

Abstract
There are currently few clinical strategies in place, which provide effective neuroprotection and repair, despite an intense international effort over the past decades. One possible explanation for this is that a deeper understanding is required of how endogenous mechanisms act to confer neuroprotection. This mini-review reports the proceedings of a recent workshop “Neuroprotection and Neurorepair: New Strategies” (Iguazu Falls, Misiones, Argentina, April 11–13, 2011, Satellite Symposium of the V Neurotoxicity Society Meeting, 2011) in which four areas of active research were identified to have the potential to generate new insights into this field. Topics discussed were i) metallothionein and other multipotent neuroprotective molecules; ii) oxidative stress and their signal mediated pathways in neuroregeneration; iii) neurotoxins in glial cells, and iv) drugs of abuse with neuroprotective effects.
Keywords: neuroprotection, oxidative stress, metallothionein, glia, cannabinoids, alcohol
1. Introduction
The central nervous system (CNS) is particularly vulnerable to injury and to a range of late onset degenerative conditions. Coupled with the limited ability of the neuronal population in the adult CNS to regenerate or to be replenished, this means that these conditions are particularly damaging to individuals and to society as a whole. It is salient to note that there are no effective treatments or cures neither for traumatic CNS injury nor for any of the major neurodegenerative diseases. It is perplexing that in some cases, the nature of the presumptive initiating agent is well known, for example, the aberrant proteins which appear to lie at the heart of familial versions of Alzheimer’s disease, Parkinson’s disease and motor neurone disease (Brundin et al. 2010). However, the way in which these proteins subsequently exert neurotoxicity is complicated by their impact on diverse downstream pathways including those associated with synaptic function, energy metabolism and cytoprotective mechanisms (Saxena and Caroni 2011). Furthermore, it is not clear why only some neuronal populations in the CNS are targeted, and why that population is different from disorder to disorder. A major gap in our current understanding is how the initiating factors, be they aberrant proteins, toxins or the sequelae of injury such as ischemia, interact with the endogenous protective mechanisms found in the CNS.
Topics related to these issues are discussed in this mini-review, which arises from the proceedings of a recent workshop “Neuroprotection and Neurorepair: New Strategies” (Iguazu Falls, Misiones, Argentina, April 11–13, 2011, Satellite Symposium of the V Neurotoxicity Society Meeting). The focus of this meeting was the interaction of neurotoxic stimuli with the endogenous protective mechanisms found in the CNS, and how mechanisms such as oxidative stress relate to disease initiation and progression. One outcome was that the complexity of the disease process may need to be matched by future therapeutic agents which have the capacity to intervene at multiple points in the protective response of neural cells.
2. Endogenous neuroprotective molecules. (AKW, GJG)
Many types of endogenous molecules which directly or indirectly contribute to neuroprotection have been identified, including growth factors, antioxidants and transcription factors. An interesting observation is that many of these molecules have a broad specificity of action and appear to modulate more than one aspect of neural biology. For example, outcomes related to a given neuroprotective molecule or pathway might differ between different types or populations of neural cells, or they might be time dependent. One example of a neuroprotective molecule with wide ranging effects is metallothionein (specifically, metallothionein subtypes MT-I and MT-II which are hereafter referred to as MT), a relatively small, endogenous peptide which is expressed in the CNS and which is increased following injury or during neurodegeneration (Hidalgo et al. 2001). Experiments based on MT-deficient mice, and on administration of MT to injury models in animals and cultured cells have shown that it is beneficial in a large range of scenarios (West et al. 2004), but its mechanism of action has been unclear. It is now known that MT has multiple strands of action, which encompass roles at intracellular loci and also following its release into the extracellular milieu. Furthermore, it is able to influence the response of both neurons and glial cells, and it may also have an effect on immune system cells. For example, MT promotes regenerative neuronal growth after injury (Chung et al. 2003) and separately, it improves neuronal survival in the face of a variety of neurotoxic insults (Ambjorn et al. 2008) – for example, following Aβ administration (Chung et al. 2010). MT appears to do this by stimulating receptors of the lipoprotein receptor-related protein (LRP) family and triggering a pathway involving Erk1 and CREB activation (Ambjorn et al. 2008). However, it also acts on glial cells to promote an environment favourable to regeneration. Extracellular MT is able to convert astrocytes to a reactive form, based on expression of GFAP and on morphology, but one which is pro-regenerative rather than the reactive form commonly associated with an inhibitory environment and the formation of glial scars (Leung et al. 2010).
Similarly, extracellular MT reduces the inflammatory response of microglia following activation (Chung et al. 2009). These findings complement earlier work by Lynes’ group, which showed that MT was able to act in a chemokine-like manner and to influence the migration of leukocytes (Yin et al. 2005). These actions are additional to the well-characterised ability of intracellular MT to protect cells against toxic levels of heavy metal and against a broad range of agents, which induce oxidative stress. Thus, a single molecule appears to possess a spectrum of activities, which impinge on most of the cell types involved in neural biology and its disorders (Fig 1). It is interesting that MT expression is highly inducible from a low basal level in the brain, and for example, its expression peaks following physical injury in the rat cortex at about the time that the lesion environment tends towards one more permissive for neuronal survival and regenerative growth.
Figure 1.

The upregulation and release of metallothionein is a component of the astrocytic response to neural injury and disease. As well as exerting a protective intracellular role within astrocytes, MT is released and has a spectrum of actions on glial and neuronal cells which, in combination, results in the formation of an environment permissive for neuroprotection and regeneration.
MT is likely to be only one of a number of molecules with a similar broad area of action, and proteins including HSP70, ACAP1, apolipoprotein E and transthyretin, amongst others, also have been shown to have multiple modes of action. Furthermore, work with MTs and other proteins has highlighted their interactions with key receptor-mediated pathways, such as those activated by the LRP receptors which appear to be able to account for at least some of the neuroprotective properties exhibited by proteins of widely differing structures and abundances. It is pertinent to note that the neuroprotective actions of a single protein, such as MT, might be relatively minor and that the concerted actions of number of agents might be necessary to reach a threshold of significant neuroprotection following major neural injury. In this context, receptors such LRPs might act as moieties which integrate the input from stress-associated molecules and when a threshold is reached, activate intracellular pathways leading to neuroprotection. One corollary of this is that a successful therapeutic strategy for neuroprotection might require the development of treatments, which similarly have broad ranges of action, or are based on a combination of agents.
Over the last decade, several studies have unveiled the Janus face of microglial cells showing that these immune brain cells are not just “basic killing and cleaning machines” (M1) but can have also major neuroprotective and neurorepair functions (M2). The concept of M1 and M2 microglia is relatively recent (Salemi et al. 2011).
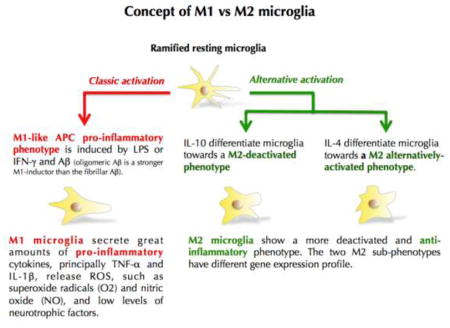
Currently, there are 2 main hypotheses to explain the microglial involvement in neurodegenerative processes (Polazzi and Monti 2010): The “classic hypothesis” utters that microglial activation induced by either genetic or environmental stimuli triggers neuroinflammation through microglia releasing neurotoxic agents such as proinflammatory cytokines and chemokines, reactive oxygen species (ROS), nitric oxide and quinolinic acid leading ultimately to neuronal death and astroglial dysfunction (Dheen et al. 2007). The second one is the “microglial dysfunction hypothesis”. Microglial neuroprotective functions decrease or disappear with ageing (Streit 2006; von Bernhardi et al. 2010), or during neurodegenerative diseases, neuronal loss mostly results from a lack of microglial neurotrophic and neuroprotective factors. These latter include various trophic factors including cytokines, antioxidants, neurotrophins and lysosomal enzymes and through the scavenging of toxic compounds such as proteinaceous aggregates and cell debris from dying neurons (Polazzi and Monti 2010).
After brain injury, microglia are activated within minutes (Nimmerjahn et al. 2005). This brings the notion of timing and switch between M1 and M2 phenotypes, thus defining a “protective time window”. Some studies on ischemia and stroke have shown that microglia protect against ischemic neuronal damage and engage in close physical cell-cell contact with neurons in the damaged brain area. Microglia are neuroprotective even when applied up to four hours after ischemia. However, pre-activated microglia (turned as M1 phenotype), or pharmacologically inhibited microglia result in a significant decrease of their neuroprotective functions (Neumann et al. 2006; Narantuya et al. 2010; Yenari et al. 2010).
In acute brain injury such as trauma or stroke, activated microglia may initially have a key neuroprotective role; early anti-inflammatory treatments within the protective time window would therefore be paradoxally harmful.
Therapeutic strategies involving M2 microglia can be potentially considered (Rock and Peterson 2006). For example, the identification and quantification of the neuroprotective factors released by microglia could potentially be used as a combination therapeutic strategy; or genetically modified autologous microglia (producing more neuroprotective factors) could represent another possible option.
3. Oxidative stresses and their signal mediated pathways in neuroregeneration. (RR-V, EDB)
The formation of free radicals is a normal physiological event, essential for the central nervous system function in healthy people. Free radicals that are formed as by-products of metabolism include superoxide anion (O2−), H2O2, nitric oxide (NO), peroxynitrite (ONOO−), nitroxyl radical (N2O2) and hydroxy radical (HO•), and are collectively referred to as reactive oxygen species (ROS) or reactive nitrogen species (RNS). Their excessive production can disable key mitochondrial respiratory chain enzymes, alter DNA and DNA-associated proteins and inhibit sodium-potassium ATPase, generating oxidative and nitrosative stress, collectively conspiring to induce the metabolic collapse and subsequent necrotic or apoptotic death of the cell (Chiueh et al. 2000). Oxidative and nitrosative stress has long been implicated in aging and age-associated disorders such as neurodegenerative diseases, for example, Parkinson’s disease (PD) (Prediger et al. 2011; Danielson and Andersen 2008; Del-Bel et al. 2011)
Free radical stress is related to many features of idiopathic PD, including overproduction and accumulation of misfolded proteins, accumulation of Lewy body-like intraneuronal inclusions and impairment of behavioural functions. The brain of PD patients displays an increase in the amount of lipid peroxidation products such as malondialdehyde (Dexter et al. 1989); evidence of protein nitration and oxidation as indicated by 3-nitrotyrosine accumulation within Lewy bodies; protein cross-linking and fragmentation as well as carbonyl group formation (Good et al. 1998); and the presence of 8-hydroxy-2-deoxyguanosine, a product of DNA oxidation (Alam et al. 1997). In PD there is a reduction in the levels of the glutathione in the substantia nigra, resulting in a decrease in complex I activity and a marked reduction in overall mitochondrial function (Martin and Teismann 2009). Iron accumulation in dopaminergic and glial cells in the substantia nigra may contribute to the generation of oxidative stress by an unclear mechanism. Consistent with these observations, an increase in the expression of an isoform of the divalent metal transporter 1 (DMT-1) was described in the substantia nigra of PD patients (Salazar et al. 2008).
Alternatively, in vitro evidence suggests that dopamine itself could be the oxidative/nitrosative stress villain, producing reactive semiquinones on the way to neuromelanin formation (Asanuma et al. 2003). Dopamine could generate hydrogen peroxide, which can be transformed to highly reactive radicals by iron-mediated Fenton reactions. However, not all dopaminergic neurons in the brain are affected in PD, suggesting that the degeneration of dopaminergic neurons in substantia nigra compacta might involve factors that are not related to dopamine metabolism. At the present time most post-mortem results were obtained after a long evolution of the neurodegenerative disease and patients receiving drug treatment during life. It is not clear how the treatments could influence results. The main point that needs to be clearly determined is if this oxidative process described in the brain is cause or consequence of the neuronal death (Prediger et al. 2011; Del-Bel et al. 2011). This will be only addressed when pre-symptomatic diagnosis of the neurodegenerative disease is possible.
4. Neurotoxins in glial cells. Neuroprotective mechanisms. (MA, GJG)
As the two major glial cell types in the brain, astrocytes and microglia play pivotal but different roles in maintaining optimal brain function. Astrocytes are metabolically coupled with neurons and this relationship is best exemplified by neuronal dependence on thiols that originate from astrocytes for the maintenance of optimal glutathione (GSH) concentrations (Dringen and Hirrlinger 2003), the major antioxidant (~90%) of intracellular non-protein thiols. The synthesis of GSH is a two-step process, commencing with the formation of γ-glutamylcysteine from cysteine and glutamate [via γ-glutamylcysteine (γ-GS) synthase] and followed by the addition of glycine (via GSH synthetase). It is noteworthy that GSH concentrations in neurons are several-fold lower vs. astrocytes (Sagara et al. 1993). Accordingly, when cellular vulnerability is considered within the context of damage induced by the generation of ROS, neurons are at significantly greater risk, as their GSH levels are depleted at a faster rate (vs. astrocytes). Toxic injury that is mediated via excitotoxic mechanisms also bears a consideration on the astrocyte-neuronal interdependence. Optimal synaptic glutamate concentrations are maintained by the glutamate aspartate transporter (GLAST) (Storck et al. 1992) and the glutamate transporter 1 (GLT1) (Lehre et al. 1995), and both these transporters are predominantly localized on astrocytes. Low synaptic extracellular glutamate concentrations are dependent upon optimal function by these transporters, and toxins that enhance astrocytic ROS levels may not only consume metabolites that are destined for GSH synthesis in neurons, but intracellular ROS will also directly inhibit glutamate transporter function. Combined, these events will feed forward in an unabated fashion, compromising GSH levels both in neurons and astrocytes and concomitantly increasing synaptic glutamate concentrations.
5. Drugs of abuse with neuroprotective effects. (YT, RM, MAC)
Drugs of abuse such as cannabinoids can be used for therapy due to their neuroprotective effects. In Alzheimer’s disease (AD), for example, cannabinoids can prevent microglial activation, which is a characteristic feature in AD. It has been shown that the phytocannabinoid, cannabidiol (CBD), which lacks psychoactivity effects, can control microglial cell function and induce neuroprotective effects in a mouse model of AD. This neuroprotective effect consists of preventing the induction of β-amyloid microglial activation, both in vivo and in vitro, by involving #33}cannabinoid (CB) receptors (Martin-Moreno et al. 2011). Similarly, in Huntington’s disease, cannabinoid receptors also take part in neuroprotection. The early downregulation of CB1 receptors in Huntington’s disease decreases striatal brain-derived neurotrophic factor expression, accelerating striatal damage (Blazquez et al. 2011). The activation of CB2 receptors, on the other hand, reduces brain edema, striatal neuronal loss, motor symptoms, and neuroinflammation. This suggests a key role of CB2 receptors in diminishing microglial activation and in preventing neurodegeneration (Palazuelos et al. 2009). Moreover, amphetamine derivatives like methamphetamine or 3,4-methylenedioxymethamphetamine (MDMA), also known as ecstasy, induce neurotoxicity accompanied by astrogliosis and microgliosis (Granado et al. 2011; Granado et al. ; Granado et al. 2010) and Delta(9)-tetrahydrocannabinol (THC), the main psychoactive component of cannabis, has been shown to counteract this neurotoxic effects induced by MDMA (Tourino et al. 2010). This neuroprotective effect is mediated by the activation of CB1 and possibly CB2 receptors, which reduces the hyperthermic and neuroinflammatory response caused by MDMA (Tourino et al. 2010). In conclusion, it has been demonstrated that cannabinoids can have neuroprotective effects, and this can be exploited for therapeutic strategies against neurodegenerative diseases such as Alzheimer’s and Huntington’s or the toxic effects of amphetamine derivatives.
One of the most common drugs of abuse, alcohol (ethanol), also appears to be a double-edged sword with regard to the brain. While chronic alcohol abuse is a major worldwide factor in causing neurodamage and dementia, recent epidemiological meta-analyses indicate that modest, responsible alcohol intake (1–2 drinks daily) among older individuals may reduce the risk of cognitive decline or even Alzheimer’s disease (Peters et al. 2008; Anstey et al. 2009). Although the consumption of wine tends to be the most effective, other alcoholic beverages are linked to lowered risks as well. Experimental studies with rat brain cultures (organotypic slices, dispersed primary cells) now provide evidence that alcohol alone in low-moderate concentrations, particularly within a pretreatment or preconditioning paradigm, can neuroprotect against inflammatory proteins such as amyloid-beta, the neurotoxin most often associated with Alzheimer’s (Collins et al. 2010). Similarly, it has been recently demonstrated that alcohol at low to moderate concentrations can protect against salsolinol, an agent implicated in Parkinson’s disease (PD), in neuroblastoma derived SH-SY5Y cells (Ramlochansingh et al. 2011). The neuroprotective signal transduction pathway stimulated by alcohol preconditioning fits the sensor→transducer→effector model elaborated initially for ischemic preconditioning (Dirnagl et al. 2003). Synaptic NMDA receptors, which can activate neuroprotective pathways, emerge as possible alcohol preconditioning sensors, since they are upregulated early; indeed, NMDA receptor antagonists block downstream transducers + effectors, and ultimately neuroprotection (Mitchell et al. 2009). Transducing the NMDA receptor signals apparently involves activation of kinases, particularly protein kinase Cε (PKCε) and focal adhesion kinase (FAK) (Sivaswamy et al. 2010). Neuroprotective effectors that are then subsequently upregulated via these kinases include heat shock proteins (HSP70 and HSP27 in both neurons and astrocytes – and blocking their upregulation suppresses neuroprotection) and, interestingly, peroxiredoxins (antioxidant enzymes known to be potentiated by synaptic NMDA receptor activation). With detailed knowledge of these preconditioning routes and players, it is conceivable that less hazardous agents could be discovered that mimic alcohol’s signaling pathways and facilitate “neuroprotective aging”. For example, dithiole-3-thione (D3T) is a cruciferous vegetable constituent that, when used as a preconditioning stimulator with brain cultures, acts like alcohol in increasing peroxiredoxin levels and antagonizing amyloid-beta induced neurotoxicity (Mitchell et al. 2011).
6. Summary
The overarching theme of this mini-review is the complexity of the endogenous response to neurotoxins, traumatic injury and the progression of neurodegenerative disease. Several issues are identified which, in part, form the basis of this complexity. Oxidative stress is common to all of these conditions, but it is not clear whether it is an initiator of the disorder or whether it is a downstream consequence of neural cell dysfunction. The synthesis of glutathione and other non-protein thiols is one response to oxidative stress, but to be effective it requires the coordinated interaction of glial cells and neurons. Like oxidative stress, neuroinflammation is observed in most neural disorders and there is vigorous debate about whether it underlies disease progression (Glass et al. 2010). It is perhaps significant that three neuroprotective agents discussed here, MT, cannabinoids and ethanol (moderate concentrations), tend to reduce neuroinflammation via direct actions on activated glial cells. Furthermore, these agents have multiple sites of action and likely modulate both glial and neuronal protective pathways. In light of the overlapping pathways thought to be triggered during neurodegeneration or injury, one suggestion is that efficacious treatments may be based on agents which have multiple effects which, although perhaps quantitatively small individually, are synergistic, particularly over the time course associated with most neurological disorders.
Acknowledgments
AKW and GJG acknowledge the support of NH&MRC, MA acknowledges the support of NIH R01 ES07331 and R01 ES10563. YT acknowledges the support of NIH/NIGMS (2SO6GM08016-39). RM acknowledges the support by grants PI071073 and PNSD from the Spanish MSPS and BFU2010-20664 from the Spanish MICINN. MAC acknowledges support from NIH RO1 AA013568 and T32 AA013527. The workshop “Neuroprotection and Neurorepair: New Strategies” (Iguazu Falls, Misiones, Argentina, April 11–13, 2011) was partially supported by CONICET-Argentina (3715/10) and ANPCYT- Argentina (RC-290/10)
The authors wish to thank Mrs. Susana Buglione for excellent bibliographic management.
Footnotes
The authors declare that they have no conflicts of interest.
Contributor Information
Gilles J. Guillemin, Email: g.guillemin@unsw.edu.au.
Rita Raisman-Vozari, Email: ritaraisman@gmail.com.
Elaine A. Del-Bel, Email: eadelbel@forp.usp.br.
Michael Aschner, Email: michael.aschner@vanderbilt.edu.
Michael A. Collins, Email: mcollin@lumc.edu.
Yousef Tizabi, Email: ytizabi@Howard.edu.
Rosario Moratalla, Email: moratalla@cajal.csic.es.
Adrian K. West, Email: Adrian.West@utas.edu.au.
References
- Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69 (3):1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- Ambjorn M, Asmussen JW, Lindstam M, Gotfryd K, Jacobsen C, Kiselyov VV, Moestrup SK, Penkowa M, Bock E, Berezin V. Metallothionein and a peptide modeled after metallothionein, EmtinB, induce neuronal differentiation and survival through binding to receptors of the low-density lipoprotein receptor family. J Neurochem. 2008;104 (1):21–37. doi: 10.1111/j.1471-4159.2007.05036.x. JNC5036 [pii] [DOI] [PubMed] [Google Scholar]
- Anstey KJ, Mack HA, Cherbuin N. Alcohol consumption as a risk factor for dementia and cognitive decline: meta-analysis of prospective studies. Am J Geriatr Psychiatry. 2009;17(7):542–555. doi: 10.1097/JGP.0b013e3181a2fd07. 00019442-200907000-00003 [pii] [DOI] [PubMed] [Google Scholar]
- Asanuma M, Miyazaki I, Ogawa N. Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox Res. 2003;5 (3):165–176. doi: 10.1007/BF03033137. [DOI] [PubMed] [Google Scholar]
- Blazquez C, Chiarlone A, Sagredo O, Aguado T, Pazos MR, Resel E, Palazuelos J, Julien B, Salazar M, Borner C, Benito C, Carrasco C, Diez-Zaera M, Paoletti P, Diaz-Hernandez M, Ruiz C, Sendtner M, Lucas JJ, de Yebenes JG, Marsicano G, Monory K, Lutz B, Romero J, Alberch J, Gines S, Kraus J, Fernandez-Ruiz J, Galve-Roperh I, Guzman M. Loss of striatal type 1 cannabinoid receptors is a key pathogenic factor in Huntington’s disease. Brain. 2011;134 (Pt 1):119–136. doi: 10.1093/brain/awq278. awq278 [pii] [DOI] [PubMed] [Google Scholar]
- Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010;11 (4):301–307. doi: 10.1038/nrm2873. nrm2873 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ, Wang K, Achille NJ, Mitchell RM, Sivaswamy S. Moderate ethanol preconditioning of rat brain cultures engenders neuroprotection against dementia-inducing neuroinflammatory proteins: possible signaling mechanisms. Mol Neurobiol. 2010;41 (2–3):420–425. doi: 10.1007/s12035-010-8138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiueh CC, Andoh T, Lai AR, Lai E, Krishna G. Neuroprotective strategies in Parkinson’s disease: protection against progressive nigral damage induced by free radicals. Neurotox Res. 2000;2 (2–3):293–310. doi: 10.1007/BF03033799. [DOI] [PubMed] [Google Scholar]
- Chung RS, Howells C, Eaton ED, Shabala L, Zovo K, Palumaa P, Sillard R, Woodhouse A, Bennett WR, Ray S, Vickers JC, West AK. The native copper- and zinc-binding protein metallothionein blocks copper-mediated Abeta aggregation and toxicity in rat cortical neurons. PLoS One. 2010;5 (8):e12030. doi: 10.1371/journal.pone.0012030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung RS, Leung YK, Butler CW, Chen Y, Eaton ED, Pankhurst MW, West AK, Guillemin GJ. Metallothionein treatment attenuates microglial activation and expression of neurotoxic quinolinic acid following traumatic brain injury. Neurotox Res. 2009;15 (4):381–389. doi: 10.1007/s12640-009-9044-y. [DOI] [PubMed] [Google Scholar]
- Chung RS, Vickers JC, Chuah MI, West AK. Metallothionein-IIA promotes initial neurite elongation and postinjury reactive neurite growth and facilitates healing after focal cortical brain injury. J Neurosci. 2003;23(8):3336–3342. doi: 10.1523/JNEUROSCI.23-08-03336.2003. 23/8/3336 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson SR, Andersen JK. Oxidative and nitrative protein modifications in Parkinson’s disease. Free Radic Biol Med. 2008;44 (10):1787–1794. doi: 10.1016/j.freeradbiomed.2008.03.005. S0891-5849(08)00149-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del-Bel E, Padovan-Neto FE, Raisman-Vozari R, Lazzarini M. Role of nitric oxide in motor control: implications for Parkinson’s disease pathophysiology and treatment. Curr Pharm Des. 2011;17(5):471–488. doi: 10.2174/138161211795164176. BSP/CPD/E-Pub/000338 [pii] [DOI] [PubMed] [Google Scholar]
- Dexter D, Wells FR, Lees A, Agid F, Agid Y, Jenner P, Marsden C. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. Journal of Neurochemistry. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14 (11):1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26(5):248–254. doi: 10.1016/S0166-2236(03)00071-7. S0166223603000717 [pii] [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384 (4):505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- Glass C, Saijo K, Winner B, Marchetto C, Gage F. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good PF, Hsu A, Werner P, Perl DP, Olanow CW. Protein nitration in Parkinson’s disease. J Neuropathol Exp Neurol. 1998;57 (4):338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- Granado N, Ares-Santos S, O’Shea E, Vicario-Abejon C, Colado MI, Moratalla R. Selective vulnerability in striosomes and in the nigrostriatal dopaminergic pathway after methamphetamine administration: early loss of TH in striosomes after methamphetamine. Neurotox Res. 2010;18 (1):48–58. doi: 10.1007/s12640-009-9106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granado N, Ares-Santos S, Oliva I, O’Shea E, Martin ED, Colado MI, Moratalla R. Dopamine D2-receptor knockout mice are protected against dopaminergic neurotoxicity induced by methamphetamine or MDMA. Neurobiol Dis. 2011;42 (3):391–403. doi: 10.1016/j.nbd.2011.01.033. S0969-9961(11)00054-4 [pii] [DOI] [PubMed] [Google Scholar]
- Granado N, Lastres-Becker I, Ares-Santos S, Oliva I, Martin E, Cuadrado A, Moratalla R. NRF2 deficiency potentiates Methamphetamine-induced dopaminergic axonal damage and gliosis in the striatum. Glia(Accepted) doi: 10.1002/glia.21229. [DOI] [PubMed] [Google Scholar]
- Hidalgo J, Aschner M, Zatta P, Vasak M. Roles of the metallothionein family of proteins in the central nervous system. Brain Res Bull. 2001;55(2):133–145. doi: 10.1016/s0361-9230(01)00452-x. S0361-9230(01)00452-X [pii] [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15 (3 Pt 1):1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung YK, Pankhurst M, Dunlop SA, Ray S, Dittmann J, Eaton ED, Palumaa P, Sillard R, Chuah MI, West AK, Chung RS. Metallothionein induces a regenerative reactive astrocyte phenotype via JAK/STAT and RhoA signalling pathways. Exp Neurol. 2010;221 (1):98–106. doi: 10.1016/j.expneurol.2009.10.006. S0014-4886(09)00423-3 [pii] [DOI] [PubMed] [Google Scholar]
- Martin-Moreno AM, Reigada D, Ramirez BG, Mechoulam R, Innamorato N, Cuadrado A, de Ceballos ML. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer’s disease. Mol Pharmacol. 2011;79 (6):964–973. doi: 10.1124/mol.111.071290. mol.111.071290 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin HL, Teismann P. Glutathione--a review on its role and significance in Parkinson’s disease. FASEB J. 2009;23 (10):3263–3272. doi: 10.1096/fj.08-125443. fj.08-125443 [pii] [DOI] [PubMed] [Google Scholar]
- Mitchell RM, Neafsey EJ, Campbell EM, Collins MA. Synaptic NMDA receptor-linked peroxiredoxin pathway is upregulated by moderate ethanol preconditioning and mimicked by cruciferous neuroprotectant, D3T. Alcohol Clin Exp Res. 2011;35:240A. [Google Scholar]
- Mitchell RM, Neafsey EJ, Collins MA. Essential involvement of the NMDA receptor in ethanol preconditioning-dependent neuroprotection from amyloid-betain vitro. J Neurochem. 2009;111 (2):580–588. doi: 10.1111/j.1471-4159.2009.06351.x. JNC6351 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narantuya D, Nagai A, Sheikh AM, Masuda J, Kobayashi S, Yamaguchi S, Kim SU. Human microglia transplanted in rat focal ischemia brain induce neuroprotection and behavioral improvement. PLoS One. 2010;5 (7):e11746. doi: 10.1371/journal.pone.0011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. 2006;20 (6):714–716. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308 (5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, Sagredo O, Benito C, Romero J, Azcoitia I, Fernandez-Ruiz J, Guzman M, Galve-Roperh I. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain. 2009;132 (Pt 11):3152–3164. doi: 10.1093/brain/awp239. awp239 [pii] [DOI] [PubMed] [Google Scholar]
- Peters R, Peters J, Warner J, Beckett N, Bulpitt C. Alcohol, dementia and cognitive decline in the elderly: a systematic review. Age Ageing. 2008;37 (5):505–512. doi: 10.1093/ageing/afn095. afn095 [pii] [DOI] [PubMed] [Google Scholar]
- Polazzi E, Monti B. Microglia and neuroprotection: from in vitro studies to therapeutic applications. Prog Neurobiol. 2010;92 (3):293–315. doi: 10.1016/j.pneurobio.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Prediger RD, Aguiar AS, Jr, Moreira EL, Matheus FC, Castro AA, Walz R, De Bem AF, Latini A, Tasca CI, Farina M, Raisman-Vozari R. The intranasal administration of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP): a new rodent model to test palliative and neuroprotective agents for Parkinson’s disease. Curr Pharm Des. 2011;17(5):489–507. doi: 10.2174/138161211795164095. BSP/CPD/E-Pub/000339 [pii] [DOI] [PubMed] [Google Scholar]
- Ramlochansingh C, Taylor RE, Tizabi Y. Toxic Effects of Low Alcohol and Nicotine Combinations in SH-SY5Y Cells are Apoptotically Mediated. Neurotox Res. 2011 doi: 10.1007/s12640-011-9239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock RB, Peterson PK. Microglia as a pharmacological target in infectious and inflammatory diseases of the brain. J Neuroimmune Pharmacol. 2006;1 (2):117–126. doi: 10.1007/s11481-006-9012-8. [DOI] [PubMed] [Google Scholar]
- Sagara JI, Miura K, Bannai S. Maintenance of neuronal glutathione by glial cells. J Neurochem. 1993;61 (5):1672–1676. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nunez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105 (47):18578–18583. doi: 10.1073/pnas.0804373105. 0804373105 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salemi J, Obregon DF, Cobb A, Reed S, Sadic E, Jin J, Fernandez F, Tan J, Giunta B. Flipping the switches: CD40 and CD45 modulation of microglial activation states in HIV associated dementia (HAD) Mol Neurodegener. 2011;6 (1):3. doi: 10.1186/1750-1326-6-3. 1750-1326-6-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71 (1):35–48. doi: 10.1016/j.neuron.2011.06.031. S0896-6273(11)00561-7 [pii] [DOI] [PubMed] [Google Scholar]
- Sivaswamy S, Neafsey EJ, Collins MA, Sivaswamy S, Neafsey EJ, Collins MA. Neuroprotective preconditioning of rat brain cultures with ethanol: potential transduction by PKC isoforms and focal adhesion kinase upstream of increases in effector heat shock proteins. European Journal of Neuroscience. 2010;32 (11):1800–1812. doi: 10.1111/j.1460-9568.2010.07451.x. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A. 1992;89 (22):10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci. 2006;29 (9):506–510. doi: 10.1016/j.tins.2006.07.001. S0166-2236(06)00144-5 [pii] [DOI] [PubMed] [Google Scholar]
- Tourino C, Zimmer A, Valverde O. THC Prevents MDMA Neurotoxicity in Mice. PLoS One. 2010;5 (2):e9143. doi: 10.1371/journal.pone.0009143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernhardi R, Tichauer JE, Eugenin J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem. 2010;112 (5):1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x. JNC6537 [pii] [DOI] [PubMed] [Google Scholar]
- West AK, Chuah MI, Vickers JC, Chung RS. Protective role of metallothioneins in the injured mammalian brain. Rev Neurosci. 2004;15 (3):157–166. doi: 10.1515/revneuro.2004.15.3.157. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Kauppinen TM, Swanson RA. Microglial activation in stroke: therapeutic targets. Neurotherapeutics. 2010;7 (4):378–391. doi: 10.1016/j.nurt.2010.07.005. S1933-7213(10)00110-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Knecht DA, Lynes MA. Metallothionein mediates leukocyte chemotaxis. BMC Immunol. 2005;6:21. doi: 10.1186/1471-2172-6-21. 1471-2172-6-21 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]