

Abstract
The cholesterol-dependent cytolysins (CDCs) are a large family of pore-forming toxins that are produced, secreted and contribute to the pathogenesis of many species of Gram-positive bacteria. The assembly of the CDC pore-forming complex has been under intense study for the past 20 years. These studies have revealed a molecular mechanism of pore formation that exhibits many novel features. The CDCs form large β-barrel pore complexes that are assembled from 35-40 soluble CDC monomers. Pore formation is dependent on the presence of membrane cholesterol, which functions as the receptor for most CDCs. Cholesterol binding initiates significant secondary and tertiary structural changes in the monomers, which lead to the assembly of a large membrane embedded β-barrel pore complex. This review will focus on the molecular mechanism of assembly of the CDC membrane pore complex and how these studies have led to insights into the mechanism of pore formation for other pore-forming proteins.
Keywords: toxin, pore, perfringolysin, membrane, cholesterol, CD59, intermedilysin, complement, perforin
1.INTRODUCTION
The cholesterol dependent cytolysins (CDCs) are a large family of pore forming toxins that are produced by many species of Gram-positive pathogens. These include species from Clostridium, Streptococcus, Listeria, Arcanobacterium, Gardnerella, Bacillus and Lactobacillus. It is likely that additional species that express a CDC will be discovered in the future. The CDC gene is probably one of the most widely disseminated toxin genes known, suggesting that many bacterial species find it useful to introduce large pores in cholesterol containing eukaryotic membranes. How these bacterial pathogens use these toxins to establish and/or contribute to the progression of an infection has been studied in only a handful of organisms. It is clear, however, that these pathogens use the CDCs in far more sophisticated ways than as simple cell lytic agents [1-4]. Many studies have shown that the CDCs can induce various cellular effects, usually at levels that do not lyse a cell [5-20], but the contribution of these effects to disease progression remains less well understood. Some disease causing pathogens express CDCs that lack the ability to form a pore [21, 22], which suggests that there may be functions of the CDCs other than pore formation that can contribute to disease.
The pore forming mechanism of these toxins has been the object of intense scrutiny for the past 20 years, which has provided significant insights into their pore forming mechanism. The CDCs exhibit several hallmark features that include a complete dependence of their pore-forming mechanism on the presence of membrane cholesterol and the formation of extraordinarily large membrane pore complexes. The study of the CDC pore complex has focused on understanding how these soluble, monomeric proteins assemble into a large membrane spanning pore complex. The subject of this review will be confined to providing a description of the assembly of the CDC pore complex. As will be discussed below the study of the CDC pore forming mechanism has revealed new paradigms in the assembly of a membrane pore complex as well as providing insight into the mechanism of other pore forming proteins.
2.THE CDC STRUCTURE
2.1. The CDC primary structure
The first primary structure of a CDC was reported in 1987 by Walker et al. [23] for the Streptococcus pneumoniae CDC pneumolysin (PLY), which was followed shortly thereafter by the primary structure for the S. pyogenes streptolysin O (SLO) structure [24]. Since that time the primary structures of over 25 different CDCs have been reported in GenBank. These structures revealed that most CDCs are secreted as soluble monomers from the bacterial cell using a typical type II signal peptide. PLY is an exception to this rule since it lacks a signal peptide: how PLY is released from S. pneumonia remains unclear. An early study [25] suggested it was released by cell lysis whereas more recent studies suggest that release is not associated with cell lysis but may be via an currently unidentified transport system [26, 27]. Ultimately, the CDCs are released into the extracellular milieu as soluble monomers that eventually bind to and form a large pore complex in cholesterol containing membranes. The mass of the secreted CDCs range from about 50 to 72 kD. The core structure responsible for pore formation is about 50 kD: several CDCs, however, exhibit amino terminal peptide extensions that range from about a dozen amino acids to a 150 residue fucose binding lectin [28]. The function of these amino terminal extensions is generally poorly understood. A short amino terminal peptide extension in the Listeria monocytogenes CDC, listeriolysin O (LLO), appears to influence its stability [29, 30]. Another short peptide extension found at the amino terminus of SLO appears to contribute to the SLO-mediated transport of another protein across the membrane of a eukaryotic cell [4, 31]. The amino terminal fucose binding lectin of the Streptococcus mitis lectinolysin (LLY) appears to modulate its cytolytic activity, but how it does so is not known [28].
The primary structures of the CDC family show a high degree of conservation (40-70% identity). A highly conserved structural motif of 11 residues (ECTGLAWEWWR) near the carboxy terminus is a signature motif for the CDCs. This motif, which is termed the undecapeptide or tryptophan rich motif, contains the only cysteine residue normally found in the secreted form of the CDCs and typically contains 3 tryptophans. As will be apparent from this review it’s role in the pore forming mechanism has yet to be established unambiguously. The primary structure of the secreted form of the CDCs is remarkably hydrophilic with no obvious hydrophobic regions that could be potential transmembrane regions.
2.2. The CDC crystal structure
The first crystal structure of the soluble secreted form of a CDC was solved in 1997 by Rossjohn et al. [32]. They crystallized and solved the structure of the secreted form of the CDC from Clostridium perfringens, perfringolysin O (PFO). In retrospect, the choice of PFO for crystallization trials was somewhat fortuitous, as the structures of the other three most well studied CDCs at that time, SLO, listeriolysin O (LLO) and PLY, have yet to be solved 14 years later. Other CDCs structures have been solved, however, and include Streptococcus intermedius intermedilysin (ILY) [33], Bacillus anthracis anthrolysin O (ALO) [34] and most recently Streptococcus suis suilysin (SLY) [35]. The structure of PFO (Fig. 1) revealed an elongated four-domain protein that is rich in β-sheet. The structures of the other CDCs mentioned above are highly similar to that of PFO with only small differences in their overall shape. Initially, we proposed that the domain 4 undecapeptide signature motif of PFO acted as a hydrophobic dagger to penetrate the membrane and form the pore [32]. This model of pore formation was based on the previous observation by Nakamura et al. [36] showing that the tryptophans of the undecapeptide entered the membrane. As described below, however, even though the undecapeptide enters the membrane it does not directly contribute to the formation of the membrane pore.
Figure 1. Molecular structure of Clostridium perfringens perfringolysin O.
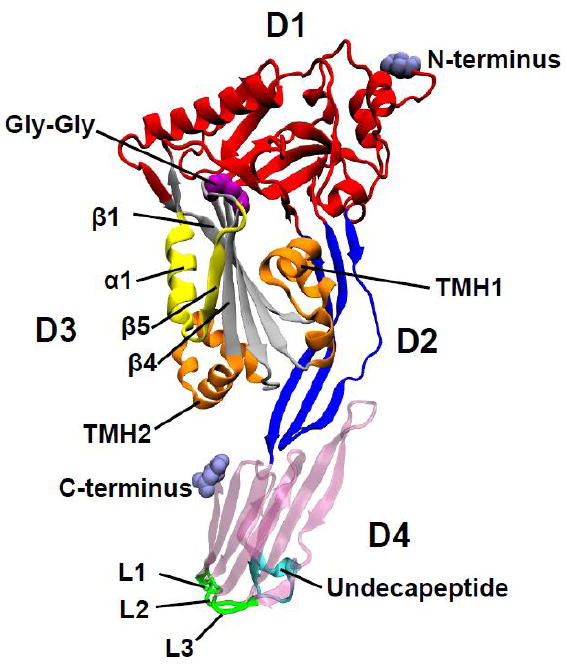
Shown is a ribbon representation of the PFO crystal structure (PDB ID: 1PFO) [32]. Specific features of the structure are designated as D1-D4, domains 1-4; TMH1 and TMH2 (orange), transmembrane hairpins 1 and 2; β1, β4 and β5, β-strands 1, 4 and 5, α1, α-strand 1. L1-L3, loops L1-L3 The double glycine motif is shown as purple space filled atoms. All structures where generated using VMD [126]
The crystal structure of PFO was an important discovery that was critical to the subsequent elucidation of the pore forming mechanism of the CDCs. A combination of the PFO monomer structure with biophysical and biochemical studies revealed significant insights into the remarkable structural transitions in the CDC monomer that are required to form the pore.
3. A BRIEF INTRODUCTION TO THE CDC PORE-FORMING MECHANISM
Before proceeding into the details of the pore forming mechanism the reader will be provided a brief description of the general features of the CDC pore forming mechanism as it relates to the PFO crystal structure in Fig. 1.
Bacteria release CDCs as soluble monomers: typically by a type II secretion pathway. The soluble monomers then bind to the eukaryotic cell surface via cholesterol, although a few CDCs have evolved to use human CD59 as their receptor [37, 38]. As described below the CD59 binding CDCs still require the presence of membrane cholesterol to function [39-41]. The initial interaction of the cholesterol binding CDC monomers with the cell surface is mediated via the tip of domain 4 (Fig. 1) where the conserved undecapeptide and loops L1-L3 have been shown to anchor the monomer to the membrane [42, 43]. The monomers are bound via the tip of domain 4 in an upright position that is perpendicular to the membrane [44, 45]. Membrane binding initiates changes within the monomer structure that lead to the formation of intermolecular contacts between membrane-bound monomers [46]. A major transition that is required for monomer-monomer contact is the rotation of the loop comprised of β5 and α1 (Fig.1) away from β4 of the domain 3 core β-sheet. This action frees up the edge of β4 to pair with β1 of another monomer [46]. The monomers continue to extend the oligomeric structure until a ring shaped structure is achieved. This structure is termed the prepore complex and is defined as the completed ring complex that has not yet inserted its β-barrel pore. The prepore state likely has several intermediate states that are currently defined by whether the ring complex is SDS-sensitive or resistant [47-49]. During assembly of the ring-shaped prepore complex two transmembrane β-hairpins (TMHs) from each monomer are derived from the two domain 3 α-helical bundles (Fig. 1) and contribute to the formation of the large β-barrel structure [50, 51]. To assemble the PFO pore approximately 36 monomers form a completed ring complex, although this number can vary by a few monomers either way [45]. Therefore, PFO assembles and coordinates the insertion of a β-barrel structure that contains approximately 144 membrane-spanning β-strands or 72 β-hairpins, which results in the formation of a pore with a diameter of 250-300 Å. The CDC pore is currently the largest known toxin pore.
4. MEMBRANE RECOGNITION
4.1. Characteristics of lipid, sterol and membrane structure that influence CDC binding
Most CDCs bind to membranes by using the cholesterol as their receptor. One of the first hallmark traits to be identified for the CDCs was the ability of added cholesterol to inhibit the hemolytic activity of these toxins, presumably by occupying a receptor binding site (reviewed in [52]). The interaction of these toxins with cholesterol and its role as the receptor has been difficult to elucidate due to the insoluble nature of cholesterol and the inability to generate cocrystal structures of the CDCs with cholesterol. Early studies by Prigent et al. [53] showed that an intact 3-β-hydroxyl group, an aliphatic sidechain of appropriate length at carbon 17 of the D ring, a methyl group at carbon 20 and an intact B ring were important to its recognition by CDCs. Sterols with a 3-α-hydroxl or modifications of the 3-β-hydroxyl were not bound by these CDCs, thus demonstrating that recognition was restricted to the 3-β-hydroxyl. Other minor changes in the ring structure, such as the saturation state of the B ring, did not affect their ability to inhibit the hemolytic activity of the CDCs. These studies primarily determined the structures of purified sterols required to bind and inhibit the CDCs, but as described below the lipid environment of intact membranes also plays a major role in whether cholesterol will be recognized and bound by the CDCs. In a subsequent study Alouf and coworkers also showed that that the stoichiometry of the interaction between the CDCs and cholesterol was 1:1 [54], suggesting a single cholesterol binding site was present on the toxins.
Ohno-Iwashita and colleagues have extensively studied the interaction of the CDC from Clostridium perfringens, PFO [36, 55-67] with membranes and cholesterol. One of their first observations was that chemical modification of the single cysteine in the conserved undecapeptide of PFO caused a significant loss of hemolytic activity [55]. The loss in activity resulted from an altered membrane binding affinity that was associated with a reversible conformational change in PFO. This was the first evidence that suggested the undecapeptide was involved in the interaction of PFO with the membrane. They also found that PFO exhibited biphasic binding to cholesterol-rich membranes: high and low affinity binding was observed [68]. Since both high and low affinity binding sites were dependent on cholesterol, they suggested that cholesterol exhibited a heterogeneous distribution in membranes and that only a fraction of the cholesterol was easily accessible and formed the high affinity binding sites. This insight was prescient as Heuck et al. [69] subsequently studied binding to cholesterol containing liposomes. Although they did not observe a biphasic binding they did find that binding of PFO to cholesterol-rich liposomes exhibits a sharp transition from undetectable binding at 40 mole% total membrane cholesterol to maximal binding at 55 mol%. Hence, not all cholesterol at the surface of the membrane forms a suitable receptor for PFO (and probably most CDCs). Typically, efficient binding of CDCs requires >30 mol% of the total membrane lipid to be cholesterol [69-71], therefore, only a fraction of the total membrane cholesterol likely serves as a suitable receptor.
The studies of Flanagan et al. [70] and Nelson et al. [72] showed that the accessibility of cholesterol to PFO is dependent on the phospholipid structure: lipids that could pack tightly with cholesterol, such as those with unsaturated acyl chains, tended to decrease binding to cholesterol. Lipids that did not pack tightly with cholesterol, such as those with unsaturated acyl chains, shifted the half maximal binding to lower cholesterol levels. They also found that the addition of epicholesterol, which contains a 3-α-hydroxyl group and is not bound by PFO, increased binding of PFO to the membrane. This apparently resulted from the displacement of cholesterol by epicholesterol from its interaction with the lipids, thus freeing up cholesterol from its association with lipid and increasing its availability to PFO. These studies suggested that when the cholesterol concentration exceeded the associative capacity of the lipid it was free to be recognized and bound by PFO. The phospholipid headgroup size also influences the availability of cholesterol at the membrane surface. Zitzer et al. [73] have shown for SLO that lipids with smaller headgroups increase the binding of SLO to membranes. In retrospect these observations makes sense since tightly packed lipid-sterol mixtures or mixtures containing lipids with larger headgroups are more likely to sterically occlude the relatively small 3-β-hydroxyl of cholesterol thereby restricting access of the CDC cholesterol binding motif (described below) to the cholesterol 3-β-hydroxy headgroup.
It was also shown by Waheed et al. [66] that PFO preferentially partitioned into the detergent-resistant membrane (DRM) fraction of cells. Since microdomain structure is often rich in cholesterol it made sense that PFO partitioned with the detergent resistant membrane fraction. This observation, however, has been complicated by the recent studies of Flanagan et al. [70]. They showed that increasing the molar fraction of sphingomyelin, a constituent of rafts, in liposomes decreased PFO binding to cholesterol. Sphingomyelin contains unsaturated acyl chains and would tend to pack more tightly with cholesterol. Since sphingomyelin is concentrated in DRMs these data suggested that PFO would not preferentially bind to sphingomyelin/cholesterol-rich microdomains in natural membranes. More problematic, however, is the observation by Flanagan et al. that the PFO oligomer alone exhibited a propensity to partition into the detergent resistant fraction, therefore detergent fractionation cannot be reliably used to determine if CDC complexes are associated with DRMs.
4.2. The CDC cholesterol recognition/binding motif (CRM)
The identification of the CDC cholesterol recognition/binding motif (CRM) has been an elusive problem for nearly three decades. Many studies have shown that regions and residues in domain 4 of the CDC structure affect binding of the CDCs to the membrane [36, 55, 62, 65, 67, 74-76]. More recent studies have narrowed the regions of domain 4 that interact with the membrane to the undecapeptide [43] and three loops (L1-L3) at the tip of domain 4 [42] (Fig. 1). The rest of domain 4 is surrounded by water throughout the assembly of the oligomeric pore complex [42]. Investigators have generally accepted that the conserved undecapeptide motif contributed to cholesterol recognition. This was not an unreasonable theory since the undecapeptide is highly conserved in the CDCs and studies showed that one or more of the tryptophans in the undecapeptide inserted into the bilayer [36], although not deeply [43]. Furthermore, Ohno-iwashita and colleagues [55, 74] showed that chemical modification of the undecapeptide cysteine of PFO decreased binding, although the modification also induced a conformation change in the structure of PFO, which could have affected binding indirectly. Furthermore, alanine mutants within nearly every residue of the PFO undecapeptide resulted in significant perturbations in its hemolytic activity and membrane binding [33]. Jacobs, et al. showed that monoclonal antibodies to the undecapeptide inhibit binding [65], however a large antibody bound at the tip of domain 4 would likely disrupt any interaction mediated by this region with the membrane. In total, these studies provided compelling evidence that the undecapeptide was the cholesterol-binding motif.
A series of relatively recent studies, however, suggested the CRM resided within loops L1-L3 rather than the undecapeptide. Soltani et al. [77, 78] showed that they could uncouple the cholesterol-dependent membrane insertion of the undecapeptide from the cholesterol binding function of the CDCs, which suggested that the CRM resided somewhere in loops L1-L3. Furthermore, the solution of the crystal structures for other cholesterol binding CDCs showed that the undecapeptide 3-dimensional structure in the crystal structures of PFO, Streptococcus intermedius ILY, Bacillus anthracis anthrolysin O (ALO) and more recently Streptococcus suis suilysin (SLY) adopt different structures (Fig. 2A), which makes it difficult to envision how they could recognize and bind to a common receptor motif. These differences were not due to crystal packing forces, but appear to be the result of differences in specific sidechain interactions between conserved residues of the undecapeptide and residues outside of the undecapeptide.
Figure 2. The structure of the undecapeptide motif and location of the CRM.
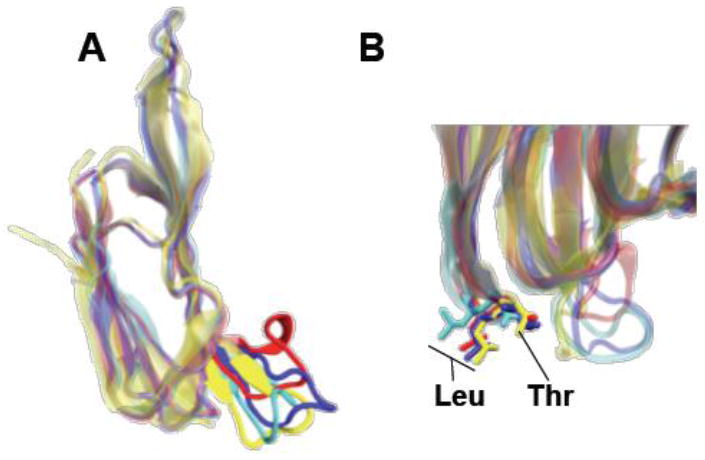
Shown in panel A is an overlay of domain 4 structures from Clostridium perfringens PFO (red), Streptococcus suis SLY (PDB ID: 3HVN) [35] (yellow), Bacillus anthracis ALO (PDB ID: 3CQF) [34] (cyan) and Streptococcus intermedius ILY (PDB ID: 1S3R) [33](blue). Shown in B is the location of the cholesterol recognition/binding motif of the same CDCs as in A (colors assignments are the same as in panel A).
Farrand et al. [40] alanine scanned the residues in loops L1-L3 of PFO and determined that two mutations affected hemolytic activity and binding to a greater extent than any other residues in these loops. These residues are a threonine-leucine pair (residues 490 and 491 of PFO) in loop L1 (Fig. 2B). These residues are conserved in all known CDCs, even those that use human CD59 rather than cholesterol as a receptor. As described below (section 4.3) the Thr-Leu CRM is also necessary to the cytolytic mechanism of the human CD59 binding CDCs, although it does not function as a receptor-binding motif in those proteins. Farrand et al. showed that alanine and/or glycine substitutions for both residues in PFO, SLO and PLY abolished binding to cholesterol rich liposomes, purified cholesterol and the cholesterol rich membranes of eukaryotic cells. They also showed conservative substitutions for either residue significantly decreased binding and activity: substitution of serine for Thr-490 and either valine or isoleucine for the leucine-491 of PFO nearly completely abolished binding. Therefore, even the most conservative substitutions are not well tolerated.
The cholesterol recognition/binding motif of the CDCs is surprisingly simple and inviolable. Neither trait should be unexpected since the headgroup of cholesterol is also comparatively simple when compared to other receptor molecules. A complex binding site on the CDCs for cholesterol would not be expected since cholesterol has a relatively limited headgroup exposed at the surface if the membrane. Only the 3-β-hydroxyl and possibly portions of the A ring of cholesterol are exposed at the surface. One possibility is that the leucine inserts into the bilayer and interact with the A ring and the threonine forms a hydrogen bond with the 3-β-hydroxyl group. The leucine may constantly sample the membrane surface and only upon the correct orientation with the cholesterol headgroup does the threonine lock in the interaction by forming a hydrogen bond with the 3-β-hydroxyl. This interaction is also stereo-specific, as the CDCs do not interact with epicholesterol, which has a 3-α-hydroxyl.
Once the Thr-Leu pair establishes an interaction with the membrane cholesterol the residues within loops 2 and 3 insert into the bilayer and firmly anchor the monomer to the membrane [40]. At some point, perhaps coincident with insertion of the L2-L3 loops, portions of the undecapeptide also enter the membrane. The undecapeptide is located near the Thr-Leu pair and so many of the previously described undecapeptide mutations [33, 55, 62] may have altered its structure in a way that interfered with the interaction of the CRM with cholesterol and/or membrane insertion of the loops L2 and L3.
4.3. Human CD59 binding CDCs
Streptococcus intermedius produces ILY, which was shown by Nagamune et al. [79] to lyse only human erythrocytes: nonhuman primate erythrocytes were 100-fold less sensitive to ILY whereas all animal erythrocytes tested were resistant to lysis. They further showed that the hemolytic activity on human erythrocytes could be decreased by pretreatment of the erythrocytes with a protease, suggesting that a protein contributed to ILY binding. This observation was in stark contrast to what was generally understood about CDCs, which were thought to use only cholesterol as their receptor. Nearly a decade after its discovery the basis for the highly selective behavior of the ILY cytolytic activity was revealed by the studies of Giddings et al. [37] who showed that human CD59 was the receptor for ILY. Since then two more CDCs have been discovered that also use human CD59 as their receptor, Gardnerella vaginalis vaginolysin (VLY) [38] and Streptococcus mitis lectinolysin (LLY) [80]. Interestingly, CD59 is a GPI-anchored surface protein and is an inhibitor of the complement membrane attack complex (MAC) [81, 82]. CD59 protects host cells from complement-mediated lysis during times of bacterial infection when complement is activated. It does so by binding to complement proteins C8α and C9, which are two proteins of the complement system that are necessary for the formation of the complement membrane attack complex (MAC) pore (reviewed in [83]). CD59 is homologously restricted [82]: i.e., it only functions to inhibit the host’s complement and not that of other species. The fact that ILY only bound and lysed human cells was shown to result from its ability to bind to the site on human CD59 that also endowed it with its species-specific inhibition of the complement MAC [37, 82].
The ILY binding site for CD59 was recently mapped by Wickham et al. [80]. This CD59 binding site is located in domain 4 and contains a signature motif of YXYX14RS that is found in VLY and was used to identify LLY as a member of the CD59 binding family of CDCs. This motif is located on the outer surface of the ring on the side of domain 4 that is predicted to face away from the lumen of the pore. As indicated above, the normal cellular function of CD59 is to inhibit the assembly of the MAC pore complex by binding to complement proteins C8α and C9. The ILY consensus sequence is not present in C8α or C9. More details on the interaction of ILY and the complement proteins with CD59 are described below in section 7.2, as the CDCs and C8α and C9 may share mechanistic and evolutionary relationships.
For most CDCs binding to the cholesterol receptor initiates changes in the monomer structure that lead to the formation of the pore complex. In ILY this function has been transferred to the CD59 binding site [77]. Binding to human CD59 rather than cholesterol initiates these same structural transitions [77]. Binding to CD59 still triggers these changes in cholesterol-depleted membranes, but without cholesterol the pore itself does not form [41]. Therefore membrane cholesterol remains important to the mechanism of ILY, even though it has switched receptors from cholesterol to CD59. Why then does ILY still require cholesterol for its pore forming activity and why did it retain the CRM if its receptor function was replaced by CD59?
These questions were addressed in two studies: the first by Lachapelle et al. [39] and the second by Farrand et al. [40]. Lachapelle et al. [39] first showed that ILY disengaged from CD59 upon prepore to pore conversion. This study raised the question, however, that if ILY disengaged from its receptor how did it maintain contact with the membrane so that it could insert its β-barrel pore? Farrand et al. [40] subsequently showed that ILY containing a CRM knockout remained bound to the membrane via its interaction with CD59 prior to conversion of the prepore to pore, but upon conversion of the prepore to the pore the oligomeric complex formed by the CRM knockout lost contact with the membrane as it disengaged from CD59. Therefore, interaction between ILY and CD59 performs two important functions: (1) it initiates the structural changes that are necessary for pore formation and (2) it positions the CRM so that it can bind to cholesterol and initiate the insertion of loops L1-L3 into the membrane. The additional membrane anchor provided by the cholesterol-dependent insertion of loops L1-L3 is necessary to keep the prepore complex attached to the membrane as it disengages from CD59 during the critical prepore to pore transition. If the CRM is knocked out then the pore complex detaches from the membrane as it disengages from CD59 and tries to insert the β-barrel pore [40].
Why does ILY disengage from CD59 during its transition to the pore complex? It appears that the adoption of CD59 by ILY as its receptor did not occur without consequences. The disengagement of ILY from CD59 is apparently necessary for the transition from the prepore to pore. Wickham et al. [80] showed that mutations in either CD59 or ILY that increased the binding affinity of the interaction decreased the rate of pore formation. Their studies strongly suggested that the increased affinity of the ILY-CD59 interaction slowed the rate at which ILY disengaged from CD59, therefore slowing the rate of the prepore to pore transition [80]. This predicted that if the binding affinity was sufficiently high that it would prevent the prepore to pore transition. Therefore the binding affinity between ILY and CD59 must be balanced such that it is not too high, which would slow the rate of pore formation, but not so low as to affect the on-rate of binding, which would also slow pore formation.
It is also interesting to note that in the absence of the CRM the residues of loops 2 and 3 do not independently insert into the membrane and anchor ILY to the membrane surface. The CRM of ILY cannot bind it to the cell surface if CD59 is absent [37], which suggests that binding to CD59 must orient the CRM near the membrane so that it can interact with cholesterol. Presumably binding to CD59 also brings loops 2 and 3 near the membrane since they are juxtaposed to the CRM (Fig. 2B). Yet, in the absence of the CRM these hydrophobic loops alone cannot insert into the membrane and anchor ILY. Therefore the interaction of the CRM with cholesterol is a necessary prerequisite for the insertion of these loops.
5. FORMATION OF THE PREPORE COMPLEX
The prepore complex is defined as a completed oligomeric complex on the verge of inserting its β-barrel pore. Whether a prepore state was an assembly intermediate of the large CDC oligomeric pore complex was initially an issue of debate. Palmer et al. [84] proposed a model of pore assembly in which a CDC dimer inserted in the membrane followed by the addition of monomers that enlarged the pore from a small pore to a large pore. They suggested that insertion of such a large β-barrel pore from a prepore complex was energetically unfavorable. In fact, the insertion of a naked amphipathic β-hairpin into a membrane is energetically unfavorable [85]. The membrane core lacks hydrogen bond donors or acceptors to which the polar atoms of the β-hairpin polypeptide backbone could hydrogen bond, therefore significantly raising the energetic cost of positioning the hairpins within the bilayer. Formation of a partial or wholly formed pre-β-barrel would be more energetically favorable for membrane insertion. It was subsequently shown in two studies by Shepard et al. [49] and Hotze et al. [48] that PFO formed a large prepore complex and that this complex then converted to the pore complex. The formation of the prepore complex may facilitate the organization of the large β-barrel pore prior to its insertion by allowing the formation of some or all of the interstrand hydrogen bonds between the transmembrane β-hairpins (TMHs). The fact that insertion of the β-barrel after prepore formation as favorable was shown by the work of Hotze et al. [48]. They engineered a disulfide between TMH1 and domain 2 (Fig. 1) that prevented the extension of TMH1 and allowed PFO to form a SDS-resistant prepore on membranes. If the disulfide was then reduced after oligomerization was completed the insertion of the β-barrel pore was rapid. These studies showed that (1) the rate limiting step in the formation of the pore complex is the assembly of the oligomeric prepore complex and (2) that once it is completed the insertion of the β-barrel pore is rapid.
More than one prepore state has been identified in PFO suggesting that monomer-monomer interactions proceed through various intermediate states during the assembly of an SDS-resistant prepore complex. Membrane binding primes the monomers for oligomerization: once a monomer has bound to the membrane its structure changes to allow monomer-monomer interactions. These interactions do not occur in solution, even at the high concentrations of protein required for crystallization [86]. The extent to which the structure of the CDC monomer changes upon binding, however, is incompletely understood. Ramachandran et al. [46] have shown that the disruption of the edge-on interaction of β5 with β4 in domain 3 of PFO is kinetically linked to membrane binding. This event frees up the edge of β4 to then pair and form interstrand backbone hydrogen bonds with β1 of another monomer. In addition to the formation of interstrand hydrogen bonds an intermolecular π-stacking interaction is also formed between a tyrosine in β1 and a phenylalanine in β4 (Tyr-181 and Phe-318 in PFO). This interaction is critical to the formation of a SDS-resistant prepore complex; loss of either residue, or moving one of these aromatics out of register on either strand results in the formation of an SDS-sensitive prepore complex that cannot form a pore [46, 47]. It was suggested [46] that the π-stacking interaction locks β1 and β4 into the correct in-register pairing of their backbone hydrogen bonds. Although the great majority of CDCs maintain these two residues, the π-stacking interaction is not present in the CD59 binding CDCs and is also missing in the cholesterol binding CDCs pneumolysin, Lactobacillus iners inerolysin and Arcanobacterium pyogenes pyolysin. Regardless of the function of the π-stacking interaction in those CDCs that maintain this aromatic pair it is obvious that some CDCs have solved this problem in another way.
Monomer-monomer contact may also help drive some of the domain 3 structural transitions necessary for pore formation. As indicated above, elimination of one of the aromatics that participate in the interstrand π-stacking interaction traps PFO in a SDS-sensitive prepore complex. If mixed with functional PFO this π-stacking mutant can be driven to form an SDS-resistant oligomeric complex and to insert its TMHs into the membrane [47]. This presumably occurs because the functional PFO monomers form cooperative interactions with the mutant PFO molecules that induce these monomers to undergo the necessary structural transitions to form a pore. Hence, membrane binding may induce some structural changes that prepare the CDC for oligomerization, but monomer-monomer interactions may propagate additional structural changes necessary for pore formation.
A second structurally important signature motif of the CDCs was also revealed by these studies: the presence of a conserved Gly-Gly pair (Gly-324 and 325 in PFO) that are located in the β-loop between β4 and β5 (Fig. 1). Residues with sidechains cannot be substituted for either glycine without blocking the disengagement of β5 from β4 [46], which suggests that sidechains at this location clash with other residues and prevent rotation of β5 away from β4. Interestingly, this motif is also positionally conserved within the recently solved crystal structures of several membrane attack/perforin (MACPF) proteins (Fig. 3). The MACPF and CDC proteins lack similarity at primary structure level, but the MACPF proteins exhibit structural similarity with domain 3 of the CDCs (described in section 7 below) [87-90].
Figure 3. The crystal structures of MACPF family proteins showing a CDC domain 3 like structure.
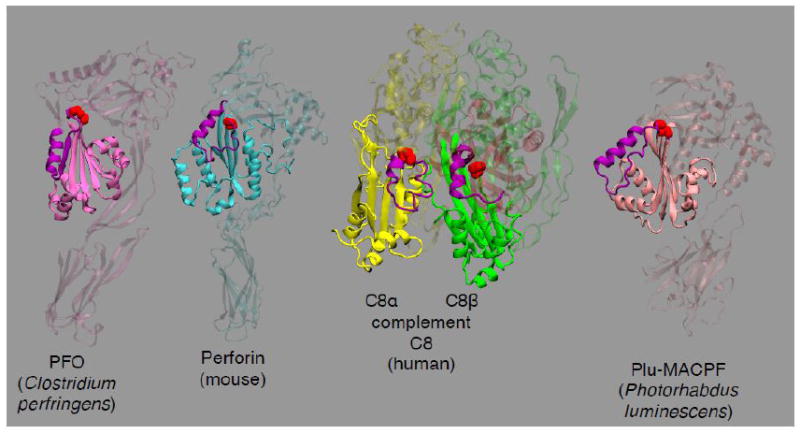
The crystal structures of the MACPF proteins mouse perforin (PDB ID: 3NSJ) [88], complement C8 (PDB ID: 3OJY) [120] and the Photorhabdus luminescens Plu-MACPF (PDB ID: 2QP2) [89] are shown with that of PFO [32]. In each MACPF structure the PFO-like domain 3-like fold is highlighted. Shown in purple ribbon structure in each MACPF structure is the equivalent structure to the PFO α1-β5 loop that rotates away from β4. Shown in red spacefilled atoms is the conserved twin glycine motif of each protein that is present at the junction between β4 and β5, which is conserved in all know CDCs [46].
6. FORMATION OF THE β-BARREL PORE
6.1. The structure of the β-barrel pore
After solution of the PFO crystal structure one of the first features of the CDC mechanism to be revealed was the structure of the membrane-spanning domain. Originally, we proposed that the domain 4 undecapeptide acted as a hydrophobic dagger that plunged into the membrane, which was based on the available data that showed the undecapeptide tryptophans entered the bilayer [62]. Palmer et al. [91], however, identified three residues within domain 3 of streptolysin O that also appeared to also enter the membrane and proposed the presence of a membrane spanning amphipathic α-helix. Shortly thereafter Shepard et al. [51] and Shatursky et al. [50] disproved both models by completely mapping the membrane spanning regions of PFO using a combination of cysteine scanning and fluorescence spectroscopy. Two sets of α-helical bundles that flank the domain 3 core β-sheet, which extend from the domain 3 core β-strands 1-4 (Fig. 1), were identified that underwent a change in secondary structure to form two amphipathic β-hairpins. Shatursky et al. [50] further showed that these twin hairpins spanned the bilayer. Since the PFO oligomeric complex is composed of 35-40 monomers (some CDCs may contain more monomers) up to 160 amphipathic β-strands are positioned in the membrane to form the CDC β-barrel pore.
The primary structure of the TMHs is some of the least conserved primary structure of the CDCs, although they maintain the typical amphipathic structure of a membrane spanning β-hairpin. Other than the amphipathic nature of the TMHs, no other conserved features are apparent in either TMH. Each TMH is approximately 30 residues long or 15 residues per β-strand. A few residues are highly conserved within each TMH between the CDCs, although no reason for their conservation has been identified. Leu-207 and Phe-211 of PFO are the most well conserved in TMH1 among the CDCs (96% identity), but can be changed to cysteine with comparatively little loss in pore forming activity [51]. Ala-293 in TMH2 is also highly conserved (96% identify) among the CDCs. Substitution of Ala-293 with cysteine did not significantly affect its pore forming activity, however, the subsequent modification of the cysteine thiol with a fluorescent probe decreased the activity to less than 10% of native PFO suggesting that a bulky group at this position is not tolerated [50].
6.2. The CDC oligomeric pore complex
CDCs are often seen to form a mixture of rings and incomplete, or arc-shaped complexes by electron microscopy (EM) [49, 92-94]. Whether these incomplete rings can insert a partial β-barrel remains unknown: the available data suggests that they do not insert into the membrane and may be artifacts of the EM process, dead end complexes or simply a matter of partially inactive protein preparations. If incomplete rings inserted into the membrane with the same probability as a complete ring then the likelihood of forming complete rings would be comparatively low. It is possible that membrane inserted fragments could associate to form a complete ring but they would require, like a puzzle, pieces that would be just the right size to assemble the ring. This type of assembly mechanism would be a comparatively inefficient method for assembling a ring, especially if formation of a ring complex was necessary. A method of quantifying the formation of the CDC prepore and pore complex was developed by Shepard et al.[49]. They used SDS-agarose electrophoresis [49], which separated the very large CDC pore complex from the monomer and intermediates in the assembly process. They showed that the only SDS-resistant form of the PFO oligomeric complex was a fairly homogeneous high molecular weight species shown to be ring complexes: intermediates in the assembly of the ring complex were rare, even at early stages of assembly when they were stabilized by crosslinking. Furthermore, Heuck et al. [43] have shown that at limiting dilutions of PFO where, on average, a single pore is formed in a liposomal membrane the release of low and high molecular weight markers remains the same as observed at higher concentrations of pores. Hence, the available data suggests that a very large complex, which is presumably a complete ring, is the functional pore-forming species.
6.3. CDC activation: disruption of the domain 2-3 interface
Disruption of the domain 2-3 interface is at the heart of the CDC pore forming mechanism. This interface primarily exists between the α-helical bundle that ultimately forms TMH1 and domain 2 (Fig. 1), although regions of domain 1 also form some contacts with the domain 3 α-helical bundle. In order to unravel this α-helical bundle to form and extend TMH1 it must pull out of its interface with domains 2 and 3. Before it was understood that these α-helical bundles formed the TMHs Rossjohn et al. [32] had noted in the crystal structure of the PFO monomer that this interface exhibited poor complementarity, which was consistent with the fact that this interface must be disrupted to extend TMH1.
How this interface is disrupted is incompletely understood. Heuck et al. [69] showed that membrane insertion of the domain 4 undecapeptide was conformationally linked to the insertion of the TMHs, demonstrating a link between structural changes in domain 3 that are necessary for insertion of the β-barrel and membrane insertion of the domain 4 undecapeptide. Based on different crystal forms of PFO Rossjohn et al. [95] showed that rotational torsion between domains 1-3 and domain 4 broke several contacts at this interface: presumably some torsion may be induced by binding of PFO to the membrane. Whether binding alone can induce sufficient torsional stress to disrupt this interface and extend TMH1 remains unknown. How this conformation signal is communicated from the tip of domain 4, which interacts with the membrane, to this interface is also unknown. As indicated above in section 5 monomer-monomer contact is also important in driving some domain 3 structural changes [47]. Hence, it is likely that a combination of membrane binding and monomer-monomer contact may be important in driving this transition.
6.4. Membrane insertion of the β-barrel pore
Shepard et al. [51] and Shatursky et al. [50] had clearly demonstrated that both TMH1 and TMH2 crossed the bilayer, but the length of the extended TMHs were calculated to be only long enough to reach the surface of the bilayer, but not cross it. This conundrum results from the fact that the bottom of domain 3 is suspended about 40Å above the membrane, which is approximately the length of the extended hairpins. Therefore, how did the β-barrel pore cross the membrane? Czajkowsky et al. [45] solved this problem by using atomic force microscopy to measure the height of the prepore and pore complexes. They first measured the height of a PFO mutant that was trapped in the prepore state by an engineered disulfide between domain 2 and the domain 3 α-helical bundle that inhibited the complete disruption of the domain 2-3 interface [48]. The prepore complex was determined to extend about 113 Å above the membrane, which closely corresponded to the length of the monomer from the tip of domain 4 to the top of domain 1. Upon reduction of the disulfide, which allowed the prepore complex to convert to the pore complex, they showed that the pore complex extended only 73 Å above the membrane. These data showed that insertion of the β-barrel was associated with a 40 Å vertical collapse of the prepore complex, the distance required to bring domain 3 sufficiently near the membrane surface to allow the hairpins to extend across the bilayer. This was confirmed shortly thereafter by Ramachandran et al. [44] who used Förster resonance energy transfer (FRET) based distance measurements between fluorescent probes within domains 1 and 3 in PFO and the membrane surface. They showed that domains 1 and 3 moved about 35 Å closer to the membrane when the prepore was unlocked and allowed to convert to the pore complex.
Czajkowsky et al. [45] also proposed that once the domain 2-3 interface was disrupted that the domain 2 structure collapsed or folded thereby allowing domains 1 and 3 to move towards the membrane. Tilley et al. [96] later supported this scenario using cryoEM data of the PLY pore, which they used to generate a 3D reconstruction of the pore complex that was fitted with the crystal structure of the PFO monomer. Their data showed that the vertical collapse of the prepore complex was associated with the appearance of a bulge on the outer ring surface that was consistent with a folded domain 2 when fitted with the PFO crystal structure.
6.5. Regulation of the listeriolysin O pore forming activity by pH
The disruption of the domain 2-3 interface is also used to control the pH dependent activity of one CDC, listeriolysin O (LLO), from Listeria monocytogenes. For nearly 25 years it’s been known that the pore-forming activity of LLO was sensitive to pH: LLO exhibits maximal activity at lower pH ranges (pH 5-6) and undetectable activity at pH values of 7 or higher [97]. LLO was also shown to be critical to the escape of L. monocytogenes from the vacuole into the cytosol, where it replicated and could undergo cell-to-cell spread without an extracellular phase, thereby protecting it from the immune system (reviewed in [98-100]). It is important that LLO cytolytic activity be tightly controlled: it must function at the low pH of the endosome to effect escape of the bacterial cell into the cytoplasm but must be switched off once escape has occurred to prevent lysis of the host cell membrane. If the host cell membrane is lysed it exposes the bacterial cells to the immune system, which results in an avirulent phenotype [1, 101-103]. The structural basis for the pH sensitivity of LLO was unknown, although it was generally assumed to be similar to other pH-sensitive toxins, such as diphtheria toxin and anthrax toxin. These toxins are inactive and stable at neutral pH, but upon being taken up into an endosome, which is acidified by the vacuolar ATPase, the low pH triggers the structural changes that allow them to insert into the membrane and translocate their enzymatic fragment [104-110].
This mechanism, however, is not utilized by LLO to control formation and insertion of its β-barrel pore. In contrast, acidic residues in domain 3 act as a pH sensor and prematurely trigger the disruption of the domain 2-3 interface in the soluble monomer at neutral pH above temperatures of 30°C [111]. The premature unfurling of the domain 3 α-helical bundles in the soluble monomers inactivates LLO and causes it to aggregate in solution. The aggregation results from the exposure of hydrophobic residues that are normally protected in the soluble monomer, but which are exposed by the disruption of the domain 2-3 interface [111]. Disulfide locking TMH1 to domain 2 significantly decreased the rate of this inactivation, showing that it was the disruption of the domain 2-3 interface that results in this inactivation. These structural changes do not occur at acidic pH, which is found in the vacuole where the LLO monomers remain stable and capable of membrane binding and insertion of the β-barrel pore. Hence, LLO is not pH activated, but is pH inactivated at neutral pH at temperatures above 30°C. This mechanism allows LLO to function in the acidic environment of the vacuole where it is required to effect release of the phagocytized bacterial cell, but then is inactivated to prevent lysis of the host cell as the bacteria cell escapes into the neutral environment of the cytoplasm.
The LLO pH sensor is composed of a triad of domain 3 acidic residues, Asp-208, Asp-320 and Glu-247. They are located in the domain 3 core β-sheet near the domain 2-3 interface and their carboxylates are predicted to point in at each other [111]. The close proximity of carboxylates is known to increase the pKa of carboxylates to pH 6-8 [112-115]: therefore at acidic pH these residues are mostly protonated and domain 3 remains stable. However, as the pH increases the carboxylates deprotonate and charge repulsion between the carboxylate groups is predicted to destabilize the domain 2-3 interface and cause its disruption, which inactivates the soluble monomer. Replacement of these LLO residues by those present in PFO, a CDC that is stable at acidic and neutral pH, eliminated the pH sensitive nature of LLO [111].
Therefore, at the acidic pH of the vacuole the LLO monomers remain stable and can bind to the membrane and follow the normal mechanism of activation to form a pore, thus facilitating the escape of the bacterial cell in to the cytoplasm. Remaining or newly synthesized soluble LLO monomers that are present after escape of L. monocytogenes from the vacuole would be rapidly inactivated in the neutral pH environment of the cytoplasm at 37°C. Hence, in LLO domain 3 plays a dual role, at acidic pH it functions normally to extend the TMHs and form the β-barrel pore after membrane binding, but at neutral pH it causes the premature unfurling of the α-helical bundles in soluble monomers, which results in the inactivation of LLO thereby preventing the unwanted lysis of the host cell.
7. THE MEMBRANE ATTACK COMPLEX/PERFORIN FAMILY PROTEINS
7.1. Structural similarities between the CDC and MACPF protein families suggest a pore forming mechanism for the MACPF proteins
The pore forming mechanism(s) of the complement membrane attack complex/perforin (MACPF) family of proteins has remained ambiguous due to the technical challenges of working with these proteins. The pore forming complement membrane attack complex (MAC) and perforin define the MACPF protein family (reviewed in [83, 116]), but there are many members that are widespread among both eukaryotes and prokaryotes [117]. The terminal pathway of complement, the MAC, is assembled from C5b, C6, C7, C8 and C9 proteins. C8 and C9 are the major proteins that form the MAC pore. This pathway is involved in the defense against invading Gram-negative bacteria by the formation of pores in their membrane [118]. Perforin is a major mediator of host cell destruction by natural killer cells and CD8+ cytotoxic T-cells. Tumor cells, cells infected with viruses or intracellular bacterial pathogens can be targeted for destruction. Perforin forms pores in the host membrane, which can be directly cytotoxic. It appears, however, that its major function may be effect the delivery of granzymes to the cytosol of the targeted cells to induce apoptosis [119]. Regardless of the mechanism by which perforin effects cell killing the perforin pore is required.
The mechanism by which the MAC and perforin form pores in the membrane has languished due to the difficulty of working with these proteins and the difficulty in expressing functional recombinant forms of these proteins. Recently, however, the crystal structures have been solved for several members of the MACPF family of proteins. These include complement protein C8 [120] and C8α [87, 90], mouse perforin [88] and a complement C9-like protein, pleurotolysin, from the bacterium Photorhabdus luminescens [89]. The MACPF crystal structures revealed that they contain a protein fold strikingly similar to domain 3 of PFO (Fig. 3). The similarities of the MACPFs with the CDCs and a description of the MACPF family of proteins has been the subject of an excellent recent review by Rosado et al. [117]. This common protein fold, which is responsible for the formation of the CDC pore [50, 51], was found only in the CDCs prior to the solution of the MACPF crystal structures. This revelation was a major breakthrough in elucidating a potential pore forming mechanism for the MACPF proteins. These investigators [87, 89, 90] suggested that like the CDCs the MACPF proteins also unravel two α-helical bundles to form two amphipathic β-hairpins that combine to form an oligomeric β-barrel pore complex.
If the MACPF α-helical bundles do form extended transmembrane β-hairpins like the CDCS then there is at least one obvious difference in their structures when compared to the CDCs: the estimated length of these hairpins is significantly longer, in most cases, than those found in the CDCs. This suggests that these hairpins may be sufficiently long to span the bilayer without a significant collapse of a prepore structure, as is found in the CDCs. Law et al. [88] recently proposed that the putative hairpins of mouse perforin were sufficiently long to reach and cross the membrane. In fact, they proposed that instead of extending out on the same side of the perforin molecule that they extend to the opposite side of the molecule. If true this would represent a remarkably deviation from the CDC mechanism.
Are there any other features of the CDCs that suggest a functional relationship with the MACPF proteins? No sequence homology exists between the MACPF proteins and the CDCs, however, the double glycine motif that exists in the loop that connects β4 and β5 in the CDC domain 3 core β-sheet is also found in the MACPFs (Fig. 3). As indicated above in section 5, these two glycines are conserved in all known CDC primary structures. They are also conspicuously present in a similar location in the MACPF monomer crystal structures (Fig. 3). We have shown that this glycine pair cannot be changed in PFO to residues with a sidechain without loss of pore forming activity [46]. If amino acids with sidechains are introduced at this location the rotation of β5 away from β4 is prevented, presumably due to steric clashes of the sidechains with nearby residues. As mentioned above in section 5, this rotation frees up the edge of β4 to pair with β1 of another monomer. The presence of a glycine pair in a conserved location in the MACPF crystal structures suggests that these residues may also be necessary for the rotation of the MACPF structure that is analogous to the CDC β5-α1 loop away from β4 [46] to allow the intermolecular pairing of β1 and β4. Although it is compelling to suggest a mechanism of pore formation for the MACPF protein family based on the structural similarities with the CDCs, they must be viewed with caution until they are rigorously demonstrated.
7.2. Human CD59: The Yin and Yang receptor
As described in section 4.3 the CDCs ILY, VLY and LLY initiate the formation of the pore by binding to human CD59. Interestingly, the primary function of CD59 on human cells is to inhibit the formation of the complement MAC pore on the host cell membranes to prevent their lysis during times of complement activation. Complement activation during a Gram-negative bacterial infection results in the deposition of the complement complex MAC onto the bacterial outer membrane, thereby forming a pore in the outer membrane of the bacterial cell. Activated complement MAC, however, can also cause collateral damage to nearby host cells. In addition to CD59 host cells can express a number of other proteins that antagonize complement assembly at different stages (reviewed in [121]). CD59 is a GPI-anchored terminal complement inhibitor that binds to MACPF proteins C8α and C9 and inhibits the formation of the terminal MAC pore [122, 123]. In contrast, the CDCs ILY, VLY and LLY initiate formation of their pores on human cells by binding to human CD59. Therefore, CD59 serves to initiate the formation of the ILY, VLY and LLY pores whereas it inhibits the formation of the complement MAC pore.
Prior to formation of the pore ILY remains bound to CD59 [39]. While bound to CD59 it blocks the site on CD59 that interacts with complement proteins C8α and C9. Therefore, CD59 cannot inhibit assembly of the MAC while ILY remains engaged with CD59 [39]. This results in the host cells being more prone to attack by their hosts own complement system. This observation and prior studies [37] suggested that both ILY and C8α and C9 bound to similar sites on CD59. As discussed in section 4.3 above Wickham et al. recently mapped the binding site for ILY on CD59. Interestingly, the binding site for ILY exhibited an deep correspondence to the site on CD59 that binds to C8α and C9 [80]. Most of the residues involved in the binding site for the CDCs were also involved in its interaction with C8α and C9. As indicated above the ILY binding site for CD59 exhibits a consensus motif of YXYX14RS that is not found in C8α or C9. In fact, neither C8α nor C9 exhibit significant similarity in their binding sites for CD59 [122-125]. Therefore the MAC proteins and the CDCs bind to a highly conserved site on CD59, yet their binding sites for human CD59 exhibit a remarkable lack of similarity [122, 123].
8. SUMMARY AND FUTURE PERSPECTIVES
The study of the CDC pore-forming mechanism has revealed new paradigms of how the large CDC oligomeric pore complex is assembled. The study of the CDCs has shown that they undergo significant secondary and tertiary structural changes in order to form the CDC pore. Furthermore, the CDCs employ a novel cholesterol-binding motif to recognize and bind cholesterol at the membrane surface. The study of the CDC mechanism has also led to new insights into the possible mechanism of pore formation for the large family of eukaryotic and prokaryotic MACPF proteins and suggests that they may be ancient ancestors of the CDCs.
Some major features of the CDC pore forming mechanism still remain elusive. For instance, we do not yet understand how the CDCs time the insertion of the large β-barrel pore. Evidence suggests that this happens upon completion of the ring shaped complex, but the nature of the mechanism that triggers/regulates the insertion remains unknown. After insertion of the β-barrel the fate of the lipid and any associated proteins from the lumen of the pore also remains unknown. Do the CDCs specifically carve out sections of membrane that reduces the level of specific membrane proteins that may have important roles in cellular physiology or signaling? Over a decade ago Caparon and colleagues revealed that the Streptococcus pyogenes CDC, SLO, specifically translocated another secreted protein from S. pyogenes, a NAD glycohydrolase enzyme (SPN), into human keratinocytes in an actin-independent process [4]. Remarkably, they recently showed that this translocation system functions in the absence of pore formation by SLO by showing that the prepore locked form of SLO translocates SPN as well or better than native SLO. Revealing the basis of this transport system will likely reveal an entirely new non-pore forming function for some CDCs. The study of the MACPF proteins also promises to reveal new insights into how they form a pore. Like the CDCs they may form a β-barrel pore from the α-helical bundle-derived β-hairpins, but are other features of the assembly of the CDC pore complex conserved in the MACPF proteins? Future studies into these various problems will reveal new paradigms that will likely change our understanding of this fascinating group of toxins and provide new insights into the mechanism of other pore forming proteins.
HIGHLIGHTS.
The cholesterol dependent cytolysins (CDCs) have a novel cholesterol-binding motif.
The CDCs assemble oligomeric membrane complexes composed of 35-40 monomers.
CDCs undergo major secondary and tertiary structural changes to form a β-barrel pore.
Membrane attack complex/perforin immunity proteins may be related to the CDCs.
Acknowledgments
This work was supported by a grant (2R01AI037657) from the National Institutes of Allergy and Infectious Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BIBLIOGRAPHY
- 1.Portnoy D, Jacks PS, Hinrichs D. The role of hemolysin for intracellular growth of Listeria monocytogenes. J Exp Med. 1988;167:1459–1471. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Awad MM, Bryant AE, Stevens DL, Rood JI. Virulence studies on chromosomal alpha-toxin and theta-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of alpha-toxin in Clostridium perfringens-mediated gas gangrene. Mol Microbiol. 1995;15:191–202. doi: 10.1111/j.1365-2958.1995.tb02234.x. [DOI] [PubMed] [Google Scholar]
- 3.Ellemor DM, Baird RN, Awad MM, Boyd RL, Rood JI, Emmins JJ. Use of genetically manipulated strains of Clostridium perfringens reveals that both alpha-toxin and theta-toxin are required for vascular leukostasis to occur in experimental gas gangrene. Infect Immun. 1999;67:4902–4907. doi: 10.1128/iai.67.9.4902-4907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madden JC, Ruiz N, Caparon M. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell. 2001;104:143–152. doi: 10.1016/s0092-8674(01)00198-2. [DOI] [PubMed] [Google Scholar]
- 5.Gekara NO, Weiss S. Lipid rafts clustering and signalling by listeriolysin O. Biochem Soc Trans. 2004;32:712–714. doi: 10.1042/BST0320712. [DOI] [PubMed] [Google Scholar]
- 6.Gekara NO, Westphal K, Ma B, Rohde M, Groebe L, Weiss S. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell Microbiol. 2007;9:2008–2021. doi: 10.1111/j.1462-5822.2007.00932.x. [DOI] [PubMed] [Google Scholar]
- 7.Hamon MA, Batsche E, Regnault B, Tham TN, Seveau S, Muchardt C, Cossart P. Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci U S A. 2007;104:13467–13472. doi: 10.1073/pnas.0702729104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rose F, Zeller SA, Chakraborty T, Domann E, Machleidt T, Kronke M, Seeger W, Grimminger F, Sibelius U. Human endothelial cell activation and mediator release in response to Listeria monocytogenes virulence factors. Infect Immun. 2001;69:897–905. doi: 10.1128/IAI.69.2.897-905.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wadsworth SJ, Goldfine H. Mobilization of protein kinase C in macrophages induced by Listeria monocytogenes affects its internalization and escape from the phagosome. Infect Immun. 2002;70:4650–4660. doi: 10.1128/IAI.70.8.4650-4660.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen BR, Duncan JL. Activation of human neutrophil metabolism by streptolysin O. J Infect Dis. 1980;141:680–685. doi: 10.1093/infdis/141.5.680. [DOI] [PubMed] [Google Scholar]
- 11.Cockeran R, Steel HC, Mitchell TJ, Feldman C, Anderson R. Pneumolysin potentiates production of prostaglandin E(2) and leukotriene B(4) by human neutrophils. Infect Immun. 2001;69:3494–3496. doi: 10.1128/IAI.69.5.3494-3496.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cockeran R, Theron AJ, Steel HC, Matlola NM, Mitchell TJ, Feldman C, Anderson R. Proinflammatory interactions of pneumolysin with human neutrophils. J Infect Dis. 2001;183:604–611. doi: 10.1086/318536. [DOI] [PubMed] [Google Scholar]
- 13.Baba H, Kawamura I, Kohda C, Nomura T, Ito Y, Kimoto T, Watanabe I, Ichiyama S, Mitsuyama M. Induction of gamma interferon and nitric oxide by truncated pneumolysin that lacks pore-forming activity. Infect Immun. 2002;70:107–113. doi: 10.1128/IAI.70.1.107-113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cockeran R, Durandt C, Feldman C, Mitchell TJ, Anderson R. Pneumolysin activates the synthesis and release of interleukin-8 by human neutrophils in vitro. J Infect Dis. 2002;186:562–565. doi: 10.1086/341563. [DOI] [PubMed] [Google Scholar]
- 15.Fickl H, Cockeran R, Steel HC, Feldman C, Cowan G, Mitchell TJ, Anderson R. Pneumolysin-mediated activation of NFkappaB in human neutrophils is antagonized by docosahexaenoic acid. Clin Exp Immunol. 2005;140:274–281. doi: 10.1111/j.1365-2249.2005.02757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernatoniene J, Zhang Q, Dogan S, Mitchell TJ, Paton JC, Finn A. Induction of CC and CXC chemokines in human antigen-presenting dendritic cells by the pneumococcal proteins pneumolysin and CbpA, and the role played by toll-like receptor 4, NF-kappaB, and mitogen-activated protein kinases. J Infect Dis. 2008;198:1823–1833. doi: 10.1086/593177. [DOI] [PubMed] [Google Scholar]
- 17.Bryant AE, Bergstrom R, Zimmerman GA, Salyer JL, Hill HR, Tweten RK, Sato H, Stevens DL. Clostridium perfringens invasiveness is enhanced by effects of theta toxin upon PMNL structure and function: the roles of leukocytotoxicity and expression of CD11/CD18 adherence glycoprotein. FEMS Immunol Med Microbiol. 1993;7:321–336. doi: 10.1111/j.1574-695X.1993.tb00414.x. [DOI] [PubMed] [Google Scholar]
- 18.Stevens DL, Bryant AE. Role of theta-toxin, a sulfhydryl-activated cytolysin, in the pathogenesis of clostridial gas gangrene. Clin Infect Dis. 1993;16:S195–S199. doi: 10.1093/clinids/16.supplement_4.s195. [DOI] [PubMed] [Google Scholar]
- 19.Stevens DL, Mitten J, Henry C. Effects of alpha and theta toxins from Clostridium perfringens on human polymorphonuclear leukocytes. J Infect Dis. 1987;156:324–333. doi: 10.1093/infdis/156.2.324. [DOI] [PubMed] [Google Scholar]
- 20.Stevens DL, Troyer BE, Merrick DT, Mitten JE, Olson RD. Lethal effects and cardiovascular effects of purified alpha- and theta-toxins from Clostridium perfringens. J Infect Dis. 1988;157:272–279. doi: 10.1093/infdis/157.2.272. [DOI] [PubMed] [Google Scholar]
- 21.Kirkham LA, Jefferies JM, Kerr AR, Jing Y, Clarke SC, Smith A, Mitchell TJ. Identification of invasive serotype 1 pneumococcal isolates that express nonhemolytic pneumolysin. J Clin Microbiol. 2006;44:151–159. doi: 10.1128/JCM.44.1.151-159.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jefferies JM, Johnston CH, Kirkham LA, Cowan GJ, Ross KS, Smith A, Clarke SC, Brueggemann AB, George RC, Pichon B, Pluschke G, Pfluger V, Mitchell TJ. Presence of nonhemolytic pneumolysin in serotypes of Streptococcus pneumoniae associated with disease outbreaks. J Infect Dis. 2007;196:936–944. doi: 10.1086/520091. [DOI] [PubMed] [Google Scholar]
- 23.Walker JA, Allen RL, Falmagne P, Johnson MK, Boulnois GJ. Molecular cloning, characterization, and complete nucleotide sequence of the gene for pneumolysin, the sulfhydryl-activated toxin of Streptococcus pneumoniae. Infection and immunity. 1987;55:1184–1189. doi: 10.1128/iai.55.5.1184-1189.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kehoe MA, Miller L, Walker JA, Boulnois GJ. Nucleotide sequence of the streptolysin O (SLO) gene: structural homologies between SLO and other membrane-damaging, thiol-activated toxins. Infect Immun. 1987;55:3228–3232. doi: 10.1128/iai.55.12.3228-3232.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson MK. Cellular location of pneumolysin. FEMS Microbiol Lett. 1977;2:243–245. [Google Scholar]
- 26.Balachandran P, Hollingshead SK, Paton JC, Briles DE. The autolytic enzyme LytA of Streptococcus pneumoniae is not responsible for releasing pneumolysin. J Bacteriol. 2001;183:3108–3116. doi: 10.1128/JB.183.10.3108-3116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price KE, Camilli A. Pneumolysin localizes to the cell wall of Streptococcus pneumoniae. Journal of bacteriology. 2009;191:2163–2168. doi: 10.1128/JB.01489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrand S, Hotze E, Friese P, Hollingshead SK, Smith DF, Cummings RD, Dale GL, Tweten RK. Characterization of a streptococcal cholesterol-dependent cytolysin with a lewis y and b specific lectin domain. Biochemistry. 2008;47:7097–7107. doi: 10.1021/bi8005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decatur AL, Portnoy DA. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science. 2000;290:992–995. doi: 10.1126/science.290.5493.992. [DOI] [PubMed] [Google Scholar]
- 30.Schnupf P, Zhou J, Varshavsky A, Portnoy DA. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect Immun. 2007;75:5135–5147. doi: 10.1128/IAI.00164-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meehl MA, Caparon MG. Specificity of streptolysin O in cytolysin-mediated translocation. Mol Microbiol. 2004;52:1665–1676. doi: 10.1111/j.1365-2958.2004.04082.x. [DOI] [PubMed] [Google Scholar]
- 32.Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. Structure of a cholesterol-binding thiol-activated cytolysin and a model of its membrane form. Cell. 1997;89:685–692. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- 33.Polekhina G, Giddings KS, Tweten RK, Parker MW. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc Natl Acad Sci. 2005;102:600–605. doi: 10.1073/pnas.0403229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourdeau RW, Malito E, Chenal A, Bishop BL, Musch MW, Villereal ML, Chang EB, Mosser EM, Rest RF, Tang WJ. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J Biol Chem. 2009;284:14645–14656. doi: 10.1074/jbc.M807631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu L, Huang B, Du H, Zhang XC, Xu J, Li X, Rao Z. Crystal structure of cytotoxin protein suilysin from Streptococcus suis. Protein Cell. 2010;1:96–105. doi: 10.1007/s13238-010-0012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura M, Sekino N, Iwamoto M, Ohno-Iwashita Y. Interaction of theta-toxin (perfringolysin O), a cholesterol-binding cytolysin, with liposomal membranes: change in the aromatic side chains upon binding and insertion. Biochemistry. 1995;34:6513–6520. doi: 10.1021/bi00019a032. [DOI] [PubMed] [Google Scholar]
- 37.Giddings KS, Zhao J, Sims PJ, Tweten RK. Human CD59 is a receptor for the cholesterol–dependent cytolysin intermedilysin. Nat Struct Mol Biol. 2004;11:1173–1178. doi: 10.1038/nsmb862. [DOI] [PubMed] [Google Scholar]
- 38.Gelber SE, Aguilar JL, Lewis KL, Ratner AJ. Functional and phylogenetic characterization of Vaginolysin, the human-specific cytolysin from Gardnerella vaginalis. J Bacteriol. 2008;190:3896–3903. doi: 10.1128/JB.01965-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LaChapelle S, Tweten RK, Hotze EM. Intermedilysin-receptor interactions during assembly of the pore complex: assembly intermediates increase host cell susceptibility to complement-mediated lysis. J Biol Chem. 2009;284:12719–12726. doi: 10.1074/jbc.M900772200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farrand AJ, LaChapelle S, Hotze EM, Johnson AE, Tweten RK. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc Natl Acad Sci U S A. 2010;107:4341–4346. doi: 10.1073/pnas.0911581107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giddings KS, Johnson AE, Tweten RK. Redefining cholesterol’s role in the mechanism of the cholesterol-dependent cytolysins. Proc Natl Acad Sci U S A. 2003;100:11315–11320. doi: 10.1073/pnas.2033520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramachandran R, Heuck AP, Tweten RK, Johnson AE. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat Struct Biol. 2002;9:823–827. doi: 10.1038/nsb855. [DOI] [PubMed] [Google Scholar]
- 43.Heuck AP, Tweten RK, Johnson AE. Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J Biol Chem. 2003;278:31218–31225. doi: 10.1074/jbc.M303151200. [DOI] [PubMed] [Google Scholar]
- 44.Ramachandran R, Tweten RK, Johnson AE. The domains of a cholesteroldependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc Natl Acad Sci. 2005;102:7139–7144. doi: 10.1073/pnas.0500556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Czajkowsky DM, Hotze EM, Shao Z, Tweten RK. Vertical collapse of a cytolysin prepore moves its transmembrane β-hairpins to the membrane. EMBO J. 2004;23:3206–3215. doi: 10.1038/sj.emboj.7600350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramachandran R, Tweten RK, Johnson AE. Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit β-strand alignment. Nat Struct Mol Biol. 2004;11:697–705. doi: 10.1038/nsmb793. [DOI] [PubMed] [Google Scholar]
- 47.Hotze EM, Heuck AP, Czajkowsky DM, Shao Z, Johnson AE, Tweten RK. Monomer-monomer interactions drive the prepore to pore conversion of a beta-barrel-forming cholesterol-dependent cytolysin. J Biol Chem. 2002;277:11597–11605. doi: 10.1074/jbc.M111039200. [DOI] [PubMed] [Google Scholar]
- 48.Hotze EM, Wilson-Kubalek EM, Rossjohn J, Parker MW, Johnson AE, Tweten RK. Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane beta-sheet from a prepore intermediate. J Biol Chem. 2001;276:8261–8268. doi: 10.1074/jbc.M009865200. [DOI] [PubMed] [Google Scholar]
- 49.Shepard LA, Shatursky O, Johnson AE, Tweten RK. The mechanism of assembly and insertion of the membrane complex of the cholesterol-dependent cytolysin perfringolysin O: Formation of a large prepore complex. Biochemistry. 2000;39:10284–10293. doi: 10.1021/bi000436r. [DOI] [PubMed] [Google Scholar]
- 50.Shatursky O, Heuck AP, Shepard LA, Rossjohn J, Parker MW, Johnson AE, Tweten RK. The mechanism of membrane insertion for a cholesterol dependent cytolysin: A novel paradigm for pore-forming toxins. Cell. 1999;99:293–299. doi: 10.1016/s0092-8674(00)81660-8. [DOI] [PubMed] [Google Scholar]
- 51.Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, Johnson AE, Tweten RK. Identification of a membrane-spanning domain of the thiolactivated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry. 1998;37:14563–14574. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- 52.Alouf JE. Cholesterol-binding cytolytic protein toxins. Int J Med Microbiol. 2000;290:351–356. doi: 10.1016/S1438-4221(00)80039-9. [DOI] [PubMed] [Google Scholar]
- 53.Prigent D, Alouf JE. Interaction of streptolysin O with sterols. Biochem Biophys Acta. 1976;433:422–428. doi: 10.1016/0005-2736(76)90511-3. [DOI] [PubMed] [Google Scholar]
- 54.Johnson MK, Geoffroy C, Alouf JE. Binding of cholesterol by sulfhydryl-activated cytolysins. Infect Immun. 1980;27:97–101. doi: 10.1128/iai.27.1.97-101.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iwamoto M, Ohno-Iwashita Y, Ando S. Role of the essential thiol group in the thiol-activated cytolysin from Clostridium perfringens. Eur J Biochem. 1987;167:425–430. doi: 10.1111/j.1432-1033.1987.tb13355.x. [DOI] [PubMed] [Google Scholar]
- 56.Nagy I, Ohno-Iwashita Y, Ohta M, Nagy V, Kitani K, Ando S, Imahori K. Effect of perfringolysin O on the lateral diffusion constant of membrane proteins of hepatocytes as revealed by fluorescence recovery after photobleaching. Biochim Biophys Acta. 1988;939:551–560. doi: 10.1016/0005-2736(88)90102-2. [DOI] [PubMed] [Google Scholar]
- 57.Ohno-Iwashita Y, Iwamoto M, Mitsui K, Ando S, Nagai Y. Protease nicked θ-toxin of Clostridium perfringens, a new membrane probe with no cytolytic effect, reveals two classes of cholesterol as toxin-binding sites on sheep erythrocytes. Eur J Biochem. 1988;176:95–101. doi: 10.1111/j.1432-1033.1988.tb14255.x. [DOI] [PubMed] [Google Scholar]
- 58.Ohno-Iwashita Y, Iwamoto M, Ando S, Mitsui K, Iwashita S. A modified q-toxin produced by limited proteolysis and methylation: a probe for the functional study of membrane cholesterol. Biochim Biophys Acta. 1990;1023:441–448. doi: 10.1016/0005-2736(90)90137-d. [DOI] [PubMed] [Google Scholar]
- 59.Ohno-Iwashita Y, Iwamoto M, Mitsui K, Ando S, Iwashita S. A cytolysin, theta-toxin, preferentially binds to membrane cholesterol surrounded by phospholipids with 18-carbon hydrocarbon chains in cholesterol-rich region. J Biochem (Tokyo) 1991;110:369–375. doi: 10.1093/oxfordjournals.jbchem.a123588. [DOI] [PubMed] [Google Scholar]
- 60.Ohno-Iwashita Y, Iwamoto M, Ando S, Iwashita S. Effect of lipidic factors on membrane cholesterol topology-mode of binding of theta-toxin to cholesterol in liposomes. Biochim Biophys acta. 1992;1109:81–90. doi: 10.1016/0005-2736(92)90190-w. [DOI] [PubMed] [Google Scholar]
- 61.Iwamoto M, Nakamura M, Mitsui K, Ando S, Ohno-Iwashita Y. Membrane disorganization induced by perfringolysin O (theta-toxin) of Clostridium perfringens - effect of toxin binding and self-assembly on liposomes. Biochim Biophys Acta. 1993;1153:89–96. doi: 10.1016/0005-2736(93)90279-9. [DOI] [PubMed] [Google Scholar]
- 62.Sekino-Suzuki N, Nakamura M, Mitsui KI, Ohno-Iwashita Y. Contribution of individual tryptophan residues to the structure and activity of theta-toxin (perfringolysin o), a cholesterol-binding cytolysin. Eur J Biochem. 1996;241:941–947. doi: 10.1111/j.1432-1033.1996.00941.x. [DOI] [PubMed] [Google Scholar]
- 63.Iwamoto M, Morita I, Fukuda M, Murota S, Ando S, Ohno-Iwashita Y. A biotinylated perfringolysin O derivative: a new probe for detection of cell surface cholesterol. Biochim Biophys Acta. 1997;1327:222–230. doi: 10.1016/s0005-2736(97)00061-8. [DOI] [PubMed] [Google Scholar]
- 64.Nakamura M, Sekino-Suzuki N, Mitsui K, Ohno-Iwashita Y. Contribution of tryptophan residues to the structural changes in perfringolysin O during interaction with liposomal membranes. J Biochem (Tokyo) 1998;123:1145–1155. doi: 10.1093/oxfordjournals.jbchem.a022054. [DOI] [PubMed] [Google Scholar]
- 65.Jacobs T, Cima-Cabal MD, Darji A, Mendez FJ, Vazquez F, Jacobs AA, Shimada Y, Ohno-Iwashita Y, Weiss S, de los Toyos JR. The conserved undecapeptide shared by thiol-activated cytolysins is involved in membrane binding. FEBS Lett. 1999;459:463–466. doi: 10.1016/s0014-5793(99)01297-1. [DOI] [PubMed] [Google Scholar]
- 66.Waheed AA, Shimada Y, Heijnen HF, Nakamura M, Inomata M, Hayashi M, Iwashita S, Slot JW, Ohno-Iwashita Y. Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts) Proc Natl Acad Sci U S A. 2001;98:4926–4931. doi: 10.1073/pnas.091090798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimada Y, Maruya M, Iwashita S, Ohno-Iwashita Y. The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur J Biochem. 2002;269:6195–6203. doi: 10.1046/j.1432-1033.2002.03338.x. [DOI] [PubMed] [Google Scholar]
- 68.Ohno-Iwashita Y, Iwamoto M, Mitsui K, Ando S, Nagai Y. Protease-nicked theta-toxin of Clostridium perfringens, a new membrane probe with no cytolytic effect, reveals two classes of cholesterol as toxin-binding sites on sheep erythrocytes. European journal of biochemistry / FEBS. 1988;176:95–101. doi: 10.1111/j.1432-1033.1988.tb14255.x. [DOI] [PubMed] [Google Scholar]
- 69.Heuck AP, Hotze E, Tweten RK, Johnson AE. Mechanism of membrane insertion of a multimeric b-barrel protein: Perfringolysin O creates a pore using ordered and coupled conformational changes. Molec Cell. 2000;6:1233–1242. doi: 10.1016/s1097-2765(00)00119-2. [DOI] [PubMed] [Google Scholar]
- 70.Flanagan JJ, Tweten RK, Johnson AE, Heuck AP. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry. 2009;48:3977–3987. doi: 10.1021/bi9002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rottem S, Cole RM, Habig WH, Barile MF, Hardegree MC. Structural characteristics of tetanolysin and its binding to lipid vesicles. J Bacteriol. 1982;152:888–892. doi: 10.1128/jb.152.2.888-892.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nelson LD, Johnson AE, London E. How interaction of perfringolysin O with membranes is controlled by sterol structure lipid structure and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction. J Biol Chem. 2008;283:4632–4642. doi: 10.1074/jbc.M709483200. [DOI] [PubMed] [Google Scholar]
- 73.Zitzer A, Westover EJ, Covey DF, Palmer M. Differential interaction of the two cholesterol-dependent membrane-damaging toxins streptolysin O and Vibrio cholerae cytolysin, with enantiomeric cholesterol. FEBS Lett. 2003;553:229–231. doi: 10.1016/s0014-5793(03)01023-8. [DOI] [PubMed] [Google Scholar]
- 74.Iwamoto M, Ohno-Iwashita Y, Ando S. Effect of isolated C-terminal fragment of theta-toxin (perfringolysin-O) on toxin assembly and membrane lysis. Eur J Biochem. 1990;194:25–31. doi: 10.1111/j.1432-1033.1990.tb19422.x. [DOI] [PubMed] [Google Scholar]
- 75.Shimada Y, Nakamura M, Naito Y, Nomura K, Ohno-Iwashita Y. C-terminal amino acid residues are required for the folding and cholesterol binding property of perfringolysin O, a pore-forming cytolysin. J Biol Chem. 1999;274:18536–18542. doi: 10.1074/jbc.274.26.18536. [DOI] [PubMed] [Google Scholar]
- 76.Tweten RK, Harris RW, Sims PJ. Isolation of a tryptic fragment from Clostridium perfringens q-toxin that contains sites for membrane binding and self-aggregation. J Biol Chem. 1991;266:12449–12454. [PubMed] [Google Scholar]
- 77.Soltani CE, Hotze EM, Johnson AE, Tweten RK. Specific protein-membrane contacts are required for prepore and pore assembly by a cholesterol-dependent cytolysin. J Biol Chem. 2007;282:15709–15716. doi: 10.1074/jbc.M701173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Soltani CE, Hotze EM, Johnson AE, Tweten RK. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc Natl Acad Sci U S A. 2007;104:20226–20231. doi: 10.1073/pnas.0708104105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagamune H, Ohnishi C, Katsuura A, Fushitani K, Whiley RA, Tsuji A, Matsuda Y. Intermedilysin a novel cytotoxin specific for human cells secreted by Streptococcus intermedius UNS46 isolated from a human liver abscess. Infect Immun. 1996;64:3093–3100. doi: 10.1128/iai.64.8.3093-3100.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wickham SE, Hotze EM, Farrand AJ, Polekhina G, Nero TL, Tomlinson S, Parker MW, Tweten RK. Mapping the intermedilysin-human CD59 receptor interface reveals a deep correspondence with the binding site on CD59 for complement binding proteins C8{alpha} and C9. The Journal of biological chemistry. 2011 doi: 10.1074/jbc.M111.237446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rollins SA, Sims PJ. The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. J Immunol. 1990;144:3478–3483. [PubMed] [Google Scholar]
- 82.Rollins SA, Zhao J, Ninomiya H, Sims PJ. Inhibition of homologous complement by CD59 is mediated by a species-selective recognition conferred through binding to C8 within C5b-8 or C9 within C5b-9. J Immunol. 1991;146:2345–2351. [PubMed] [Google Scholar]
- 83.Plumb ME, Sodetz JM. Proteins of the membrane attack complex. In: Volanakis JE, Frank MM, editors. The Human Complement System in Health and Disease. Vol. 1. Marcel Dekker, Inc.; New York: 1998. pp. 119–148. [Google Scholar]
- 84.Palmer M, Harris R, Freytag C, Kehoe M, Tranum-Jensen J, Bhakdi S. Assembly mechanism of the oligomeric streptolysin O pore: the early membrane lesion is lined by a free edge of the lipid membrane and is extended gradually during oligomerization. EMBO J. 1998;17:1598–1605. doi: 10.1093/emboj/17.6.1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 86.Feil SC, Rossjohn J, Rohde K, Tweten RK, Parker MW. Crystallization and preliminary X-ray analysis of a thiol-activated cytolysin. FEBS Lett. 1996;397:290–292. doi: 10.1016/s0014-5793(96)01200-8. [DOI] [PubMed] [Google Scholar]
- 87.Hadders MA, Beringer DX, Gros P. Structure of C8alpha-MACPF reveals mechanism of membrane attack in complement immune defense. Science. 2007;317:1552–1554. doi: 10.1126/science.1147103. [DOI] [PubMed] [Google Scholar]
- 88.Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, D’Angelo ME, Orlova EV, Coulibaly F, Verschoor S, Browne KA, Ciccone A, Kuiper MJ, Bird PI, Trapani JA, Saibil HR, Whisstock JC. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468:447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 89.Rosado CJ, Buckle AM, Law RH, Butcher RE, Kan WT, Bird CH, Ung K, Browne KA, Baran K, Bashtannyk-Puhalovich TA, Faux NG, Wong W, Porter CJ, Pike RN, Ellisdon AM, Pearce MC, Bottomley SP, Emsley J, Smith AI, Rossjohn J, Hartland EL, Voskoboinik I, Trapani JA, Bird PI, Dunstone MA, Whisstock JC. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317:1548–1551. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 90.Slade DJ, Lovelace LL, Chruszcz M, Minor W, Lebioda L, Sodetz JM. Crystal structure of the MACPF domain of human complement protein C8 alpha in complex with the C8 gamma subunit. J Mol Biol. 2008;379:331–342. doi: 10.1016/j.jmb.2008.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Palmer M, Saweljew P, Vulicevic I, Valeva A, Kehoe M, Bhakdi S. Membrane-penetrating domain of streptolysin O identified by cysteine scanning mutagenesis. J Biol Chem. 1996;271:26664–26667. doi: 10.1074/jbc.271.43.26664. [DOI] [PubMed] [Google Scholar]
- 92.Sekiya K, Satoh R, Danbara H, Futaesaku Y. A ring-shaped structure with a crown formed by streptolysin-O on the erythrocyte membrane. J Bacteriol. 1993;175:5953–5961. doi: 10.1128/jb.175.18.5953-5961.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mitsui K, Sekiya T, Okamura S, Nozawa Y, Hase J. Ring formation of perfringolysin O as revealed by negative stain electron microscopy. Biochim Biophys Acta. 1979;558:307–313. doi: 10.1016/0005-2736(79)90265-7. [DOI] [PubMed] [Google Scholar]
- 94.Jacobs T, Darji A, Frahm N, Rohde M, Wehland J, Chakraborty T, Weiss S. Listeriolysin O: cholesterol inhibits cytolysis but not binding to cellular membranes. Mol Microbiol. 1998;28:1081–1089. doi: 10.1046/j.1365-2958.1998.00858.x. [DOI] [PubMed] [Google Scholar]
- 95.Rossjohn J, Polekhina G, Feil SC, Morton CJ, Tweten RK, Parker MW. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J Mol Biol. 2007;367:1227–1223. doi: 10.1016/j.jmb.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell. 2005;121:247–256. doi: 10.1016/j.cell.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 97.Geoffroy C, Gaillard JL, Alouf JE, Berche P. Purification characterization and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect Immun. 1987;55:1641–1646. doi: 10.1128/iai.55.7.1641-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cossart P. Molecular and cellular basis of the infection by Listeria monocytogenes: an overview. Int J Med Microbiol. 2002;291:401–409. doi: 10.1078/1438-4221-00146. [DOI] [PubMed] [Google Scholar]
- 99.Portnoy DA, Auerbuch V, Glomski IJ. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol. 2002;158:409–414. doi: 10.1083/jcb.200205009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kayal S, Charbit A. Listeriolysin O: a key protein of Listeria monocytogenes with multiple functions. FEMS Microbiol Rev. 2006;30:514–529. doi: 10.1111/j.1574-6976.2006.00021.x. [DOI] [PubMed] [Google Scholar]
- 101.Bielecki J, Youngman P, Connelly P, Portnoy DA. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature. 1990;345:175–176. doi: 10.1038/345175a0. [DOI] [PubMed] [Google Scholar]
- 102.Portnoy DA, Tweten RK, Kehoe M, Bielecki J. The capacity of of listeriolysin O, streptolysin O and perfringolysin O to mediate growth of Bacillus subtilis within mammalian cells. Infect Immun. 1992;60:2710–2717. doi: 10.1128/iai.60.7.2710-2717.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jones S, Portnoy DA. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun. 1994;62:5608–5613. doi: 10.1128/iai.62.12.5608-5613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Draper RK, Simon MI. The entry of diphtheria toxin into the mammalian cell cytoplasm: evidence for lysosomal involvement. The Journal of cell biology. 1980;87:849–854. doi: 10.1083/jcb.87.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sandvig K, Olsnes S. Diphtheria toxin entry into cells is facilitated by low pH. The Journal of cell biology. 1980;87:828–832. doi: 10.1083/jcb.87.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Donovan JJ, Simon MI, Draper RK, Montal M. Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc Natl Acad Sci U S A. 1981;78:172–176. doi: 10.1073/pnas.78.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dorland RB, Middlebrook JL, Leppla SH. Effect of ammonium chloride on receptor-mediated uptake of diphtheria toxin by Vero cells. Exp Cell Res. 1981;134:319–327. doi: 10.1016/0014-4827(81)90432-8. [DOI] [PubMed] [Google Scholar]
- 108.Kagan BL, Finkelstein A, Colombini M. Diphtheria toxin fragment forms large pores in phospholipid bilayer membranes. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:4950–4954. doi: 10.1073/pnas.78.8.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blewitt MG, Chung LA, London E. Effect of pH on the conformation of diphtheria toxin and its implications for membrane penetration. Biochemistry. 1985;24:5458–5464. doi: 10.1021/bi00341a027. [DOI] [PubMed] [Google Scholar]
- 110.Collier RJ. Mechanism of membrane translocation by anthrax toxin: insertion and pore formation by protective antigen. J Appl Microbiol. 1999;87:283. doi: 10.1046/j.1365-2672.1999.00889.x. [DOI] [PubMed] [Google Scholar]
- 111.Schuerch DW, Wilson-Kubalek EM, Tweten RK. Molecular basis of Listeriolysin O pH-dependence. Proc Natl Acad Sci. 2005;102:12537–12542. doi: 10.1073/pnas.0500558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morrill JA, MacKinnon R. Isolation of a single carboxyl-carboxylate proton binding site in the pore of a cyclic nucleotide-gated channel. J Gen Physiol. 1999;114:71–83. doi: 10.1085/jgp.114.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tozawa K, Ohbuchi H, Yagi H, Amano T, Matsui T, Yoshida M, Akutsu H. Unusual pKa of the carboxylate at the putative catalytic position of the thermophilic F1-ATPase beta subunit determined by 13C-NMR. FEBS Lett. 1996;397:122–126. doi: 10.1016/s0014-5793(96)01155-6. [DOI] [PubMed] [Google Scholar]
- 114.Kawaminami S, Takahashi H, Ito S, Arata Y, Shimada I. A multinuclear NMR study of the active site of an endoglucanase from a strain of Bacillus. Use of Trp residues as structural probes. J Biol Chem. 1999;274:19823–19828. doi: 10.1074/jbc.274.28.19823. [DOI] [PubMed] [Google Scholar]
- 115.Davoodi J, Wakarchuk WW, Campbell RL, Carey PR, Surewicz WK. Abnormally high pKa of an active-site glutamic acid residue in Bacillus circulans xylanase. The role of electrostatic interactions. Eur J Biochem. 1995;232:839–843. [PubMed] [Google Scholar]
- 116.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 117.Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, Buckle AM, Voskoboinik I, Bird PI, Trapani JA, Whisstock JC, Dunstone MA. The MACPF/CDC family of pore-forming toxins. Cell Microbiol. 2008;10:1765–1774. doi: 10.1111/j.1462-5822.2008.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Esser AF. The membrane attack complex of complement. Assembly, structure and cytotoxic activity. Toxicology. 1994;87:229–247. doi: 10.1016/0300-483x(94)90253-4. [DOI] [PubMed] [Google Scholar]
- 119.Baran K, Dunstone M, Chia J, Ciccone A, Browne KA, Clarke CJ, Lukoyanova N, Saibil H, Whisstock JC, Voskoboinik I, Trapani JA. The molecular basis for perforin oligomerization and transmembrane pore assembly. Immunity. 2009;30:684–695. doi: 10.1016/j.immuni.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 120.Lovelace LL, Cooper CL, Sodetz JM, Lebioda L. Structure of human C8 protein provides mechanistic insight into membrane pore formation by complement. The Journal of biological chemistry. 2011 doi: 10.1074/jbc.M111.219766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 122.Huang Y, Qiao F, Abagyan R, Hazard S, Tomlinson S. Defining the CD59-C9 binding interaction. J Biol Chem. 2006;281:27398–27404. doi: 10.1074/jbc.M603690200. [DOI] [PubMed] [Google Scholar]
- 123.Lockert DH, Kaufman KM, Chang CP, Husler T, Sodetz JM, Sims PJ. Identity of the segment of human complement C8 recognized by complement regulatory protein CD59. J Biol Chem. 1995;270:19723–19728. doi: 10.1074/jbc.270.34.19723. [DOI] [PubMed] [Google Scholar]
- 124.Chang CP, Husler T, Zhao J, Wiedmer T, Sims PJ. Identity of a peptide domain of human C9 that is bound by the cell-surface complement inhibitor, CD59. J Biol Chem. 1994;269:26424–26430. [PubMed] [Google Scholar]
- 125.Husler T, Lockert DH, Kaufman KM, Sodetz JM, Sims PJ. Chimeras of human complement C9 reveal the site recognized by complement regulatory protein CD59. J Biol Chem. 1995;270:3483–3486. doi: 10.1074/jbc.270.8.3483. [DOI] [PubMed] [Google Scholar]
- 126.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]