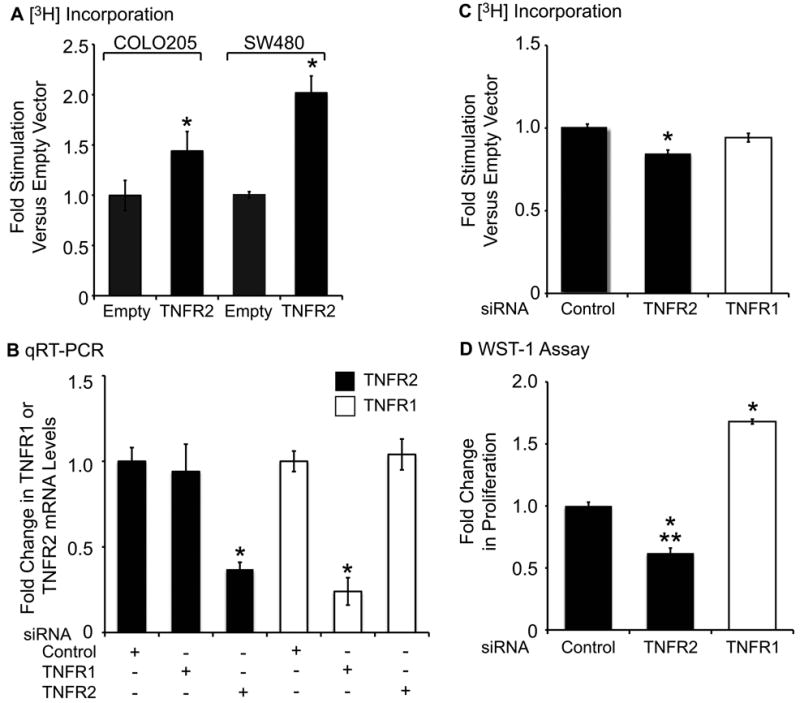
Effects of TNFR2 over-expression or siRNA-mediated knockdown on colon cancer cell proliferation. (A) Histogram of [3H]-thymidine incorporation into DNA as a measure of COLO205 or SW480 cell proliferation after 24-hour over-expression of TNFR2. TNFR2 over-expression significantly increased cell proliferation (*p ≤ 0.05 compared to empty vector). (B) SW480 cells were treated with control, TNFR1, or TNFR2 siRNA for 24 hours. Histogram shows fold change in expression of TNFR1 or TNFR2 mRNA. TNFR2 siRNA led to a 60% decrease in TNFR2 mRNA, with no effect on TNFR1 mRNA (*p ≤ 0.05 compared to other treatments). (C) Untreated SW480 cells transfected with TNFR2 siRNA exhibit a modest, but significant (*p ≤ 0.05) decrease in [3H]-thymidine incorporation into DNA. (D) Cytokine-treated SW480 cells with TNFR2-knockdown exhibited a significant decreases cell number measured by WST-1 assay, while TNFR1 knockdown significantly increased cell number (*p ≤ 0.05 compared to control siRNA, **p≤ 0.05 compared to cells treated with TNFR1 siRNA). (n = 3).
SOCS3 inhibits cytokine-induced TNFR2 expression, STAT3 binding to the -1578 consensus site, and proliferation and anchorage-independent growth of colon cancer cells
Negative regulation of STAT3 by SOCS3 is well established (43-45). To test whether SOCS3 inhibits TNFR2 expression, we treated COLO205 and SW480 cells with SOCS3 adenovirus or empty vector control and examined TNFR2 mRNA. As anticipated, SOCS3 over-expression significantly inhibited cytokine-induced TNFR2 in both cell lines (Fig. 5A). ChIP assay also revealed that SOCS3 over-expression dramatically inhibited cytokine-induced STAT3 binding to the -1578 site, but had variable and non-significant effects on STAT3 binding to the -364 site (Fig. 5B).
FIGURE 5.
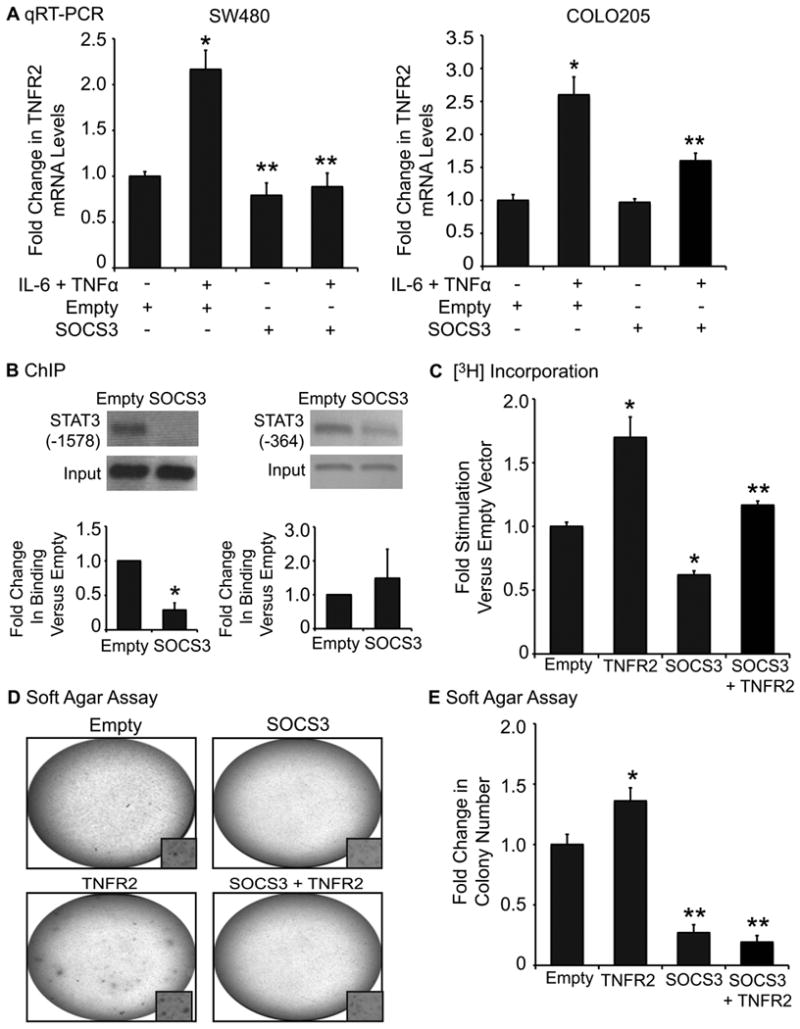
SOCS3 over-expression decreases TNFR2 mRNA, STAT3 binding to the -1578 binding site on theTNFR2 promoter, cell proliferation, and anchorage-independent growth. (A) Histogram shows levels of TNFR2 mRNA in SW480 or COLO205 cells in the absence (-) or presence (+) of IL-6 and TNFα and/or SOCS3 adenovirus. Cytokine treatment significantly increased TNFR2 mRNA, and SOCS3 over-expression attenuated this effect. (*p ≤ 0.05 versus vehicle, Empty vector; **p ≤ 0.05 versus Empty vector plus cytokine treatment). (n ≥ 3 independent experiments performed in duplicate). (B) Upper panel shows PCR products from ChIP assays of SW480 cells stimulated with IL-6 and TNFα for 30-60 minutes. Histograms indicate fold change in STAT3 binding with SOCS3 over-expression compared to empty vector. SOCS3 decreased STAT3 binding to the TNFR2 promoter at the -1578 site and had variable, non-significant effects on binding at the -364 site. (*p ≤ 0.05 compared to empty vector). (n = 3). (C) Histogram of [3H]-thymidine incorporation into DNA as a measure of SW480 cell proliferation after 24-hour over-expression of TNFR2, SOCS3 or both. TNFR2 over-expression significantly increased cell proliferation, and SOCS3 over-expression limited this effect. (*p ≤ 0.05 compared to empty vector; **p ≤ 0.05 compared to TNFR2 over-expressing cells). (D) Representative photographs of individual wells of COLO205 cells grown in 0.3% soft agar and over-expressing TNFR2 and/or SOCS3. Images are representative of at least three independent experiments. (E) Colonies were stained with MTT and quantified using NIH ImageJ. Cells treated with TNFR2 retrovirus exhibited a small, but significant increase in colony number (*p <0.05 versus empty vector), and treatment with SOCS3, or SOCS3 combined with TNFR2, caused a dramatic (>70% decrease) in colony number. (**p ≤ 0.05 compared to empty vector or cells over-expressing TNFR2 alone).
We have previously reported that SOCS3 over-expression reduces proliferation of SW480 cells (22). We used these cells to test whether SOCS3 could prevent the increase in proliferation resulting from TNFR2 over-expression. SOCS3 over-expression reduced [3H]-thymidine incorporation in SW480 cells transfected with empty vector or TNFR2 expression construct (Fig. 5C). However, the magnitude of the increase in [3H]-thymidine incorporation resulting from TNFR2 over-expression was similar in SOCS3 over-expressing and control cells suggesting that SOCS3 over-expression cannot reverse TNFR2-associated increases in DNA synthesis.
SOCS3 over-expression had more potent effects on anchorage-independent growth of COLO205 cells. COLO205 cells over-expressing TNFR2 showed a small, but significant increase in colony formation when compared to empty vector controls. SOCS3 over-expression dramatically decreased (>70%) colony formation in both empty vector or TNFR2 over-expressing cells (Fig. 5D & E).
Increased TNFR2 immunostaining in colon tumors of mice with intestinal epithelial cell (IEC)-specific deletion of SOCS3
Previously published work showed a dramatic increase in AOM/DSS- induced tumor number and size in mice with villin-cre (VC) mediated SOCS3 gene disruption in IEC (VC-SOCS3Δ/Δ) compared with controls with floxed, but intact SOCS3 genes (WT-SOCS3fl/fl). We therefore assessed TNFR2 expression in colon of these AOM/DSS treated mice. In normal colon tissue TNFR2 immunostaining was weak in both WT-SOCS3fl/fl and VC-SOCS3Δ/Δ mice (Fig. 6A-F). Only VC-SOCS3Δ/Δ mice showed histologically detectable tumors and these tumors showed dramatic increases in TNFR2 immunostaining (Fig. 6G-I). Thus, the increase in tumor load in VC-SOCS3Δ/Δ mice is associated with increased expression of TNFR2 within the tumors.
FIGURE 6.
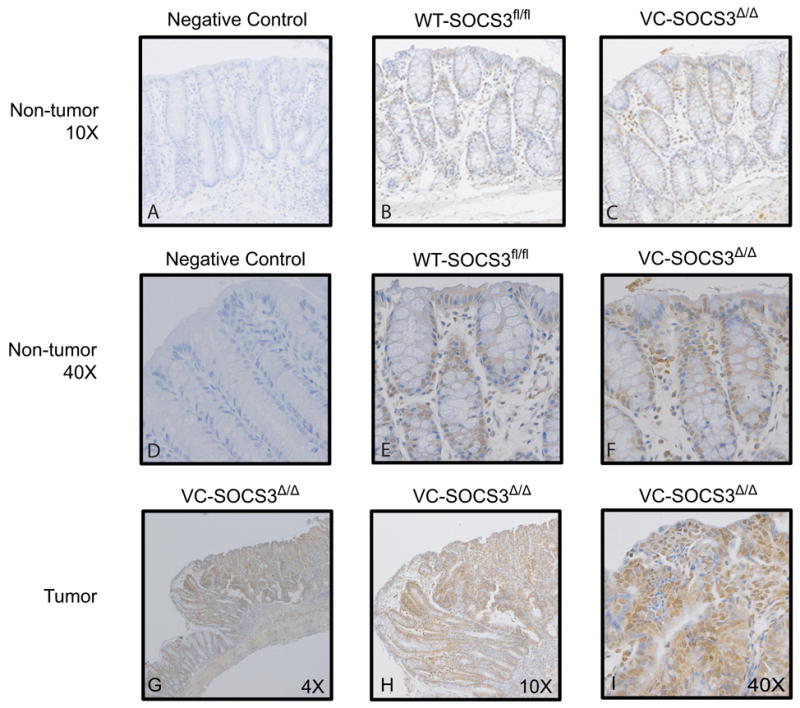
Increased TNFR2 immunostaining in colon tumors from mice with villin-Cre (VC) mediated SOCS3 gene deletion in IEC cells. Immunohistochemical analysis of TNFR2 in non-tumor tissue from WT-SOCS3fl/fl (B, E) and VC-SOCS3Δ/Δ (C, F) mice compared to negative control, which corresponds to no primary antibody control (A, D). (A-C 10× magnification, D-F 40× magnification) Note that TNFR2 staining is weak in non-tumor tissue from colon of both VC-SOCS3Δ/Δ mice and WT- SOCS3fl/fl mice. TNFR2 immunostaining is enhanced in tumor tissue from VC-SOCS3Δ/Δ mice (G-4×, H-10×, I-40× magnification).
Short-term SOCS3 knockdown is not sufficient to up-regulate TNFR2
We used Caco2 colon cancer cells, which are known to have higher basal SOCS3 expression than SW480 and COLO205 cells, and show cytokine mediated up-regulation of SOCS3 (30, unpublished data) to test if SOCS3 knockdown was sufficient to increase basal or cytokine-stimulated TNFR2 expression. Caco2 cells were transfected with control or SOCS3 siRNA followed by cytokine treatment for 24 hours. As shown in Fig. S1A, SOCS3 siRNA treatment reduced the basal and cytokine-induced levels of SOCS3 mRNA, but we were unable to completely abolish the increase in SOCS3 mRNA resulting from cytokine treatment. SOCS3 siRNA did not enhance, but rather reduced TNFR2 expression in these cells (Fig. S1B). Thus while SOCS3 over-expression is sufficient to inhibit basal and TNFR2 over-expression, short-term knockdown of SOCS3 expression was itself not sufficient to increase basal or cytokine-stimulated TNFR2 expression.
Discussion
The etiology of inflammation-associated CRC is based strongly on the model that chronically up-regulated cytokines drive excessive proliferation of intestinal epithelial cells, tumor initiation and progression. TNFR2 has recently emerged as a pro-proliferative factor that is up-regulated in IBD and in the AOM/DSS model of IBD-associated cancer (14, 16). Mechanisms regulating TNFR2 expression in IBD or CRC are not fully defined, although prior studies suggest that combined effects of IL-6 and TNFα promote TNFR2 expression (16). The current study provides novel evidence that IL-6 and TNFα act predominantly through STAT3 and a consensus STAT3 binding site in the TNFR2 promoter to induce TNFR2 in CRC. We also demonstrate that SOCS3 over-expression inhibits cytokine induction of TNFR2 and STAT3 binding to this STAT3 consensus site, and can dramatically decrease anchorage-independent growth of colon cancer cells, even those over-expressing TNFR2.
Mizoguchi and colleagues provided the first evidence that TNFR2 was up-regulated during acute DSS-colitis and that this was preceded by IL-6/STAT3 activation. They also demonstrated that mice with global TNFR2 gene disruption exhibited decreased IEC proliferation in the T-cell receptor α (TCRα) null model of colitis (16). Furthermore, TCRα mice with disruption of both IL-6 alleles showed reduced colitis severity and decreased TNFR2 expression compared to TCRα mice with intact IL-6 (16). While these studies suggested an association between IL-6/STAT3 and TNFR2, the ability of STAT3 to directly regulate TNFR2 expression has not been previously reported. Prior in vitro studies indicated that both IL-6 and TNFα were required to induce TNFR2 in CRC cells, suggesting that TNFR2 induction requires a specific micro-environment of multiple cytokines, as found in IBD or IBD-associated CRC. The current study confirmed induction of TNFR2 mRNA and protein by combined IL-6 and TNFα in two different colon cancer cell lines. Importantly we provide novel evidence for a model of IL-6 and TNFα interaction in regulating TNFR2 in colon cancer cells (Fig. 7). Our studies demonstrate a predominant role of STAT3 in TNFR2 induction by IL-6 and TNFα, which involves cooperative effects of IL-6 and TNFα to induce STAT3 binding to a consensus element within the TNFR2 promoter. Our studies also reveal that TNFα acts through NFκB to induce endogenous IL-6 and promote TNFR2 induction via autocrine effects of IL-6 (Fig. 7).
FIGURE 7.
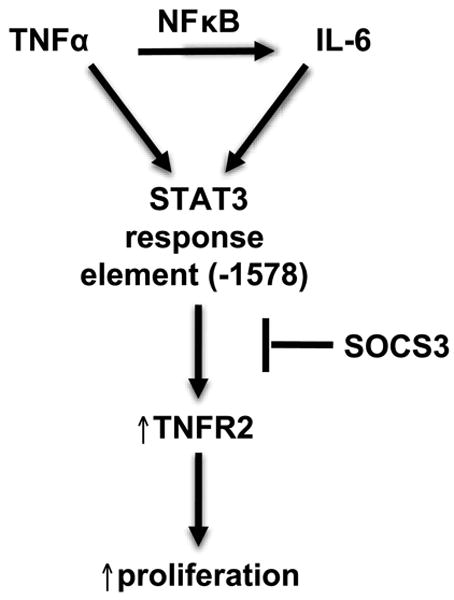
Model of interactions between IL-6 and TNFα in regulating TNFR2 expression in colon cancer cells. IL-6 and TNFα cooperatively induce STAT3 binding to the -1578 site of the TNFR2 promoter to induce TNFR2 expression. TNFα also induces endogenous IL-6 expression through NFκB. SOCS3 over-expression limits STAT3 binding to the TNFR2 promoter, TNFR2 expression, and proliferation of colon cancer cells.
The TNFR2 promoter contains two putative STAT binding sequences and NFκB binding sequences (20), but functional effects of these binding elements have not been reported. Since TNFα typically activates NFκB and IL-6 typically activates STAT3, we hypothesized that IL-6 and TNFα induction of TNFR2 would be mediated by activation of both of these transcription factors. We provide several independent pieces of evidence to indicate that STAT3, rather than NFκB, is the predominant mediator of TNFR2 induction by IL-6 and TNFα. Specifically, STAT3 inhibition reduced basal TNFR2 expression and completely reversed the induction of TNFR2 by IL-6 and TNFα. In contrast, NFκB inhibition had no effect on basal TNFR2 expression and only non-significantly reduced induction of TNFR2 by combined IL-6 and TNFα. This was despite data verifying that the NFκB inhibitor potently and completely reversed cytokine-induction of ICAM-1 mRNA, whose expression is known to be dependent on NFκB. Importantly, combined STAT3 and NFκB inhibitors did not further reduce basal or cytokine-induced TNFR2 expression compared with STAT3 inhibitor alone. Constitutively activated STAT3 was able to induce TNFR2 to a similar extent as IL-6 and TNFα. Together these findings indicate a predominant role of STAT3 in mediating TNFR2 induction and demonstrate that STAT3 activation alone is sufficient to mimic cytokine effects on TNFR2 expression.
ChIP assays also confirmed that IL-6 and TNFα induced STAT3 binding to two putative STAT3 binding sites on TNFR2 promoter, but had no effect on NFκB binding. This was despite the fact that TNFα is known to induce phosphorylation of NFκB in this same cell system (22). It is also notable that combined IL-6 and TNFα more potently induced STAT3 binding to the -1578 binding site in the TNFR2 promoter than the -364 site. Interestingly, the -1578 STAT3 binding site also showed dramatic cooperative effects of IL-6 and TNFα to induce STAT3 binding, while either cytokine alone only modestly induced STAT3 binding to this site. These combinatorial effects of IL-6 and TNFα to dramatically enhance STAT3 binding to the -1578 site were not associated with effects of combined IL-6 and TNFα to increase tyrosine phosphorylation or nuclear levels of STAT3. Collectively, these observations provide compelling evidence that IL-6 and TNFα interact to promote maximal STAT3 binding to the TNFR2 promoter and TNFR2 induction, and that this cooperative effect appears to occur primarily at the -1578 STAT binding site. The specific mechanisms by which TNFα promotes STAT3 binding to the TNFR2 promoter remain undefined and will require further study. We cannot rule out the possibility that NFκB binds to other regions in the TNFR2 gene than the consensus sites tested, but observations that IL-6 and TNFα did not induce NFκB binding to these NFκB consensus sites, and the minimal effects of NFκB inhibitor on basal or IL-6 and TNFα-induced TNFR2 expression support a novel mechanism of TNFα and IL-6 interaction to induce TNFR2 by predominant effects on STAT3.
Recent and increasing evidence implicates TNFR2 as a mediator of colitis-associated cancer. TNFR2 has been shown to increase migration of colon cancer cell lines and is up-regulated in mouse models and patients with inflammatory bowel diseases (16, 17). Additionally, disruption of TNFR2 genes was associated with decreased proliferation of crypt epithelial cells in colitis models (16, 17). Recent studies in the AOM/DSS model of inflammation-associated CRC revealed that TNFR2 is preferentially up-regulated over TNFR1 and that treatment with the anti-TNFα mAb MP6-XT22 reduced the number and size of tumors, although colitis severity was unchanged (14). In a separate study, anti-TNFα antibodies given at late stages of the AOM/DSS model reduced tumor load (9). To our knowledge, a direct effect of TNFR2 on CRC proliferation or transformed phenotype has not been previously demonstrated. Our current study demonstrates in two independent colon cancer cell lines that TNFR2 over-expression directly enhances proliferation. In COLO205 cells, TNFR2 over-expression increased anchorage independent growth. Although effects of TNFR2 over-expression on proliferation and anchorage-independent growth might be considered relatively modest, it is important to emphasize that the colon cancer cell lines used exhibit high rates of basal proliferation and are generally refractory to increases in growth in response to exogenous stimuli. Indeed, we have examined effects of serum deprivation and serum supplementation on these cells and serum, typically a potent growth inducer, does not significantly increase proliferation (unpublished observations). Thus even the small increases in proliferation and anchorage-independent growth support a direct effect of TNFR2 to promote colon cancer cell proliferation and the concept that TNFR2 is a pro-tumorigenic factor. This is further supported by data showing that siRNA-mediated TNFR2 knockdown modestly decreased proliferation of multiple colon cancer cell lines. A limitation of our study is that the impact of TNFR2 knockdown on cancer cell proliferation was modest. This may reflect the fact that despite testing of multiple siRNAs, the maximum knockdown achieved was 60%, which may reflect the existence of multiple TNFR2 splice variants (34). Future experiments will be required to definitively establish the role of normal levels of endogenously expressed TNFR2 in intestinal epithelial cell or colon cancer cell proliferation.
SOCS3 is induced by cytokines, is a known endogenous negative feedback inhibitor of STAT3 and is epigenetically silenced in lung, liver, and squamous cell cancers or cancer cell lines (25, 46-49). Our previous study showed that IEC-specific deletion of SOCS3 led to an increase in tumor load in the AOM/DSS model, supporting the hypothesis that SOCS3 may act as a suppressor of colitis-associated cancer (22). This same study revealed that loss of IEC-SOCS3 resulted in enhanced activation of both STAT3 and NFκB. We report here that SOCS3 over-expression limits TNFR2 expression in colon cancer cell lines and limits STAT3 binding to -1578 consensus element in the TNFR2 promoter. A predominant effect of SOCS3 on the -1578 site versus -364 STAT3 binding site further supports a critical role of this region of the TNFR2 promoter in regulation of TNFR2 expression levels. SOCS3 over-expression reduced proliferation of colon cancer cells and dramatically decreased anchorage-independent growth. Also, increased TNFR2 was observed in tumors that develop in AOM/DSS treated mice with specific SOCS3 gene disruption in IEC. Together, these findings support a role of SOCS3 as a potent inhibitor of colon cancer cell growth and suggest that these effects may at least in part involve the ability of SOCS3 to limit cytokine-induced TNFR2 expression. The findings also indicate that loss of SOCS3 in IEC may promote colon tumors by facilitating increases in cytokine-induced TNFR2 expression in a setting of chronic intestinal inflammation. However, short-term siRNA-mediated reductions in basal or cytokine-induced SOCS3 mRNA did not enhance TNFR2 expression, indicating that SOCS3 knockdown alone or in the short term is itself not sufficient to cause increased TNFR2 expression.
Anti-TNFα therapies are widely used in the treatment of human IBD (50-54). The effect of anti-TNFα therapy on risk of colitis-associated cancer or other cancers is not well defined and is a topic of intensive investigation (55, 56). Our findings that TNFα cooperates with IL-6 to induce TNFR2, an inducer of colon cancer cell growth, suggests that TNFR2 may prove useful as a biomarker of the potential effects of anti-TNFα on colitis-associated cancer risk, or could represent a specific target to decrease colon cancer risk in IBD. Indeed, increased circulating TNFR2 was recently reported to be associated with enhanced CRC risk in humans, and those with higher plasma TNFR2 exhibit reduced CRC risk with use of anti-inflammatory drugs (57). Furthermore, the finding that STAT3 is a mediator of TNFR2 induction by combined TNFα and IL-6 adds to the growing evidence for STAT3 as a key mediator of colitis-associated cancer and supports further investigation of STAT3 inhibitors as potential cancer therapies.
Supplementary Material
Supplementary Figure 1. SOCS3 knockdown does not enhance cytokine-induction of TNFR2 in Caco2 cells. Histograms show effects of SOCS3 siRNA on (A) SOCS3 mRNA levels and (B) TNFR2 mRNA levels (*p ≤ 0.05 for cytokine versus no treatment; **p ≤ 0.05 effect of SOCS3 siRNA versus control siRNA). (n=3). SOCS3 siRNA reduced the levels of SOCS3 mRNA in untreated and cytokine-treated cells, but could not prevent cytokine-induction of SOCS3 mRNA. SOCS3 siRNA-treated cells showed reduced levels of basal or cytokine-induced TNFR2 mRNA versus control siRNA-treated cells.
Supplementary Table 1. Percent change in [3H] thymidine incorporation with TNFR2 shRNA plasmid compared to control in cytokine-treated SW480 and COLO205 cells. TNFR2 knockdown. Values expressed as percent change from scramble control ± SEM.
Supplementary Table 2. Percent change in [3H] thymidine incorporation with TNFR1 or TNFR2 siRNA compared to control in Caco2 or HIECs. Values expressed as percent change from scramble control ± SEM.
Supplementary Table 3. Percent change in proliferation with TNFR2 siRNA compared to control in cytokine-treated cells. WST-1 assay performed on Caco2 or HIECs. Values expressed as percent change from scramble control ± SEM.
Acknowledgments
We thank Drs. Richard Furlanetto and Peter Nissley for providing the human SOCS3 plasmid, Dr. Daniela Männel for the human TNFR2-expressing retroviral construct, Dr. Christian Jobin for CA-STAT3 adenovirus, and Dr. Albert Baldwin for NFκB inhibitors. We thank Kirk McNaughton and Kimberly Etzel for help with TNFR2 staining. We would also like to express thanks to members of KEH doctoral dissertation committee Drs. James Anderson, Susan Henning, John Rawls, and Robert Sandler for useful input. We acknowledge Agostina Santoro, Amanda Mah, Eric Blue, Lauren Parker, Drs. Rachael Rigby, Marcus Muehlbauer, and Paul Campbell for technical assistance. We thank Dr. Jean-Francois Beaulieu (University of Sherbrooke, Quebec, Canada) for the human intestinal epithelial cell line (HIECs). Finally, we express thanks to Dr. Anil Rustgi for helpful suggestions. The National Institute of Diabetes and Digestive and Kidney Disease Grant DK-47769 (PKL), a grant from the UNC University Cancer Research Fund (PKL), and a Howard Hughes Medical Institute-sponsored Med-Into-Grad pre-doctoral training grant (KEH) supported this work. Further assistance was obtained from the UNC CGIBD Immunotechnologies Core DK-24987 and UNC Vector Core.
References
- 1.Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–33. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- 2.Ekbom A, Helmick C, Zack M, Adami HO. Increased risk of large-bowel cancer in Crohn's disease with colonic involvement. Lancet. 1990;336:357–9. doi: 10.1016/0140-6736(90)91889-i. [DOI] [PubMed] [Google Scholar]
- 3.Munkholm P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Alimentary Pharmacology & Therapeutics. 2003;18 2:1–5. doi: 10.1046/j.1365-2036.18.s2.2.x. [DOI] [PubMed] [Google Scholar]
- 4.Becker C, Fantini MC, Wirtz S, Nikolaev A, Lehr HA, Galle PR, Rose-John S, Neurath MF. IL-6 signaling promotes tumor growth in colorectal cancer. CellCycle. 2005;4:217–20. [PubMed] [Google Scholar]
- 5.Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huber LA, Zatloukal K, Beug H, Ohlschlager P, Schutz A, Halbhuber KJ, Friedrich K. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia (New York, NY) 2005;7:545–55. doi: 10.1593/neo.04571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin Q, Lai R, Chirieac LR, Li C, Thomazy VA, Grammatikakis I, Rassidakis GZ, Zhang W, Fujio Y, Kunisada K, Hamilton SR, Amin HM. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. The American Journal of Pathology. 2005;167:969–80. doi: 10.1016/S0002-9440(10)61187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawada M, Seno H, Uenoyama Y, Sawabu T, Kanda N, Fukui H, Shimahara Y, Chiba T. Signal transducers and activators of transcription 3 activation is involved in nuclear accumulation of beta-catenin in colorectal cancer. Cancer research. 2006;66:2913–7. doi: 10.1158/0008-5472.CAN-05-3460. [DOI] [PubMed] [Google Scholar]
- 8.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, Nebelsiek T, Lundgren-May T, Canli O, Schwitalla S, Matthews V, Schmid RM, Kirchner T, Arkan MC, Ernst M, Greten FR. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–13. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lind DS, Hochwald SN, Malaty J, Rekkas S, Hebig P, Mishra G, Moldawer LL, Copeland EM, 3rd, Mackay S. Nuclear factor-kappa B is upregulated in colorectal cancer. Surgery. 2001;130:363–9. doi: 10.1067/msy.2001.116672. [DOI] [PubMed] [Google Scholar]
- 11.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 12.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 13.Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–70. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onizawa M, Nagaishi T, Kanai T, Nagano Ki, Oshima S, Nemoto Y, Yoshioka A, Totsuka T, Okamoto R, Nakamura T, Sakamoto N, Tsuchiya K, Aoki K, Ohya K, Yagita H, Watanabe M. Signaling pathway via TNF-{alpha}/NF-{kappa}B in intestinal epithelial cells may be directly involved in colitis-associated carcinogenesis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G850–9. doi: 10.1152/ajpgi.00071.2008. [DOI] [PubMed] [Google Scholar]
- 15.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 16.Mizoguchi E, Mizoguchi A, Takedatsu H, Cario E, De J, Jin Ooi C, Xavier RJ, Terhorst C, Podolsky DK, Bhan AK. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology. 2002;122:134–44. doi: 10.1053/gast.2002.30347. [DOI] [PubMed] [Google Scholar]
- 17.Corredor J, Yan F, Shen CC, Tong W, John SK, Wilson G, Whitehead R, Polk DB. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. AJP - Cell Physiology. 2003;284:C953–61. doi: 10.1152/ajpcell.00309.2002. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez A, Smith F, Wang Q, Wang X, Evers BM. Assessment of differential gene expression patterns in human colon cancers. Ann Surg. 2000;232:576–85. doi: 10.1097/00000658-200010000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR. IFN-[gamma]-Induced TNFR2 Expression Is Required for TNF-Dependent Intestinal Epithelial Barrier Dysfunction. Gastroenterology. 2006;131:1153–63. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santee SM, Owen-Schaub LB. Human tumor necrosis factor receptor p75/80 (CD120b) gene structure and promoter characterization. J Biol Chem. 1996;271:21151–9. doi: 10.1074/jbc.271.35.21151. [DOI] [PubMed] [Google Scholar]
- 21.Greenhalgh CJ, Miller ME, Hilton DJ, Lund PK. Suppressors of cytokine signaling: Relevance to gastrointestinal function and disease. Gastroenterology. 2002;123:2064–81. doi: 10.1053/gast.2002.37068. [DOI] [PubMed] [Google Scholar]
- 22.Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26:4833–41. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]
- 23.Sutherland KD, Lindeman GJ, Choong DY, Wittlin S, Brentzell L, Phillips W, Campbell IG, Visvader JE. Differential hypermethylation of SOCS genes in ovarian and breast carcinomas. Oncogene. 2004;23:7726–33. doi: 10.1038/sj.onc.1207787. [DOI] [PubMed] [Google Scholar]
- 24.Oshimo Y, Kuraoka K, Nakayama H, Kitadai Y, Yoshida K, Chayama K, Yasui W. Epigenetic inactivation of SOCS-1 by CpG island hypermethylation in human gastric carcinoma. Int J Cancer. 2004;112:1003–9. doi: 10.1002/ijc.20521. [DOI] [PubMed] [Google Scholar]
- 25.He B, You L, Uematsu K, Zang K, Xu Z, Lee AY, Costello JF, McCormick F, Jablons DM. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100:14133–8. doi: 10.1073/pnas.2232790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, de Haar C, Chen M, Deuring J, Gerrits MM, Smits R, Xia B, Kuipers EJ, van der Woude CJ. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59:227–35. doi: 10.1136/gut.2009.184176. [DOI] [PubMed] [Google Scholar]
- 27.Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–9. [PubMed] [Google Scholar]
- 28.Yasuda S, Yogosawa S, Izutani Y, Nakamura Y, Watanabe H, Sakai T. Cucurbitacin B induces G2 arrest and apoptosis via a reactive oxygen species-dependent mechanism in human colon adenocarcinoma SW480 cells. Mol Nutr Food Res. 54:559–65. doi: 10.1002/mnfr.200900165. [DOI] [PubMed] [Google Scholar]
- 29.Mori N, Yamada Y, Ikeda S, Yamasaki Y, Tsukasaki K, Tanaka Y, Tomonaga M, Yamamoto N, Fujii M. Bay 11-7082 inhibits transcription factor NF-kappa B and induces apoptosis of HTLV-I-infected T-cell lines and primary adult T-cell leukemia cells. Blood. 2002;100:1828–34. doi: 10.1182/blood-2002-01-0151. [DOI] [PubMed] [Google Scholar]
- 30.Wang L, Walia B, Evans J, Gewirtz AT, Merlin D, Sitaraman SV. IL-6 Induces NF-kB Activation in the Intestinal Epithelia. The Journal of Immunology. 2003;171:3194–201. doi: 10.4049/jimmunol.171.6.3194. [DOI] [PubMed] [Google Scholar]
- 31.Smith DR, Hoyt EC, Gallagher M, Schwabe RF, Lund PK. Effect of age and cognitive status on basal level AP-1 activity in rat hippocampus. Neurobiol Aging. 2001;22:773–86. doi: 10.1016/s0197-4580(01)00240-8. [DOI] [PubMed] [Google Scholar]
- 32.Hoentjen F, Sartor RB, Ozaki M, Jobin C. STAT3 regulates NF-{kappa}B recruitment to the IL-12p40 promoter in dendritic cells. Blood. 2005;105:689–96. doi: 10.1182/blood-2004-04-1309. [DOI] [PubMed] [Google Scholar]
- 33.Theiss AL, Simmons JG, Jobin C, Lund PK. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. Journal of Biological Chemistry. 2005;280:36099–109. doi: 10.1074/jbc.M505291200. [DOI] [PubMed] [Google Scholar]
- 34.Scherubl C, Schneider-Brachert W, Schutze S, Hehlgans T, Mannel D. Colocalization of endogenous TNF with a functional intracellular splice form of human TNF receptor type 2. Journal of Inflammation. 2005;2:7. doi: 10.1186/1476-9255-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puhr M, Santer FR, Neuwirt H, Susani M, Nemeth JA, Hobisch A, Kenner L, Culig Z. Down-regulation of suppressor of cytokine signaling-3 causes prostate cancer cell death through activation of the extrinsic and intrinsic apoptosis pathways. Cancer Res. 2009;69:7375–84. doi: 10.1158/0008-5472.CAN-09-0806. [DOI] [PubMed] [Google Scholar]
- 36.Simmons JG, Pucilowska JB, Keku TO, Lund PK. IGF-I and TGF-beta1 have distinct effects on phenotype and proliferation of intestinal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;283:G809–18. doi: 10.1152/ajpgi.00057.2002. [DOI] [PubMed] [Google Scholar]
- 37.Girish V, Vijayalakshmi A. Affordable image analysis using NIH Image/ImageJ. Indian J Cancer. 2004;41:47. [PubMed] [Google Scholar]
- 38.Szlosarek PW, Grimshaw MJ, Kulbe H, Wilson JL, Wilbanks GD, Burke F, Balkwill FR. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol Cancer Ther. 2006;5:382–90. doi: 10.1158/1535-7163.MCT-05-0303. [DOI] [PubMed] [Google Scholar]
- 39.Ono M, Kohda H, Kawaguchi T, Ohhira M, Sekiya C, Namiki M, Takeyasu A, Taniguchi N. Induction of Mn-superoxide dismutase by tumor necrosis factor, interleukin-1 and interleukin-6 in human hepatoma cells. Biochem Biophys Res Commun. 1992;182:1100–7. doi: 10.1016/0006-291x(92)91845-h. [DOI] [PubMed] [Google Scholar]
- 40.Koo AS, Armstrong C, Bochner B, Shimabukuro T, Tso CL, deKernion JB, Belldegrum A. Interleukin-6 and renal cell cancer: production, regulation, and growth effects. Cancer Immunol Immunother. 1992;35:97–105. doi: 10.1007/BF01741856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Legrand-Poels S, Schoonbroodt S, Piette J. Regulation of interleukin-6 gene expression by pro-inflammatory cytokines in a colon cancer cell line. Biochem J. 2000;349:765–73. doi: 10.1042/bj3490765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10:2327–34. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K, Akira S, Matsumoto S, Toyonaga A, Sata M, Yoshimura A. CIS3/SOCS3/SSI3 Plays a Negative Regulatory Role in STAT3 Activation and Intestinal Inflammation. The Journal of Experimental Medicine. 2001;193:471–82. doi: 10.1084/jem.193.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lang R, Pauleau AL, Parganas E, Takahashi Y, Mages J, Ihle JN, Rutschman R, Murray PJ. SOCS3 regulates the plasticity of gp130 signaling. Nature immunology. 2003;4:546–50. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 45.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nature immunology. 2003;4:540–5. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 46.Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer Rp BTK, Fausto N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. The Journal of Experimental Medicine. 2008;205:91–103. doi: 10.1084/jem.20070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ogata H, Kobayashi T, Chinen T, Takaki H, Sanada T, Minoda Y, Koga K, Takaesu G, Maehara Y, Iida M, Yoshimura A. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131:179–93. doi: 10.1053/j.gastro.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 48.Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T, Yoshikawa H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005;24:6406–17. doi: 10.1038/sj.onc.1208788. [DOI] [PubMed] [Google Scholar]
- 49.Weber A, Hengge UR, Bardenheuer W, Tischoff I, Sommerer F, Markwarth A, Dietz A, Wittekind C, Tannapfel A. SOCS-3 is frequently methylated in head and neck squamous cell carcinoma and its precursor lesions and causes growth inhibition. Oncogene. 2005;24:6699–708. doi: 10.1038/sj.onc.1208818. [DOI] [PubMed] [Google Scholar]
- 50.Sands BE, Anderson FH, Bernstein CN, Chey WY, Feagan BG, Fedorak RN, Kamm MA, Korzenik JR, Lashner BA, Onken JE, Rachmilewitz D, Rutgeerts P, Wild G, Wolf DC, Marsters PA, Travers SB, Blank MA, van Deventer SJ. Infliximab maintenance therapy for fistulizing Crohn's disease. N Engl J Med. 2004;350:876–85. doi: 10.1056/NEJMoa030815. [DOI] [PubMed] [Google Scholar]
- 51.Rutgeerts P, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, Hanauer SB. Comparison of scheduled and episodic treatment strategies of infliximab in Crohn's disease. Gastroenterology. 2004;126:402–13. doi: 10.1053/j.gastro.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 52.Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, de Villiers WJ, Present D, Sands BE, Colombel JF. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–76. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 53.Osterman MT, Lichtenstein GR. Current and Future Anti-TNF Therapy for Inflammatory Bowel Disease. Curr Treat Options Gastroenterol. 2007;10:195–207. doi: 10.1007/s11938-007-0013-3. [DOI] [PubMed] [Google Scholar]
- 54.Osterman MT, Lichtenstein GR. Infliximab in fistulizing Crohn's disease. Gastroenterol Clin North Am. 2006;35:795–820. doi: 10.1016/j.gtc.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 55.Esposito E, Cuzzocrea S. TNF-Alpha as a Therapeutic Target in Inflammatory Diseases, Ischemia- Reperfusion Injury and Trauma. Current Medicinal Chemistry. 2009;16:3152–67. doi: 10.2174/092986709788803024. [DOI] [PubMed] [Google Scholar]
- 56.Lakatos PL, Miheller P. Is there an increased risk of lymphoma and malignancies under anti-TNF therapy in IBD? Curr Drug Targets. 11:179–86. doi: 10.2174/138945010790309867. [DOI] [PubMed] [Google Scholar]
- 57.Chan AT, Ogino S, Giovannucci EL, Fuchs CS. Inflammatory markers are associated with risk of colorectal cancer and chemopreventive response to anti-inflammatory drugs. Gastroenterology. 140:799–808. doi: 10.1053/j.gastro.2010.11.041. quiz e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. SOCS3 knockdown does not enhance cytokine-induction of TNFR2 in Caco2 cells. Histograms show effects of SOCS3 siRNA on (A) SOCS3 mRNA levels and (B) TNFR2 mRNA levels (*p ≤ 0.05 for cytokine versus no treatment; **p ≤ 0.05 effect of SOCS3 siRNA versus control siRNA). (n=3). SOCS3 siRNA reduced the levels of SOCS3 mRNA in untreated and cytokine-treated cells, but could not prevent cytokine-induction of SOCS3 mRNA. SOCS3 siRNA-treated cells showed reduced levels of basal or cytokine-induced TNFR2 mRNA versus control siRNA-treated cells.
Supplementary Table 1. Percent change in [3H] thymidine incorporation with TNFR2 shRNA plasmid compared to control in cytokine-treated SW480 and COLO205 cells. TNFR2 knockdown. Values expressed as percent change from scramble control ± SEM.
Supplementary Table 2. Percent change in [3H] thymidine incorporation with TNFR1 or TNFR2 siRNA compared to control in Caco2 or HIECs. Values expressed as percent change from scramble control ± SEM.
Supplementary Table 3. Percent change in proliferation with TNFR2 siRNA compared to control in cytokine-treated cells. WST-1 assay performed on Caco2 or HIECs. Values expressed as percent change from scramble control ± SEM.