

Abstract
The role played by the beta2-adrenergic receptor (β2AR) in regulating the level of T and B lymphocyte function has been studied for over half a century. During this time, we have learned that T and B lymphocytes express almost exclusively the β2AR, and that the level of expression on a specific lymphocyte subset differs due to epigenetic regulation by histone and DNA methylation. We have also learned that engagement of the β2AR on lymphocytes, by either norepinephrine or a selective pharmacologic ligand, regulates the level of lymphocyte activity differentially, depending on the time of receptor engagement in relation to the activation and differentiation state of the cell, the molecular signaling pathway activated, and the cytokine microenvironment. The challenge now is to determine if we understand enough about how this receptor functions on lymphocytes to predict the relevance of such regulation to overall immune homeostasis and the development/progression of human disease.
Keywords: Norepinephrine, beta2-adrenergic receptor, sympathetic nervous system, immune system, CD4+ T cell, Th1 cell, Th2 cell, B cell, antibody, cytokine
It was Norman Cousins who challenged all of us to determine the mechanism by which he survived two major illnesses, ankylosing spondylitis and a near-fatal heart attack, simply by harnessing the power of human emotions. When he was taken to the hospital for the heart attack, he said, “…I want you to know that you're looking at the darnedest healing machine that's ever been wheeled into this hospital”. He believed that the mind and body were connected somehow and that each could help the other to heal. Exactly how this communication occurred to bring about healing was a mystery to him, but he knew it was real and that an understanding of the mechanism would lead to cures that were unimaginable. For the past 50 years, a number of researchers have accepted this challenge to study this connection between the mind and body, in particular, to determine the mechanism responsible for mediating the effect on health.
A fine balance exists in the body to maintain health and overall homeostasis. This balance is maintained by the proper functioning of every organ system. One participant in this balance equation involves the immune system, which evolved to protect us not only from the environment around us, which is filled with infection-causing microorganisms, allergens, and cancer-promoting agents, but also from the environment within us, which can develop transformed cells that cause cancer and autoimmune disease (Figure 1). It is essential that a mechanism exists to coordinate these organ systems to respond immediately to a threat and to bring the organ systems back to normal after the crisis subsides. One key mechanism responsible for such coordination involves the autonomic nervous system, which serves as the messenger from the mind to the body for all organ systems, including the immune system [Figure 2; (Ader et al., 1990; Nance and Sanders, 2007)]. Another key mechanism responsible for such coordination involves cytokines, which serve as the messenger to the brain from the activated immune cells that are responding to an external or internal threat (Besedovsky et al., 1983). These two mechanisms of communication between the brain and immune system are now known to play a major role in maintaining a protective balance in the body to maintain health and homeostasis.
Fig. 1.
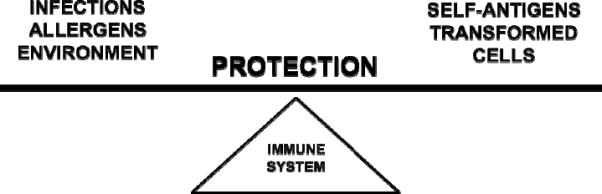
Immune system protection is a balancing act. The immune system keeps our health in balance by protecting us against not only external threats such as infectious microorganisms that cause disease, allergens that precipitate allergy and asthma, and environmental agents that cause disease/cancer, but also internal threats such as self-antigens that cause autoimmune disease and transformed cells that cause cancer.
Fig. 2.

The brain-immune communication pathway. Activation of the immune system allows for communication with the brain via the release of cytokines from activated immune cells and/or the trafficking of activated immune cells into the brain. Activation of selective regions in the brain allows for communication with the immune system via activation of the sympathetic nervous system centrally and the release of the neurotransmitter norepinephrine from sympathetic nerve terminals that penetrate lymphoid tissue in the periphery.
EXPRESSION OF THE β2AR ON T AND B LYMPHOCYTES
The autonomic nervous system is composed of two distinct systems, namely the symapathetic and parasympathestic nervous systems that secrete norepinephrine and acetylcholine, respectively. The parenchyma of lymphoid organs are innervated primarily by sympathetic nerve fibers that release norepinephrine within hours of antigen recognition by immune cells (Felten et al., 1985). The released norepinephrine binds to either alpha- or beta-adrenergic receptors that are expressed by immune cells, although T and B lymphocytes express the beta2-adrenergic receptor (β2AR) almost exclusively (Sanders et al., 2001). Engagement of the β2AR activates a cascade of signaling intermediates, including cAMP and protein kinase A, which leads to the phosphorylation of cellular proteins, including transcription factors that mediate gene expression. For the purposes of this review, only B cells and CD4+ T cells will be discussed. Murine B cells express the β2AR, as do naïve CD4+ T cells and effector Th1 cells, while effector Th2 cells do not [Figure 3; (Sanders et al., 1997)]. Differential expression in human effector CD4+ T cells remains unclear, mainly due to the difficulty in generating and isolating a homogenous population of cells that secrete just one cytokine profile. Nonetheless, the differential expression found in murine cells is due to epigenetic factors involving histone modifications and DNA methylation. Epigenetic mechanisms do not cause any change in the DNA itself, but involve postsynthetic modifications to DNA and/or DNA-associated histones that remodel chromatin, are heritable, and occur during and after early development. Th1 cells that develop from a naïve CD4+ T cell increase the level of β2AR expression as compared to naïve cells via a mechanism that involves histone-3 & -4 acetylation and histone-3 lysine-4 methylation (McAlees and Sanders, 2009). In contrast, Th2 cells that develop from a naïve CD4+ T cell dramatically repress the level of β2AR expression as compared to naïve cells via a mechanism that involves histone-3 & -4 acetylation, histone-3 Lysine-4 methylation, histone-3 lysine-9 & -27 methylation, and DNA methylation. Thus, an epigenetic mechanism exists to explain the difference in effector T cell expression of the β2AR, although the clinical relevance for differential expression remains unclear, but will be discussed below.
Fig. 3.

Epigenetic factors influence beta2-adrenergic receptor expression by murine CD4+ T cell subsets. Naïve CD4+ T cells express the β2AR protein on the surface and a level of histone acetylation and methylation within the beta2-adrenergic receptor gene promotor. As naïve cells differentiate to a Th1 or Th2 cell that express and repress expression, respectively, the level of histone acetylation and methylation change. The pattern and level of specific histones involved in expression vs. repression is determined by which T cell subset develops. DNA methylation is also involved in beta2-adrenergic receptor repression in Th2 cells.
EFFECT OF β2AR ENGAGEMENT ON CD4+ T CELL FUNCTION
The effect of β2AR engagement on an activated naïve CD4+ T cell versus an effector Th1 cell was found to be dependent on different factors. β2AR engagement on an activated naïve T cell cultured in the presence of IL-12 induced more IFN-γ to be produced in comparison to naïve cells activated alone without β2AR engagement [Figure 4; (Swanson et al., 2001)]. This increase in IFN-γ was due to a higher level of IFN-γ being secreted per cell by the resulting Th1 cells that developed, as opposed to more Th1 cells being made. As the concentration of IL-12 increased in the presence of β2AR engagement, so did the amount of IFN-g secreted per Th1 cell that developed. A similar result was caused by β2AR engagement on an activated naïve T cell cultured in the presence of IL-4, which caused a change in the amount of IL-4 secreted per cell by the Th2 cells that developed. However, in contrast, β2AR engagement on an activated naïve T cell cultured with low levels of IL-4 resulted in Th2 cells that secreted a higher amount of IL-4, while naïve cells cultured with moderate or high levels of IL-4 resulted in Th2 cells that secreted normal or a lower amount of IL-4, respectively [Figure 4, unpublished results]. If the β2AR was engaged on an activated effector Th1 cell, the amount of IFN-γ secreted, in comparison to Th1 cells that were activated alone without β2AR engagement, depended on the time of β2AR engagement in relation to the time of Th1 cell activation. If β2AR engagement occurred before, during, or after cell activation, respectively, IFN-γ was less, unchanged, or higher than control cells that were activated alone. [Figure 5 (Ramer-Quinn et al., 1997; Ramer-Quinn et al., 2000) and Unpublished results]. As predicted, when an effector Th2 cell was activated in the presence of a β2AR ligand, the level of IL-4 produced was the same as that produced by Th2 cells that were activated alone. Thus, the effect of β2AR engagement on CD4+ T cells is not the same for each CD4+ T cell subset or culture condition.
Fig. 4.

Beta2-adrenergic receptor engagement on a naïve CD4+ T cells during differentiation influences the level of cytokine produced by the resulting Th1 or Th2 cell. The level of IFN-g produced by the resulting Th1 cells increases in a manner that is not only dependent on the concentration of IL-12 that was available during naïve T cell differentiation, but also depends on an increase in the amount of IFN-g produced per cell as opposed to an increase in the number of Th1 cells that develop. The level of IL-4 produced by the resulting Th2 cells appears to increase when the concentration of IL-4 available during naïve T cell differentiation is low, but decreases as the concentration of IL-4 available during naïve T cell differentiation is elevated.
Fig. 5.

Beta2-adrenergic receptor engagement on a differentiated Th1 cell influences the level of cytokine produced depending on the time of beta2-adrenergic receptor engagement in relation to the time of cell exposure to antigen. Th1 cell exposure to antigen prior to beta2-adrenergic receptor engagement decreases the amount of IFN-g produced, while Th1 cells receiving concurrent exposure/engagement or engagement after antigen exposure yields either no change or increased IFN-g production, respectively.
POTENTIAL CLINICAL RELEVANCE FOR DIFFERENTIAL β2AR EXPRESSION ON CD4+ T CELLS
Does this mean that these findings cannot be translated clinically? The answer is that they will be translatable after we understand more about the mechanisms responsible for these differences so that we can apply these findings to clinical situations in a rational manner. For example, diseases that appear to be mediated by changes in either Th1 cell IFN-γ production, including autoimmune diseases such as multiple sclerosis, and infectious diseases such as those caused by viruses, might be either prevented or delayed by β2AR engagement or blockade during either naïve CD4+ T cell differentiation and/or the effector phase of a Th1 response. We will also need to understand if β2AR engagement either by norepinephrine or a drug agonist is responsible for contributing to a change in either naïve CD4+ T cell differentiation and/or effector Th1 cell function that precipitates a change in disease development and/or progression. Likewise, although Th2 cell activity is unaffected by β2AR ligand exposure due to the absence of receptor expression, an understanding of the epigenetic mechanisms responsible for β2AR repression in Th2 cells, as well as for enhanced expression in Th1 cells, may be useful for application to clinical situations in which expression of more or less of the β2AR might be helpful. We can only speculate that a Th2 cell that expresses the β2AR might respond similarly to Th2 cells exposed to cAMP-elevating drugs, which have been reported to increase IL-4 production. Such a manipulation of β2AR expression in a Th2 cell might be relevant during a peripheral motor nerve injury response where Th2 cells and the IL-4 they secrete have been shown to be essential for the maintenance of motoneuron survival in the brain so that effective nerve repair can proceed (Serpe et al., 2003). An understanding of the epigenetic mechanism responsible for β2AR repression might also be applied to repress this receptor on naïve CD4+ T cells during differentiation to prevent the development of Th1 cells that produce too much IFN-γ. Alternatively, it is possible that a disease process itself might change the level of β2AR expression on a naïve or effector CD4+ T cell and that this change contributes to a change in cell activity and disease development/progression. In such a situation, an understanding of how to regulate β2AR expression back to normal using epigenetic approaches would be useful. Thus, a deeper understanding of β2AR expression on CD4+ T cells during health and disease, as well as how engagement of the receptor affects T cell activity, is needed before we can truly apply this knowledge in a rational manner to clinical situations.
EFFECT OF β2AR ENGAGEMENT ON B CELL FUNCTION
Interestingly, the lack of β2AR expression on a Th2 cell provided a means to determine the direct effect of β2AR engagement on a B cell involved in a Th2 cell-dependent IgG1 or IgE response, without the complication of a simultaneous direct effect on the Th2 cell (Figure 6). Although a number of reports exist as to the effect of β2AR ligand exposure on a T cell-dependent antibody response (Kohm and Sanders, 2001), the mechanism by which β2AR engagement directly affected either T or B cell activity could not be ascertained. However, using the Th2 model system, whatever change occurred in B cell function would be due to a direct effect of β2AR engagement on the B cell alone. An in vivo study in which mice were depleted of norepinephrine prior to the adoptive transfer of antigen-specific Th2 and B cells showed that the loss of norepinephrine prevented the expression of a normal IgG1 after antigen administration (Kohm and Sanders, 1999). Similar findings using intact mice administered an alpha- or beta-adrenergic antagonist showed that the βAR was responsible for mediating the norepinephrine effect. Histological examination of the spleens from the norepinephrine-depleted mice revealed that there was less cellular expansion and that the germinal centers in these mice, which are essential for IgG1 production to occur, did not develop in comparison to norepinephrine-intact mice. When the spleen cells from these mice were phenotyped, only CD86 (also known as B7-2) was found to be expressed at a lower level in the norepinephrine-depleted mice administered antigen. Thus, since CD86 has been reported to be involved in germinal center formation, it became important to now understand how the expression of this molecule on a B cell was regulated by norepinephrine and β2AR engagement, especially since this antibody isotype plays a major role in the clearance of bacterial antigens such as Streptococcus pneumoniae.
Fig. 6.

The effect of beta2-adrenergic receptor agonist exposure on a Th2 cell-dependent antibody response is due to the direct effect of β2AR engagement on the B cell alone. The B cell expresses the β2AR, while the Th2 cell does not. The resulting antibody isotype produced by a B cell that receives help from the Th2 cell will be either IgG1, which is important for the neutralization of antigens, or IgE, which is involved in allergy/asthma. The key cell surface molecules involved in the physical interaction between the B cell and Th2 cell are shown, as is the IL-4 that is produced by the Th2 cell to direct B cell switching to either IgG1 or IgE production.
MECHANISM FOR THE β2AR-INDUCED UPREGULATION OF CD86 AND IgG1 IN B CELLS
In the resting state, B cells express a very low level of CD86, which increases after antigen exposure and crosslinking of the B cell receptor. A β2AR agonist was also found to increase CD86 expression on B cells and, in combination with antigen, increased CD86 expression in an additive manner (Kasprowicz et al., 2000; Kohm et al., 2002). The relevance of this effect was shown in vivo using the norepinephrine-depleted antigen-specific adoptive transfer model (Kohm et al., 2002). In these mice, serum IgG1 increased after the administration of CD40L and IL-4 to activate B cells, and was further enhanced after the administration of an anti-CD86 antibody or terbutaline alone. When both CD86 and the β2AR were engaged on an activated B cell, the level of serum IgG1 was enhanced in an additive manner. These results suggested that CD86 delivered a signal directly to the B cell that may be a key regulatory molecule for mediating the effect of β2AR engagement on a B cell on the IgG1 response, even though CD86 direct signaling has been thought to be impossible due to the short, tyrosine-deficient cytoplasmic domain. It was possible that the direct effect of β2AR engagement on an activated B cell to increase CD86 expression on the B cell surface might have enhanced the IgG1 response indirectly, by allowing for increased B cell interaction with Th2 cells through CD86-CD28 interaction, resulting in Th2 cells becoming activated to secrete a higher level of IL-4 that would increase IgG1 switching. This possibility was tested by the introduction of an antibody against CD86 to prevent the CD86-CD28 interaction. The blockade prevented the IL-4 increase that was dependent on CD86 engagement of CD28 on the Th2 cell surface, but was unable to prevent the effect on the B cell to produce a higher level of IgG1 (Kasprowicz et al., 2000), suggesting that the engagement of CD86 on a B cell induced a signal in the B cell to directly affect the level of IgG1 produced via a mechanism that was independent of it's effect on the Th2 cell. Thus, it became important to understand the signaling pathways that were activated by both the β2AR and CD86, as well as how the signaling pathways mediated the increase in IgG1.
The β2AR- and CD86-mediated increase in IgG1 was due to an increase in the amount of IgG1 secreted per B cell, and not to an increase in the number of cells secreting IgG1, ruling out an effect on switching (Kasprowicz et al., 2000; Podojil and Sanders, 2003). The increase in IgG1 per cell was due to an increase in 3'-IgH enhancer activity and the rate of IgG1 mRNA produced (Podojil et al., 2004; Podojil and Sanders, 2003). The signaling pathways activated by β2AR engagement involved cAMP-PKA-CREB, while CD86 engagement involved the activation of two pathways, namely Lyn-CD19-Akt-NF-kB p50–p65 and PLCγ2α-PKC-p65 phosphorylation [Figure 7; and (Kin and Sanders, 2007; Kin and Sanders, 2006)]. Both pathways were found to converge when CREB increased the level of phosphorylated co-activator protein OCA-B and when NFkB p50–p65 increased the transcription of the B cell-specific transcription factor Oct-2, which then interacted with OCA-B. The increased level of OCA-B/Oct-2 resulted in more complexes being translocated to the nucleus to bind to the 3'-IgH enhancer, which increased the rate of enhancer activity and the rate of IgG1 mRNA production. Thus, the additive increase in IgG1 induced by simultaneous β2AR and CD86 engagement on a primed B cell was due to the convergence of the two separate signaling pathways to mediate the activation of a protein complex that regulated 3'-IgH enhancer activity.
Fig. 7.

The beta2-adrenergic receptor and CD86 signaling pathways converge to induce an increase in the level of IgG1 produced by an activated B cell. The β2AR engagement on an activated B cell activates cAMP/PKA/ CREB, while CD86 engagement on an activated B cell activates lyn kinase, CD19, Akt, IkB, and NFkB. The signaling pathway activated by β2AR and CD86 engagement cause an increase in the co-activator protein OCA-B and the transcription factor Oct-2, respectively. OCA-B and Oct-2 interact and translocate to the nucleus and bind to the 3'IgH-enhancer to increase the rate of IgG1 transcription, which increases the level of IgG1 produced by the cell. The increase in IgG1 is not due to an increase in the number of naïve B cell switching to the production of IgG1.
Yet, understanding the signaling intermediates activated by both the β2AR and CD86 to enhance the IgG1 response was incomplete because the link between CD86 and Lyn/ PLCγ2α activation was unclear. Both Lyn and PLCγ2α activation requires their interaction with protein tyrosine residues. CD86 is devoid of such tyrosine residues and is unable to directly activate Lyn and PLCγ2α. Therefore, an adaptor/scaffolding protein must exist to allow for a link between CD86 engagement and Lyn/ PLCγ2α activation. Recent data show that two proteins appear to be involved, namely prohibitin-1 and prohibitin-2 (Manuscript In Preparation). In addition, the cytoplasmic domain of CD86 appears to also be necessary for CD86 function, likely due to the presence of serine residues that may need to be phosphorylated for the docking of some other proteins involved as adaptor/scaffolding proteins. In summary, β2AR engagement on an primed B cell activates a series of signaling intermediates that lead to increased OCA-B and CD86 expression. CD86 engagement on a primed B cell activates signaling intermediates that lead to increased Oct-2 expression. When concurrent β2AR and CD86 engagement occurs on a primed B cell, the increased level of OCA-B and Oct-2 interact and bind to the 3'-IgH enhancer to increase enhancer activity and the rate of IgG1 mRNA expression. Thus, the increase in IgG1 measured after β2AR engagement on a B cell is due to an increase in the rate of mature IgG1 transcription.
POTENTIAL CLINICAL RELEVANCE FOR THE β2AR-INDUCED UPREGULATION OF CD86 AND IgG1 IN B CELLS
These findings will be translatable after we understand more about the mechanisms responsible for these differences so that we can apply these findings to clinical situations in a rational manner. For example, the level of antibody produced in vivo by a B cell in response to a T cell-dependent antigen is critical for providing protection against pathogens and assuring host survival. One report has shown that the level of antibody produced by individuals immunized with Streptococcus pneumoniae positively correlated with the level of protection afforded against the microorganism, i.e., a 2- to 3-fold increase in total serum IgG levels allowed for a 3- to 9-fold increase in protection against infection. This finding in humans supports the premise that CD86 signaling in a B cell may be relevant clinically. If an increase in CD86 expression on the B cell functions as part of a normal IgG response to promote an increase in the level of IgG produced, then individuals that fail to upregulate expression of CD86 on their B cells might be at risk for infection. If true, the ability of a B cell to increase expression of CD86 upon activation might predict the potential effectiveness of vaccination protocols. Conversely, over expression of CD86 on a B cell may lead to an exacerbated IgG response, increasing the potential to develop an autoimmune reaction. Thus, the ability of CD86 to directly regulate the level of an IgG1 response 2- to 3-fold may be relevant clinically and, therefore, understanding the mechanism by which CD86 signals within a B cell to regulate the level of an IgG1 response will provide us with potential molecular targets for therapeutic interventions to selectively regulate the IgG1 response positively or negatively.
MECHANISM FOR THE β2AR-INDUCED UPREGULATION OF IgE IN B CELLS
During a Th2-dependent response, some B cells will switch to IgG1 and some to IgE, which is the antibody isotype that contributes to the development of allergy and asthma. Engagement of the β2AR on an activated B cell in the presence of IL-4 leads to an increase in IgE that is cAMP and PKA-dependent. As opposed to the β2AR-induced increase in IgG1, which is also cAMP and PKA-dependent, the increase in IgE is not dependent on CREB activation, but is dependent on the activation of p38MAPK (Pongratz et al., 2006). However, the link between PKA and p38MAPK had to be via an indirect mechanism since p38MAPK activation requires tyrosine phosphorylation, which cannot be mediated by PKA that phosphorylates only serines and threonines. In T and B cells, unphosphorylated p38MAPK interacts with the phosphatase hematopoietic protein tyrosine phosphatase (HePTP), which regulates the available pool of p38MAPK that can be activated within the cell (McAlees and Sanders, 2009). Phosphorylation of a specific serine residue on HePTP causes the release of p38MAPK, making it available for phosphorylation and activation by tyrosine kinases activated by cell activation, e.g., the MAPK pathway activated by CD40 engagement on a B cell. Therefore, the presence of HePTP in a B cell provides the mechanism by which β2AR-induced PKA activation allows for an increase in the level of p38MAPK phosphorylation, which is linked to the β2AR-induced increase in IgE (Figure 8).
Fig. 8.

Beta2-adrenergic receptor engagement on a B cell that will make IgE uses a unique signaling pathway to increase the level of IgE produced. The β2AR engagement on an activated B cell that will switch to IgE production activates a signaling pathway involving cAMP/PKA and hematopoietic protein tyrosine phosphatase (HePTP), which leads to an increase in the level of p38MAPK phosphorylation. Downstream events affected by the β2AR-induced increase in p38mAPK involve an increase in the synthesis of CD23 and an increase in the level of CD23 cleaved to produce soluble CD23 (sCD23), which feedback on the B cell to increase the rate of IgE transcription and amount of IgE produced per cell. The increase in CD23 cleavage may be mediated via a β2AR-induced effect to either increase the localization of the CD23 sheddase ADAM10 within the plasma membrane to be in close association with CD23 and/or increase the shuttling of both CD23 and ADAM10 into exosomes that are secreted from the cell to release cleaved CD23. The increase in IgE is not due to an increase in the number of naïve B cell switching to the production of IgE.
The increase in p38MAPK activation after β2AR engagement in a B cell is known to be responsible for increasing the level of CD23 and soluble CD23 (sCD23), which is directly related to the regulation of the amount of IgE produced. However, although β2AR engagement increased the level of CD23 mRNA/protein intracellularly and the level of sCD23, an increase in the level of CD23 expressed on the cell membrane was not found. This finding suggested that β2AR engagement might affect the mechanism responsible for regulating CD23 cleavage, which can occur either at the cell membrane or on exosomes. The primary enzyme responsible for CD23 cleavage is the sheddase ADAM10 (Mathews et al.), which may serve as a target for signaling intermediates activated after β2AR engagement on a B cell. This possibility is currently being investigated, and preliminary results suggest that β2AR engagement enhances the level of sCD23 by increasing the localization of CD23 and ADAM10 to exosomes, which creates a microenvironment in which CD23 can be cleaved and released extracellularly (Manuscript In Preparation). Thus, the pathway linking β2AR engagement to an increase in IgE involves an increase in the activation of at least two key signaling intermediates, HePTP and ADAM10, which allows for the increase in p38MAPK activation and CD23 cleavage, respectively.
POTENTIAL CLINICAL RELEVANCE FOR β2AR-INDUCED UPREGULATION OF IgE IN B CELLS
The clinical application for these findings relate primarily to conditions of allergy and allergic asthma, which are both linked to an increase in IgE. Not only does β2AR engagement on a B cell occur when norepinephrine is released in response to allergens under normal physiological conditions, but also the released norepinephrine participates in the generation of a normal level of IgE and lung pathology, as evidenced in mice depleted of norepinephrine prior to antigen exposure (Pongratz et al., 2006). Also, norepinephrine release and β2AR engagement occurs at a higher level when an asthmatic individual either experiences stress and/or uses an inhaler containing a β2AR agonist drug. Individuals with asthma use a β2AR agonist drug to stimulate the β2AR on the smooth muscle cells of the lung to relieve bronchoconstriction. However, the beneficial effect attained from using these drugs lessens over time. The findings discussed above suggest a potential mechanism to explain the loss of β2AR agonist efficacy over time, namely that β2AR engagement on B cells in the lung would increase the level of IgE produced over time, which in turn would exacerbate allergic asthma symptoms and intensify bronchoconstriction to a point where β2AR agonist drugs were no longer effective. It is interesting that corticosteroids are co-administered with a β2AR agonist in an effort to suppress immune cell activity and lessen the level of inflammation in the lung. The findings described above would suggest that the corticosteroids may suppress the ability of B cells to produce excess IgE that is induced by β2AR overstimulation on a B cell. Also, allergic asthma patients under high levels of stress during an asthmatic attack are reported to experience exacerbated asthma symptoms, which we predict is precipitated by a similar mechanism involving higher levels of IgE that are produced subsequent to β2AR overstimulation on a B cell by a higher level of norepinephrine released by stress.
SUMMARY
In summary, the role played by the β2AR in regulating the level of T and B lymphocyte function is difficult to summarize simply because the mechanisms involved in mediating the effects are different and complex. The challenge now is to determine if we understand enough about how this receptor functions on T and B lymphocytes to predict the relevance of such regulation to overall immune homeostasis and the development/progression of human disease. Norman Cousins wrote in his book Anatomy of an Illness, “Some people don't really know enough to make a pronouncement of doom on a human being.” Perhaps we still do not know enough to doom individuals with autoimmunity, infectious diseases, allergies, and asthma to limitations on living their life to the fullest without first exploring the many diverse mechanisms that might be responsible for the development and exacerbation of these clinical conditions. The ability of neurotransmitters and hormones to affect immune system function, which is so critically linked to the development and exacerbation of these clinical conditions, is one of these diverse mechanisms that needs to be actively examined so that we can rationally use this knowledge to help improve the quality of life.
RESEARCH HIGHLIGHT.
Various mechanisms are responsible for the regulation of T and B lymphocyte activity and function following beta2-adrenergic receptor engagement.
ACKNOWLEDGEMENTS
I am forever indebted to the dedication of the graduate and postdoctoral trainees, as well as my esteemed colleagues and collaborators, who have participated in the research presented in this article. Without them, as well as the generous funding from the NIH (AI37326 and AI47420), none of these discoveries would have been possible.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES CITED
- Ader R, Felten D, Cohen N. Interactions between the brain and the immune system. Ann.Rev.Pharmacol.Toxicol. 1990;30:561–602. doi: 10.1146/annurev.pa.30.040190.003021. [DOI] [PubMed] [Google Scholar]
- Besedovsky H, Del Rey A, Sorkin E, Da Prada M, Burri R, Honegger C. The immune response evokes changes in brain noradrenergic neurons. Science. 1983;221:564–566. doi: 10.1126/science.6867729. [DOI] [PubMed] [Google Scholar]
- Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S. Noradrenergic and peptidergic innervation of lymphoid tissue. J.Immunol. 1985;135(2):755s–765s. [PubMed] [Google Scholar]
- Kasprowicz DJ, Kohm AP, Berton MT, Chruscinski AJ, Sharpe AH, Sanders VM. Stimulation of the B cell receptor, CD86 (B7-2), and the beta 2-adrenergic receptor intrinsically modulates the level of IgG1 and IgE produced per B cell. J.Immunol. 2000;165:680–690. doi: 10.4049/jimmunol.165.2.680. [DOI] [PubMed] [Google Scholar]
- Kin N, Sanders V. CD86 regulates IgG1 production via a CD19-dependent mechanism. J Immunol. 2007;179:1516–1523. doi: 10.4049/jimmunol.179.3.1516. [DOI] [PubMed] [Google Scholar]
- Kin NW, Sanders VM. CD86 Stimulation on a B Cell Activates the Phosphatidylinositol 3-Kinase/Akt and Phospholipase C{gamma}2/Protein Kinase C{alpha}beta Signaling Pathways. J Immunol. 2006;176:6727–6735. doi: 10.4049/jimmunol.176.11.6727. [DOI] [PubMed] [Google Scholar]
- Kohm AP, Mozaffarian A, Sanders VM. B cell receptor- and beta-2-adrenergic receptor-induced regulation of B7-2 (CD86) expression in B cells. J.Immunol. 2002;168:6314–6322. doi: 10.4049/jimmunol.168.12.6314. IgG1. [DOI] [PubMed] [Google Scholar]
- Kohm AP, Sanders VM. Suppression of antigen-specific Th2 cell-dependent IgM and IgG1 production following norepinephrine depletion in vivo. J.Immunol. 1999;162:5299–5308. [PubMed] [Google Scholar]
- Kohm AP, Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. 2001;53:487–525. [PubMed] [Google Scholar]
- Mathews JA, Gibb DR, Chen BH, Scherle P, Conrad DH. CD23 Sheddase A disintegrin and metalloproteinase 10 (ADAM10) is also required for CD23 sorting into B cell-derived exosomes. J Biol Chem. 285:37531–37541. doi: 10.1074/jbc.M110.141556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlees JW, Sanders VM. Hematopoietic protein tyrosine phosphatase mediates beta2-adrenergic receptor-induced regulation of p38 mitogen-activated protein kinase in B lymphocytes. Mol Cell Biol. 2009;29:675–686. doi: 10.1128/MCB.01466-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007) Brain Behav Immun. 2007;21:736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podojil JR, Kin NW, Sanders VM. CD86 and beta2-adrenergic receptor signaling pathways, respectively, increase Oct-2 and OCA-B Expression and binding to the 3'-IgH enhancer in B cells. J.Biol.Chem. 2004;279:23394–23404. doi: 10.1074/jbc.M313096200. [DOI] [PubMed] [Google Scholar]
- Podojil JR, Sanders VM. Selective regulation of mature IgG1 transcription by CD86 and beta2-adrenergic receptor stimulation. J.Immunol. 2003;170:5143–5151. doi: 10.4049/jimmunol.170.10.5143. [DOI] [PubMed] [Google Scholar]
- Pongratz G, McAlees JW, Conrad DH, Erbe RS, Haas KM, Sanders VM. The Level of IgE Produced by a B Cell Is Regulated by Norepinephrine in a p38 MAPK- and CD23-Dependent Manner. J Immunol. 2006;177:2926–2938. doi: 10.4049/jimmunol.177.5.2926. [DOI] [PubMed] [Google Scholar]
- Ramer-Quinn DS, Baker RA, Sanders VM. Activated Th1 and Th2 cells differentially express the beta-2-adrenergic receptor: A mechanism for selective modulation of Th1 cell cytokine production. J.Immunol. 1997;159:4857–4867. [PubMed] [Google Scholar]
- Ramer-Quinn DS, Swanson MA, Lee WT, Sanders VM. Cytokine production by naive and primary effector CD4(+) T cells exposed to norepinephrine. Brain Behav.Immun. 2000;14:239–255. doi: 10.1006/brbi.2000.0603. [DOI] [PubMed] [Google Scholar]
- Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta-2-adrenergic receptor by Th1 and Th2 clones: Implications for cytokine production and B cell help. J.Immunol. 1997;158:4200–4210. [PubMed] [Google Scholar]
- Sanders VM, Kasprowicz DJ, Kohm AP, Swanson MA. In: Neurotransmitter receptors on lymphocytes and other lymphoid cells. Ader R, Felten D, Cohen N, editors. Psychoneuroimmunology Academic Press; San Diego,CA: 2001. pp. 161–196. [Google Scholar]
- Serpe CJ, Coers S, Sanders VM, Jones KJ. CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun. 2003;17:393–402. doi: 10.1016/s0889-1591(03)00028-x. [DOI] [PubMed] [Google Scholar]
- Swanson MA, Lee WT, Sanders VM. IFN-gamma Production by Th1 Cells Generated from Naive CD4(+) T Cells Exposed to Norepinephrine. J. Immunol. 2001;166:232–240. doi: 10.4049/jimmunol.166.1.232. [DOI] [PubMed] [Google Scholar]