

Abstract
AIM: To investigate the anti-fibrosis effect of IκB kinase-beta inhibitor (IKK2 inhibitor IMD0354) in liver fibrosis.
METHODS: Twenty male C57BL6 mice were divided into four groups. Five high-fat fed mice were injected with lipopolysaccharide (LPS, 10 mg/kg) intraperitoneally and five high-fat fed mice were without LPS injection to build models of liver injury, and the intervention group (five mice) was injected intraperitoneally with IKK2 inhibitor (IMD 30 mg/kg for 14 d), while the remaining five mice received a normal diet as controls. Hepatic function, pathological evaluation and liver interleukin-6 (IL-6) expression were examined. Western blotting and real-time polymerase chain reaction were used to detect the expressions of nuclear factor-κB (NF-κB), alpha-smooth muscle actin (α-SMA), tumor growth factor-beta1 (TGF-β1), tumor necrosis factor-alpha (TNF-α), typeIand type III collagen proteins and mRNA.
RESULTS: A mouse model of liver injury was successfully established, and IMD decreased nuclear translocation of NF-κB p65 in liver cells. In the IMD-treated group, the levels of alanine aminotransferase (103 ± 9.77 μ/L vs 62.4 ± 7.90 μ/L, P < 0.05) and aminotransferase (295.8 ± 38.56 μ/L vs 212 ± 25.10 μ/L, P < 0.05) were significantly decreased when compared with the model groups. The histological changes were significantly ameliorated. After treatment, the expressions of IL-6 (681 ± 45.96 vs 77 ± 7.79, P < 0.05), TGF-β1 (Western blotting 5.65% ± 0.017% vs 2.73% ± 0.005%, P < 0.05), TNF-α (11.58% ± 0.0063% vs 8.86% ± 0.0050%, P < 0.05), typeIcollagen (4.49% ± 0.014% vs 1.90% ± 0.0006%, P < 0.05) and type III collagen (3.46% ± 0.008% vs 2.29% ± 0.0035%, P < 0.05) as well as α-SMA (6.19 ± 0.0036 μ/L vs 2.16 ± 0.0023 μ/L, P < 0.05) protein and mRNA were downregulated in the IMD group compared to the fibrosis control groups (P < 0.05).
CONCLUSION: IKK2 inhibitor IMD markedly improved non-alcoholic fatty liver disease in mice by lowering NF-κB activation, which could become a remedial target for liver fibrosis.
Keywords: Liver fibrosis, IKK2 inhibitor, Nuclear factor-kappa B, Tumor growth factor-beta1, Interleukin-6, Alpha-smooth muscle actin, C57BL mouse
INTRODUCTION
The incidence rate of non-alcoholic fatty liver disease (NAFLD) has increased annually. Simple steatosis in the early stage may gradually develop into fatty hepatitis[1,2], and subsequently develop towards hepatic fibrosis and liver cirrhosis[3]. In an advanced stage, the incidence rate of liver cancer, multiple organ failure and other fatal complications reaches up to 0.6%-3%[4]. Based on the World Health Organization prognostication, chronic liver disease is the ninth leading cause of death in western countries and this situation will not be improved in the coming decades[5]. Among patients with non-alcoholic steatohepatitis (NASH), there were 10%-25% of patients that developed hepatic fibrosis or even liver cirrhosis[6-9]. The nosogenesis of NASH remains unclear, but the hypothesis of “secondary strike” has been widely accepted[10,11]. Unfortunately, some treatments initially gradually improve liver adipose degeneration, but are unable to achieve long-term control[9,12].
IκB kinase (IKK) is a large protein complex that is 700-900 kDa, including the two kinase subunits IKKα (IKK1) and IKKβ (IKK2) and one regulatory subunit that is either nuclear factor-kappa B (NF-κB) essential modifier or IKKγ. IKK2 is part of the inhibitor of the κB (IκB) IKK complex, which activates NF-κB through phosphorylation of the IκBs, leading to a series of inflammatory reactions[13-16]. Many effective drugs can reduce the inflammatory reaction in the liver by inhibiting the nuclear factor IKK2-NF-κB pathway and reducing insulin resistance in the liver[17]. Mathers et al[5] have demonstrated that application of the IKK2 inhibitor reduces fat accumulation in the liver and body weight gain in the mice. It has been reported that antioxidants inhibit the activity of NF-κB and can reduce inflammatory reactions[18] or even change fibrotic tissue[19].
We speculated that specific inhibition of NF-κB activation by an IKK2 inhibitor could effectively suppress the expression of inflammatory factors and even improve hepatic fibrosis. Therefore, in our study, a NASH model was established by a high-fat diet in mice and intraperitoneal injection of lipopolysaccharides (LPS) promoted acute hepatic injury and the expression of inflammatory factors. An IKK2 inhibitor (IMD0354) was used to suppress the NF-κB signaling pathway. Liver function and histological changes were observed and expression levels of interleukin-6 (IL-6), tumor growth factor-beta1 (TGF-β1), tumor necrosis factor-alpha (TNF-α), as well as other pro-inflammatory and pro-fibrosis factors, were determined. We focused on measurement of the fibrosis index, which represented hepatic stellate cells (HSCs), alpha-smooth muscle actin (α-SMA) and the expression levels of collagenI, collagen III and mRNA, which showed fibrotic hepatic changes. Accordingly, we investigated potential therapeutic prospects of the IKK2-NF-κB signaling pathway for reversing fibrosis in NAFLD.
MATERIALS AND METHODS
Experimental protocol and animal model
Twenty-four-week-old male C57BL6 mice, weighing approximately 12-16 g, were purchased from the Shanghai Experimental Animal Center of the Chinese Academy of Science. Mice were housed in a clean grade barrier systems laboratory in the Medical Laboratory Animal Center of Shanghai Jiao Tong University. Animals were randomized into four groups: the control group (n = 5), high-fat (HF) diet group (n = 5), HF + LPS group (n = 5), and HF + LPS + IMD (IKK2 inhibitor) group (n = 5). The mice in the control group were given a normal diet (ND) and the HF group animals were fed with an HF diet for 10 wk. The ND chow was supplied by the Animal Center of the Medical College of Subsidiary Basic Medical of Shanghai Jiao Tong University. The HF diet (50% fat, pork fat 18%, yolk 12%, sugar 8% and basal diet 62%) was supplied by SLAC Precision Equipment Inc. Mice in the intervention group were intraperitoneally injected with 30 mg/kg IKK2 IMD 0354 (Tocris Bioscience, Bristol, United Kingdom) for 14 d, and at the end of 12 wk this was combined with intraperitoneal injection of 10 mg/kg LPS (Sigma-Aldrich, St Louis, MO, United States). Mice were then sacrificed after fasting for 12 h. Subsequently, 1 mL eyeball blood was obtained and all mice were killed by cervical dislocation. The liver tissue was fast fixed and lightly washed in ice-cold phosphate buffered solution (PBS). Then, part of the liver was placed into 10% formalin fixation solution, while the other part of the liver was quickly stored at -70 °C for cryopreservation. The blood was sent to the laboratory of Renji Hospital for liver enzyme assays. Some liver tissue was embedded in paraffin for 24 h and was then observed by hematoxylin and eosin (HE) staining, Masson staining and immunohistochemistry (IHC). Other liver tissue was saved under an ultra low temperature for Western blotting and polymerase chain reaction (PCR) procedures.
Biochemical and liver enzymes assays
Serum was collected to analyze alanine aminotransferase (ALT) and aspartate aminotransferase (AST) using automatic biochemical instrumentation at Renji Hospital Lab, Shanghai, China.
Histopathological staining and analysis
For HE and Masson staining, 4-μm liver tissue sections were cut from the same position and embedded in paraffin after being stabilized in 10% formalin. Changes in liver tissues were observed under a light microscope.
Evaluation of inflammation activity: scores of inflammation activity were in accordance with chronic liver disease activity[20], and were divided into four parts; i.e., portal area inflammation (P), lobular inflammation (L), patch necrosis (PN) and bridging necrosis (BN, including lobular necrosis). Every item was recorded as 1, 2, 3 or 4 based on degrees of pathological changes. The scores counting formula was: P + L + 2 × (PN + BN).
Fatty hepatic fibrosis: fibrosis scores were divided into four stages according to the degree of fibrosis in three areas of the liver; i.e., the liver acinus, the portal vein, and bridging fibrosis, as well as the presence or absence of liver cirrhosis. S1 indicated perisinusoidal space fibrosis of three areas of the local or extensive liver acinus; S2 indicated the above pathological changes with local or extensive periportal fibrosis; S3 indicated S2 pathological changes with local or extensive bridging fibrosis; and S4 indicated fatty liver cirrhosis, forming fibrous septa that divided lobuli hepatic and central veins to the portal area and formed false lobules.
Immunohistochemical analysis
Liver tissue sections (4 μm) were prepared for IL-6 immunohistochemical study. The glass was treated by polylysine to promote cell attachment. Microwave antigen repairing was carried out with 0.01 mol/L citrate buffer solution (pH 6.0). After blocking with rabbit serum, the sections were incubated overnight at 4 °C with monoclonal primary antibody against mouse IL-6 (PPMX, Tokyo, Japan). On the next day, liver sections were taken out and washed with PBS three times and were incubated with the second antibody for 1 h at room temperature. Coloration with freshly prepared diaminobenzidine (DAB) was performed, and then the tissues were counterstained with hematoxylin, dewatered, and then mounted with neutral gum. The second antibody in the ElivisionTM plus Polyer HRP (Mouse/Rabbit) IHC Kit and DAB developer were supplied by Manxin Bio Co®, Fuzhou, China. PBS taking the place of the primary antibody was considered as the negative control. We chose 10 views of each section under light microscopy to obtain the average positive absorbance using ImageProplus2.0.
Enzyme linked immunosorbent assay
Liver (1 g) was placed in PBS with 0.1 mmol/L phenylmethyl sulfonylfluoride (PMSF, Sigma) and was then manually homogenized, centrifuged at 100 000 r/min for 15 min at 4 °C, and the supernatant was removed. Double antibody enzyme linked immunosorbent assay (ELISA) (ELISA kit TNF-α, BD Bioscience, Franklin Lakes, NJ, United States) was used for detection, and the procedure was strictly based on the guidelines provided by the manufacturer.
Western blotting analysis of NF-κB p65, TGF-β1 and α-SMA
Liver tissue was saved in a refrigerator at -80 °C after homogenization, and the tissue protein extract solution was prepared by centrifugation. Based on the operating instructions of the bicinchoninic acid protein quantitation kit (Sunbio, Beijing, China), the concentration of protein was detected. After denaturation, each tissue protein was sampled at 50 μg, and reducibility was performed with sodium dodecyl sulfate polyacrylamide gel electrophoresis cataphoresis in an 8% polyacrylamide gel. Sampling after denaturation was performed and electrophoresis was started. The electrophoresis voltage of the condensed glue and separation gel was 80 V and 120 V, respectively. The electrophoresis terminated after bromophenol blue electrophoresis moved to the bottom of the glue, and a damp-dry transmembrane (polyvinylidene fluoride membrane) was applied with a constant 50 mA current for 90 min. The membrane was sealed at room temperature with 5% defatted milk powder prepared with Tris-buffered saline Tween-20 (TBST) and primary antibodies (NF-κB p65, TGF-β1, α-SMA and β-actin, 1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, United States) and was incubated overnight in a swing bed at 4 °C. After the film was washed with TBST buffer solution in the swing bed, the second antibody 1:3000 (rabbit polyclonal antibody, Manxin Bio, Fuzhou, China) was incubated at room temperature for 1 h. After being repeatedly washed in TBST buffer solution, DAB staining was performed. After proper staining, the reaction was terminated by water. In a dark room, a nitrocellulose filter was put into a brightening agent with sufficient contact, and was then exposed to light with an X-ray device. The image was developed and fixed. Simultaneous determination of the expression level of β-actin in the same filter was carried out as an internal control. Separate analyses were performed for each sample and the experiment was repeated three times. We obtained the integrated density value by Microsoft BandScan, and the ratio of the target bands to β-actin substantiated the presence of the proteins NF-κB p65, TGF-β1 and α-SMA.
RNA extraction and analysis of mRNA expression of typesIand III collagen, α-SMA and TGF-β1
Total RNA was isolated from snap-frozen liver tissue using Trizol reagent (Invitrogen, Carlsbad, CA, United States) and the ratio between the absorbance values at 260 nm and 280 nm gave an estimate of RNA purity. Real-time (RT)-PCR was performed using a one-step RT-PCR kit from the Shanghai Daweike Biotechnology Company. Two micrograms of the total RNA was chosen and reverse transcription was performed. Its reaction product was placed into a 50-μL PCR reaction system. α-SMA, TGF-β1, typesIand III collagen, and the specific primer of the internal reference β-actin was used in PCR amplification, and agarose electrophoresis was performed. Electrophoresis results were scanned with a BioSens GelImaging System. For the PCR primer sequences and fragment lengths (Table 1). The real-time survey meter (7500 Sequence Detection System) was obtained from ABI, United States. The PCR conditions were: predegeneration for 2 min at 50 °C, denaturation for 20 s at 95 °C, annealing for 45 s at 60 °C, and extension for 30 s at 72 °C, with a total of 40 cycles; an internal reference of β-actin was used with predegeneration for 3 min at 94 °C, denaturation for 20 s at 95 °C, annealing for 20 s at 60 °C, extension for 30 s at 72 °C, with a total of 35 cycles, and extension again for 10 min after the cycles, and then termination at 4 °C.
Table 1.
Pathological scores in liver tissues
| Groups | P + L + 2 × (PN + BN) | S0 S1 S2 S3 S4 Z |
| Control | 0 | 5 0 0 0 0 |
| HF | 10.70 ± 2.62a | 0 3 2 0 0 |
| HF + LPS | 13.30 ± 3.83a | 0 1 3 1 0 |
| HF + LPS + IMD | 4.60 ± 3.83ac | 0 4 1 0 0 -2.35e |
The score of inflammation is given by P + L + 2 × (PN + BN).
P < 0.01 vs the control group;
P < 0.01 vs the HF and HF + LPS groups;
Z = -2.35, P = 0.018, P < 0.05 vs the HF + LPS group. P: Portal area inflammation; L: Lobular inflammation; PN: Patch necrosis; BN: Bridging necrosis; HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor.
Statistical analysis
Data were expressed as mean ± SD. Statistical analysis was performed with a one-way analysis of variance using SPSS17.0 software, followed by Scheffe’s test, and comparisons between groups was performed using the Mann-Whitney test. P < 0.05 was considered to be statistically significant.
RESULTS
Effects of IKK2 inhibitor (IMD0354) on serum ALT and AST
The levels of ALT and AST for each group are shown in Figure 1. In Figure 1A, the ALT levels of the HF and HF + LPS groups were significantly increased compared to the control group (P < 0.05). After treatment with the IKK2 inhibitor (IMD0354), the level of serum ALT in the mice was significantly decreased compared to the HF group (P < 0.01), as well as in the HF + LPS group (P < 0.05), but was still higher than that of the control group (P < 0.05). Figure 1B shows that the change in serum AST was not as significant as that of ALT. The level of serum AST in the HF + LPS group was significantly increased compared to the control group (P < 0.01). The level of serum AST when treated with IMD0354 was significantly decreased compared to that of the HF + LPS group (P < 0.05).
Figure 1.
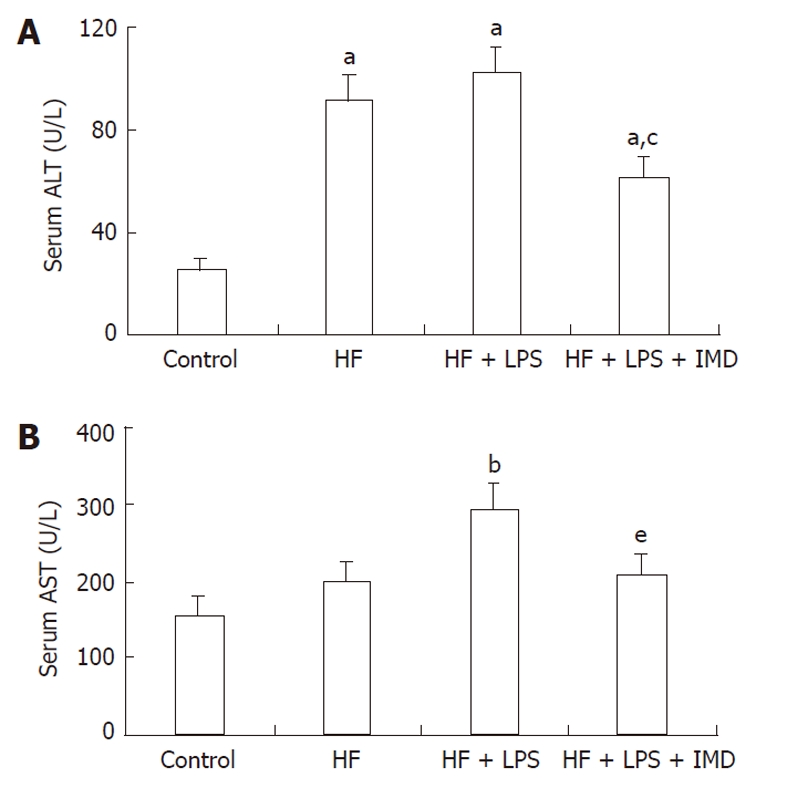
IKK2 inhibitor prevented HF + LPS-induced liver injury, as determined by serum ALT and AST levels. The normal values for ALT and AST were 45 U/L and 160 U/L. Serum ALT (A) and AST (B) were measured in different groups (control group, HF group, LPS-induced HF group and IMD-treated group), data are expressed as mean ± SD. A: aP < 0.05 vs control group, cP < 0.05 vs the HF and LPS + HF groups; B: bP < 0.01 vs the control group, eP < 0.05 vs the LPS + HF group. HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase.
Effects of IKK2 inhibitor on liver inflammation and fibrosis during liver injury development
HE staining results showed that pathological changes in the mice were in line with the diagnostic gold standard of chronic NASH (Figure 2A). At the same time, typical hepatic fibrosis was observed with Masson staining (Figure 2B). In the control group, the structure of the hepatic lobules was clear without inflammatory cell infiltration in the portal area and without fibrotic tissue hyperplasia. The liver sections of the HF and HF + LPS groups showed that the normal structure of the hepatic lobules was lost, the structure of the blood vessels in the liver was disordered with severe liver cell degeneration, patch necrosis and bridging necrosis, and there were many inflammatory cell infiltrates in the portal area. There was also light to moderate hyperplasia of the broglia fibrils and fibrous septa were formed occasionally. Inflammation and fibrosis scores showed significant differences compared with the control group (P < 0.05). In the IMD-treated group, the structure of the hepatic lobules was normal, liver cell degeneration was significantly decreased, and liver cell necrosis and inflammatory cell infiltration were significantly improved. Fibrillar collagen sediment still existed, which was significantly reduced compared with the controls, and its inflammation score was also significantly decreased (P < 0.05), but was still higher than that of the control group (P < 0.05, Table 1). The results of the fibrosis scores were analyzed by Mann-Whitney statistical methods, and the results showed that there was a significant difference between the HF + LPS + IMD and HF + LPS groups (Z = -2.35, P = 0.018, P < 0.05, Table 1). There were no differences among the other groups.
Figure 2.

Hematoxylin and eosin stain and Masson staining in sections of (a) control group; (b) HF group; (c) HF + LPS group; and (d) HF + LPS + IMD group. A: Hematoxylin and eosin stain. Macrovesicular steatosis, lobular inflammation and balloon degeneration of hepatocytes were observed in liver sections of HF-treated mice and HF + LPS + treated mice with a significantly large amount of inflammatory cell infiltration surrounding the centrilobular veins of the liver. Significant amelioration was observed in the group treated with IMD (d); B: Masson staining. A thin lining of collagen was observed in the HF group, HF + LPS group and HF + LPS + IMD group. With LPS treatment, there was an increase in the amount of collagen accumulated along the central vein with the presence of collagen in the pericellular area. Treatment with IMD reduced LPS-induced collagen accumulation. HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor.
Immunohistochemistry assay for the changes in IL-6 expression in the livers of mice
Previous studies have demonstrated that many cell factors, such as TNF-α and IL-6, play important roles in the NF-κB dependent signaling pathway[21]. Some evidence has indicated that TNF-α, as well as IL-6, participated in the formation of hepatic fibrosis and had a positive correlation with the level of serum hyaluronic acid, laminin type IV, for example, suggesting that TNF-α, as well as IL-6, not only mediated inflammatory reactions but also participated in the formation of hepatic fibrosis during the promotion of extracellular matrix (ECM) synthesis. In this study, the average absorbance values (A) of liver cell positive immunity of mice in each group were analyzed by Image-pro plus 6.0 and statistical analysis was performed, showing that there was a small amount of IL-6 expression in the control and HF groups, and the expression of IL-6 was significantly increased in the HF group after being activated by LPS (P = 0.013, P < 0.05). IL-6 expression was mainly concentrated in the liver cell cytoplasm around the central veins and portal area, appearing as brown and grainy, and was also expressed in the sinus hepaticus and parts of monocytes. In the HF + LPS group treated with the intervention of IMD, the expression of IL-6 was significantly decreased (P = 0.012, P < 0.05). There was no significant difference when compared with the HF group (P = 0.70, P > 0.05), and the expression of IL-6 was higher than the control group (Figure 3).
Figure 3.

Interleukin-6 expression was assessed by immunohistochemistry. A: Positive staining was observed in hepatocytes in the control group (a), HF group (b), LPS-induced HF group (c) and IMD-treated group (d). B: The optical density (OD) of interleukin-6 (IL-6)-positive areas was measured with ImageProplu6.0 (aP < 0.05). HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor.
IKK2 inhibitor (IMD) inhibited an LPS-induced increase in the pro-inflammatory cytokine levels of TNF-α in mice livers
Recent research has shown that the IKK2-NF-κB signal pathway participate in insulin resistance, and cell factors of TNF-α and IL-6 play important roles dependent on NF-κB signals[21], especially in the pathological process of hepatic fibrosis[17,22]. Therefore, TNF-α was the key pro inflammatory factor, which was likely to induce the formation and development of hepatic fibrosis. The protein levels of TNF-α in mouse livers were detected by ELISA. With the development of liver injury, an increased expression of TNF-α was shown in the liver[23]. The levels of TNF-α in mouse livers of the HF and HF + LPS groups were significantly increased compared to the control group (P < 0.05). After intervention with the IKK2 inhibitor, the level of TNF-α was significantly reduced compared to the non-intervention group (P < 0.05, Figure 4).
Figure 4.
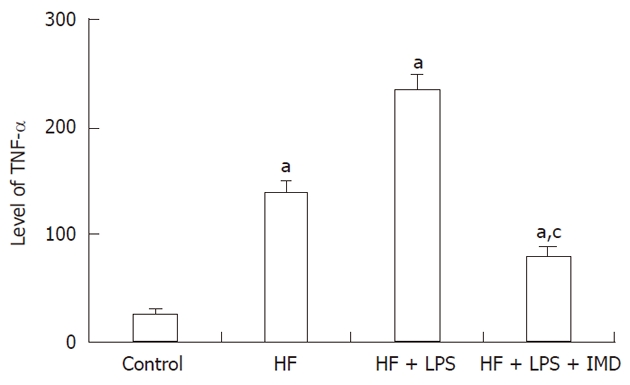
The levels of nuclear factor-κB-dependent pro inflammatory cytokines and tumor necrosis factor-alpha were measured in livers obtained from the control group, HF group, HF + LPS group and HF + LPS + IMD group. aP < 0.05, compared with the control group. cP < 0.05, significant compared with both the HF and LPS + HF exposed groups. HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor; TNF-α: Tumor necrosis factor-alpha.
IKK2 inhibitor (IMD) decreased nuclear translocation of NF-κB p65 in mice livers in response to LPS and HF exposure
When combined with IκBα in the cytoplasm, NF-κB was inactive. IKK2, as the major subunit of promotion, activated NF-κB and its subunit with phosphorylation, while IMD could inhibit the activation of NF-κB p65 and its nuclear transcription[24]. Therefore, expressions of NF-κB p65 and P-IκBα in the livers in each group was detected to determine the inhibitory action of IMD. Western blotting showed that the expression of NF-κB p65 in the HF group was increased compared to that of the normal diet group. In the HF group, LPS promoted the expression of NF-κB p65, and P-IκBα increased simultaneously, while intervention by the IKK2 inhibitor reduced the pro-inflammatory role of LPS and significantly reduced the expression of NF-κB p65 and its subunit (Figure 5).
Figure 5.
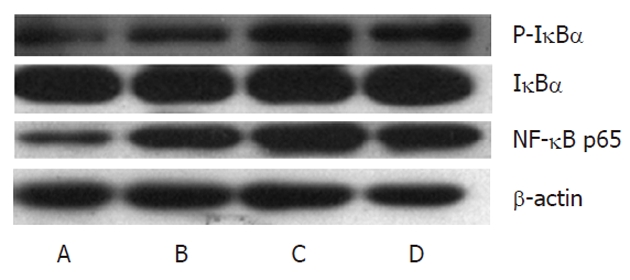
IKK2 inhibitor decreased lipopolysaccharide-induced nuclear translocation of nuclear factor-κB p65 and P-IκBα in livers. Nuclear levels of the p65 subunit of nuclear factor-κB (NF-κB) were measured by Western blotting in different groups (A: Control; B: HF group; C: HF + LPS group; D: HF + LPS + IMD group). β-actin was used as a loading control. Administration of IMD at 30 mg/kg doses decreased the DNA binding activity of NF-κB, which was induced by HF and LPS in mice livers. HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor.
IKK2 inhibitor (IMD) decreased protein levels of TGF-β1 and α-SMA related to fibrosis in livers
TGF-β1 is an important inflammatory factor that stimu-lates accumulation in the ECM and tissue fibrosis. Our results showed that TGF-β1 expression in the liver was increased under the condition of the HF diet and stimulation of LPS. While α-SMA was a marker of HSC activation, its variation tendency was similar to TGF-β1. In the mouse liver model group, the expression of α-SMA was significantly increased compared to the control group. After application of the IKK2 inhibitor, the protein expression of TGF-β1 in livers decreased, and the expression of α-SMA was reduced accordingly. Correspondingly, activation of HSCs and the formation of collagen decreased, leading to effectively preventing hepatic fibrosis (Figure 6).
Figure 6.
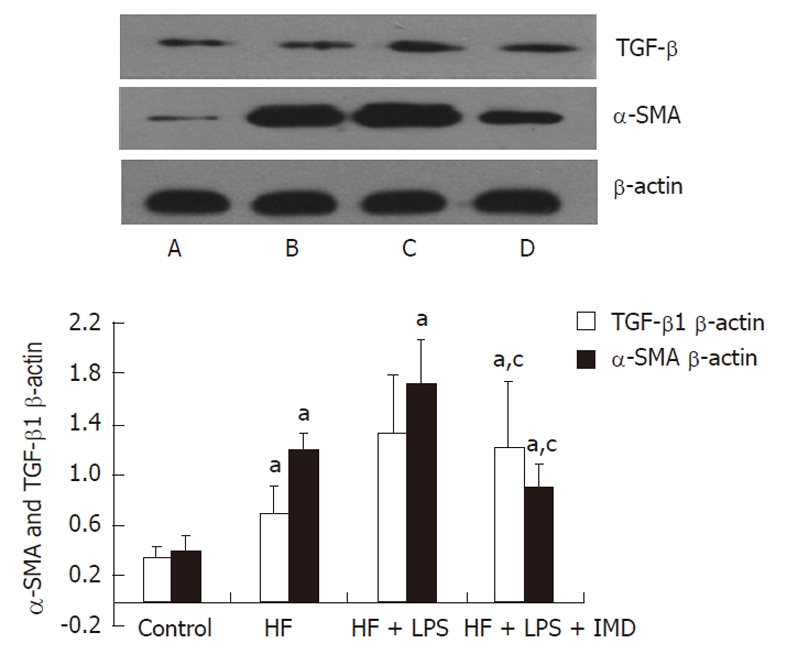
Western blotting analysis of tumor growth factor-beta1, alpha-smooth muscle actin proteins were measured that were involved in IKK2-nuclear factor-κB pathways in the liver in different groups (A: Control; B: HF group; C: HF + LPS group; D: HF + LPS + IMD group). β-actin was used as a loading control. The levels of tumor growth factor-beta1 (TGF-β1) and alpha-smooth muscle actin (α-SMA) measured in livers were increased in the HF and HF + LPS groups. IKK2 inhibitor significantly inhibited LPS and HF-induced expression of TGF-β1 and α-SMA in mouse livers. The ratio of TGF-β1 and α-SMA/β-actin in the liver was increased in other groups, compared with the control group, aP < 0.05. IKK2 inhibitor normalized TGF-β1 and α-SMA significantly compared with the HF or LPS + HF groups. cP < 0.05. HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor.
IKK2 inhibitor IMD inhibited α-SMA, TGF-β1, typesI(αI) and III collagen and mRNA expression in LPS-stimulated mice
The formula described previously was used to measure mRNA relative expression (amount = 2-Δct × 100%), and the relative expression levels of α-SMA, typeI(αI) collagen, type III collagen and TGF-β1 mRNA were obtained. RT-PCR was performed with the mouse primers shown in Table 2. The results showed that the expression of TGF-β1 mRNA in the HF group and the HF + LPS group was higher than the control group (P < 0.05), which was significantly decreased after intervention of IMD. In addition, the fibrosis indexes of α-SMA, typeIcollagen and type III collagen in the model group were also increased. In addition, the contents of typeIcollagen in the HF diet group and type III collagen in the HF + LPS group were significantly increased, independently, compared to the control group (P < 0.01, P < 0.05, respectively). After intervention with IMD, the levels of α-SMA, typeI(αI) collagen and III collagen were significantly decreased compared to the HF + LPS group (P < 0.05). However, there was no significant difference in α-SMA and type III collagen with intervention of IMD and the normal group (P > 0.05, Figure 7).
Table 2.
Oligonucleotide sequences used in real-time polymerase chain reaction
| mRNA | Sequence | Length (bp) |
| TypeI(Iα) collagen | F: ACAGTGGTGAACCTGGTGCT | 151 |
| R: CTCCTTTGGCACCAGTGTCT | ||
| Type III collagen | F: GGAGCCCCTGGACTAATAG | 193 |
| R: ATCCATCTTTGCCATCTTCG | ||
| α–SMA | F: TGCTGTCCCTCTATGCCTCT | 185 |
| R: GAAGGAATAGCCACGTCAG | ||
| TGF-β1 | F: CTTGCCCTCTACAACCAACA | 189 |
| R: CTTGCGACCCACGTAGTAGA | ||
| β-actin | F: TGTGTCCGTCGTGGATCTGA | 126 |
| R: CTTGCGACCCACGTAGTAGA |
α–SMA: Alpha-smooth muscle actin; TGF-β1: Tumor growth factor-beta1.
Figure 7.

The IKK2 inhibitor inhibited LPS and HF-induced increases in pro inflammatory cytokine levels in mouse livers. The level of tumor growth factor-beta1 (TGF-β1) was measured in the livers of mice in the control, HF, HF + LPS and HF + LPS + IMD groups. Also, expression of the fibrosis index, such as alpha-smooth muscle actin (α-SMA), typeIcollagen and type III collagen, were detected in the four groups by real-time polymerase chain reaction. The level of TGF-β1 measured in livers was increased in the HF and HF + LPS groups, compared with the control group, aP < 0.05. The mRNA content of typeIcollagen in the HF group and type III collagen in the HF + LPS group were significantly higher, aP < 0.05, compared with the control group. Intraperitoneally administered IKK2 inhibitor normalized the expression of TGF-β1, as well as the contents of α-SMA, typeIand type III collagen mRNA, compared with the HF or LPS + HF groups. aP < 0.05, compared with the control group. cP < 0.05, compared with both HF group and LPS + HF exposed group. HF: High-fat; LPS: Lipopolysaccharide; IMD: IKK2 inhibitor.
DISCUSSION
NAFLD includes simple liver steatosis, NASH and liver cirrhosis, while NASH has become a central issue of chronic liver disease with worldwide attention[7]. Currently, the pathogenesis of NAFLD is not clear, and the hypothesis of “secondary strike” has been widely accepted. It is well known that insulin resistance is involved in the process, and inflammatory reaction, lipid peroxidation, and oxidative stress also play important roles[25-27]. The best measure for preventing the progression of hepatic fibrosis is to prevent or reverse the initial cascade reactions of fibrosis[28]. Therefore, inhibition of the generation of inflammatory factors effectively reduces HSC activation, decreases accumulation in the ECM, and fundamentally reverses fibrosis[29]. The expression of NF-κB is significantly increased in NASH patients, and TNF-α, IL-6, TGF-β, and other inflammatory factors also showed high expression levels[23]. Further research has found that activation of NF-κB is the key step in regulating gene expression of various kinds of pro inflammatory factors in NASH patients[23]. TNF-α is the key pro inflammatory factor and it induces the formation and development of hepatic fibrosis. TGF-β1 mediates the synthesis of different kinds of collagen with time dependence. However, inhibition of TGF-β1 significantly reduces the synthesis of collagen and sedimentation of the ECM[30].
It has been reported that blockage of IKKβ (IKK2) significantly reduces the incidence rate of liver steatosis and improves NASH pathologically[31]. Is it possible that IKKβ (IKK2)-NF-κB is also a key in improving and even reversing hepatic fibrosis? Various macromolecular protein-joining enzymes, including IKKβ (IKK2), IKK or NF-κB inhibitors, have become new types of anti-inflammatory agents; therefore, many researchers have tried to inhibit NF-κB-mediated proinflammatory responses based on these agents. The IKK2 inhibitor played a specific anti-inflammatory role through inhibition of the major subunit IKKβ, which served as a promotor in the IKK protein kinase complex center. In our study, the IKK2 inhibitor (IMD 0354) was used in liver injury in mice. We detected its inhibitory effect on liver NF-κB-dependent inflammatory factors, changes in liver function, histological changes and, at the same time, the expression of TGF-β1 involving fibrosis and relevant fibrosis indexes, such as α-SMA, typeIcollagen and type III collagen. It is warranted to investigate the potential therapeutic effect of IMD on hepatic fibrosis.
In our study, during HF-diet-induced chronic non-alcoholic hepatic injury in mice, the serology index of ALT was doubly higher than that of the control group. As for pathological changes, moderate to severe steatosis was observed, inflammatory infiltration was found in the lobules, local inflammatory infiltration was detected in the portal area, Masson staining showed fibrous tissue hyperplasia, Western blotting and RT-PCR results demonstrated that TGF-β1 expression increased, α-SMA content was raised, and there was sedimentation of typesIand III collagen. Therefore, a hepatic injury model with typical inflammation and fibrosis pathological manifestation was successfully established by an HF diet and intraperitoneal injection of LPS, stimulating activation of NF-κB and promoting an inflammatory reaction[32-34]. When chronic hepatic injury occurs, different initial cau-sative agents trigger the activation of HSCs, activation of the Janus kinase-signal transducers and activators of transcription signal transduction pathway[35], promotion of α-SMA expression in sinus hepaticus cells, further proliferation and activation, and synthesis and secretion of the ECM and collagen, which finally enhances the occurrence of hepatic fibrosis. Thus, expression of α-SMA has been considered as one of the dominant features of HSC activation, and has become an important evaluation index for hepatic fibrosis. In our study, when the mice were stimulated with LPS and an HF diet, the expression of α-SMA increased in the liver. At the same time, sedimentation of typesIand III collagen also occurred, suggesting that HSC was triggered and activated, and then started the process of hepatic fibrosis. However, the results for the group treated with IMD showed that expression of α-SMA, as well as sedimentation of typesIand III collagen, were significantly reduced, suggesting that inhibition of inflammatory factor expression also effectively suppressed HSC activation, and accordingly blocked the occurrence of hepatic fibrosis from the source.
Fatty tissue was the principle source of cell factors, liver steatosis promoted macrophage infiltration, and activation of HSC promoted the cascading release of many kinds of cell factors with an intensive pro inflammatory role, which further made the pathological changes and insulin resistance more serious[36,37]. We found that inflammatory cell and inflammatory factor expression after stimulation with LPS significantly increased compared to that in the HF group. Liver steatosis in the mice treated with intraperitoneal injection of the IKK2 inhibitor was significantly improved, and inflammatory factors released from the hepatic cell fat were correspondingly reduced compared to that of the HF and LPS groups. In addition, it was shown that the IKK2 inhibitor could significantly reduce pro inflammatory stimulation by LPS, and even reverse the fibrosis process in a mouse hepatic injury model. Western blots demonstrated that NF-κB p65 activation was significantly inhibited, and NF-κB-dependent pro inflammatory factors, such as IL-6, were simultaneously suppressed. Separation of NF-κB and its suppressor factor IκBα resulted in continuous activation of intercellular adhesion molecule-1 and other cell factors, and finally the up regulation of IL-6. The increase in IL-6 stimulated HSC proliferation, induced production of multiple acute-phase proteins, and promoted ECM sedimentation by facilitating matrix degeneration or interaction with its adhesion receptor, leading to significant hepatic fibrosis[38,39]. Similarly, the high expression of IL-6 significantly promoted liver apoptosis. In our study, it was found that after intervention with the IKK2 inhibitor, NF-κB p65 activation was significantly inhibited, IL-6 expression in the livers of LPS model mice was significantly decreased with the improvement of serology indexes and histological changes, and blocking IL-6 expression significantly improved hepatic injury, which could demonstrate that the IKK2 inhibitor could improve hepatic fibrosis by inhibition of the inflammatory factor IL-6.
It is known that TNF-α promotes insulin resistance and the development of liver inflammation, which is related to multiple cell factors, and induces the synthesis of IL-1, IL-6 and C-reactive protein, including caspase 3 and growth arrest and DNA-damage-inducible beta. Adiponectin inhibits expression of TNF-α, as well as other inflammatory factors, with positive feedback[40,41]. A peroxisome proliferator-activated receptor antagonist blocked TNF-α mediated insulin resistance, and it also had intensive anti-inflammatory action[42,43], which made the inflammation signal transduction pathway become a multiple cross and participated in the process of hepatic fibrosis[44]. The level of TNF-α could better reflect the regulatory condition of the inflammatory reaction in hepatic fibrosis. Hepatocellular carcinoma (HCC) invariably develops within a setting of chronic inflammation caused by metabolic liver disease or autoimmunity. Mechanisms that link these two processes are not completely understood, but transcription factors of the NF-κB family have been suggested to be involved. Cytokines such as IL-6 are clearly pivotal players, and high levels of serum IL-6 correlate positively with tumor size and with poor prognosis in HCC patients[45]. Our results showed that the levels of IL-6 and TNF-α in mouse livers in which hepatic fibrosis existed increased, while the IKK2 inhibitor reversed such an imbalance, and α-SMA expression in the liver and sedimentation of collagenIand collagen III decreased, illustrating that TNF-α improved liver pathological changes in hepatic fibrosis in mice, relieved inflammatory cell infiltration, and reduced fiber hyperplasia by the IKK2-NF-κB-dependent signaling pathway.
During the process of HSCs activation, TGF-β1 plays a major role as a fibroblast growth factor[46], and the major stimulating factor promoting HSCs to accumulate ECM[47]. TGF-β1 receptors on the surface of HSC complete the signaling pathway combined with Smads[48], which continuously stimulates HSC activation, and finally transcribes target gene expression in the HSC nucleolus, mainly including typeIcollagen, which plays a key regulatory role in ECM metabolism and function. However, there is evidence demonstrating that NF-κB does not directly activate HSCs[39]. In our experiment, when injury and hepatic fibrosis occurred, protein and gene expression of TGF-β1 increased. While inhibiting the activation of NF-κB certainly reduced the activation of HSCs, TGF-β1 expression was decreased, and the expressions of various kinds of fibrosis factors, such as α-SMA and typeIand type III collagen, decreased. Therefore, we suggest that indirect correlation or other pathways exist between NF-κB and HSCs, which indirectly reduce TGF-β1 expression, decrease continuous activation of HSCs, and then improve the fibrosis process.
In brief, our in vivo experiment demonstrated that an IKK2 inhibitor could significantly decrease the expression of various inflammatory factors in mouse livers after exposure to LPS, which could play an anti-fibrosis role in inhibiting inflammation, reducing collagen content in liver tissues, and decreasing the expression of hepatic fibrosis correlation factors. IMD inhibited the phosphorylation activation of IκBα and NF-κB stimulated by LPS. Meanwhile, the expression of NF-κB-dependent inflammatory cytokines were suppressed, illustrating that inflammatory factors inducing hepatic injury were effectively reduced by inhibition of the NF-κB signaling pathway. In addition, we found that various fibrosis markers, such as TGF-β1 and α-SMA, as well as typeIcollagen and type III collagen, were decreased in the liver, demonstrating that HSC activation was decreased and ECM accumulation was reduced. Therefore, it was presumed that the mechanism of the IKK2 inhibitor in anti hepatic fibrosis might be relevant, with the inhibition of cell factors promoting HSC activation, indirect inhibition of HSC proliferation, suppression of continuous activation of HSCs by TGF-β1, and then secretory volume and expression levels of α-SMA being reduced. Finally, the extent of hepatic fibrosis was decreased. The major pathological changes of chronic fatty liver disease are insulin resistance and inflammatory reactions[49,50]. In summary, the NF-κB signaling pathway participates with the IKK2 inhibitor playing an influential role in NASH and hepatic fibrosis. Inhibiting the production of a variety of inflammatory factors could effectively reduce HSC activation, decrease accumulation in the ECM, and then reduce fibrosis formation.
COMMENTS
Background
The nuclear factor-κB (NF-κB) signaling pathway improves insulin resistance and fat accumulation in the development of nonalcoholic fatty liver disease (NAFLD). Inhibition of NF-κB activation by an IKK2 inhibitor could effectively suppress the expression of many kinds of inflammatory factors, and even improve hepatic fibrosis. Therefore, the authors of this study investigated the potential therapeutic prospects of the IKK2-NF-κB signaling pathway in the reversion of fibrosis in NAFLD.
Research frontiers
The study is believed to be the first to evaluate the role of IKK2-NF-κB signals in NAFLD. The potential effect of an IKK2 inhibitor is likely to block inflammation through inhibiting NF-κB activation. The IKK2 inhibitor may play an important role in the occurrence and development of NAFLD.
Innovations and breakthroughs
This study explored one of the possible mechanisms of inflammation, which could produce a potentially facilitative effect in the occurrence and development of NAFLD.
Applications
This study provides an experimental basis for future studies on the role of IKK2 in NAFLD. Control of the expression level of IKK2 in the liver may become a new possibility for therapy of NAFLD.
Terminology
In the present study, the authors tested the effect of IKK2 in NAFLD in mice, and found a facilitative effect on the occurrence and development of NAFLD.
Peer review
The paper should be accepted with some minor revisions. IκB kinase-beta inhibitor attenuates hepatic fibrosis in mice liver injury and its potential mechanisms.
Footnotes
Supported by Shanghai Municipal Health Bureau Youth Grant, No. 2008Y032
Peer reviewer: Rui Marinho, Professor, Hospital Santa Maria, Rua Prof. Aires de Sousa, 1 r/c A, 1600-590 Lisboa, Portugal
S- Editor Gou SX L- Editor Kerr C E- Editor Li JY
References
- 1.Laleman W, Verbeke L, Meersseman P, Wauters J, van Pelt J, Cassiman D, Wilmer A, Verslype C, Nevens F. Acute-on-chronic liver failure: current concepts on definition, pathogenesis, clinical manifestations and potential therapeutic interventions. Expert Rev Gastroenterol Hepatol. 2011;5:523–537; quiz 537. doi: 10.1586/egh.11.47. [DOI] [PubMed] [Google Scholar]
- 2.Kim WR, Brown RS, Terrault NA, El-Serag H. Burden of liver disease in the United States: summary of a workshop. Hepatology. 2002;36:227–242. doi: 10.1053/jhep.2002.34734. [DOI] [PubMed] [Google Scholar]
- 3.Leon DA, McCambridge J. Liver cirrhosis mortality rates in Britain from 1950 to 2002: an analysis of routine data. Lancet. 2006;367:52–56. doi: 10.1016/S0140-6736(06)67924-5. [DOI] [PubMed] [Google Scholar]
- 4.Roberts SE, Goldacre MJ, Yeates D. Trends in mortality after hospital admission for liver cirrhosis in an English population from 1968 to 1999. Gut. 2005;54:1615–1621. doi: 10.1136/gut.2004.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 7.Clark JM, Brancati FL, Diehl AM. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649–1657. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 8.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 9.Adams LA, Lindor KD. Nonalcoholic fatty liver disease. Ann Epidemiol. 2007;17:863–869. doi: 10.1016/j.annepidem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Diehl AM, Li ZP, Lin HZ, Yang SQ. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut. 2005;54:303–306. doi: 10.1136/gut.2003.024935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987–1000. doi: 10.1002/hep.20920. [DOI] [PubMed] [Google Scholar]
- 12.Nobili V, Manco M, Devito R, Di Ciommo V, Comparcola D, Sartorelli MR, Piemonte F, Marcellini M, Angulo P. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: a randomized, controlled trial. Hepatology. 2008;48:119–128. doi: 10.1002/hep.22336. [DOI] [PubMed] [Google Scholar]
- 13.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 14.Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–6874. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 15.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- 16.Zandi E, Chen Y, Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]
- 17.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 18.Qian F, Deng J, Cheng N, Welch EJ, Zhang Y, Malik AB, Flavell RA, Dong C, Ye RD. A non-redundant role for MKP5 in limiting ROS production and preventing LPS-induced vascular injury. EMBO J. 2009;28:2896–2907. doi: 10.1038/emboj.2009.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kishore N, Sommers C, Mathialagan S, Guzova J, Yao M, Hauser S, Huynh K, Bonar S, Mielke C, Albee L, et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J Biol Chem. 2003;278:32861–32871. doi: 10.1074/jbc.M211439200. [DOI] [PubMed] [Google Scholar]
- 20.Farrell GC, Chitturi S, Lau GK, Sollano JD. Guidelines for the assessment and management of non-alcoholic fatty liver disease in the Asia-Pacific region: executive summary. J Gastroenterol Hepatol. 2007;22:775–777. doi: 10.1111/j.1440-1746.2007.05002.x. [DOI] [PubMed] [Google Scholar]
- 21.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, et al. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest. 2001;108:437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu SQ, Yu JP, Chen HL, Luo HS, Chen SM, Yu HG. Therapeutic effects and molecular mechanisms of Ginkgo biloba extract on liver fibrosis in rats. Am J Chin Med. 2006;34:99–114. doi: 10.1142/S0192415X06003679. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29:72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 26.Kojima H, Sakurai S, Uemura M, Fukui H, Morimoto H, Tamagawa Y. Mitochondrial abnormality and oxidative stress in nonalcoholic steatohepatitis. Alcohol Clin Exp Res. 2007;31:S61–S66. doi: 10.1111/j.1530-0277.2006.00288.x. [DOI] [PubMed] [Google Scholar]
- 27.Duvnjak M, Lerotic I, Barsic N, Tomasic V, Virovic Jukic L, Velagic V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol. 2007;13:4539–4550. doi: 10.3748/wjg.v13.i34.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gudbrandsen OA, Wergedahl H, Berge RK. A casein diet added isoflavone-enriched soy protein favorably affects biomarkers of steatohepatitis in obese Zucker rats. Nutrition. 2009;25:574–580. doi: 10.1016/j.nut.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 29.Benyon RC, Iredale JP. Is liver fibrosis reversible? Gut. 2000;46:443–446. doi: 10.1136/gut.46.4.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chuang HY, Ng LT, Lin LT, Chang JS, Chen JY, Lin TC, Lin CC. Hydrolysable tannins of tropical almond show antifibrotic effects in TGF-β1-induced hepatic stellate cells. J Sci Food Agric. 2011;91:2777–2784. doi: 10.1002/jsfa.4521. [DOI] [PubMed] [Google Scholar]
- 31.Beraza N, Malato Y, Vander Borght S, Liedtke C, Wasmuth HE, Dreano M, de Vos R, Roskams T, Trautwein C. Pharmacological IKK2 inhibition blocks liver steatosis and initiation of non-alcoholic steatohepatitis. Gut. 2008;57:655–663. doi: 10.1136/gut.2007.134288. [DOI] [PubMed] [Google Scholar]
- 32.Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, Catalano D, Mandrekar P, Dolganiuc A, Kurt-Jones E, et al. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433–G441. doi: 10.1152/ajpgi.00163.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–144. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, Roskams T, Trautwein C, Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 35.Lakner AM, Moore CC, Gulledge AA, Schrum LW. Daily genetic profiling indicates JAK/STAT signaling promotes early hepatic stellate cell transdifferentiation. World J Gastroenterol. 2010;16:5047–5056. doi: 10.3748/wjg.v16.i40.5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clin Chim Acta. 2006;364:33–60. doi: 10.1016/j.cca.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 38.Spinozzi F, Rambotti P, Gerli R, Cernetti C, Rondoni F, Frascarelli A, Bertotto A, Grignani F. Immunoregulatory T cells in alcoholic liver disease: phenotypical dissection of circulating Leu3+/T4+ inducer T-lymphocytes. J Clin Lab Immunol. 1987;23:161–167. [PubMed] [Google Scholar]
- 39.Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Umemura A, Fujii H, Okanoue T, Yoshikawa T. Blockade of IL-6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline-deficient diet-fed db/db mice. Lab Invest. 2011;91:609–618. doi: 10.1038/labinvest.2011.2. [DOI] [PubMed] [Google Scholar]
- 40.Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46–54. doi: 10.1002/hep.20280. [DOI] [PubMed] [Google Scholar]
- 41.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 42.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 43.Romics L, Kodys K, Dolganiuc A, Graham L, Velayudham A, Mandrekar P, Szabo G. Diverse regulation of NF-kappaB and peroxisome proliferator-activated receptors in murine nonalcoholic fatty liver. Hepatology. 2004;40:376–385. doi: 10.1002/hep.20304. [DOI] [PubMed] [Google Scholar]
- 44.Kallwitz ER, McLachlan A, Cotler SJ. Role of peroxisome proliferators-activated receptors in the pathogenesis and treatment of nonalcoholic fatty liver disease. World J Gastroenterol. 2008;14:22–28. doi: 10.3748/wjg.14.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pang XH, Zhang JP, Zhang YJ, Yan J, Pei XQ, Zhang YQ, Li JQ, Zheng L, Chen MS. Preoperative levels of serum interleukin-6 in patients with hepatocellular carcinoma. Hepatogastroenterology. 2011;58:1687–1693. doi: 10.5754/hge10799. [DOI] [PubMed] [Google Scholar]
- 46.Shek FW, Benyon RC. How can transforming growth factor beta be targeted usefully to combat liver fibrosis? Eur J Gastroenterol Hepatol. 2004;16:123–126. doi: 10.1097/00042737-200402000-00001. [DOI] [PubMed] [Google Scholar]
- 47.Lang Q, Liu Q, Xu N, Qian KL, Qi JH, Sun YC, Xiao L, Shi XF. The antifibrotic effects of TGF-β1 siRNA on hepatic fibrosis in rats. Biochem Biophys Res Commun. 2011;409:448–453. doi: 10.1016/j.bbrc.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 48.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 49.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 50.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]