

Conspectus
RNA represents a prominent class of biomolecules present in all living systems that plays many essential roles in gene expression, regulation, and development. Accordingly, many biological processes depend on the accurate enzymatic processing, modification, and cleavage of RNA. Understanding the catalytic mechanisms of these enzymes therefore represents an important goal in defining living systems at the molecular level. In this context, RNA molecules bearing 3′- or 5′-S-phosphorothiolate linkages comprise what are arguably among the most incisive mechanistic probes available. They have been instrumental in showing that RNA splicing systems are metalloenzymes and in mapping the ligands that reside within RNA active sites. These models have in turn verified the functional relevance of crystal structures. In other cases, phosphorothiolates have offered mechanistic enzymologists an experimental strategy to circumvent the classic problem of kinetic ambiguity and assign precise roles to catalytic groups as general acids or bases. These insights into macromolecular function are enabled by the synthesis of nucleic acids bearing phosphorothiolate linkages and the unique chemical properties they impart.
Here, the synthesis, properties, and applications of oligonucleotides and oligodeoxynucleotides containing an RNA dinucleotide phosphorothiolate linkage are reviewed. Phosphorothioate substitutions, in which sulfur replaces one or both non-bridging oxygens within a phosphodiester linkage, are now widely available and are used routinely in numerous biochemical and medicinal applications. In contrast, phosphorothiolate oligonucleotides, in which sulfur replaces a specific 5′ or 3′ bridging oxygen, have presented a more difficult synthetic challenge, requiring chemical alterations to the attached sugar moiety. In this review, we outline the synthetic strategies used to access these RNA analogues and summarize their responses to chemical and enzymatic cleavage agents, as well as mechanistic insights their use has engendered.
1. Introduction
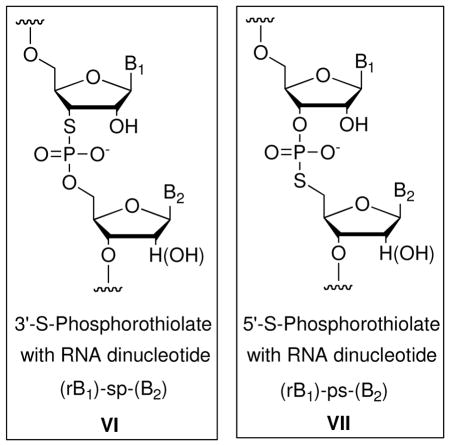
Nucleic acid analogs that resemble naturally occurring DNA and RNA are widely employed in biochemical research and medicine. In particular, oxygen-to-sulfur substitutions within the phosphodiester backbone (Figure 1) have received much attention. Phosphorothioate oligonucleotides (I – III), in which sulfur replaces one or both of the non-bridging phosphodiester oxygens within a linkage, are used widely in fields ranging from enzymology1 to therapeutics.2 Recently, the modification has also been shown to occur naturally in bacterial genomic DNA,3 where it appears to impart greater chemical stability. Synthetic phosphorothioate linkages are introduced readily via a sulfurization step that can be programmed into automated solid-phase oligonucleotide synthesizers.4 Phosphorothiolate linkages, in which sulfur replaces the 3′ or 5′ bridging oxygen connected to furanose, pose a more difficult synthetic challenge. Nevertheless, they also represent important synthetic targets for biochemical and potential therapeutic applications. Phosphorothiolate oligonucleotides have been indispensable for studying the mechanisms of RNA and protein enzymes that chemically manipulate nucleic acids. In addition, the effects of phosphorothiolates on oligonucleotide sugar conformation make them ideal modifications for fine-tuning the potency of antisense DNA and small interfering RNAs (siRNAs). The synthesis and applications of oligodeoxynucleotide (ODN) phosphorothiolates (IV and V) have been reviewed elsewhere.5 This review focuses specifically on the synthesis, properties, and applications of oligoribonucleotides (ORN) and ODN containing an RNA dinucleotide phosphorothiolate linkage (VI and VII).
Figure 1.

Structures of oligonucleotides containing bridging or non-bridging sulfur atoms.
2. Synthesis
Perhaps inspired by Eckstein’s work demonstrating the broad utility of phosphorothioates for biochemical investigations of nucleic acids, Reese and colleagues pioneered the synthesis of 3′-S- and 5′-S-phosphorothiolate (3′- and 5′-PS, respectively) RNA dinucleotides.6,7 In the context of DNA, Cosstick and colleagues made an important advance by adapting phosphoramidite chemistry to the construction of 3′-PS linkages.8 Figure 2 summarizes the relevant synthetic approaches. 3′- or 5′-PS dinucleotides have been prepared by Arbusov reactions of 3′ or 5′ disulfides with the appropriate nucleoside H-phosphonate salt. 5′-PS dinucleotides have also been prepared by SN2 reactions of 3′-phosporothioates with 5′-iodoribonucleosides. Solid-phase strategies have employed 3′-S-thioribonucleoside phosphoramidites or 5′-S-tritylthio-2′-deoxyribonucleoside phosphoramidites. Enzymatic ligation methods have also enabled construction of 5′-PS oligonucleotides.
Figure 2.

Summary of approaches used for the preparation of dinucleotides or oligonucleotides containing phosphorothiolate linkages.
2.1 Oligonucleotides containing an RNA 3′-PS linkage
2.1.1 RNA dinucleotides r(B1-sp-B2) via Arbusov reaction
Liu and Reese7,9 synthesized the RNA dinucleotide r(UspU) (3) via Arbusov reaction of disulfide 1 and 5′-H-phosphonate 2 (Scheme 1), both of which were prepared from uridine in nine and four steps, respectively. Weinstein et al.10 synthesized r(IspU) (6) in 9% yield via Arbusov reaction of inosine disulfide 4, prepared from 3′-β-iodoxanthosine in 21% overall yield (four steps), and the 5′-H-phosphonate 5, prepared from 2′,3′-di-O-TBS-uridine in 65% yield.
Scheme 1.

Synthesis of dinucleotides r(UspU) (3) and r(IspU) (6).
2.1.2 Oligonucleotides containing an RNA dinucleotide 3′-PS linkage via phosphoramidite chemistry
Sun et al.11 prepared the 2′-O-TBS-3′-S-phosphoramidites 8a–d (U, CBz, GiBu, I) from the corresponding 3′-S-thiol-nucleosides 7a–d in high yields (Scheme 2). The 3′-S-thiol-nucleoside derivatives 7a–c were prepared by nucleobase glycosylation using a 3-thioribofuranosylation agent. The inosine derivative 7d was prepared from a 2′,3′-epoxide inosine derivative in eight steps (8% overall yield). 8c (Scheme 2) could also be prepared in eleven steps starting from a guanosine derivative.12 This approach gave a higher overall yield of 8c (12.5%)12 compared to the glycosylation approach (11 steps, 1.1%).11 Additionally, Lu et al.13 used an analogous strategy to prepare 2′-O-methyl-3′-S-thioguanosine phosphoramidite 11 (Scheme 2) in eight steps starting from N2-isobutyryl-2′-O-methylguanosine (10) with 10.4% overall yield.
Scheme 2.

Synthesis of 3′-S-thioribonucleoside phosphoramidites 8a–d and 2′-O-methyl-3′-S-thioguanosine phosphoramidite 11.
The 3′-S-thionucleoside phosphoramidites (8a–d and 11) are incorporated into RNAs or DNAs via solid-phase synthesis. Although coupling of 2′-deoxy-3′-S-thiopyrimidine phosphoramidites can be accomplished via a fully automated protocol,14 3′-S-thioribonucleoside phosphoramidites 8a–d and 11 are less reactive and require manual coupling (using p-nitrophenyltetrazole as an activator).11,13 A fully automated protocol for solid-phase oligonucleotide synthesis that incorporates 3′-S-thioribonucleoside phosphoramidites has not been reported.
2.2 Oligonucleotides containing an RNA 5′-PS linkage
2.2.1 RNA dinucleotides r(B1-ps-B2) via Arbusov reaction or SN2 reaction
Liu and Reese6 used the Arbusov reaction to synthesize r(UpsU) (14) from 2′-O-THP-5′-O-(9-phenylxanthen-9-yl)-uridine 3′-H-phosphonate (13) and the disulfide 12 (Scheme 3).
Scheme 3.

Synthesis of dinucleotide r(UpsU) 14 via Arbusov reaction.
Thomson et al.15 synthesized 14 and its 2′-amino analogue 17 using the commercially available phosphoramidites 15a and 15b (Glenn Research) (Scheme 4). Reaction with 3-hydroxypropionitrile in the presence of tetrazole followed by sulfurization and deprotection gave the nucleoside 3′-phosphorothioates 16a and 16b. Nucleophilic displacement of 5′-iodo-5′-deoxyuridine by 16a and 16b, followed by desilylation with triethylamine trihydrofluoride, gave r(UpsU) (14) and the 2′-amino dinucleotide U2′-NH2 - ps -rU 17, respectively.
Scheme 4.

Synthesis of r(UpsU) (14) and U2′-NH2 - ps - rU (17) via phosphoramidite chemistry.
2.2.2 Oligonucleotides containing an RNA dinucleotide 5′-PS linkage
In 1962, Michelson16 prepared ORNs containing multiple 5′-PS linkages (19) via polymerization of 5′-S-thiouridine-2′,3′-cyclic phosphate (18) (Scheme 5). The 2′,3′-cyclic phosphate 18 was prepared from uridine-2′,3′-cyclic phosphate in four steps.
Scheme 5.

Synthesis of poly 5′-S-thiouridylyl phosphate (19).
Kuimelis and McLaughlin,17 using phosphoramidite-based solid-phase methods, synthesized an ODN containing cytidine linked via a 5′-PS to a subsequent adenosine deoxynucleotide (rCps-dA). The 5′-S-thioadenosine 3′-O-phosphoramidite 21 was synthesized in 46% overall yield starting from the 2′-deoxyadenosine derivative 20 (Scheme 6). Sproat et al.18 have also synthesized the 5′-S-thioadenosine 3′-O-phosphoramidite 23 from 2′-deoxyadenosine derivative 22 in six steps with 21% overall yield (Scheme 6).
Scheme 6.

Synthesis of 5′-S-thioadenosine 3′-O-phosphoramidites 21 and 23.
Kuimelis and McLaughlin17 used 21 and the 2′-O-Cee-cytidine 3′-phosphoramidite 24 to prepare via solid-phase synthesis an ODN containing the RNA dinucleotide 5′-S-phosphorothiolate rC-ps-dA (Figure 3). After coupling, oxidizing, and capping 21, the 5′ terminal thiol was deprotected manually before coupling 24. Following synthesis and release from the support, the oligonucleotide was isolated and purified by ion exchange HPLC in low yield. The 8-mer fragment d(ACGGTCT)rC and the disulfide [5′-S-d(ACGAGC)]2 were detected and likely formed from cleavage of the full-length product. The oligonucleotide was subsequently used to investigate the mechanism of the hammerhead ribozyme reaction.19
Figure 3.

Solid-phase synthesis of d(ACGGTCT)rC-ps-d(ACGAGC), a substrate for the hammerhead ribozyme reaction.
Using solid-phase methods, Das and Piccirilli20 synthesized in low yield a hepatitis delta virus (HDV) ribozyme substrate containing rC2′-O-NBn-ps-dG (Figure 4). The photosensitive 2′-O-o-nitrobenzyl group21 on cytidine protects the base-labile 5′-PS from premature cleavage by the adjacent 2′-OH. The 2′-O-o-nitrobenzylcytidine 3′-phosphoramidite 25 and the 5′-triphenylthio-2′-deoxyguanosine 3′-phosphoramidite 26a were incorporated into the oligonucleotide according to the protocol developed by Kuimelis and McLaughlin.17 The phenoxyacetyl groups protecting the exocyclic amines of 25 and 26a permitted nucleobase deprotection of the oligonucleotide under mild conditions. The corresponding all-RNA substrate r(UUC2′-O-NBn-ps-GGGUCGGC) has also been prepared in low yield via solid-phase synthesis using phosphoramidites 25 and 26b.
Figure 4.

Solid-phase synthesis of r[UUC2′-O-NBn-ps-(dG)GGUCGGC] and r(UUC2′-O-NBn-ps-GGGUCGGC), substrates for the HDV ribozyme reaction.
To investigate general acid catalysis by the hammerhead ribozyme, Thomas and Perrin22 used enzymatic ligation to construct an ODN substrate containing rCps-dC at the cleavage site (Figure 5). The bridging thiophosphate at the 5′-end of oligo-2 was installed by iodination of the protected, support-bound ODN, followed by treatment with aqueous Na3SPO3. Deprotection, gel purification, and desalting gave the 5′-thiophosphorylated oligo-2 in low yield (~10%). To produce the full-length substrate, 5′-32P-labeled oligo-1 and 5′-thiophosphorylated oligo-2 were annealed to a biotinylated DNA template and joined using T4 DNA ligase (Figure 5). The resulting biotinylated duplex was bound to streptavidin-tagged magnetic particles. Following removal of unreacted oligonucleotides, the 5′-S-linked substrate was liberated from the particle-bound DNA template under mild denaturing conditions.
Figure 5.

Construction of an ODN containing rCps-dC via enzymatic ligation.
Recently, we developed a general semisynthetic strategy to obtain ORNs containing a protected 5′-PS linkage reliably and relatively efficiently.23 The approach begins with chemical synthesis of 5′-PS RNA dinucleotide 29, whose adjacent 2′-hydroxyl group is protected as the photolabile 2′-O-o-nitrobenzyl derivative21 (Scheme 7). Following enzymatic phosphorylation of the 5′-hydroxyl group of 29 to give 30 (Scheme 8), enzymatic ligation to 5′ and 3′ flanking RNAs yields the full-length ORN 34, a modified substrate for the VS ribozyme reaction.24
Scheme 7.

Synthesis of dinucleotide r(G2′-O-NBn-ps-A) 29.
Scheme 8.

Ligation scheme for constructing the 29-nucleotide VS ribozyme substrate 34.
3. Properties
3.1 Oligonucleotides containing a 3′-PS linkage
3′-PS linkages endow nucleic acids with unique chemical properties, summarized in Figure 6 for r(IspU) (6) and r(UspU) (3).9,10 Incubation with T4 polynucleotide kinase and [γ-32P]ATP readily converts r(IspU) to p*r(IspU),10 but with ~100-fold slower rate compared to r(IpU). Silver ion induces cleavage of p*IspU to give p*-I3′-SH and uridine 5′-monophosphate. Iodine cleaves the P-S bond of p*IspU to yield several products. p*r(IspU) undergoes base-catalyzed cleavage ~2000-fold faster than does p*r(IpU) over pH 10–14 (10 °C, I = 1.0 M). The observed rate acceleration likely reflects both increased delocalization of the negative charge of the dianionic transition state onto the more polarizable sulfur atom, and reduced ring strain due to the longer P-S bond. Similarly, r(UspU) undergoes hydrolysis at pH 10.06 (50 °C, t1/2 = 25 min) ~200-fold faster than does r(UpU).7,9 In glacial acetic acid solution at 30 °C, r(UspU) decomposes more than an order of magnitude more rapidly (t1/2 = 4 min) than does r(UpU) (t1/2 = 60 min). Both r(IspU) and r(UspU) are resistant to cleavage by nuclease P1 and bovine spleen phosphodiesterase. Sun et al.11 found that the 3′-PS linkage of 5′-(C2′-OMe)3UCUspA-3′ resists cleavage by S1 nuclease. Consistent with the reactivities observed for r(IspU) and r(UspU), the UspA linkage cleaves in the presence of sodium hydroxide, iodine, or silver ion.
Figure 6.

Summary of the unique properties of r(B1)-sp-r(B2).
Elzagheid et al.25,26 studied the reaction kinetics for intramolecular 2′-O-transphosphorylation of r(IspU) (6) over a wide pH range (Scheme 9). Figure 7 summarizes the pH-dependent first-order rate constants for cleavage (k1) and isomerization (k2) of r(IspU) and r(UpU). In the base-catalyzed reaction (Route A) at pH 8.3, r(IspU) degrades ~350-fold faster than r(UpU). The pH-dependent isomerization of r(IspU) (Route B) between pH 3 and 6 occurs 50-fold faster than the corresponding reaction of r(UpU). Below pH 2, r(IspU) undergoes both acid-catalyzed cleavage and isomerization with a total reactivity close to that of r(UpU). Under alkaline conditions, r(IspU) undergoes base-catalyzed cleavage exclusively (Route A), forming uridine and inosine 2′-O,3′-S-cyclic phosphorothiolate 36, which subsequently hydrolyzes to 3′-S-phosphorothiolate 37a exclusively. Below pH 7, isomerization (Route B) competes with cleavage (Route A) and predominates at pH values between 4 and 6. The isomerization reaction generates the thiol 35, which subsequently oxidizes to the disulfide. Below pH 3, acid-catalyzed depurination of inosine (Route C) competes with hydrolysis and isomerization. Acid-catalyzed hydrolysis yields 3′-S-thioinosine 2′-O-phosphate (37b), presumably via formation of the intermediate 2′-O,3′-S-cyclic phosphorothiolate 36, although accumulation of 36 could be verified by HPLC only during alkaline hydrolysis. The reasons underlying the distinct regioselective hydrolysis reactions of 36 under acidic and basic conditions remain unknown.
Scheme 9.

Figure 7.

The pH-dependent first-order rate constants for cleavage (k1, left) and isomerization (k2, right) of r(UpU) (red dots) and r(IspU) (blue dots) at 90 °C (0.1 M NaCl).26,27
3.2 Oligonucleotides containing a 5′-PS linkage
Like 3′-PS linkages, 5′-PS linkages undergo cleavage specifically at the phosphorothiolate linkage in the presence of silver ion.20,23 However, in contrast to RNA 3′-PS linkages, RNA 5′-PS linkages resist cleavage by snake venom phosphodiesterase.6
RNA 5′-PS linkages also undergo 2′-O-transphosphorylation reactions via cleavage and isomerization pathways (Scheme 10). Liu and Reese6 reported that hydrolysis of r(UpsU) (14) resembles that of r(UpU) in acidic conditions (Scheme 10). At pH 1.0 and 30 °C, the half-life of 14 is estimated to be ~35 h. After various time intervals, the constituents of the reaction mixture include starting material (14), 5′-S-thiouridine (38) and its disulfide, uridine 2′,3′-cyclic phosphate (39) (trace), uridine 3′-phosphate (40a), uridine 2′-phosphate (40b), and uridylyl-(2′→5′)-(5′-S-thiouridine) (41). Above pH 2, cleavage dominates over isomerization due to the superior leaving ability of sulfur over oxygen. This “hyperactivation” of the P-S bond manifests more fully under neutral to basic conditions, where r(UpsU) undergoes cleavage four orders of magnitude faster than does r(UpU) at pH 9.0 and 30 °C.6 Sund and Chattopadhyaya28 also report that a partially protected r(ApsU) dinucleotide decomposes to adenosine 2′,3′-cyclic phosphate in 58–75% yield when exposed to silica gel (pH ~5).
Scheme 10.

Pathways for cleavage and isomerization of r(UpsU) (14) under aqueous conditions.6
3.3 Transition state structures from model compounds
Studies employing phosphorothiolates assume that they react similarly to phosphate diesters with respect to charge transfer and transition state structure. Experiments show that this is largely the case for intermolecular reactions with nucleophiles, although important differences exist. Brønsted analyses suggest that aryl-substituted phosphate and phosphorothiolate diesters react with pyridine nucleophiles via concerted, dissociative transition states involving similar charge accumulations on both the nucleophile and the phenolate leaving group.29 However, in the corresponding intermolecular nucleophilic displacement reactions involving nucleic acids, a ribofuranosylthiolate leaving group likely would give rise to an even more dissociative transition state than the corresponding ribofuranosylalkoxide leaving group, based on the larger pKa difference between ethanol and ethanethiol (~5 units) compared to that between phenol and thiophenol (~ 0.9 units) leaving groups. In addition, the larger volume of sulfur and the longer P-S bond likely contribute to a more open transition state for a phosphorothiolate diester compared to a phosphate diester.
RNA strand scission via 2′-O-transphosphorylation can occur via a concerted pathway or a stepwise pathway involving a phosphorane intermediate. Kinetic isotope effects (KIE) provide a highly sensitive measure of bonding changes that occur as reactants proceed from ground state to rate-limiting transition state. While KIEs by themselves generally do not reveal unambiguously whether the transition state forms via concerted or stepwise pathways, they help to define the transition state structures of phosphates and phosphorothiolates undergoing 2′-O-transphosphorylation. KIE analysis of the nucleophile and leaving group for alkaline cleavage of r(UpG) (Figure 8) suggests a late rate-limiting transition state in which both formation of the P-O bond to the nucleophile and fission of the P-O bond to the leaving group are far advanced.30
Figure 8.

Model compounds30–32 simulating 2′-O-transphosphorylation, with measured nucleophile and leaving group kinetic isotope effects (ND, not determined).
For alkaline cleavage of phosphorothiolates, a different picture emerges from reactions of model phosphorothiolate diesters that simulate nucleic acid 2′-O-transphosphorylation.31 Attack of the hydroxyl group of S-3′mNB (Figure 8) on phosphorus mimics the reaction of 6 to form 36 (Scheme 9), while the analogous reaction of S-5′mNB mimics the reaction of 14 to form 39 (Scheme 10). The similar, large leaving group KIEs for r(UpG) and U-3′mNB suggest that m-nitrobenzylalcohol provides a reasonable mimic of the nucleoside leaving group. Compared to r(UpG), S-3′mNB shows a relatively large normal nucleophile KIE (18knuc = 1.1188, not corrected for equilibrium isotope effects) and a relatively small leaving group KIE (18klg = 1.0118), consistent with an earlier transition state with minimal bonding changes to both the nucleophile and leaving group. In contrast, S-5′mNB shows a less normal nucleophile KIE (18knuc = 1.0245) than S-3′mNB, and an exceedingly small leaving group KIE (34klg = 1.0009) in comparison to known KIEs for sulfuryl transfer reactions. Reaction of S-5′mNB therefore proceeds through a transition state having a larger degree of bonding to the oxygen nucleophile than S-3′mNB, but with minimal bonding changes to the leaving group. Mechanistic inferences derived from experiments using 3′-S- and 5′-S-phosphorothiolates must take into account these differences in transition state structure, as well as potential differences in the reaction pathway (concerted versus stepwise), between modified and unmodified substrates.
4. Applications of oligonucleotides containing phosphorothiolate linkages
4.1 3′-S-phosphorothiolates
As probes of metal ion-dependent phosphotransesterification
Compared to oxygen, sulfur does not interact strongly with magnesium, the key metal ion cofactor for numerous RNA and protein enzymes involved in phosphotransesterification. In the context of an inhibitory oxygen-to-sulfur substitution, restoration (“rescue”) of enzymatic activity upon the addition of a relatively more thiophilic metal ion (Mn2+, Cd2+, or Zn2+) constitutes strong evidence for a functional metal ion-ligand interaction. Because the 3′ oxygen of DNA and RNA often acts as a nucleophile or leaving group, 3′-PS linkages are especially attractive substitutions for identifying catalytic metal ions via rescue experiments. 3′-PS substrates were instrumental in demonstrating that metal ion-leaving group,33 metal ion-nucleophile,34 and metal ion-nonbridging oxygen interactions35,36 occur during catalysis by the group I intron. Metal ion rescue of these substrates formed part of the basis for subsequent investigations37–39 that produced the current model of the group I intron transition state (Figure 9). X-ray crystal structures40–42 of various group I introns are largely concordant with the biochemical data, although discrepancies remain regarding the number of metal ions within the active site. Similar analyses have implicated specific metal ion-ligand contacts as important for catalysis by the group II intron,43 the spliceosome,44 Klenow exonuclease,45 and human topoisomerase IIα.46
Figure 9.

Model of the group I intron transition state showing metal ion-ligand interactions. Hashed lines represent hydrogen bonds, black dotted lines represent experimentally confirmed metal ion interactions, purple and blue dotted lines represent metal ion interactions still under investigation, and dashed lines represent the bonds broken and formed during the reaction. MA, MB, and MC represent the three distinct metal ions implicated from functional data. MB is not observed in the crystal structures of group I ribozymes, and it has been proposed, based on spatial proximity, that MC forms an additional interaction with the 3′ oxygen of the nucleophilic guanosine in the transition state.
As probes for rate-limiting chemistry
In some instances, the enhanced reactivity of 3′-PS diesters may provide a signature to determine whether the chemical step limits the rate of an enzymatic reaction. As an example, under optimal conditions, the ribotoxin restrictocin cleaves a specific phosphodiester bond of a 27-nt sarcin-ricin loop mimic at a rate approaching the diffusion-limit maximum.47 Substitution of the scissile phosphate with a 3′-PS increases the reaction rate to a fully diffusion-limited rate, suggesting that the chemical step limits the reaction rate for the unmodified substrate.
As stabilizers of nucleic acid structures
The 3′-PS linkage strongly influences the conformation of the attached sugar moiety. In particular, 3′-S-phosphorothiolates reinforce the “north” conformation (Figure 10) not only for their own sugar, but also (to a lesser extent) for the sugar of the subsequent nucleotide. This transmission is thought to be caused by favorable stacking of the heterocyclic bases.48
Figure 10.

Equilibrium between C3′-endo and C2′-endo conformations in RNA.
The preference of 3′-PS sugars for the north conformation has sparked interest in these substitutions as stabilizers of A-form helical geometry for biochemical and therapeutic applications. For example, by forcing antisense DNA to adopt an RNA-like conformation, 3′-S-phosphorothiolates increase the Tm of DNA:RNA duplexes by about 1–2° C per modification.49 They are also potential modulators of transcriptional repression induced by siRNAs. RNA interference requires A-form double helical RNA with characteristic 3′ overhangs, of which one strand guides subsequent transcriptional repression by the RNAi-induced silencing complex (RISC).50 Experiments have established that the strand whose 5′ end is less tightly bound within the RNA:RNA duplex ultimately becomes the guide sequence. This observation has prompted investigations into whether 3′-S-phosphorothiolates could increase the efficacy of RNA silencing by selectively stabilizing one of the two 5′ ends of an siRNA. Methods for synthesizing oligonucleotides containing multiple 2′-deoxy-3′-S-thiouridine residues have already been published51,52 (the RNA interference apparatus tolerates some 2′-deoxy substitutions, which stabilize the 3′-PS linkage against endonucleolytic cleavage). The ability of such oligonucleotides to function as siRNAs still awaits testing.
4.2 5′-S-phosphorothiolates
As probes of reaction mechanism
RNA 5′-PS linkages are less dependent on acid-catalyzed hydrolysis than native linkages, since the 5′-sulfur anion is a good leaving group. This unique property makes them ideal substrates for studying the mechanisms of ribozymes that cleave phosphodiester bonds via acid-base catalysis (Figure 11). These “nucleolytic” ribozymes, which include the hammerhead, hairpin, HDV, VS, and glmS ribozymes, produce 2′,3′-cyclic phosphate and 5′-OH products, and do not require divalent metal ions for activity.53 Instead, they appear to utilize a general acid-base mechanism involving specific nucleobases and metal ion-bound water molecules. In the case of the HDV ribozyme, evidence pointed to the involvement of a key cytosine nucleobase in the reaction mechanism (C76). Substitution of this cytosine with uracil (C76U) abolished catalytic activity, which could be restored by the addition of imidazole,54 whose pKa lies near that of cytosine. However, the precise role of cytosine in the mechanism could not be determined unequivocally. The available data could be explained equally well by models in which the cytosine acted as either the general acid or as the general base.
Figure 11.

Mechanism of the general acid-catalyzed ribozyme (HDV and VS) reactions. A 5′-sulfur substitution at the cleavage site provides a ‘hyperactivated’ substrate that is less dependent on leaving group protonation.
To resolve this kinetic ambiguity, Das and Piccirilli20 synthesized an HDV substrate with a cleavage site 5′-PS linkage. Because cleavage of this linkage is less dependent on the presence of a general acid (possibly because the P-S bond is largely intact in the transition state, as suggested by kinetic isotope effect analysis31 of model compounds), mutations of a putative general acid in the ribozyme would not be expected to affect the cleavage rate of the modified substrate. To prevent premature hydrolysis of the extremely base-labile 5′-PS, the adjacent 2′-OH was protected with an o-nitrobenzyl group,21 which was removed by UV light exposure prior to experiments. The sulfur-modified substrate suppressed the catalytic defect of the C76U HDV ribozyme, as well as defects associated with atomic substitutions of C76 such as 3-deazacytosine. These results supported a model in which the N3 imino nitrogen of C76 functions as the general acid in the HDV ribozyme mechanism. Subsequent analyses using Raman spectroscopy and X-ray crystallography have supported this model.
Recently, Wilson et al. used substrates with cleavage site 5′-PS linkages to reveal the role of catalytic nucleobases responsible for general acid-base catalysis by the VS ribozyme.24 Previous work had implicated residues A756 and G638 as likely candidates for the general acid-base pair. However, as with the HDV ribozyme, the precise roles of the nucleobases could not be assigned based solely on pH-rate profile data. Accordingly, Wilson et al. synthesized a 29-nucleotide RNA substrate containing a 5′-PS linkage at the scissile phosphate (Scheme 8). The 2′-OH group was protected with an o-nitrobenzyl group to prevent premature cleavage, similarly to the HDV substrate. Again, because the thio-modified substrate does not require a general acid for activation, mutation of the ribozyme’s putative general acid would not be expected to affect reactivity. In fact, the thio-modified substrate restored the activity of the mutant A756G VS ribozyme, which cleaves the all-oxygen substrate 10,000-fold more slowly than the wild-type ribozyme. This result provided strong evidence that A756 functioned as the general acid in the wild-type reaction. Furthermore, the pH dependences of both the wild-type and A756G ribozyme reaction rates with the thio-modified substrate were linear up to pH 6.5, implying a pKa of at least 7 for the general base. Since the pKa of A756 had been previously determined55 to be 5.2, this new finding excluded A756 as the general base, leaving G638 as the most probable alternative.
5. Limitations, conclusions and outlook
Despite the demonstrated value of 3′-S- and 5′-S-phosphorothiolates in elucidating the mechanisms of enzymatic and non-enzymatic phosphoryl transfer reactions, their application poses significant practical challenges for the user. ORNs containing these linkages remain difficult to acquire and manipulate. As described herein, chemists have succeeded in constructing the desired ORNs, but in comparison to unmodified ORNs, their synthesis generally requires much more effort and yields significantly less oligonucleotide. Anecdotally, we tried unsuccessfully for more than one year to prepare a 5′-S-modified VS ribozyme substrate using solid-phase synthetic approaches and ultimately resorted to a semisynthetic approach involving enzymatic ligations. Once in hand, 3′- and 5′-PS oligonucleotides demand special precautions during experiments to avoid cleavage at the modified linkages due to their greater lability. Finally, researchers who use the relative reactivity of S- versus O-modified substrates as a strategy to infer mechanistic features operative in protein and RNA enzymes must consider the data in the context of possible differences in transition state structure and reaction pathway induced by sulfur substitution.
Phosphorothiolate oligonucleotides are important for biochemical and potential therapeutic applications that range from enzymology to RNAi. Both chemical and enzymatic approaches for preparing these oligonucleotides have improved greatly over the nearly 50 years since the linkages were first described. However, significant synthetic challenges remain, notably the lack of an efficient, fully automated solid-phase protocol for synthesizing all-RNA 5′-S-phosphorothiolates. Ongoing research should continue to expand the repertoire of available synthetic strategies, reaction pathways, and applications for these unique modified oligonucleotides.
Acknowledgments
This work was supported by an N.I.H. grant to J.A.P. (AI081987).
Biographies
Nan-Sheng Li received a bachelor’s degree in chemistry (1987) from Jiangxi Normal University (Nanchang, China) and a Ph.D. in organic chemistry (1993) under the supervision of Profs. Min-Zhi Deng and Yao-Zeng Huang of the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. From 1994 to 1998, he worked with Prof. George W. Kabalka at the University of Tennessee as a postdoctoral research associate. In 1999, he joined Prof. Joseph A. Piccirilli’s laboratory as a research specialist in the Howard Hughes Medical Institute. Since 2009, he has been a research professor in the Department of Biochemistry and Molecular Biology at the University of Chicago. His current research focuses on the chemical and enzymatic synthesis of modified nucleic acids for mechanistic studies of ribozyme-catalyzed reactions.
John K. Frederiksen received a bachelor’s degree in chemistry (1998) from Stanford University. He received both an M.D. (2009) and a Ph.D. (2007) from the University of Chicago. He completed his thesis work in the Department of Biochemistry and Molecular Biology under the supervision of Prof. Piccirilli. He is now a resident in the Department of Pathology and Laboratory Medicine at the University of Rochester Medical Center. His research interests include hematopathology, catalytic RNA, and miRNAs in health and disease.
Joseph A. Piccirilli received a bachelor’s degree in chemistry (1982) from the University of Scranton and a Ph.D. in chemistry (1989) from Harvard University under the supervision of Prof. Steven A. Benner. From 1989 to 1993, he was a Howard Hughes postdoctoral research fellow in Prof. Thomas R. Cech’s laboratory at the University of Colorado, Boulder. He received Rheinisch Westfalische Technische Hochschule Aachen, Fulbright Scholar in 1983 and was a Harvard Traveling Scholar at the Swiss Federal Institute of Technology from 1986–1989. He was appointed as an assistant professor in the departments of Biochemistry and Molecular Biology, and Chemistry, at the University of Chicago in 1993. He became an assistant investigator in the Howard Hughes Medical Institute in 1994, and in 2000 was appointed as an associate professor at the University of Chicago and associate Investigator in the Howard Hughes Medical Institute. In 2004, he became a full Investigator in the Howard Hughes Medical Institute. His current research focuses on chemical and enzymatic cleavage and modification of RNA, and the mechanisms of RNA catalysis and RNA-protein interactions.
References
- 1.Eckstein F. Nucleoside phosphorothioates. Annu Rev Biochem. 1985;54:367–402. doi: 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- 2.Agrawal S, Zhao Q. Antisense therapeutics. Curr Opin Chem Biol. 1998;2:519–528. doi: 10.1016/s1367-5931(98)80129-4. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Chen S, Xu T, Taghizadeh K, Wishnok JS, Zhou X, You D, Deng Z, Dedon PC. Phosphorothioation of DNA in bacteria by dnd genes. Nat Chem Biol. 2007;3:709–710. doi: 10.1038/nchembio.2007.39. [DOI] [PubMed] [Google Scholar]
- 4.Iyer RP, Egan W, Regan JB, Beaucage SL. 3H-1,2-Benzodithiole-3-One 1,1-Dioxide as an Improved Sulfurizing Reagent in the Solid-Phase Synthesis of Oligodeoxyribonucleoside Phosphorothioates. J Am Chem Soc. 1990;112:1253–1254. [Google Scholar]
- 5.Gaynor JW, Cosstick R. Synthesis, properties and application of nucleic acids containing phosphorothiolate linkages. Curr Org Chem. 2008;12:291–308. [Google Scholar]
- 6.Liu XH, Reese CB. Uridylyl-(3′→5′)-(5′-thiouridine) - an exceptionally base-labile di-ribonucleoside phosphate analog. Tetrahedron Lett. 1995;36:3413–3416. [Google Scholar]
- 7.Liu XH, Reese CB. 3′-Thiouridylyl-(3′→5′)-uridine. Tetrahedron Lett. 1996;37:925–928. [Google Scholar]
- 8.Cosstick R, Vyle JS. Solid-phase synthesis of oligonucleotides containing 3′-thiothymidine. Tetrahedron Lett. 1989;30:4693–4696. [Google Scholar]
- 9.Liu X, Reese CB. Preparation and cleavage reactions of 3′-thiouridylyl-(3′→5′)-uridine. J Chem Soc Perkin Trans. 2000;1:2227–2236. [Google Scholar]
- 10.Weinstein LB, Earnshaw DJ, Cosstick R, Cech TR. Synthesis and characterization of an RNA dinucleotide containing a 3′-S-phosphorothiolate linkage. J Am Chem Soc. 1996;118:10341–10350. [Google Scholar]
- 11.Sun S, Yoshida A, Piccirilli JA. Synthesis of 3′-thioribonucleosides and their incorporation into oligoribonucleotides via phosphoramidite chemistry. RNA. 1997;3:1352–1363. [PMC free article] [PubMed] [Google Scholar]
- 12.Matulic-Adamic J, Beigelman L. A practical synthesis of 3′-thioguanosine and of its 3′-phosphoramidothioite (a thiophosphoramidite) Helv Chim Acta. 1999;82:2141–2150. [Google Scholar]
- 13.Lu J, Li NS, Sengupta RN, Piccirilli JA. Synthesis and biochemical application of 2′-O-methyl-3′-thioguanosine as a probe to explore group I intron catalysis. Bioorg Med Chem. 2008;16:5754–5760. doi: 10.1016/j.bmc.2008.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaynor JW, Bentley J, Cosstick R. Synthesis of the 3′-thio-nucleosides and subsequent automated synthesis of oligodeoxynucleotides containing a 3′-S-phosphorothiolate linkage. Nat Protoc. 2007;2:3122–3135. doi: 10.1038/nprot.2007.451. [DOI] [PubMed] [Google Scholar]
- 15.Thomson JB, Patel BK, Jimenez V, Eckart K, Eckstein F. Synthesis and properties of diuridine phosphate analogues containing thio and amino modifications. J Org Chem. 1996;61:6273–6281. doi: 10.1021/jo960795l. [DOI] [PubMed] [Google Scholar]
- 16.Michelson AM. Polynucleotides. Part IV. Synthesis of oligonucleotide analogues substituted in the sugar portion. J Chem Soc. 1962:979–982. [Google Scholar]
- 17.Kuimelis RG, McLaughlin LW. Cleavage properties of an oligonucleotide containing a bridged internucleotide 5′-phosphorothioate RNA linkage. Nucleic Acids Res. 1995;23:4753–4760. doi: 10.1093/nar/23.23.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sproat BS, Beijer B, Rider P, Neuner P. The synthesis of protected 5′-mercapto-2′,5′-dideoxyribonucleoside-3′-O-phosphoramidites; uses of 5′-mercapto-oligodeoxyribonucleotides. Nucleic Acids Res. 1987;15:4837–4848. doi: 10.1093/nar/15.12.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuimelis RG, Mclaughlin LW. Hammerhead Ribozyme-Mediated Cleavage of a Substrate-Analog Containing an Internucleotidic Bridging 5′-Phosphorothioate - Implications for the Cleavage Mechanism and the Catalytic Role of the Metal Cofactor. J Am Chem Soc. 1995;117:11019–11020. [Google Scholar]
- 20.Das SR, Piccirilli JA. General acid catalysis by the hepatitis delta virus ribozyme. Nat Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 21.Chaulk SG, MacMillan AM. Caged RNA: photo-control of a ribozyme reaction. Nucleic Acids Res. 1998;26:3173–3178. doi: 10.1093/nar/26.13.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas JM, Perrin DM. Probing general acid catalysis in the hammerhead ribozyme. J Am Chem Soc. 2009;131:1135–1143. doi: 10.1021/ja807790e. [DOI] [PubMed] [Google Scholar]
- 23.Li NS, Frederiksen JK, Koo SC, Lu J, Wilson TJ, Lilley DM, Piccirilli JA. A general and efficient approach for the construction of RNA oligonucleotides containing a 5′-phosphorothiolate linkage. Nucleic Acids Res. 2011;39:e31. doi: 10.1093/nar/gkq1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson TJ, Li NS, Lu J, Frederiksen JK, Piccirilli JA, Lilley DM. Nucleobase-mediated general acid-base catalysis in the Varkud satellite ribozyme. Proc Natl Acad Sci USA. 2010;107:11751–11756. doi: 10.1073/pnas.1004255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elzagheid M, Oivanen M, Colin B, Jones NM, Cosstick R, Lönnberg H. Hydrolytic reactions of an RNA dinucleotide containing a 3′-S-phosphorothiolate linkage. Nucleosides Nucleotides. 1999;18:1265–1266. [Google Scholar]
- 26.Elzagheid M, Oivanen M, Klika KD, Jones BCNM, Cosstick R, Lönnberg H. Hydrolytic reactions of 3′-deoxy-3′-thioinosylyl-(3′→5′)-uridine; an RNA dinucleotide containing a 3′-S-phosphorothiolate linkage. Nucleosides Nucleotides. 1999;18:2093–2108. [Google Scholar]
- 27.Järvinen P, Oivanen M, Lönnberg H. Interconversion and Phosphoester Hydrolysis of 2′,5′-Dinucleoside and 3′,5′-Dinucleoside Monophosphates - Kinetics and Mechanisms. J Org Chem. 1991;56:5396–5401. [Google Scholar]
- 28.Sund C, Chattopadhyaya J. Intra- and intermolecular nucleophilic phosphorus-sulfur bond cleavage. The reaction of fluoride ion with O-aryl-O,S-dialkylphosphorothioates, & the degradation of phosphorothioate linkage in di-ribonucleotides by the vicinal 2′-hydroxyl group. Tetrahedron. 1989;45:7523–7544. [Google Scholar]
- 29.Ye JD, Barth CD, Anjaneyulu PS, Tuschl T, Piccirilli JA. Reactions of phosphate and phosphorothiolate diesters with nucleophiles: comparison of transition state structures. Org Biomol Chem. 2007;5:2491–2497. doi: 10.1039/b707205h. [DOI] [PubMed] [Google Scholar]
- 30.Harris ME, Dai Q, Gu H, Kellerman DL, Piccirilli JA, Anderson VE. Kinetic isotope effects for RNA cleavage by 2′-O-transphosphorylation: nucleophilic activation by specific base. J Am Chem Soc. 2010;132:11613–11621. doi: 10.1021/ja103550e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iyer S, Hengge AC. The effects of sulfur substitution for the nucleophile and bridging oxygen atoms in reactions of hydroxyalkyl phosphate esters. J Org Chem. 2008;73:4819–4829. doi: 10.1021/jo8002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerratana B, Sowa GA, Cleland WW. Characterization of the transition-state structures and mechanisms for the isomerization and cleavage reactions of uridine 3 ′-m-nitrobenzyl phosphate. J Am Chem Soc. 2000;122:12615–12621. [Google Scholar]
- 33.Piccirilli JA, Vyle JS, Caruthers MH, Cech TR. Metal ion catalysis in the Tetrahymena ribozyme reaction. Nature. 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- 34.Weinstein LB, Jones BCNM, Cosstick R, Cech TR. A second catalytic metal ion in a group I ribozyme. Nature. 1997;388:805–808. doi: 10.1038/42076. [DOI] [PubMed] [Google Scholar]
- 35.Shan S, Kravchuk AV, Piccirilli JA, Herschlag D. Defining the catalytic metal ion interactions in the Tetrahymena ribozyme reaction. Biochemistry. 2001;40:5161–5171. doi: 10.1021/bi002887h. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida A, Sun S, Piccirilli JA. A new metal ion interaction in the Tetrahymena ribozyme reaction revealed by double sulfur substitution. Nat Struct Biol. 1999;6:318–321. doi: 10.1038/7551. [DOI] [PubMed] [Google Scholar]
- 37.Shan S, Yoshida A, Sun S, Piccirilli JA, Herschlag D. Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc Natl Acad Sci USA. 1999;96:12299–12304. doi: 10.1073/pnas.96.22.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hougland JL, Kravchuk AV, Herschlag D, Piccirilli JA. Functional identification of catalytic metal ion binding sites within RNA. PLoS Biol. 2005;3:e277. doi: 10.1371/journal.pbio.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forconi M, Lee J, Lee JK, Piccirilli JA, Herschlag D. Functional identification of ligands for a catalytic metal ion in group I introns. Biochemistry. 2008;47:6883–6894. doi: 10.1021/bi800519a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams PL, Stahley MR, Kosek AB, Wang JM, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature. 2004;430:45–50. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- 41.Golden BL, Kim H, Chase E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat Struct Mol Biol. 2005;12:82–89. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- 42.Guo F, Gooding AR, Cech TR. Structure of the Tetrahymena ribozyme: Base triple sandwich and metal ion at the active site. Mol Cell. 2004;16:351–362. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 43.Gordon PM, Fong R, Piccirilli JA. A second divalent metal ion in the group II intron reaction center. Chem Biol. 2007;14:607–612. doi: 10.1016/j.chembiol.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 44.Sontheimer EJ, Sun S, Piccirilli JA. Metal ion catalysis during splicing of premessenger RNA. Nature. 1997;388:801–805. doi: 10.1038/42068. [DOI] [PubMed] [Google Scholar]
- 45.Curley JF, Joyce CM, Piccirilli JA. Functional evidence that the 3 ′-5 ′ exonuclease domain of Escherichia coli DNA polymerase I employs a divalent metal ion in leaving group stabilization. J Am Chem Soc. 1997;119:12691–12692. [Google Scholar]
- 46.Deweese JE, Burgin AB, Osheroff N. Human topoisomerase IIα uses a two-metal-ion mechanism for DNA cleavage. Nucleic Acids Res. 2008;36:4883–4893. doi: 10.1093/nar/gkn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korennykh AV, Plantinga MJ, Correll CC, Piccirilli JA. Linkage between substrate recognition and catalysis during cleavage of sarcin/ricin loop RNA by restrictocin. Biochemistry. 2007;46:12744–12756. doi: 10.1021/bi700931y. [DOI] [PubMed] [Google Scholar]
- 48.Jayakumar HK, Buckingham JL, Brazier JA, Berry NG, Cosstick R, Fisher J. NMR studies of the conformational effect of single and double 3′-S-phosphorothiolate substitutions within deoxythymidine trinucleotides. Magn Reson Chem. 2007;45:340–345. doi: 10.1002/mrc.1977. [DOI] [PubMed] [Google Scholar]
- 49.Beevers AP, Fettes KJ, Sabbagh G, Murad FK, Arnold JR, Cosstick R, Fisher J. NMR and UV studies of 3′-S-phosphorothiolate modified DNA in a DNA: RNA hybrid dodecamer duplex; implications for antisense drug design. Org Biomol Chem. 2004;2:114–119. doi: 10.1039/b311923h. [DOI] [PubMed] [Google Scholar]
- 50.Gaynor JW, Campbell BJ, Cosstick R. RNA interference: a chemist’s perspective. Chem Soc Rev. 2010;39:4169–4184. doi: 10.1039/b920362c. [DOI] [PubMed] [Google Scholar]
- 51.Gaynor JW, Brazier J, Cosstick R. Synthesis of 3′-S-phosphorothiolate oligonucleotides for their potential use in RNA interference. Nucleosides Nucleotides Nucleic Acids. 2007;26:709–712. doi: 10.1080/15257770701490753. [DOI] [PubMed] [Google Scholar]
- 52.Gaynor J, Fisher J, Campbell B, Cosstick R. Incorporation of 3′-S-phosphorothiolates into RNA: potential applications in RNAi. Nucleic Acids Symp Ser (Oxf) 2008:319–320. doi: 10.1093/nass/nrn161. [DOI] [PubMed] [Google Scholar]
- 53.Fedor MJ, Williamson JR. The catalytic diversity of RNAs. Nat Rev Mol Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 54.Perrotta AT, Shih I, Been MD. Imidazole rescue of a cytosine mutation in a self-cleaving ribozyme. Science. 1999;286:123–126. doi: 10.1126/science.286.5437.123. [DOI] [PubMed] [Google Scholar]
- 55.Wilson TJ, McLeod AC, Lilley DM. A guanine nucleobase important for catalysis by the VS ribozyme. EMBO J. 2007;26:2489–2500. doi: 10.1038/sj.emboj.7601698. [DOI] [PMC free article] [PubMed] [Google Scholar]