

Abstract
α,β-Unsaturated carbonyls are an important class of chemicals involved in environmental toxicity and disease processes. Whereas adduction of cysteine residues on proteins is a well-documented reaction of these chemicals, such a generic effect cannot explain the molecular mechanism of cytotoxicity. Instead, more detailed information is needed regarding the possible specificity and kinetics of cysteine targeting and the quantitative relationship between adduct burden and protein dysfunction. To address these datagaps, purified human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was incubated with acrylamide (ACR), acrolein or methylvinyl ketone (MVK). Results show that these α,β-unsaturated carbonyl toxicants inhibited GAPDH activity in a concentration-and time-dependent manner. The rank order of enzyme inhibition (KI); i.e., ACR << MVK < acrolein, was related to the calculated electrophilic reactivity of each compound and to the corresponding kinetics of cysteine adduct formation. Tandem mass spectrometry revealed that adduct formation was selective at lower concentrations; i.e., ACR preferentially formed adducts with Cys152 (residues 146-162). At higher concentrations, ACR also formed adducts with Cys156 and Cys247 (residues 235-248). Adduct formation at Cys152 was correlated to enzyme inhibition, which is consistent with the regulatory role of this residue in enzyme function and its location within the GAPDH active site. Further analyses indicated that Cys152 was present in a pKa-lowering microenvironment (pKa = 6.03) and, at physiological pH, the corresponding sulfhydryl group exists in the highly reactive nucleophilic thiolate-state. These data suggest a general cytotoxic mechanism where electrophilic α,β-unsaturated carbonyls selectively form adducts with reactive nucleophilic cysteine residues specifically associated with the active sites of proteins. These specialized cysteine residues are toxicologically relevant molecular targets, since chemical derivitization causes loss of protein function.
Keywords: α,β-unsaturated carbonyl chemicals; protein adduct formation; cytotoxicity; electrophile toxicity; nucleophile targets
INTRODUCTION
Unsaturated carbonyl chemicals such as acrylamide (ACR), acrolein and methylvinyl ketone (MVK) are classified as type-2 alkenes (Fig. 1). Chemicals in this class are characterized by a conjugated structure that is formed when an electron-withdrawing group (e.g., a carbonyl) is linked to an alkene. Type-2 alkenes have extensive industrial applications (textile manufacturing, agriculture and polymer production) and are recognized as significant environmental pollutants (e.g., acrolein, acrylonitrile). In addition, these chemicals are dietary contaminants (e.g., acrylamide, acrolein) and prominent components of cigarette smoke (e.g., acrolein, acrylamide, acrylonitrile, crotonaldehyde). Human environmental exposure to the type-2 alkenes is therefore pervasive and has been associated with major organ system toxicity1,2. There is also growing evidence that endogenously generated unsaturated carbonyl derivatives such as acrolein, 4-hydroxy-2-nonenal (HNE) and 4-oxononenal (ONE) are involved in the pathogenesis of diseases that have a common molecular etiology of oxidative stress1-4.
Figure 1.

(A) Line structures are presented for several conjugated α,β-unsaturated carbonyl derivatives of the type-2 alkene class. For each derivative, the electrophilic index (ω) is presented parenthetically (see Materials and Methods). Abbreviations: MVK = methylvinyl ketone, ACR = acrylamide.
Data from recent proteomic and chemical studies indicate that type-2 alkene toxicity is mediated by a common mechanism involving the inactivation of key cellular proteins through the formation of covalent adducts. Specifically, the conjugated α,β-unsaturated carbonyl structure of ACR, acrolein and other type-2 alkenes is an electrophile that forms Michael-type adducts via second order addition to nucleophilic side chains of amino acids on peptides5-7 (reviewed in LoPachin et al.,1,3,4). Relative to the nucleophilic nitrogens of lysine ε-amino groups and the imidazole side chain of histidine, the unsaturated alkenes react considerably faster with cysteine sulfhydryl groups5,7-10. These kinetic differences indicate that cysteine residues are the preferred sites of type-2 alkene adduct formation. However, most proteins contain several cysteines and it is unlikely that these residues all have structural and/or functional relevance. Therefore, it cannot be assumed that adduct formation at a given cysteine residue has toxicological significance. This ambiguity makes it difficult to distinguish primary cysteine targets from toxicologically irrelevant residues. More experimentally refined studies have shown that exposure of proteins to unsaturated alkenes causes dysfunction mediated by adduction of specific cysteine residues 11-18; e.g., in vitro HNE adduction of Cys280 is responsible for inhibition of mitochondrial sirtuin 3 activity11. Although these studies provide additional mechanistic insight, critical information is missing regarding the molecular basis of cysteine selectivity. Understanding this selectivity is complicated by the fact that the sulfhydryl thiol is non-nucleophilic and therefore unreactive5-7,19,20. Furthermore, the mechanism by which cysteine-specific adduct formation causes protein dysfunction is unknown, as is the quantitative relationship between the adduct burden and protein dysfunction.
Reactive and therefore toxicologically relevant cysteine targets of the type-2 alkenes can be identified based on electronic (e.g., nucleophilicity, softness) and kinetic characteristics1,4,21. Specifically, second order reaction rates for the formation of Michael adducts are dependent not only on the relative concentrations of each reactant, but also on the electrophilicity of the electron acceptor (type-2 alkene) and the relative nucleophilicity of the electron donor (cysteine sulfhydryl groups). The nucleophilic reactivity of sulfhydryl groups on select cysteine residues can vary depending upon their respective microenvironments. Thus, although the unreactive thiol state (pKa = 8.3) predominates at physiological pH, specialized pKa-lowering microenvironments such as catalytic triads can enhance sulfhydryl ionization and thereby increase the effective concentration of the anionic thiolate. This highly nucleophilic state mediates the biochemical activities of cysteine sulfhydryl groups and, as such, the adduct reaction with type-2 alkenes is kinetically favored and has significant functional consequences. Therefore, to assess the differential targeting of cysteine residues by type-2 alkenes, we used purified human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a model protein for adduct formation. GAPDH is a cysteine-directed glycolytic enzyme that catalyzes the conversion of glyceraldehyde 3-phosphate (G3P) to D-glycerate 1,3-bisphosphate. Although multiple proteins are probable type-2 alkene targets (reviewed in1,4,21), substantial evidence suggests that the adduction of GAPDH cysteine residues is importantly involved in mediating cytotoxicity22-25. Furthermore, GAPDH has been well characterized with respect to crystalline structure, enzyme function and redox modulation26. In the present study, GAPDH was exposed in vitro to graded concentrations of ACR, MVK or acrolein and the corresponding enzyme kinetics were determined. Quantum mechanical calculations6,7 indicate that the selected type-2 alkene vary in electrophilicity (ω) from low (ACR) to relatively high reactivity (acrolein). To identify reactive amino acid residues, GAPDH was exposed to graded ACR concentrations and then analyzed by tandem mass spectrometric analysis and the concentration-dependent changes in adduct burden were correlated to kinetic parameters of GAPDH inhibition. ACR, as a weak electrophile, was able to discriminate reactive cysteines from less reactive residues that were not likely to be involved in the pathophysiological process. In addition, the quantitative relationship between the adduct burden and protein dysfunction was defined. These results provide molecular-level insight into the cytotoxic mechanisms of ACR and structurally related type-2 alkene derivatives.
EXPERIMENTAL PROCEDURES
Chemicals
Unless otherwise indicated, all reagents were high-performance liquid chromatography grade or better, and water was doubly distilled and deionized. ACR, acrolein, MVK and human recombinant GAPDH were purchased from the Sigma/Aldrich Chemical Company (Bellefonte, PA). The following chemicals are hazardous and should be handled carefully: ACR, acrolein and MVK.
Calculations of HSAB mechanical parameters
The Lowest Unoccupied Molecular Orbital (LUMO) energy (ELUMO) and Highest Occupied Molecular Orbital (HOMO) energy (EHOMO), were determined using Spartan08 (version 1.1.1) software (Wavefunction Inc., Irvine CA). For each structure, ground state equilibrium geometries were calculated with Density Functional B3LYP 6-31G* in water starting from 6-31G* geometries. Global (whole molecule) hardness (η) was calculated as η = (ELUMO-EHOMO)/2 and softness (σ) was calculated as the inverse of hardness or σ= 1/η. The electrophilicity index (ω) was calculated as ω = μ2/2η, where μ is chemical potential of the electrophile and was calculated as μ = (ELUMO+EHOMO)/2. The nucleophilicity index (ω–) was calculated as ω– = ηA(μA - μB)2/2(ηA+ηB)2, where A = reacting nucleophile and B = reacting electrophile (see LoPachin et al.,6,7 for more detailed discussion). Linear regression analysis was used to assess the relationship between the calculated HSAB parameters (σ, ω) and the kinetics of enzyme inactivation (k2, KI). Corresponding coefficients of determination (r2) were calculated from the Pearson correlation coefficient (InStat 3.0, GraphPad Software).
Time-dependent inactivation of GAPDH
The effects of type-2 alkenes on GAPDH activity in vitro were determined using an inactivation assay previously described by Bertelsen et al.27. In brief, ACR (1-500mM), MVK (50-250μM) or acrolein (10-100 μM) solutions were prepared in enzyme activity buffer (400 μL) containing Tris-acetate (5mM) and sodium arsenate (13.6mM) at pH 7.4 or 8.5 and 30°C. Toxicant-enzyme exposure was initiated by adding 100 μL of human erythrocyte GAPDH (133nM final protein concentration). At selected times during incubation (0-30 minutes), aliquots (30μL) of experimental solutions were removed and corresponding enzyme activities (remaining GAPDH activities) were determined using a modification of the method described by Tanii and Hashimoto24. Specifically, an aliquot was added to Tris-acetate buffer (180 μL final volume) containing 3-nicotinamide adenine dinucleotide (β-NAD; 0.8mM) and glyceraldehyde-3-phosphate (1.5mM) at pH 7.4 and 30°C. The reduction of NAD+ was monitored kinetically over two minutes as increasing absorbance at 340nm using a SpectraMax 250 spectrophotometer (Molecular Devices, Sunnyvale, CA) and SoftMax Pro software (Life Sciences Edition v.4.0). The slopes of these lines were used to compute the remaining GAPDH activities (V0 – V30) at selected times (t0 – t30) during the toxicant exposure period.
For each concentration of a given type-2 alkene, the apparent rate of GAPDH inactivation (kobs) is the slope of the graph ln(Vt/Vo) where Vo is velocity at time 0 vs. time (min). Double-reciprocal replots of 1/kobs vs. 1/[alkene] were used to determine several kinetic parameters of GAPDH inhibition28. Specifically, the y-intercept of these replots yields kinact, the maximum rate of GAPDH inactivation at saturating concentrations of a given type-2 alkene, while the slope is KI/kinact where KI is the concentration of a type-2 alkene that produces a half-maximal rate of inactivation. Finally, kinact/KI is the apparent second-order rate constant (k2) for inactivation of GAPDH by a type-2 alkene.
LC-MS/MS identification of ACR-modified GAPDH peptides
In these studies we used ACR to identify reactive cysteine residues on GAPDH based on the chemical principle of reactivity-selectivity29; i.e., as a weak electrophile (Table 2), ACR will selectively from adducts with the most reactive protein residues30,31. More electrophilic aldehyde or ketone analogs (e.g., acrolein, HNE) would produce a concentration-dependent flood of adducts that could obscure the most reactive cysteine5,9,18. In addition, because amides do not form Schiff bases, the only product of ACR-amino acid interactions are Michael adducts. ACR (0.01-1M)-exposed, trypsin-digested and lyophilized GAPDH peptides were re-suspended in buffer A (10ml; 25% acetonitrile/75% 10mM ammonium formate, pH 3.0) and were then loaded onto two back-to-back OPTI-PAK 3μl SCX Packed Stem Trap Columns. Samples were desalted online and SCX chromatography was performed with a LC Packings Ultimate capillary liquid chromatography system interfaced with Leap Technologies’ HTS PAL at a final flow rate of 5μl/min. The gradient was maintained for 5 mins in buffer A followed by buffer B (25% acetonitrile/75% 200mM ammonium formate, pH 8.0) for 30 mins and finally buffer C (25% acetonitrile/75% 500mM ammonium formate, pH 8.0) for 10 mins. The gradient was returned to buffer A for 10 mins. Fractions (n = 17) were collected (4 mins) automatically and individual fractions were analyzed using an electrospray ion trap mass spectrometer (LTQ-Orbitrap, Thermo, San Jose, CA) coupled to a nanoflow liquid chromatograph (U3000, Dionex, Sunnyvale, CA) for tandem mass spectrometry (MS/MS) peptide sequencing. Samples were loaded onto a pre-column (5mm × 300μm ID packed with C18 reversed-phase resin, 5μm, 100A) and washed (8 mins) with aqueous 2% acetonitrile and 0.04% trifluoroacetic acid. Trapped peptides were eluted onto the analytical column (C18 resin, 75 μm ID × 15 cm) and the 120 min gradient was programmed as follows: 95% solvent A (2% acetonitrile + 0.1% formic acid) for 8 mins, solvent B (90% acetonitrile + 0.1% formic acid) from 5%-50% in 90mins, then solvent B from 50%-90% in 7 mins and then held at 90% for 5 mins, followed by solvent B from 90% to 5% in 1min and re-equilibrated for 10 mins. The analytical column flow rate was 300 nl/min and, following each survey scan, five tandem mass spectra were collected in a data-dependent manner. The MS scans were performed in Orbitrap to obtain accurate peptide mass measurement and the MS/MS scans were performed in linear ion trap using 60s exclusion for previously sampled peptide peaks.
Table 2.
Parameters for Type-2 Alkene Electrophiles.
| Electrophile | σ (× 10-3 ev-1) | ω (ev) | log k2 |
|---|---|---|---|
| Acrolein | 371 | 3.82 | 2.47 |
| MVK | 363 | 3.38 | 2.11 |
| ACR | 315 | 2.61 | -1.28 |
HSAB (σ, ω) and kinetic parameters (k2) were calculated as described in Materials and Methods section. Based on the HSAB parameters, acrolein and MVK are significantly softer and more reactive electrophiles than ACR; i.e., larger values of σ and ω, respectively. The rank orders of respective σ and ω values for each type-2 alkene were closely correlated to the corresponding rate constants (vs log k2; r2 = 0.9981 and 0.9225, respectively) for inhibition of GAPDH activity.
Database search for GAPDH adducts
All MS/MS spectra were analyzed using Sequest (ThermoFinnigan, San Jose, CA) set to search the SwissProt human protein database assuming trypsin digestion and a fragment ion mass tolerance of 1.00 Da and a parent ion tolerance of 1.2 Da. Oxidation of methionine, iodoacetamide derivation of cysteine and ACR adduction of cysteine, lysine and histidine were specified as variable modifications. Scaffold (version Scaffold_2.1.03, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at greater than 80% probability as specified by the Peptide Prophet algorithm32. Protein identifications were accepted if they could be established at greater than 50% probability and contained at least one identified peptide. Protein probabilities were assigned by the Protein Prophet algroithm33. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
RESULTS
Kinetic Characterization of GAPDH Inhibition by Type-2 Alkenes
GAPDH was used as a model protein to quantitate the inhibitory effects of ACR and structurally related α,β-unsaturated carbonyl derivatives on the activity of a cysteine-directed enzyme. Results indicate that the maximal rate of ACR-induced GAPDH inhibition (kinact) was 1.31 × 10-2 sec-1 at pH 7.4, which is approximately the same value as those determined for both acrolein (kinact = 1.13 × 10-2 sec-1) and MVK (kinact = 0.770 × 10-2 sec-1). The equivalent kinact values reflect a common mechanism of GAPDH inactivation, which supports previous suggestions5-7 that the molecular mechanism of type-2 alkene toxicity is mediated by the characteristic α,β-unsaturated carbonyl structure (Fig. 1) of this chemical class. Moreover, variations in the corresponding kinetic parameters of GAPDH inactivation indicated a predictable rank order of potency for these toxicants. Specifically, a Kitz-Wilson transformation of the temporal data (Figure 2 A, C, D; Table 1) revealed that the KI for ACR inhibition of GAPDH activity at pH 7.4 (247mM) was substantially higher than those of either MVK (60μM) or acrolein (38μM). Calculations of the respective apparent second-order rate constants (k2; Table 1) indicated a parallel rank-order of GAPDH inhibition; i.e., the ACR-enzyme reaction was substantially slower (0.053 M-1 sec-1) than the corresponding reactions of MVK (128 M-1 sec-1) or acrolein (297 M-1 sec-1). This rank order among kinetic parameters for inhibition of a purified protein is consistent with previous studies that indicated stratified potencies for type-2 alkene interactions with cell-level processes (e.g., synaptosomal membrane transport; LoPachin et al.5-7). Furthermore, conventional substrate-velocity analysis of GAPDH inhibition indicated that the α,β-unsaturated carbonyl derivatives reduced Vmax with no effect on Km (data not shown; see LoPachin et al.5,6,34,35). The change in Vmax reflects the noncompetitive loss of enzyme activity presumably through adduct formation with retention of substrate affinity (no change in Km). Together, these data indicate that inactivation of GAPDH by ACR and the other type 2 alkenes involve the formation of Michael-type adducts with cysteine residues (see ahead). Finally, GAPDH inactivation by ACR was shown to be pH sensitive. Whereas kinact by ACR was comparable at pH 8.5 and 7.4, the concentration required to produce half-maximal rates of inactivation (KI) was substantially lower at pH 8.5 resulting in a 5-fold higher second order rate constant (k2) at pH 8.5 (Fig. 2A vs. 2B; Table 1). Enhanced potency at high pH is expected for a reaction involving cysteine residues, since a proportionately higher concentration of the ionized sulfur species (thiolate), required for adduct formation, is present as the pH approaches the pKa of the cysteine sulfur (8.3). These data confirm previous studies that demonstrated the pH-dependency of the type-2 alkene cysteine adduct reaction5,6.
Figure 2.

Kitz-Wilson double-reciprocal replots of the temporal data for GAPDH inactivation by selected type-2 alkenes: ACR at pH 7.4 (A) or pH 8.5 (B), MVK (C) and acrolein (D). The y-intercept of these replots yields kinact, the maximum rate of GAPDH inactivation at saturating concentrations of a given type-2 alkene. The slope is KI/kinact where KI is the concentration of a type-2 alkene that produces a half-maximal rate of inactivation and kinact/KI is the apparent second-order rate constant (k2) for inactivation of GAPDH by a type-2 alkene. Data are presented as mean ± SD.
Table 1.
Kinetic Parameters for Type-2 Alkene Inactivation of Human GAPDH Activity
| Electrophile (pH) | Kinact (× 10-2 sec-1) | KI (M) | k2 (M-1sec-1) |
|---|---|---|---|
| ACR (7.4) | 1.31±0.173 | 0.247±0.056 | 0.053 |
| ACR (8.5) | 1.20±0.160 | 0.045±0.008 | 0.267 |
| MVK (7.4) | 0.770±0.250 | 6.02±1.07 × 10-5 | 128 |
| Acrolein (7.4) | 1.13±0.136 | 3.81±0.83 × 10-5 | 297 |
Values for each kinetic parameter were calculated from Kitz-Wilson replots of the inactivation data (see Materials and Methods section). The value of Kinact represents the maximal rate of enzyme inactivation, KI is the concentration of the compound that produces a half maximal rate of inhibition and k2 is the second order rate constant. Respective values (except k2) are presented as mean ± SD (n=3).
Concentration-Dependent Formation of ACR Adducts With Cysteine Residues
Results indicate that ACR selectively formed adducts with cysteine residues of human GAPDH (NCBI protein accession NP_002037, 335 amino acids in length); i.e., neither histidine nor lysine adducts were detected. Among numerous peptides that resulted from trypsin digestion of GAPDH, three cysteine residues (Cys152, Cys156 and Cys247) were localized to two specific peptides: IISNASCTTNCLAPLAK (residues 146-162) and VPTANVSVVDLTCR (residues 235-248). Our findings show that ACR incubation produced a concentration-dependent rise in the number of cysteine-adducted GAPDH peptides (Fig. 3). The graded rise in percent adducts (relative to unmodified tryptic peptides) was directly correlated to reductions in enzyme activity (Fig. 3). Tandem mass spectrometry analyses revealed that the concentration-dependent increase in adducts was primarily due to preferential formation of adducts at Cys152. At relatively low ACR concentration, Cys152 modification, contributed as mono-adducts or di-adducts of the IISNASCTTNCLAPLAK peptide, were substantially more prevalent than either Cys156 adducts on the same peptide or Cys247 adducts on the VPTANVSVVDLTCR peptide (Fig. 4). For example, at 250 mM ACR, 81% of the total peptide adducts consisted of modifications at Cys152, compared to 7% that contained modifications at both Cys152 and Cys156 or negligible amounts of modified Cys247. At higher ACR concentrations, IISNASCTTNCLAPLAK peptides with both cysteine residues modified increased in relative abundance, as did peptides modified at Cys247 (Fig. 4). As a consequence, the percentage of peptides with Cys152 as the only modified residue decreased, so that mono-adducts of Cys152 constituted 70% of the total peptide adducts at 500 mM ACR, whereas di-adducts rose to 26% (the percentage of Cys247 adducts also increased slightly). However, it should be emphasized that at this ACR concentration all of the available Cys152 residues were modified (mono-adducts + di-adducts of IISNASCTTNCLAPLAK). Thus, our combined MS data clearly demonstrate an enhanced reactivity for Cys152. This conclusion is supported by regression analysis (% adduct formation at individual cysteine residues vs. GAPDH activity; SPSS Statistics v 17.0), which indicated that decreased enzyme activity caused by ACR was primarily a function of adduct formation at Cys152 (p < 0.001), as opposed to Cys156 (p = 0.056) or Cys247 (p = 0.328). Representative mass spectra for peptides containing ACR modified adducts of Cys152 and Cys152+Cys156 are presented in Figs. 5 and 6.
Figure 3.

This graph demonstrates the close inverse relationship between the concentration-dependent ACR inhibition of GAPDH activity and corresponding percent of adducted GAPDH peptides. Purified human GAPDH was incubated with graded ACR concentrations for 30 mins (30°C). Remaining enzyme activity was measured as the reduction of NAD+ monitored at 340nm following addition of glyceraldehyde-3-phosphate. Data (filled circle) are presented as mean (± SD) GAPDH activity (mOD/min). Tandem mass spectrometry analysis was used to quantitate GAPDH peptides exhibiting ACR cysteine adducts. Data (open square) are expressed as mean (± SD) percent of total peptides exhibiting cysteine adducts.
Figure 4.

This graph depicts the percent of total cysteine adducts on GAPDH peptides as a function of ACR concentration. Purified human GAPDH was incubated with graded ACR concentrations for 30 mins (30°C). Tandem mass spectrometry analysis (MS/MS) was used to determine the percent of individual cysteine residues exhibiting ACR adducts; i.e., Cys152 - filled circle, Cys152 + Cys156 - open circle, Cys247 – filled triangle. Percent adduction was calculated from the respective peak areas for adducted and unadducted peptides.
Figure 5.

Mass spectrometry spectra of a GAPDH peptide containing an ACR adduct on Cys152. The inset panel in the upper right corner is the MS spectra of the doubly charged precursor ion with an m/z of 924.4707 and a mass accuracy of 1.1ppm. The lower panel is the MS/MS spectra of this precursor ion demonstrating the presence of carbamoylcysteine (C+71) at position 152.
Figure 6.

MS spectra of a peptide containing an ACR adduct on Cys152 and Cys 156 of GAPDH. The inset panel in the upper right corner is the MS spectra of the doubly charged precursor ion with an m/z of 931.4786 and a mass accuracy of 0.94 ppm. The lower panel is the MS/MS spectra of this precursor ion demonstrating the presence of carbamoylcysteine (C+71) at positions 152 and 156.
HSAB Description of the Type-2 Alkene-Cysteine Adduct Reactions
Kinetic (Fig. 2; Table 1) and proteomic (Figs. 3 and 4) data indicate that ACR and other electrophilic type-2 alkenes inhibit GAPDH activity by forming adducts with nucleophilic side chain sulfhydryl groups on cysteine residues. Information regarding the chemical characteristics of these adduct reactions can be generated through calculations of Hard-Soft Acid-Base (HSAB) parameters such as softness (σ), electrophilicity (ω) and nucleophilicity (ω-); all of which, have been used in previous studies of biological systems (reviewed in 1,4,36). Specifically, softness refers to the ease with which electron redistribution occurs during covalent bond (adduct) formation. Consequently, the softness of a nucleophile reflects the relative ability to transfer electron density to form a bond and, the softer the electrophile, the more readily it will accept the transfer. According to the selectivity principle of the HSAB theory4,37, relevant kinetic and thermodynamic forces are most favorable when like molecules interact; e.g., soft electrophiles preferentially react with soft nucleophiles. The electrophilic index (ω) is a higher order parameter that combines softness (σ) with chemical potential (μ; the propensity of a species to undergo chemical change) and can be used as a general measure of reactivity for the type 2 alkenes. As presented in Table 2, the corresponding σ and ω values indicate that acrolein and MVK are softer, more reactive (larger ω value) electrophiles than ACR. The rank orders of the σ and ω values for these type-2 alkenes are related to the second order rate constants (k2) for GAPDH inhibition (Table 2; see also5-7).
Calculations of nucleophilic softness (Table 3) indicate that sulfhydryl groups in the anionic thiolate state (-1) are substantially softer than the nitrogen groups of either the histidine (0) or lysine (0) sidechains. The protonated (thiol) sulfhydryl groups (0) and cationic lysine residues (+1) are also substantially harder moieties than the sulfhydryl thiolate state. Thus, based on soft-soft compatibility, thiolate groups will react preferentially with type-2 alkene electrophiles. The extent to which a given nucleophile will potentially react with, for example, ACR can be predicted by calculating an index of nucleophilicity (ω–). This HSAB derived parameter considers the hardness (η) and chemical potential (μ) of both the nucleophile (cysteine, histidine, or lysine) and the relevant type-2 alkene reactant27. The respective ω– values presented in Table 3 indicate that cysteine thiolate groups are the favored targets of ACR, MVK and acrolein.
Table 3.
Calculated HSAB Parameters for Amino Acid Nucleophiles and Their Interactions with Type-2 Alkenes
| Residue | Sidechain Group | σ × 10-3 ev-1 | ACR ω- × 10-3 ev (relative) | MVK ω- × 10-3 ev (relative) | Acrolein ω- × 10-3 ev (relative) |
|---|---|---|---|---|---|
| CYS (-1) | -CH2S- | 382 | 146 (1.00) | 214 (1.00) | 266 (1.00) |
| LYS (0) | -(CH2)4NH2 | 285 | 56.6 (0.39) | 92.7 (0.43) | 126 (0.47) |
| HIS (0) |
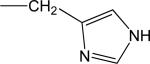
|
313 | 48.5 (0.33) | 82.2 (0.38) | 114 (0.43) |
| CYS (0) | -CH2SH | 282 | 40.0 (0.27) | 69.8 (0.33) | 98.4 (0.37) |
| LYS (+1) | -(CH2)4NH3+ | 271 | 35.3 (0.24) | 62.9 (0.29) | 90.0 (0.34) |
HSAB parameters were calculated as described in the Materials and Methods section. For each nucleophile, parameters were calculated based on selected ionization-states (in parentheses). Data show that the sulfhydryl thiolate-state is a significantly softer (σ) nucleophile than either the corresponding thiol state or the other amino acid residues; i.e., histidine or lysine. This characteristic indicates that the thiolate-state will react selectively with comparably soft electrophiles such as acrolein (see Table 2). The nucleophilic index (ω-), which reflects the propensity of adduct formation, indicates that the sulfhydryl thiolate-state is the preferential target of the type-2 alkenes. Relative to the thiolate state (1.00), thiol all and the lysine and histidine residues are substantially less competitive targets for type-2 alkene adduct formation (mean relative value = 0.35).
The combined HSAB parameters provide a detailed molecular description of adduct formation and thereby complement information derived from initial kinetic and proteomic studies; i.e., the type-2 alkenes are soft electrophiles of predictably varying potency that, as a group, selectively form adducts with soft, highly nucleophilic cysteine thiolate groups. In contrast, the side chain nitrogen groups of histidine (imidazole ring) and lysine (ε-amino group) are harder, less reactive nucleophiles and are, as our mass spectrometric analyses indicate (Figs. 5 and 6), unlikely sites of adduct formation.
DISCUSSION
Our findings demonstrate that ACR inhibition of GAPDH is directly related to the selective targeting of Cys152. This is consistent with a broader developing concept that ACR, acrolein and other type2 alkenes produce cytotoxicity via a common mechanism involving the relatively rapid formation of Michael adducts at specific cysteine residues (e.g., Cys280 of sirtuin 3) 5,11-18. However, in physiological conditions (pH 7.4) cysteine sulfhydryl groups (pKa 8.4) exist mostly in the non-nucleophilic thiol (0) state (Table 3) and are, therefore, kinetically unfavorable sites for adduct formation with soft type-2 alkene electrophiles. Experimental evidence5-7,9,10 and calculated HSAB (Table 3)5-7 parameters indicate that the nitrogen groups of histidine (imidazole ring) and lysine (ε-amino groups) are also kinetically unfavorable adduct targets since these are harder, relatively weak nucleophiles. In contrast, ionization of cysteine sulfhydryl groups yields the anionic thiolate (-1), which is a soft, highly nucleophilic anionic state that reacts correspondingly faster (higher ω- values) with soft type-2 alkene electrophiles (Table 3)5-7. This predilection for thiolate groups is not surprising, since sulfhydryl ionization (RSH → RS- + H+) is the first step in the well established reaction mechanism between thiols and α,β-unsaturated alkenes in aqueous solutions19. Therefore, the relatively rapid kinetics for the reaction of ACR with the highly nucleophilic thiolate is the theoretical basis of selective adduct formation at Cys152. This supposition is supported by direct experimental evidence. Specifically, based on the pH-dependency of thiolate formation (pKa = 8.3), we showed that at physiological pH, where the thiolate concentration is relatively low (<10%) , the rates of GAPDH inhibition (Figs. 2A and 2B) and cysteine adduct formation5,6 were substantially slower than at elevated pH values (8.5-8.8) where the thiolate concentrations were correspondingly higher (50-60%). When the second-order rate constants (k2) for the reactions of ACR with several cysteine analogs were corrected for the pH-specific thiolate concentrations (k)19 and subsequently compared to ω- and other quantum mechanical parameters, the high correspondence of the data indicated that the anionic (-1) sulfhydryl state was the preferred target of the type-2 alkenes5-7.
The preceding data suggest that the sulfhydryl thiolate state of Cys152 is the preferential site of adduct formation for ACR9,10,13,14. As also indicated, sulfhydryl groups exist mostly in the non-nucleophilic thiol state (0; Table 3) at intracellular pH ranges (7.0-7.4). However, highly nucleophilic sulfhydryl thiolate groups can be found in cysteine-centered catalytic triads and other microenvironments that significantly lower sidechain pKa values (reviewed in1,21). The selective binding of Cys152 at low ACR concentrations is consistent with previous in vitro studies that revealed adduction of specific protein cysteine residues by other type-2 alkene electrophiles5,14-17,38. This selectivity suggests that Cys152 and the other targeted cysteines are present within pKa-lowering microenvironments where ionization of the corresponding sulfhydryl side chains yields a higher percentage of residues in the thiolate vs thiol state39-43. The pKa values for Cys152 and other cysteine residues on human GAPDH (MMDB ID: 34532) can be calculated using the PROPKA program (http://propka.ki.ku.dk/). Corresponding calculations estimate a pKa of 6.03 for Cys152, which supports the existence of this cysteine within a low pKa microenvironment. Finally, GAPDH is an NAD-dependent enzyme26, and it is possible that cofactor binding further lowers the pKa of Cys152 through tertiary structural changes in the triad microenvironment. Our kinetic measurements might, therefore, underestimate the rates of GAPDH inhibition by the type-2 alkenes since, for methodological reasons, we did not pre-incubate this enzyme with cofactor.
Whether the adduction of a given cysteine by a type-2 alkene has toxicological relevance is dependent upon the role of the modified residue in protein function1,4,21. Thus, it is significant that cysteine-centered catalytic triads are often located within the active sites of enzymes (e.g., N-ethylmaleimide sensitive factor, vesicular H+-ATPase) where they play a role in modulating catalytic function21. Clearly, adduction of the corresponding anionic sulfhydryl group has substantial cytotoxicological implications. With specific reference to GAPDH, this enzyme is a homotetramer (150kDa), where each subunit consists of an N-terminal NAD+-binding domain (residues 1-150, 314-335) and a C-terminal catalytic domain (149-313). As indicated (Introduction), GAPDH catalyzes the conversion of G3P to D-glycerate 1,3-bisphosphate. The initial step of this reaction is dependent upon Cys152 within the GAPDH active site (Fig. 7), which mediates nucleophilic attack on the G3P carbonyl to yield a hemithioacetal intermediate. The IISNASCTTNCLAPLAK peptide identified in our study is located within the C-terminal catalytic domain of GAPDH44 and contained ACR adducts on Cys152 and Cys156. Because Cys152 plays a critical role in GAPDH enzyme function, ACR adduction of this residue likely mediates dysfunction. This conclusion is evidenced by the exclusive adduction of Cys152 at lower concentrations of ACR that produced significant inhibition of GAPDH activity (Fig. 4). Therefore, based on the catalytic role of Cys152, this residue represents the toxicologically relevant GAPDH target for ACR and other α,β-unsaturated carbonyl derivatives.
Figure 7.

The three-dimensional structure of a human GAPDH subunit active site is presented57. Purple indicates the helix structure and brown represents the beta-sheet and coil structure. Also highlighted in red are the cysteine residues (Cys152, 156 and 247) and in green, a vicinal histidine (H179). The structure was drawn using Swiss-PdbViewer software and the PDB coordinate, 1U8F, for human GAPDH was obtained from the NCBI Structure Database (MMDB ID: 34532). Analyses of respective pKa values (PROPKA program) suggested that Cys152 resides within a pKa-lowering catalytic triad, possibly involving H179 (<4Å distance; yellow dashed line) as the basic amino acid component. The resulting pKa of Cys152 is 6.0, which indicates that at physiological pH (7.4) this residue exists mostly (90%) in the highly nucleophilic thiolate-state (-1). This is in contrast to Cys156 and 247, which have significantly higher pKa values (9.6 and 11.8, respectively) and consequently exist primarily in the significantly less reactive thiol (0) state at pH 7.4 (compare ω- values in Table 3). ACR, acrolein and other α,β-unsaturated carbonyls are electrophiles of varying softness (compare ω and σ values in Table 2) that can preferentially form Michael-type adducts with the soft nucleophilic thiolate state of Cys152. This residue is contained within the active site of GAPDH, where it participates as a nucleophile in the enzymatic conversion of glyceraldehyde 3-phosphate (G3P) to D-glycerate 1,3-bisphosphate. Consequently, the formation of covalent type-2 alkene adducts at Cys152 leads to irreversible inhibition of enzyme function. In combination with our previous proteomic data5,22,35,52, the present findings suggest that this mechanism of GAPDH inhibition is a generic molecular process for α,β-unsaturated carbonyls whereby selective adduct formation at regulatory cysteine thiolate sites leads to dysfunction of numerous cellular proteins (e.g., N-ethylmaleimide sensitive factor, v-ATPase, membrane dopamine transporter). Because these thiolate targets are acceptors for NO and other modulatory electrophiles, we have proposed that toxicity induced by α,β-unsaturated carbonyl exposure is mediated by the irreversible inhibition of cellular redox signaling pathways1-4.
As a second order reaction, the formation of Michael adducts is governed not only by the relative concentrations of toxicant and protein, but also by the inherent electrophilicity (ω) of the toxicant and nucleophilic strength (ω-) of the amino acid target. Either of the latter electronic parameters can affect the energy of the transition state and hence, the magnitude of the rate constant. As we have indicated, thiolates are soft nucleophiles that undergo facile adduct-forming reactions with the soft electrophiles used in our studies. Thus, acrolein and MVK are very soft (Table 2), fast reacting (k2 = 297 M-1s-1 and 128 M-1s-1, respectively) and therefore potent toxicants. These type-2 alkenes impaired in vitro GAPDH activity at concentrations almost four orders of magnitude lower (KI = 38 μM and 60 μM) than the relatively harder and slower reacting ACR (k2 = 0.053 M-1s-1; KI = 247 mM). Also of considerable importance, our study showed that all type-2 alkenes tested inactivated GAPDH at the same maximal rate (kinact ~ 0.01sec-1; Table 1), which is comparable to rates of inactivation determined in previous studies of enzyme inhibition by unsaturated carbonyl toxicants12. Equivalent maximal rates of inhibition are expected for a family of chemical toxicants that vary in potency yet act via a common mechanism5-7. However, it is also notable that the KI concentration for ACR-induced GAPDH inhibition exceeded the predicted tissue levels for intoxicated animals45. This in vivo-in vitro difference is consistent, however, with the lower electrophilicity of ACR (Table 2). As indicated above, ACR is a weak electrophile that slowly forms adducts with cysteine residues and, therefore, higher concentrations are necessary to drive the adduct reaction during acute (≤30 mins) in vitro experiments that contain low protein concentrations (133nM)7,20. This is in contrast to the more potent electrophiles, acrolein and MVK, that rapidly formed cysteine adducts and thereby impaired GAPDH activity at much lower in vitro concentrations (Table 1).
Higher in vitro concentrations are, in fact, typical for neurotoxicants that are weak electrophiles. For example, the γ-diketone neurotoxicant, 2,5-hexanedione (HD), is a weak electrophile46 that forms adducts slowly and requires relatively high in vitro exposure conditions to impair protein function47,48 (reviewed in LoPachin and DeCaprio49). It should be emphasized that higher ACR or HD concentrations used in these in vitro studies do not invalidate the experiment or limit the in vivo relevance of the data. Instead, the underlying reactions are second order and, therefore, higher toxicant concentrations not only reflect lower electrophilicity, but also low target concentrations in the sample. With respect to in vivo toxicity, lower electrophilicity is the basis for the cumulative effects of ACR and HD, where daily exposure to relatively high dose-rates for extended durations is required; e.g., ACR – 21 mg/kg/d × 42 days; HD – 175 mg/kg/d × 88days50. Corroborative proteomic analyses of nervous tissue from ACR- or HD-intoxicated animals have shown that the formation of respective protein adducts in relevant neuronal regions (nerve terminals or axons, respectively) is cumulative and correlated to the development of neurotoxicity51,52. The preceding evidence indicates that the low electrophilicity of ACR determines both the in vitro and in vivo toxicodynamic characteristics of this toxicant. Therefore, our in vitro findings provide a toxicologically relevant description of the ACR interactions with cellular proteins7,22,34,35.
The present research provides confirmatory evidence that certain cysteine residues on proteins are specifically targeted by ACR and other α,β-unsaturated carbonyl derivatives. More importantly, our findings offer novel insight regarding the previously unexplained molecular basis of this selectivity; i.e., ACR inhibits human GAPDH activity by preferentially forming Michael-type adducts with the thiolate sulfhydryl group of Cys152 located in the enzyme active site. Identification of this specific residue target was facilitated by the use of ACR; i.e., as a weak electrophile only reactive cysteine residues were modified at lower concentrations and only Michael adducts need be considered since amides do not form Schiff bases. Based on these data, we propose the following unified molecular mechanism of enzyme inhibition by other α,β-unsaturated carbonyl derivatives. Cysteine residues that are selectively targeted by these chemicals exist in pKa-lowering microenvironments within the active sites of enzymes, where ionization of the sulfhydryl sidechain yields highly nucleophilic sulfhydryl thiolates (Fig. 7). However, adduct formation is a second order reaction and accordingly our study showed that, excluding steric hindrance and assuming uniform thiolate nucleophilicity, the rate (k2) of this reaction varied as a function of the inherent electrophilicity of individual type-2 alkenes. Sulfhydryl thiolate groups can function as “receptors” for electrophilic transmitters such as nitric oxide (NO) and hydrogen peroxide (H2O2) that reversibly inhibit protein activity53-55. Indeed, previous studies have shown that GAPDH is an NO-regulated enzyme and that nitrosylation of active site cysteine residues (Cys149 rabbit muscle isozyme) decreased corresponding activity40,41,56. Our earlier research showed that, similar to NO and H2O2, ACR and other type-2 alkene electrophiles have an inhibitory effect on NO-directed proteins; e.g., the dopamine transporter, N-ethylmaleimide sensitive factor, vesicular ATPase22,35,52. Accordingly, we hypothesize that ACR and related α,β-unsaturated carbonyl derivatives mimic the cellular effects of redox signaling by reacting with corresponding thiolate acceptors. However, unlike NO and H2O2, these type-2 alkenes form thiolate adducts irreversiblely in cellular conditions. Cytotoxicity results from the ensuing loss of reversible enzyme regulation mediated by redox signaling (reviewed in LoPachin et al.1-4).
Acknowledgments
FUNDING SUPPORT
This research was supported by a grant from the National Institute of Environmental Health Sciences to R.M.L. [R01 ES03830-24]
Abbreviations
- ACR
acrylamide
- MVK
methylvinyl ketone
- HNE
4-hydroxy-2-nonenal
- ONE
4-oxononenal
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- G3P
glyceraldehyde 3-phosphate
- LUMO
Lowest Unoccupied Molecular Orbital
- ELUMO
Lowest Unoccupied Molecular Orbital energy
- HOMO
Highest Occupied Molecularf Orbital
- EHOMO
Highest Occupied Molecular Orbital energy
- β-NAD
3-nicotinamide adenine dinucleotide
- MS/MS
tandem mass spectrometry
- HSAB
Hard-Soft Acid-Base
- HD
2,5-hexanedione
- NO
nitric oxide
REFERENCES
- 1.LoPachin RM, Barber DS, Gavin T. Molecular mechanisms of the conjugated α,β-unsaturated carbonyl derivatives: relevance to neurotoxicity and neurodegenerative diseases. Tox Sci. 2008;104:235–249. doi: 10.1093/toxsci/kfm301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LoPachin RM, Gavin T. Acrylamide-induced nerve terminal damage: relevance to neurotoxic and neurodegenerative mechanisms. J. Agri Food Chem. 2008;56:5994–6003. doi: 10.1021/jf703745t. [DOI] [PubMed] [Google Scholar]
- 3.LoPachin RM, Gavin T, Barber DS. Type-2 alkenes mediate synaptotoxicity in neurodegenerative diseases. NeuroToxicology. 2008;29:871–882. doi: 10.1016/j.neuro.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 4.LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem. Res. Toxicol. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LoPachin RM, Barber DS, Geohagen BC, Gavin T, He D, Das S. Structure-toxicity analysis of Type-2 alkenes: in vitro neurotoxicity. Tox. Sci. 2007;95:136–146. doi: 10.1093/toxsci/kfl127. [DOI] [PubMed] [Google Scholar]
- 6.LoPachin RM, Gavin T, Geohagen BC. Synaptosomal toxicity and nucleophilic targets of 4-hydroxy-2-nonenal. Tox. Sci. 2009;107:171–181. doi: 10.1093/toxsci/kfn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LoPachin RM, Gavin T, Geohagen BC, Das S. Neurotoxic mechanisms of electrophilic type-2 alkenes: soft-soft interactions described by quantum mechanical parameters. Tox. Sci. 2007;98:561–570. doi: 10.1093/toxsci/kfm127. [DOI] [PubMed] [Google Scholar]
- 8.Cai J, Bhatnagar A, Pierce WM. Protein modification by acrolein: formation and stability of cysteine adducts. Chem. Res. Toxicol. 2009;22:708–716. doi: 10.1021/tx800465m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doorn JA, Petersen DR. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxynonenal and 4-oxo-2-nonenal. Chem. Res. Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- 10.Doorn JA, Petersen DR. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem. Bio. Interact. 2003;143-144:93–100. doi: 10.1016/s0009-2797(02)00178-3. [DOI] [PubMed] [Google Scholar]
- 11.Fritz KS, Galligan JJ, Smathers RL, Roede JR, Shearn CT, Peigan P, Petersen DR. 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification . Chem. Res. Toxicol. 2011;24:651–662. doi: 10.1021/tx100355a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seiner DR, LaButti JN, Gates KS. Kinetics and mechanism of protein tyrosine phosphatase B inactivation by acrolein. Chem. Res. Toxicol. 2007;20:1315–1320. doi: 10.1021/tx700213s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalle-Donne I, Vistoli G, Gamberoni L, Giustarini D, Colmbo R, Facino RM, Rossi R, Milzani A, Aldini G. Actin. Cys374 as a nucleophilic target of α,β-unsaturated aldehydes. Free Rad. Biol. Med. 2007;42:583–598. doi: 10.1016/j.freeradbiomed.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 14.Nerland DE, Cai J, Benz FW. Selective covalent binding of acrylonitrile to Cys 186 in rat liver carbonic anhydrase III in vivo. 2003. pp. 583–589. [DOI] [PubMed]
- 15.Crabb JW, O'Neil J, Miyagi M, West K, Hoff HF. Hydroxylnonenal inactivates cathepsin B by forming Michael adducts with active site residues. Protein Sci. 2002;11:831–940. doi: 10.1110/ps.4400102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Y, Forgac M. Cysteine 254 of the 73-kDa A subunit is responsible for inhibition of the coated vesicle (H+)-ATPase upon modification of sulfhydryl reagents. J. Biol. Chem. 1992;267:5817–5822. [PubMed] [Google Scholar]
- 17.Stewart BJ, Doorn JA, Petersen DR. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 2007;20:1111–1119. doi: 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- 18.Eliuk SM, Renfrow MB, Shonsey EM, Barnes S, Kim H. Active site modifications of the brain isoform of creatine kinase by 4-hydroxy-2-nonenal correlate with reduced enzyme activity: mapping of modified sites by fourier transform-ion cyclotron resonance mass spectrometry. Chem. Res. Toxicol. 2007;20:1260–1268. doi: 10.1021/tx7000948. [DOI] [PubMed] [Google Scholar]
- 19.Friedman M, Cavins JF, Wall JS. Relative nucleophilic reactivities of amino groups and mercaptide ions in addition reactions with α,β-unsaturated compounds. J. Am. Chem. Soc. 1965;87:3672–3682. [Google Scholar]
- 20.Cavins JF, Friedman M. Specific modification of protein sulfhydryl groups with a,b-unsaturated compounds. J. Biol. Chem. 1968;243:3357–3360. [PubMed] [Google Scholar]
- 21.LoPachin RM, Barber DS. Synaptic cysteine sulfhydryl groups as targets of electrophilic neurotoxicants. Tox. Sci. 2006;94:240–255. doi: 10.1093/toxsci/kfl066. [DOI] [PubMed] [Google Scholar]
- 22.Barber DS, LoPachin RM. Proteomic analysis of acrylamide-protein adduct formation in rat brain synaptosomes. Toxicol Appl Pharmacol. 2004;201:120–136. doi: 10.1016/j.taap.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Furuhata A, Nakamura M, Osawa T, Uchida K. Thiolation of protein-bound carcinogenic aldehyde. J. Biol. Chem. 2002;277:27919–27926. doi: 10.1074/jbc.M202794200. [DOI] [PubMed] [Google Scholar]
- 24.Tanni H, Hashimoto K. Effect of acrylamide and related compounds on glycolytic enzymes of rat brain. Toxicol. Letters. 1985;26:79–84. doi: 10.1016/0378-4274(85)90188-2. [DOI] [PubMed] [Google Scholar]
- 25.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 26.Sirover MA. Role of the glycolytic protein, glyceraldehdye-3-phosphate dehydrogenase, in normal cell function and in cell pathology. J. Cell Biochem. 1997;66:133–140. [PubMed] [Google Scholar]
- 27.Bertelsen KM, Venkatakrishnan K, von Moltke LL, Obach RS, Greenblatt DJ. Apparent mechanism-based inhibition of human Cyp2D6 in vitro by paroxetine: comparison with fluoxetine and quinidine. Drug Met. Disp. 2003;31:289–293. doi: 10.1124/dmd.31.3.289. [DOI] [PubMed] [Google Scholar]
- 28.Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 29.Bruice PY. Organic Chemistry. 6th Edition 2011. p. 478.
- 30.Britto PJ, Knipling L, Wolff J. The local electrostatic environment determines cysteine reactivity of tubulin. J. Biol. Chem. 2002;277:29018–29027. doi: 10.1074/jbc.M204263200. [DOI] [PubMed] [Google Scholar]
- 31.Peskin AV, Winterbourn CC. Taurine chloramine is more selective than hypochlorous acid at targeting critical cysteines and inactivating creatine kinase and glyceraldehyde-3-phopphate dehydrogenase. Free Rad. Biol. Med. 2006;40:45–53. doi: 10.1016/j.freeradbiomed.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 32.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 33.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 34.LoPachin RM, Schwarcz AI, Gaughan CL, Mansukhani S, Das S. In vivo and in vitro effects of acrylamide on synaptosomal neurotransmitter uptake and release. NeuroToxicology. 2004;25:349–363. doi: 10.1016/S0161-813X(03)00149-9. [DOI] [PubMed] [Google Scholar]
- 35.LoPachin RM, Barber DS, He D, Das S. Acrylamide inhibits dopamine uptake in rat striatal synaptic vesicles. Tox Sci. 2006;89:224–234. doi: 10.1093/toxsci/kfj005. [DOI] [PubMed] [Google Scholar]
- 36.Chattaraj PK, Sarkar U, Roy DR. Electrophilicity index. Chem. Res. Toxicol. 2006;106:511–513. doi: 10.1021/cr040109f. [DOI] [PubMed] [Google Scholar]
- 37.Pearson RG. Hard and soft acids and bases – the evolution of a chemical concept. Coord. Chem. Rev. 1990;100:403–425. [Google Scholar]
- 38.Carbone DL, Doorn JA, Kiebler Z, Sampey BP, Petersen DR. Inhibition of Hsp72-mediated protein reflolding by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2004;17:1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brune B, Mohr S. Protein thiol modification of glyceraldehyde-3-phosphate dehydrogenase and caspase-2 by nitric oxide. Curr. Protein Pept. Sci. 2001;2:61–72. doi: 10.2174/1389203013381206. [DOI] [PubMed] [Google Scholar]
- 40.Mohr S, Stamler JS, Brune B. Posttranslational modification of glyceraldehyde-3-phosophate dehydrogenase by S-nitrosylation and subsequent NADH attachment. J. Biol. Chem. 1996;271:4209–4214. doi: 10.1074/jbc.271.8.4209. [DOI] [PubMed] [Google Scholar]
- 41.Padgett CM, Whorton AR. S-nitroglutathione reversibly inhibits GAPDH by S-nitrosylation. Am. J. Physiol. 1995;269:C739–C749. doi: 10.1152/ajpcell.1995.269.3.C739. [DOI] [PubMed] [Google Scholar]
- 42.Mercer WD, Winn SI, Watson HC. Twinning in crystals of human skeletal muscle D-glyceraldehyde-3-phosphate dehydrogenase. J. Mol. Biol. 1976;104:277–283. doi: 10.1016/0022-2836(76)90013-9. [DOI] [PubMed] [Google Scholar]
- 43.Thomas JA, Poland B, Honzatko R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch. Biochem. Biophys. 1995;319:1–9. doi: 10.1006/abbi.1995.1261. [DOI] [PubMed] [Google Scholar]
- 44.Ismail SA, Park HW. Structural analysis of human liver glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallog. D Biol. Crystallog. 2005;61:1508–1513. doi: 10.1107/S0907444905026740. [DOI] [PubMed] [Google Scholar]
- 45.Barber DS, Hunt JR, Ehrich M, Lehning EJ, LoPachin RM. Metabolism, toxicokinetics and hemoglobin adduct formation in rats following subacute and subchronic acrylamide dosing. NeuroToxicology. 2001;22:341–353. doi: 10.1016/s0161-813x(01)00024-9. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Gavin T, DeCaprio AP, LoPachin RM. γ-Diketone axonopathy: analysis of cytoskeletal motors and highways in CNS myelinated axons. Tox. Sci. 2011;117:180–189. doi: 10.1093/toxsci/kfq176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeCaprio AP, Olajos EJ, Weber P. Covalent binding of a neurotoxic n-hexane metabolite: conversion of primary amines to substituted pyrrole adducts by 2,5-hexanedione. Toxicol. Appl. Pharmacol. 1982;65:440–450. doi: 10.1016/0041-008x(82)90389-1. [DOI] [PubMed] [Google Scholar]
- 48.Graham DG, Anthony DC, Boekelheide K. In vitro and in vivo studies of the molecular pathogenesis of n-hexane neuropathy. Neurobehav.Toxicol. Teratol. 1982;4:629–634. [PubMed] [Google Scholar]
- 49.LoPachin RM, DeCaprio AP. Protein adduct formation as a molecular mechanism in neurotoxicity. Tox. Sci. 2005;86:214–225. doi: 10.1093/toxsci/kfi197. [DOI] [PubMed] [Google Scholar]
- 50.LoPachin RM, Ross JF, Reid ML, Das S, Mansukhani S, Lehning EJ. Neurological evaluation of toxic axonopathies in rats: acrylamide and 2,5-hexanedione. NeuroToxicology. 2002;23:95–110. doi: 10.1016/s0161-813x(02)00003-7. [DOI] [PubMed] [Google Scholar]
- 51.Karlsson J-E, Rosengren LE, Haglid KG. Quantitative and qualitative alterations of neuronal and glial intermediate filaments in rat nervous system after exposure to 2,5-hexanedione. J. Neurochem. 1991;57:1437–1444. doi: 10.1111/j.1471-4159.1991.tb08311.x. [DOI] [PubMed] [Google Scholar]
- 52.Barber DS, Stevens S, LoPachin RM. Proteomic analysis of rat striatal synaptosomes during acrylamide intoxication at a low dose-rate. Toxicol. Sci. 2007;100:156–167. doi: 10.1093/toxsci/kfm210. [DOI] [PubMed] [Google Scholar]
- 53.Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- 54.Hess DT, Matsumoto A, Kim S-O, Marshal HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 55.Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 56.Mohr S, Stamler JS, Brune B. Mechanism of covalent modification of glyceraldehyde-3-phosphate dehydrogenase at its active site thiol by nitric oxide, peroxynitrite and related nitrosating agents. FEBS Letters. 1994;348:223–227. doi: 10.1016/0014-5793(94)00596-6. [DOI] [PubMed] [Google Scholar]
- 57.Meyer-Siegler K, Mauro DJ, Seal G, Wurez J, deRiel JK, Sirover MA. A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc. Acad. Sci. 1991;88:8460–8464. doi: 10.1073/pnas.88.19.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]