

Abstract
Injury-induced cytokines act through gp130 in sympathetic neurons to suppress expression of tyrosine hydroxylase (TH) and other genes associated with noradrenergic transmission. These cytokines also trigger the local loss of TH in peri-infarct sympathetic axons after myocardial infarction, but altered gene expression cannot explain the selective loss of TH enzyme in one region of the heart. We hypothesized that inflammatory cytokines, which are highest near the infarct, stimulated local degradation of TH protein. We used cultured sympathetic neurons and neuroblastoma cells to test this hypothesis. The cytokines ciliary neurotrophic factor (CNTF) and leukemia inhibitory factor (LIF) suppressed TH content in both neurons and neuroblastoma cells. CNTF suppressed TH in a gp130-dependent manner, and decreased the half-life of TH protein by approximately 50%. CNTF stimulated the ubiquitination of TH in both neurons and neuroblastoma cells, and the proteasome inhibitors MG-132 and lactacystin prevented the CNTF-induced loss of TH protein. Inhibiting activation of extracellular signal regulated kinases 1&2 (ERK1/2) with U0126 prevented the CNTF-induced ubiquitination of TH and the associated decrease in protein half-life. Likewise, inhibiting ERK1/2 activation blunted the cytokine-stimulated loss of TH protein in sympathetic neurons, despite enhancing the loss of TH mRNA. These data suggest that gp130 cytokines stimulate proteasomal degradation of TH through an ERK1/2 dependent pathway, and may have important implications for local regulation of neurotransmission at sites of inflammation.
Introduction
Inflammatory cytokines acting through the gp130 receptor suppress noradrenergic function in a subset of sympathetic neurons during development (Stanke et al. 2006), and more broadly in adult sympathetic neurons after nerve injury (Pellegrino et al. 2011;Rao et al. 1993;Zigmond et al. 1996). Activation of gp130 decreases the expression of genes involved in noradrenergic transmission in sympathetic neurons, including that of tyrosine hydroxylase (TH), the rate-limiting enzyme for norepinephrine (NE) synthesis (Fann and Patterson 1993;Li et al. 2003;Nawa et al. 1991;Pellegrino et al. 2011). Similar changes have been modeled in vitro by treating cultured sympathetic neurons with gp130 cytokines such as ciliary neurotrophic factor (CNTF) or leukemia inhibitory factor (LIF) (Li et al. 2003;Nawa et al. 1991;Patterson and Chun 1977;Saadat et al. 1989;Yamamori et al. 1989).
Several gp130 cytokines are elevated in the left ventricle after myocardial infarction (Aoyama et al. 2000;Frangogiannis et al. 2002;Gwechenberger et al. 1999;Kreusser et al. 2008), and activation of gp130 results in the loss of TH in the peri-infarct region of the left ventricle (Parrish et al. 2010). Suppression of the TH gene cannot explain the selective loss of TH enzyme in peri-infarct neurons, because activation of cardiac sympathetic nerves after myocardial infarction increases TH mRNA (Parrish et al., 2008). Furthermore, TH protein content is normal in cardiac sympathetic axons farther away from the site of damage (Li et al. 2004;Parrish et al. 2008). This raises the possibility that cytokines have direct effects on TH degradation in addition to their well-characterized effects on TH gene expression and protein synthesis.
Targeted degradation of proteins occurs primarily through the ubiquitin-proteasome system (Ciechanover 2005). Tyrosine hydroxylase is a substrate for degradation by the ubiquitin-proteasome system (Doskeland and Flatmark 2002;Nakashima et al. 2011), and its ubiquitination degradation can be stimulated by angiotensin (1–7) (Lopez Verrilli et al. 2009). We tested the hypothesis that gp130 cytokines stimulate the ubiquitination and proteasomal degradation of TH using cultured sympathetic neurons and M17 neuroblastoma cells. Our results support the notion that cytokine activation of gp130 stimulates proteasomal degradation of TH in sympathetic neurons.
Experimental procedures
Animal
Pregnant adult Sprague Dawley rats were obtained from Charles River. Wild-type C57BL/6J mice were obtained from Jackson Laboratories. The gp130DBH-Cre/lox mice were generated as previously described (Stanke et al. 2006). All animals were housed individually with a 12 hr:12 hr light dark cycle and ad libitum access to food and water. All procedures were approved by the Institutional Animal Care and Use Committee, and comply with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996).
Cell culture
All cells were grown under sterile conditions in a humidified 5% CO2 incubator at 37°C. Superior cervical ganglia (SCG) from newborn rats or mice (P0-P1) were dissociated and grown in cell culture as previously described (Dziennis and Habecker 2003;Li et al. 2003) using C2 medium supplemented with 50ng/mL nerve growth factor (NGF, Alomone Labs), and 3% fetal bovine serum (ATCC)(Pellegrino et al. 2011). Sympathetic neurons were grown in 12, 24, or 96-well plates pre-coated with 100μg/mL poly-L-lysine (Sigma) and 10μg/mL collagen (BD bioscience). Non-neuronal cells were eliminated by treating the cultures with the anti-mitotic agent cytosine arabinoside (Ara C, 1μM, Sigma) for 2 days. SK-N-BE(2)M17 human neuroblastoma cells (M17 cells) were grown in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum. M17 cells were plated at 1 × 105 cells per well in 12-well plates.
Cytokines and other reagents were diluted in culture medium before addition to the culture plates. Cells were treated with 100 ng/ml CNTF or LIF (Pepro Tech), 100 nM MG-132 (Calbiochem), 20 μM STAT3i V (STAT3 Inhibitor V, Calbiochem), 10 μM U0126 (Sigma), and 2 μM JAK (Janus tyrosine kinase) inhibitor (Calbiochem). The duration and timing of treatments is noted for each experiment. All conditions were assayed in triplicate and experiments repeated at least 3 times.
Immunoblot Analysis
Cells were washed with ice-cold PBS and lysed at 4°C in RIPA lysis buffer (1% Triton-X100, 1% sodium deoxycholate, 0.2% SDS, 125 mM NaCl, 50 mM Tris pH 8.0, 10% glycerol, 25 mM β-glycerolphosphate, 1 mM EDTA, 25 mM NaF, protease inhibitor cocktail (Roche) and 1 mM sodium orthvanadate). Lysates were fractionated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were incubated in 5% low fat milk in Tris-buffered saline (pH 7.4) containing 0.1% tween-20 (TBST) at room temperature. Membranes were subsequently incubated at 4°C overnight with rabbit anti-TH (1:5000, Millipore), mouse anti-ubiquitin (1:500, Santa Cruz), rabbit anti-pERK1/2 (phospho-extracellular signal-regulated kinases 1 and 2, 1:1000, Cell Signaling), rabbit anti-total ERK (1:1000, Cell Signaling), rabbit anti-pSTAT3 (phospho-Tyr705 Signal Transducer and Activator of Transcription 3, 1:1000, Cell Signaling), or rabbit anti-total STAT3 (1:1000, Cell Signaling) antibodies. The immunoblots were incubated with horseradish peroxidase (HRP)- conjugated secondary antibody immune-complexes were visualized by chemiluminescence (Super Signal Dura, Pierce) and quantified using Labworks software.
Immunoprecipitation
After treatment, neurons were washed with ice-cold PBS and lysed at 4°C in RIPA lysis buffer. Aliquots of cell lysates (150 μg total protein) were incubated with Protein G-Sepharose conjugated beads (Invitrogen) for 2h to eliminate non-specific binding. Pre-cleared cell lysates were incubated with a saturating amount of primary antibody (1.5 μg rabbit anti-TH per condition) overnight at 4°C. Protein G beads were added in excess to each sample and incubated for 2h. After incubation, beads were washed with ice-cold PBS 5 times to remove non-specific binding. Proteins were released from immune-complexes by incubation with SDS sample buffer (Dziennis and Habecker 2003) at 95° C for 5 min then analyzed by western blot.
Estimation of TH half-life
1) Pulse Chase
Neurons were cultured as described above, and 5 days after plating metabolic labeling was carried out using an approach similar to that previously used to determine TH half-life (Franklin and Johnson 1998;Wu and Cepko 1994). Prior to labeling, neurons were incubated for 1h at 37° C in culture medium without L-methionine or L-cysteine supplemented with 5% fetal bovine serum. To label newly synthesized protein, [35S] methionine/cysteine (20 μCi/mL; 73% L-methionine, 22% L-cysteine, Perkin Elmer) was added for 1 hr. Cells were washed with PBS to remove unincorporated isotope, and chase medium (DMEM) with methionine supplemented to 150 mg/L was added to stop labeling. Cultures were lysed in RIPA lysis buffer with Triton-X100 at 0, 4, 8, 16, 24 hours later. TH was immunoprecipitated from samples and immune complexes were resolved by SDS-PAGE gel electrophoresis. Labeled TH was quantified using a Bio-Rad Phosphoimager.
2) Protein synthesis inhibition
M17 neuroblastoma cells were treated with 20 μg/mL cycloheximide with or without 100 ng/mL CNTF or 0.1% ethanol vehicle. For inhibition of MEK/ERK signaling, M17 cells were pretreated with vehicle or 10 μM U0126 for 30min, and then incubated with 20 μg/mL cycloheximide in the presence or absence of CNTF. 0.1 % DMSO and 0.1% ethanol were added as vehicle controls. Cells were lysed at 0, 8, 12, and 24 hours, and 8 μg aliquots were assayed for TH content by western blot.
For half-life calculations, TH levels were plotted as the percentage of TH remaining, normalized to initial TH levels, on a natural log scale versus time. The data were fitted to a first-order decay function to estimate the degradation rate constant (kd), which then was used to calculate a half-life (t1/2). TH half-life was calculated from t1/2= ln2/kd.
Transient Transfection and Reporter Assays
DNA used for transfection was purified using the Qiagen Maxiprep kit. M17 cells were plated at a density of 1.0 × 105 cells per well in 6-well plates and immediately treated with vehicle or 100ng/mL CNTF. 48 hours after plating, media was removed and cells were transfected using the CaPO4 method. Cells were transfected with 1μg of 4.5TH-fLuc and 100ng pRL-null as a control for transfection efficiency. After 4 hour incubation with DNA, cells were shocked with 15% glycerol/PBS, washed with PBS, and placed back into culture media containing media alone or media + CNTF. Firefly luciferase activity from 4.5TH-fLuc and Renilla luciferase activity from the pRL-null internal control were assayed 48 hours after transfection using the Dual-Luciferase Reporter Assay system. Firefly luciferase values were normalized to Renilla luciferase values.
Real Time PCR
Neurons were harvested using the Cells-to-cDNA II kit from Ambion. Cell lysates were treated with DNase and reverse transcribed. Real-time PCR was performed with the ABI TaqMan Universal PCR Master Mix in the ABI 7500, using ABI pre-validated TaqMan gene expression assays for TH and GAPDH. For the PCR amplification, 2 μl of RT reactions were used in a total volume of 20 μl, and each sample was assayed in triplicate. Standard curves were generated with known amounts of RNA (0.5 ng – 100 ng) and TH mRNA was normalized to GAPDH mRNA in the same sample.
Statistical analyses
Data are presented as mean values± SEM. Significant differences were assessed by one-way analysis of variance (ANOVA) using GraphPad Prism 5 (GraphPad Software, Inc.). Tukey’s post hoc test was used to compare to all conditions. P values <0.05 were considered significant.
Results
In order to investigate the mechanisms that underlie the gp130-dependent loss of tyrosine hydroxylase in the left ventricle after myocardial infarction (Parrish et al. 2010), we first confirmed that similar changes occurred in cell culture. Treatment of wild type sympathetic neurons with the cytokine CNTF decreased TH content and stimulated phosphorylation of ERK1/2 and STAT3 (Fig 1). In contrast, CNTF did not decrease TH or increase phosphorylation of ERK1/2 or STAT3 in sympathetic neurons lacking gp130 (Fig 1). To determine if CNTF had an effect on TH protein turnover, we carried out pulse chase experiments in wild type sympathetic neurons. CNTF was added during the “chase” phase, and samples were collected over 24 hours after CNTF addition. CNTF decreased TH half-life by approximately 50% compared to control neurons (control 28.3±0.7 hr vs. CNTF 12.8±2.2 hrs, n=3 experiments, p<0.05) (Fig 2B). Additional experiments confirmed that TH mRNA was unchanged 24 hours after CNTF treatment (Fig. 2A), consistent with a normal level of TH synthesis.
Figure 1. CNTF suppresses TH via gp130.
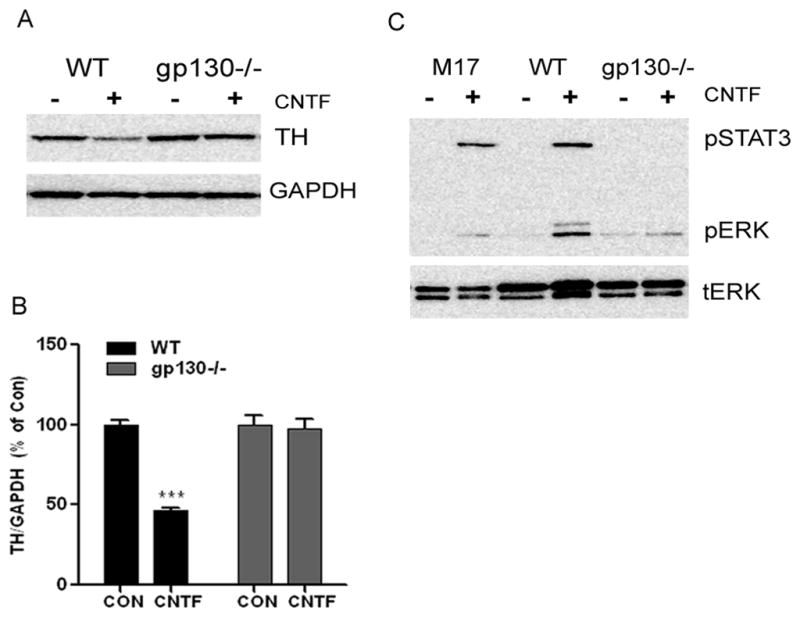
A. A representative western blot showing TH content in wild type (WT) and gp130−/− sympathetic neurons following 5 days in 100ng/ml CNTF. The blot was stripped and re-blotted for GAPDH to control for protein loading. B. Quantification of TH content assayed in triplicate by immunoblot. TH was decreased in WT but not gp130−/− neurons. Data are mean ± SEM, averaged from 3 independent experiments; ***p<0.001. C. Phospho-ERK1/2 (p-ERK1/2) and phospho-STAT3 (p-STAT3) were identified by immunoblot in M17 neuroblastoma cells, WT mouse neurons and gp130−/− neurons treated with CNTF for 15 minutes. Membranes were stripped and re-probed for total ERK as a control. Data shown are representative of three experiments assayed in triplicate.
Figure 2. CNTF decreases TH mRNA and protein half-life in sympathetic neurons.
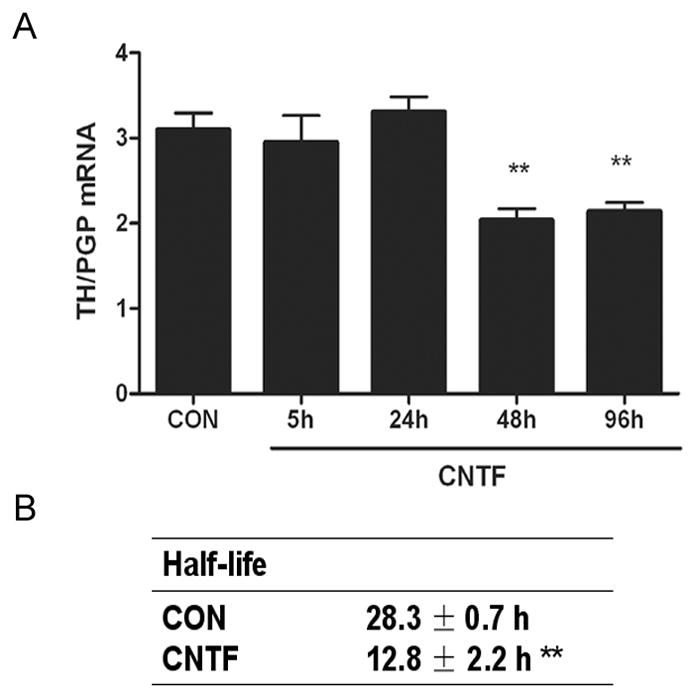
A. Time course of TH mRNA changes following treatment with vehicle (CON) or CNTF. TH mRNA was normalized to the pan-neuronal marker PGP. Data are mean ± SEM, n=3, and are representative of 3 experiments. (** p<0.01 vs. CON). B. TH half-life was quantified by pulse-chase analysis in neurons treated with vehicle (CON) or CNTF. Data are the mean ± SEM averaged from 3 experiments (** p<0.01 vs. CON).
Although primary cultures of sympathetic neurons faithfully reproduce neurotransmitter changes that are observed in vivo, the large number of cells required for half-life experiments led us to establish a cell line model system for further investigation of mechanisms. We previously found that CNTF and LIF activate the same signaling pathways and suppress dopamine beta hydroxylase expression in M17 neuroblastoma cells and sympathetic neurons (Dziennis and Habecker 2003). Therefore, we examined TH expression in M17 cells to determine if they would provide a suitable model for half-life experiments. Treatment of M17 cells with CNTF or LIF stimulated STAT3 phosphorylation (Fig. 3A), inhibited TH gene transcription (Fig. 3B), and decreased TH protein content (Fig. 3C). We then used M17 cells to determine if cytokines altered TH protein stability by measuring the effect of CNTF on TH half-life. We were unable to attain equilibrium using the pulse-chase method, and therefore inhibited protein synthesis in order to obtain the degradation rate for TH. The estimated half-life for TH in control cells was 36.8±1.9h (kd= 0.021 h−1; n=3) while the estimated half-life after CNTF treatment was 15.8±1.7h (kd = 0.046 h−1; n=3) (Fig. 4). CNTF decreased TH protein half-life by approximately 50%, similar to the change we observed in sympathetic neurons.
Figure 3. CNTF and LIF suppress TH in M17 neuroblastoma cells.
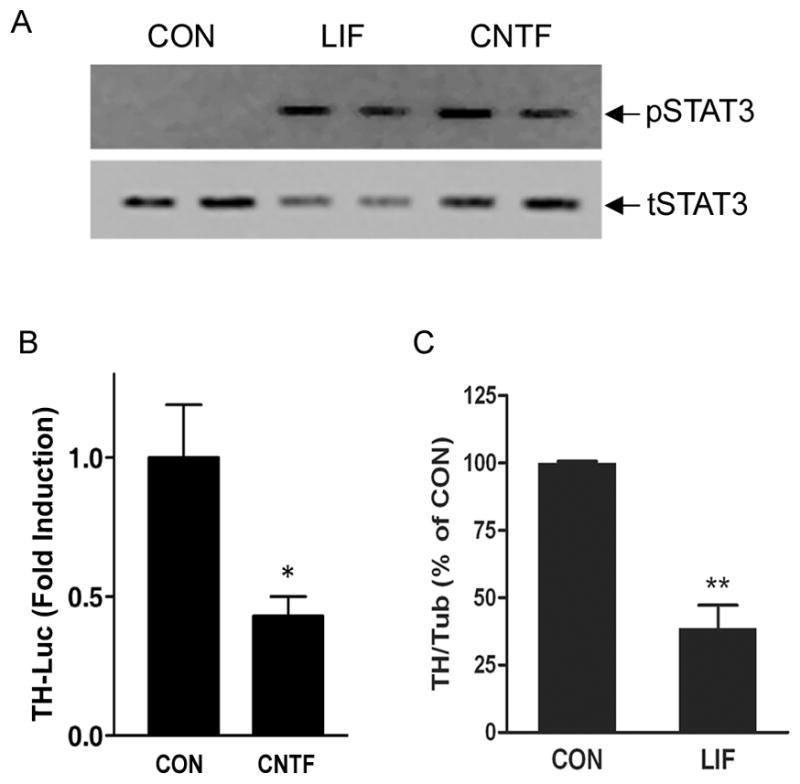
A. Representative blot of phospho-STAT3 (pSTAT3) and total STAT3 (tSTAT3) in M17 cells treated with LIF or CNTF for 15 minutes. B. TH transcription was assessed by transfecting (CON) or CNTF-treated M17 cells with the 4.5TH-fLuc reporter construct and pRL-null internal control. Cells were maintained for 2 more days in vehicle or CNTF prior to analysis of luciferase. Data are a ratio of firefly luciferase/renilla luciferase, and are mean ± SEM, n=3 (representative of 3 independent experiments, *p <0.05). C. TH content was quantified in control (CON) and LIF-treated M17 cells by immunoblot and normalized to tubulin. Data are mean ± SEM, averaged from 3 independent experiments. (** p<0.01).
Figure 4. CNTF decreases TH half-life.
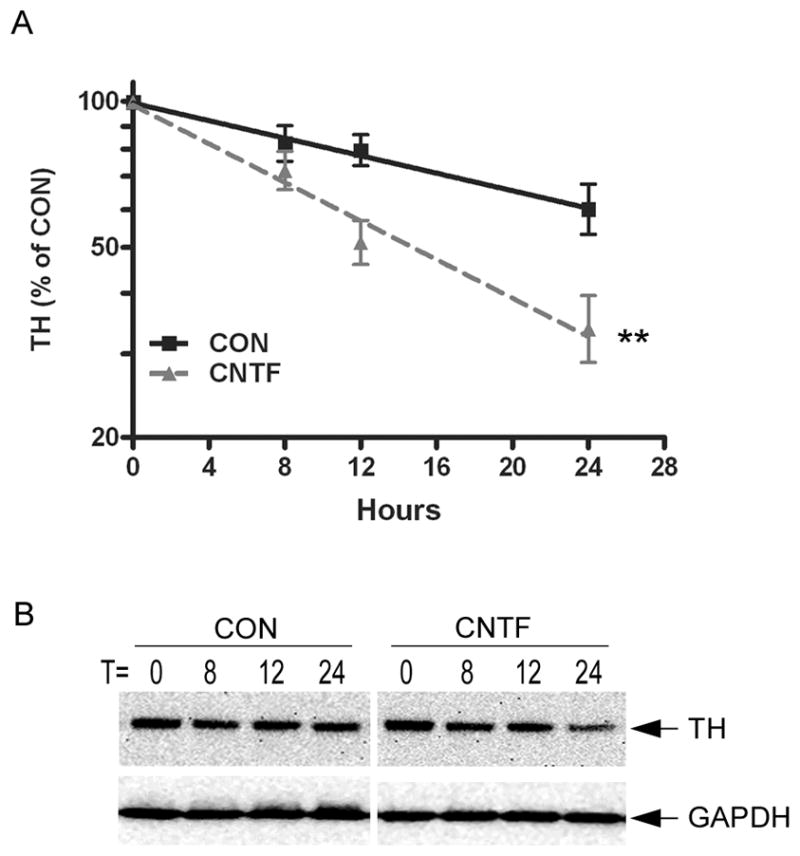
A. Natural log plot of TH content in control and CNTF treated M17 cells, graphed as percentage of the TH present at time 0. Data shown are the mean of 3 independent experiments ± SEM; ** p<0.01. B. A representative blot showing TH content at different time points. The blot was stripped and re-probed for GAPDH as a control for protein loading. One replicate is shown, but each time point was assayed in triplicate.
The half-life data suggest that CNTF stimulates TH degradation. TH can be broken down by the ubiquitin-proteasome system (Doskeland and Flatmark 2002;Lopez Verrilli et al. 2009), so we asked if CNTF stimulated the ubiquitination and proteasomal degradation of TH. Sympathetic neurons or neuroblastoma cells were treated for 30 minutes with vehicle or CNTF, and TH was immunoprecipitated (IP) with an anti-TH antibody. Samples were then fractionated and blotted with an anti-ubiquitin antibody. Ubiquitin conjugated TH was detected as a high molecular weight smear and was increased after CNTF exposure in neurons and neuroblastoma cells (Fig 5A,B). To confirm that CNTF-stimulated TH ubiquitination was a direct effect of CNTF activating its receptor, we examined ubiquitinated TH in neurons lacking the gp130 receptor. CNTF did not increase the accumulation of ubiquitinated TH in neurons lacking gp130, although TH was abundant in the immunoprecipitated samples, and absent in the post-IP lysates (Fig. 5C). Many ubiquitin-conjugated proteins were detected in the post-IP lysates present on the same blot (Fig. 5C). To determine if TH ubiquitination was associated with increased proteasomal degradation, neurons were treated with vehicle or CNTF for 4 days. During the last 2 days, cells were treated with the proteasome inhibitors MG-132 (100 nM; Fig. 5D) or lactacystin (100 nM; Fig. 5E), or 0.1% DMSO as a vehicle control. The cytokine-induced loss of TH was prevented by both proteasome inhibitors but not the DMSO control treatment, consistent with proteasomal degradation. Cell death was monitored in sister cultures and no significant cell death was observed with 2 day MG-132 and lactacystin treatment compared to control (data not shown). These data suggest that cytokines stimulate TH turnover in sympathetic neurons by targeting the enzyme for proteasomal degradation.
Figure 5. CNTF stimulates ubiquitination and proteasomal degradation of TH.

A–C. Ubiquitinated TH in wild type sympathetic neurons (A, SCG), M17 neuroblastoma cells (B, M17 cells), and sympathetic neurons lacking gp130 (C, gp130−/− neurons). All cells were treated for 30 min with 100 ng/ml CNTF. TH was immunoprecipitated and blotted with an anti-ubiquitin antibody. Blotting for total TH confirmed similar amounts of TH protein pulled down in the different treatment conditions, while no TH was detected in the post-IP lysate (C). Ubiquitin-conjugated TH is increased in wild type neurons and M17 cells, but not gp130−/− neurons. D, E. Sympathetic neurons were treated with 100 ng/mL CNTF for 4 days. During the last 2 days cells were also treated with 0.1% DMSO vehicle or the proteasome inhibitor (D) MG-132 (100 nM) or (E) lactacystin (Lac, 100 nM). TH was quantified in triplicate by western blot and the membrane stripped and blotted for GAPDH. Representative blots are shown along with their quantification (mean ± SEM, n=3, *p<0.05). Each inhibitor was tested in at least 3 independent experiments.
In order to determine which signaling pathways were involved in cytokine-stimulated ubiquitination of TH, we used a pharmacological approach to inhibit key signaling pathways. Cytokine activation of gp130 stimulates phosphorylation of JAK kinases and the downstream signaling molecules ERK1/2 and STAT3 (Dziennis and Habecker 2003;Fischer and Hilfiker-Kleiner 2008). We first examined the role of ERK1/2, and found that the MEK (MAPK/ERK kinase) inhibitor U0126 prevented both ERK1/2 phosphorylation and CNTF-stimulated TH ubiquitination (Fig. 6). We then treated sympathetic neurons with CNTF in the presence or absence of drugs that blocked activation of Jak, ERK1/2, or STAT3. Once again, blocking ERK phosphorylation prevented CNTF-stimulated ubiquitination of TH. Likewise, blocking JAK prevented phosphorylation of both ERK1/2 and STAT3, and inhibited cytokine-stimulated TH ubiquitination (Fig. 7). In contrast, treating neurons with STAT3iV blocked STAT3 phosphorylation but increased TH ubiquitination independent of CNTF (Fig. 7). The STAT3 inhibitor AG490 did not inhibit STAT3 phosphorylation at concentrations consistent with cell survival (data not shown).
Figure 6. ERK1/2 activation is required for CNTF stimulated TH ubiquitination.
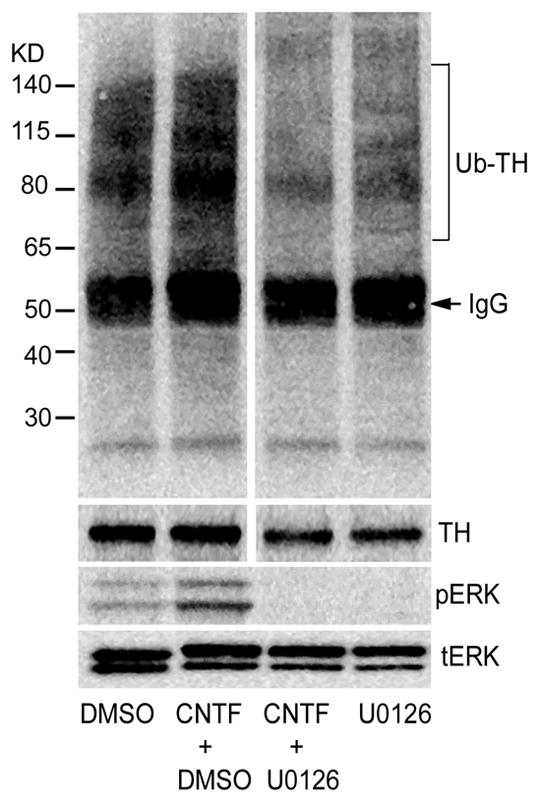
Sympathetic neurons were treated with CNTF for 30 min, with or without 10 μM U0126 to prevent ERK1/2 activation. TH was immunoprecipitated and immunoblotted with anti-ubiquitin antibody. Total TH levels were identified by immunoblot as a control. CNTF-induced phosphorylation of ERK1/2 was blocked by U0126, but total ERK1/2 was unchanged. Representative of at least 3 independent experiments.
Figure 7. Intracellular signaling involved in TH ubiquitination.

Cultured sympathetic neurons were treated with CNTF for 30 min in the presence or absence of JAK inhibitor (JAKI, 2 μM), U0126 (10 μM, UO), or STAT3I (20 μM) to block phosphorylation of STAT3. TH was immunoprecipitated and blotted with an anti-ubiquitin antibody. Additional aliquots from the same cells were blotted for phospho-STAT (pSTAT3Y705) and phospho-ERK1/2 (pERK). Total TH, ERK1/2, and STAT3 were blotted to control for protein loading. Data shown are representative of at least 3 independent experiments.
CNTF decreased the half-life of TH, and our ubiquitination experiments suggested that ERK1/2 signaling was required for cytokine-induced TH degradation. However, the time course for ubiquitination was 30 minutes, and several days of cytokine exposure were required to observe a significant proteasome-dependent loss of TH. To determine if cytokines stimulated degradation of TH via an ERK1/2-dependent pathway, we measured the half-life of TH in control and in CNTF-treated M17cells with or without U0126 to prevent ERK activation. CNTF decreased TH half-life by approximately 50%, consistent with our previous results, but the decrease in half-life was blocked by U0126 (CON 33.3± 3.6hr, UO 28.3± 2.4hr, CNTF 16± 0.5hr*, CNTF+UO 27.0± 3.5hr, n=3 experiments, *p<0.05 vs. CON) (Fig. 8). Thus, ERK1/2 activation is required for the cytokine-induced degradation of TH.
Figure 8. ERK1/2 activation is required for cytokine-induced TH protein turnover.
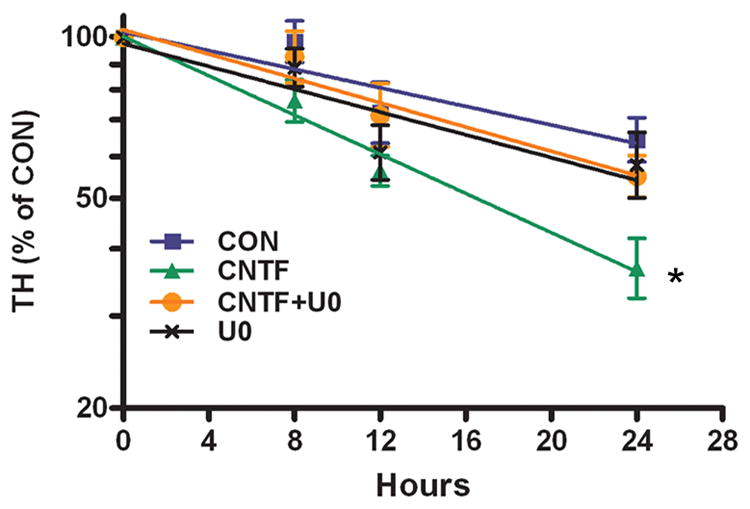
The natural log plot of TH content in M17 cells, graphed as percentage of the TH present at time 0, is shown. Data are the mean of 3 independent experiments ± SEM; * p<0.05 vs. control. Each condition was assayed in triplicate for each experiment. Calculated half-life: Control 33.3±3.6 hr, CNTF 16.3±0.5* hr, CNTF+UO 27.0±3.5 hr, UO 28.3±2.4 hr, n=3 experiments.
Since ERK1/2 was involved in cytokine-stimulated TH degradation, we tested if inhibiting ERK1/2 activation could prevent the loss of TH protein in CNTF-treated neurons. Neurons were treated with CNTF for 5 days with or without U0126, and assayed for either TH protein or TH mRNA. Blocking ERK1/2 activation in CNTF-treated cells partially rescued TH protein levels, but exacerbated the loss of TH mRNA (Fig 9). To ensure that ERK-dependent TH degradation was a general effect of gp130 activation and was not specific to CNTF, we repeated these experiments with the cytokine LIF and the MEK inhibitor PD98059, and obtained similar results. LIF decreased TH protein and mRNA in sympathetic neurons, as expected. Blocking ERK activation blunted the loss of TH protein while exacerbating the loss of TH mRNA (data not shown).
Figure 9. Blocking ERK activation blunts CNTF suppression of TH protein but exacerbates loss of TH mRNA.
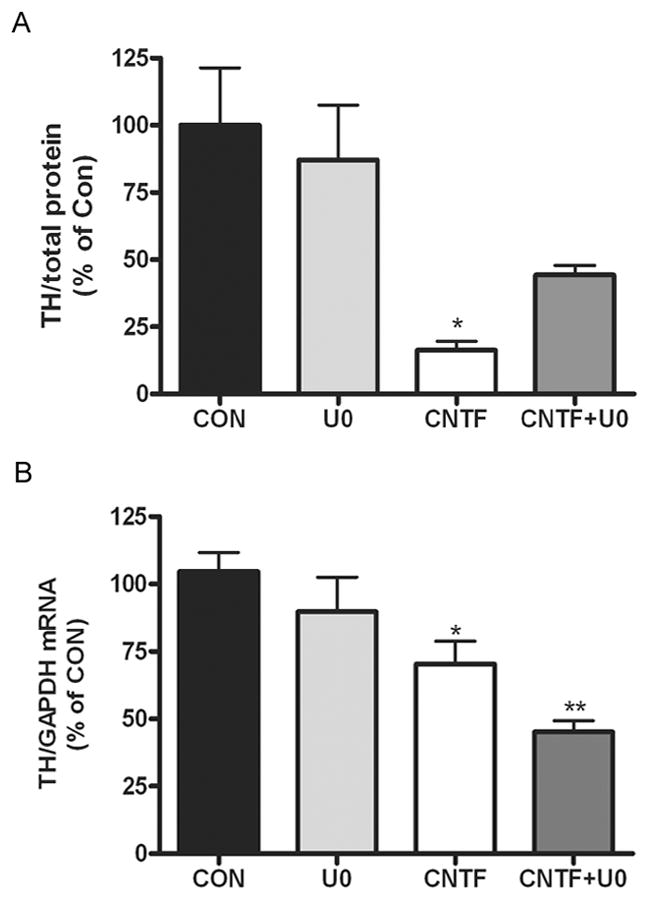
Sympathetic neurons were treated with 100 ng/ml CNTF for 5 days with or without U0126 (10 μM). A. TH protein was quantified by immunoblot and normalized to total protein. Data are from a representative experiment, mean ± SEM, n=3, * p<0.05 vs. control. B. TH mRNA was quantified by real-time PCR and normalized to GAPDH mRNA. Data are from a representative experiment, mean ± SEM, n=3, ** p<0.01, * p<0.05. All data are representative of three independent experiments.
Discussion
It is well established that inflammatory cytokines such as CNTF and LIF suppress sympathetic noradrenergic neurotransmission (Patterson and Chun 1977;Saadat et al. 1989;Yamamori et al. 1989). This suppression was thought to be mediated by reducing the expression of noradrenergic genes (Fann and Patterson 1993;Li et al. 2003;Nawa et al. 1991;Pellegrino et al. 2011). Recently, however, we found that TH content was reduced locally in sympathetic axons in the damaged left ventricle after cardiac ischemia-reperfusion, but not in axon branches projecting to unaffected regions of the heart (Parrish et al. 2008). Moreover, TH mRNA was actually increased in the cell bodies of sympathetic neurons projecting to the heart following myocardial infarction (Parrish et al. 2008). Cytokine activation of gp130 receptors triggered the local loss of TH (Parrish et al. 2010), suggesting that cytokines might directly affect the stability of TH in a portion of the target field of these sympathetic neurons.
TH is a substrate for ubiquitination (Doskeland and Flatmark 2002;Nakashima et al. 2011;Lopez Verrilli et al. 2009) which targets cells for degradation through the proteasome, the major protein degradation pathway in mammalian cells (Ciechanover 2005). Cytokines stimulate the ubiquitin-proteasome system after nerve injury (Jho et al. 2004) making this system an ideal candidate for triggering the loss of TH in the peri-infarct left ventricle. We found that cytokine activation of gp130 increased the ubiquitination of TH in cultured sympathetic neurons and neuroblastoma cells. In addition, the proteasome inhibitors MG-132 and lactacystin prevented the loss of TH in CNTF-treated cells, while the lysosome inhibitor chloroquine did not (data not shown). Together our data suggest that gp130 cytokines stimulate degradation of TH through the ubiquitin-proteasome system in sympathetic neurons.
The cytokine CNTF stimulated ubiquitination of TH within 30 minutes, consistent with targeting the enzyme for proteasomal degradation. However, significant decreases in TH enzyme levels were not observed until 4 days after cytokine addition. TH is a stable protein with reported half-life ranging from 17 hr to 34 hr in various catecholamine-producing cells (Fernandez and Craviso 1999;Tank et al. 1986;Wu and Cepko 1994), and a half-life of several days in adrenal gland (Chuang et al. 1975;Mueller et al. 1969). The long half-life is consistent with a relatively long lag time to observe the loss of TH protein in cultured neurons. Cytokines also decrease TH mRNA, and ultimately protein synthesis, in sympathetic neurons (Fann and Patterson 1993;Li et al. 2003;Nawa et al. 1991). However, TH mRNA levels were not significantly decreased until 48 hours after CNTF addition, also consistent with the delay in decreased neuronal TH content. Thus, CNTF stimulates ubiquitination of TH within minutes and decreases enzyme half-life within hours, but depletion of neuronal TH stores is not apparent until several days after exposure.
Our experiments generated half-life values for TH in control cells ranging from 28 to 37 hours, which is consistent with previous reports. Cycloheximide can stabilize TH protein in some cell types (Fernandez and Craviso 1999), but our use of cycloheximide resulted in half-life values consistent with those obtained using other methods. A limitation of our half-life experiments is that we were unable to measure TH levels beyond 24 hours due to cell viability issues. We believe the data are reliable because: the resulting half-life values are in line with values in the literature, similar results were obtained using pulse-chase and protein synthesis inhibitors, and CNTF decreased the half-life by a similar amount in both neurons and neuroblastoma cells.
Cytokines such as CNTF activate two primary signaling pathways in sympathetic neurons: JAK/STAT3 and ERK1/2. Activation of both STAT3 and ERK1/2 can regulate protein degradation through the ubiquitin-proteasome system (Niu et al. 1998;Ozawa et al. 2008). We found that cytokine activation of ERK1/2 was required for both TH ubiquitination and subsequent degradation. Blockade of ERK1/2 activation was sufficient to prevent CNTF-stimulated ubiquitination and degradation of TH as measured by protein half-life. However, inhibiting ERK1/2 activation provided only a partial rescue of TH protein during 5 days of CNTF or LIF treatment, despite completely rescuing TH half-life. CNTF and LIF suppress TH gene expression in addition to their effect on protein turnover, and inhibiting ERK1/2 activation enhanced the loss of TH mRNA. Thus, decreased synthesis of new enzyme likely offset the absence of protein degradation, resulting in a partial rescue of TH protein in cytokine treated cells. In contrast to the clear role for ERK1/2 revealed by our experiments, it is difficult to draw any specific conclusions about the role of STAT3 phosphorylation in TH ubiquitination and degradation. The STAT3 inhibitor STAT3iV increased TH ubiquitination and ERK1/2 phosphorylation (Fig 7), but that effect was independent of CNTF. Other putative STAT3 inhibitors we tested did not prevent phosphorylation of STAT3. Together our data identify a clear role for ERK signaling, but cannot rule out a role for STAT3.
This study revealed that inflammatory cytokines acting through gp130 target TH for proteasomal degradation, leading to the loss of neurotransmitter production in affected neurons. This has important implications for pathologies that involve inflammation and increased production of gp130 inflammatory cytokines. For example, cytokines are elevated in the heart after myocardial infarction (Aoyama et al. 2000;Frangogiannis et al. 2002;Gwechenberger et al. 1999) where they cause local depletion of TH and NE in the left ventricle (Parrish et al. 2010). This results in heterogeneity of sympathetic transmission that contributes to development of arrhythmias and sudden cardiac death (Rubart and Zipes 2005). Diet-induced obesity and metabolic dysfunction also result in peripheral inflammation (Elmarakby and Imig 2010;Hotamisligil 2006), which may be tied to the sympathetic dysfunction that occurs in those diseases (Grassi et al. 2010;Morgan and Rahmouni 2010). Inflammatory cytokines including IL-6 are also elevated in Parkinson’s disease, which is characterized by the loss of TH (Ciesielska et al. 2007). Recent studies indicate that dysfunction in the ubiquitin-proteasome system contributes to dopaminergic neuron degeneration in Parkinson’s disease and inhibiting proteasome activity is protective for dopaminergic neurons (McNaught et al. 2006;Oshikawa et al. 2009). Thus, our study provides an important link between inflammation and the loss of TH in disease states.
In summary, gp130 cytokines increase the degradation of TH through the ubiquitin-proteasome system via an ERK-dependent signaling pathway. Our findings suggest that local effects of inflammatory cytokines on TH stability may modulate neurotransmitter synthesis in a variety of pathological conditions.
Acknowledgments
This work was supported by National Institutes of Health Grant HL068231 (to B. A. H.) and a Tartar Trust Fellowship (to X. S.). The authors have no financial conflict of interest. The authors thank Drs. Philip Stork, Dennis Koop, and William Woodward for helpful discussions, Dr. Suzan Dziennis for technical assistance, Dr. Kwang-Soo Kim (McLean Hospital/Harvard Medical School, Waltham, MA) for SK-N-BE(2)M17 human neuroblastoma cells, and Dr. Dona Chikaraishi (Duke University Medical Center, Durham, NC) for the 4.5TH-fLuc TH promoter construct.
The abbreviations used are
- TH
tyrosine hydroxylase
- CNTF
ciliary neurotrophic factor
- JAK
Janus kinase
- STAT3
signal transducer and activator of transcription 3
- ERK 1&2
extracellular signal regulated kinases 1&2
- LIF
leukemia inhibitory factor
- NGF
nerve growth factor
- MEK
ERK kinase
- GAPDH
glyceraldehydes-3-phosphate dehydrogenase
- PBS
phosphate-buffer saline
Reference List
- Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, Yui Y, Sasayama S. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol. 2000;32:1821–1830. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- Chuang D, Zsilla G, Costa E. Turnover rate of tyrosine hydroxylase during Trans-synaptic induction. Mol Pharmacol. 1975;11:784–794. [PubMed] [Google Scholar]
- Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- Ciesielska A, Joniec I, Kurkowska-Jastrzebska I, Przybylkowski A, Gromadzka G, Czlonkowska A, Czlonkowski A. Influence of age and gender on cytokine expression in a murine model of Parkinson’s disease. Neuroimmunomodulation. 2007;14:255–265. doi: 10.1159/000113432. [DOI] [PubMed] [Google Scholar]
- Doskeland AP, Flatmark T. Ubiquitination of soluble and membrane-bound tyrosine hydroxylase and degradation of the soluble form. Eur J Biochem. 2002;269:1561–1569. doi: 10.1046/j.1432-1033.2002.02808.x. [DOI] [PubMed] [Google Scholar]
- Dziennis S, Habecker BA. Cytokine suppression of dopamine-beta-hydroxylase by extracellular signal-regulated kinase-dependent and -independent pathways. J Biol Chem. 2003;278:15897–15904. doi: 10.1074/jbc.M212480200. [DOI] [PubMed] [Google Scholar]
- Elmarakby AA, Imig JD. Obesity is the major contributor to vascular dysfunction and inflammation in high-fat diet hypertensive rats. Clin Sci (Lond) 2010;118:291–301. doi: 10.1042/CS20090395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fann MJ, Patterson PH. A novel approach to screen for cytokine effects on neuronal gene expression. J Neurochem. 1993;61:1349–1355. doi: 10.1111/j.1471-4159.1993.tb13628.x. [DOI] [PubMed] [Google Scholar]
- Fernandez E, Craviso GL. Protein synthesis blockade differentially affects the degradation of constitutive and nicotinic receptor-induced tyrosine hydroxylase protein level in isolated bovine chromaffin cells. J Neurochem. 1999;73:169–178. doi: 10.1046/j.1471-4159.1999.0730169.x. [DOI] [PubMed] [Google Scholar]
- Fischer P, Hilfiker-Kleiner D. Role of gp130-mediated signalling pathways in the heart and its impact on potential therapeutic aspects. Br J Pharmacol. 2008;153(Suppl 1):S414–S427. doi: 10.1038/bjp.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Johnson EM. Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J Cell Biol. 1998;142:1313–1324. doi: 10.1083/jcb.142.5.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Dell’oro R. Sympathetic activation in obesity: a noninnocent bystander. Hypertension. 2010;56:338–340. doi: 10.1161/HYPERTENSIONAHA.110.156596. [DOI] [PubMed] [Google Scholar]
- Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Jho DH, Engelhard HH, Gandhi R, Chao J, Babcock T, Ong E, Espat NJ. Ciliary neurotrophic factor upregulates ubiquitin-proteasome components in a rat model of neuronal injury. Cytokine. 2004;27:142–151. doi: 10.1016/j.cyto.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Kreusser MM, Buss SJ, Krebs J, Kinscherf R, Metz J, Katus HA, Haass M, Backs J. Differential expression of cardiac neurotrophic factors and sympathetic nerve ending abnormalities within the failing heart. J Mol Cell Cardiol. 2008;44:380–387. doi: 10.1016/j.yjmcc.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol. 2004;286:H2229–H2236. doi: 10.1152/ajpheart.00768.2003. [DOI] [PubMed] [Google Scholar]
- Li W, Knowlton D, Woodward WR, Habecker BA. Regulation of noradrenergic function by inflammatory cytokines and depolarization. J Neurochem. 2003;86:774–783. doi: 10.1046/j.1471-4159.2003.01890.x. [DOI] [PubMed] [Google Scholar]
- Lopez Verrilli MA, Pirola CJ, Pascual MM, Dominici FP, Turyn D, Gironacci MM. Angiotensin-(1-7) through AT receptors mediates tyrosine hydroxylase degradation via the ubiquitin-proteasome pathway. J Neurochem. 2009;109:326–335. doi: 10.1111/j.1471-4159.2009.05912.x. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jackson T, JnoBaptiste R, Kapustin A, Olanow CW. Proteasomal dysfunction in sporadic Parkinson’s disease. Neurology. 2006;66:S37–S49. doi: 10.1212/wnl.66.10_suppl_4.s37. [DOI] [PubMed] [Google Scholar]
- Morgan DA, Rahmouni K. Differential effects of insulin on sympathetic nerve activity in agouti obese mice. J Hypertens. 2010;28:1913–1919. doi: 10.1097/HJH.0b013e32833c2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller RA, Thoenen H, Axelrod J. Inhibition of trans-synaptically increased tyrosine hydroxylase activity by cycloheximide and actinomycin D. Mol Pharmacol. 1969;5:463–469. [PubMed] [Google Scholar]
- Nakashima A, Mori K, Kaneko YS, Hayashi N, Nagatsu T, Ota A. Phosphorylation of the N-terminal portion of tyrosine hydroxylase triggers proteasomal digestion of the enzyme. Biochem Biophys Res Commun. 2011;407:343–347. doi: 10.1016/j.bbrc.2011.03.020. [DOI] [PubMed] [Google Scholar]
- Nawa H, Nakanishi S, Patterson PH. Recombinant cholinergic differentiation factor (leukemia inhibitory factor) regulates sympathetic neuron phenotype by alterations in the size and amounts of neuropeptide mRNAs. J Neurochem. 1991;56:2147–2150. doi: 10.1111/j.1471-4159.1991.tb03479.x. [DOI] [PubMed] [Google Scholar]
- Niu H, Ye BH, la-Favera R. Antigen receptor signaling induces MAP kinase-mediated phosphorylation and degradation of the BCL-6 transcription factor. Genes Dev. 1998;12:1953–1961. doi: 10.1101/gad.12.13.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikawa T, Kuroiwa H, Yano R, Yokoyama H, Kadoguchi N, Kato H, Araki T. Systemic administration of proteasome inhibitor protects against MPTP neurotoxicity in mice. Cell Mol Neurobiol. 2009;29:769–777. doi: 10.1007/s10571-009-9402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa Y, Nakao K, Kurihara T, Shimazaki T, Shimmura S, Ishida S, Yoshimura A, Tsubota K, Okano H. Roles of STAT3/SOCS3 pathway in regulating the visual function and ubiquitin-proteasome-dependent degradation of rhodopsin during retinal inflammation. J Biol Chem. 2008;283:24561–24570. doi: 10.1074/jbc.M802238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish DC, Alston EN, Rohrer H, Nkadi P, Woodward WR, Schutz G, Habecker BA. Infarction-induced cytokines cause local depletion of tyrosine hydroxylase in cardiac sympathetic nerves. Exp Physiol. 2010;95:304–314. doi: 10.1113/expphysiol.2009.049965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish DC, Gritman K, Van Winkle DM, Woodward WR, Bader M, Habecker BA. Postinfarct sympathetic hyperactivity differentially stimulates expression of tyrosine hydroxylase and norepinephrine transporter. Am J Physiol Heart Circ Physiol. 2008;294:H99–H106. doi: 10.1152/ajpheart.00533.2007. [DOI] [PubMed] [Google Scholar]
- Patterson PH, Chun LL. The induction of acetylcholine synthesis in primary cultures of dissociated rat sympathetic neurons. I. Effects of conditioned medium. Dev Biol. 1977;56:263–280. doi: 10.1016/0012-1606(77)90269-x. [DOI] [PubMed] [Google Scholar]
- Pellegrino MJ, Parrish DC, Zigmond RE, Habecker BA. Cytokines inhibit norepinephrine transporter expression by decreasing Hand2. Mol Cell Neurosci. 2011;46:671–680. doi: 10.1016/j.mcn.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Sun Y, Escary JL, Perreau J, Tresser S, Patterson PH, Zigmond RE, Brulet P, Landis SC. Leukemia inhibitory factor mediates an injury response but not a target-directed developmental transmitter switch in sympathetic neurons. Neuron. 1993;11:1175–1185. doi: 10.1016/0896-6273(93)90229-k. [DOI] [PubMed] [Google Scholar]
- Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadat S, Sendtner M, Rohrer H. Ciliary neurotrophic factor induces cholinergic differentiation of rat sympathetic neurons in culture. J Cell Biol. 1989;108:1807–1816. doi: 10.1083/jcb.108.5.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Duong CV, Pape M, Geissen M, Burbach G, Deller T, Gascan H, Otto C, Parlato R, Schutz G, Rohrer H. Target-dependent specification of the neurotransmitter phenotype: cholinergic differentiation of sympathetic neurons is mediated in vivo by gp 130 signaling. Development. 2006;133:141–150. doi: 10.1242/dev.02189. [DOI] [PubMed] [Google Scholar]
- Tank AW, Ham L, Curella P. Induction of tyrosine hydroxylase by cyclic AMP and glucocorticoids in a rat pheochromocytoma cell line: effect of the inducing agents alone or in combination on the enzyme levels and rate of synthesis of tyrosine hydroxylase. Mol Pharmacol. 1986;30:486–496. [PubMed] [Google Scholar]
- Wu DK, Cepko CL. The stability of endogenous tyrosine hydroxylase protein in PC-12 cells differs from that expressed in mouse fibroblasts by gene transfer. J Neurochem. 1994;62:863–872. doi: 10.1046/j.1471-4159.1994.62030863.x. [DOI] [PubMed] [Google Scholar]
- Yamamori T, Fukada K, Aebersold R, Korsching S, Fann MJ, Patterson PH. The cholinergic neuronal differentiation factor from heart cells is identical to leukemia inhibitory factor. Science. 1989;246:1412–1416. doi: 10.1126/science.2512641. [DOI] [PubMed] [Google Scholar]
- Zigmond RE, Hyatt-Sachs H, Mohney RP, Schreiber RC, Shadiack AM, Sun Y, Vaccariello SA. Changes in neuropeptide phenotype after axotomy of adult peripheral neurons and the role of leukemia inhibitory factor. Perspect Dev Neurobiol. 1996;4:75–90. [PubMed] [Google Scholar]