

Summary
Nearly all human beings, by the time they reach adolescence, are infected with multiple herpesviruses. At any given time, this family of viruses accounts for 35–40 billion human infections worldwide, making herpesviruses among the most prevalent pathogens known to exist. Compared to most other viruses, herpesviruses are also unique in that infection lasts the life of the host. Remarkably, despite their prevalence and persistence, little is known about how these viruses interact with their hosts, especially during the clinically asymptomatic phase of infection referred to as latency. This review explores data in human and animal systems that reveal the ability of latent herpesviruses to modulate the immune response to self and environmental antigens. From the perspective of the host, there are both potentially detrimental and surprisingly beneficial effects of this lifelong interaction. The realization that latent herpesvirus infection modulates immune responses in asymptomatic hosts forces us to reconsider what constitutes a ‘normal’ immune system in a healthy individual.
Keywords: herpesvirus, latency, mutualistic symbiosis, reactivation, immune modulation, autoimmunity
Introduction
Humans are densely populated with microorganisms, collectively referred to as the microbiome. While the physiologic importance of the gastrointestinal bacterial microbiota is well established, far less is known about the impact of viral symbionts (the ‘virobiota’). Among the most prevalent and persistent members of the virobiota are the herpesviruses. There are eight known human herpesviruses: herpes simplex viruses 1 and 2 (HSV1, HSV2), varicella zoster virus (VZV), human cytomegalovirus (HCMV), human herpesviruses 6 and 7 (HHV6, HHV7), Epstein Barr virus (EBV), and Kaposi sarcoma-associated herpesvirus (KSHV). These viruses are divided into three subfamilies (α, β, γ) based on genetic and biological similarities (1) (Table 1). Although most adults harbor multiple herpesviruses, severe disease attributable to herpesvirus infection is rare in immune competent individuals.
Table 1.
The Human Herpesviruses
| Subfamily | Human members |
Seroprevalence (adults) |
Associated diseases in the immunocompetent hosta |
Lytic cell targets |
Latent cell targets | Naturally evolved rodent virus models |
|---|---|---|---|---|---|---|
| α | HSV1 HSV2 VZV |
50–90% 15–95%b 90–100%c |
HSV1/2: Recurrent oral and genital ulcers; VZV: chicken pox, zoster (shingles) |
HSV1/2: mucosal epithelium. VZV: respiratory epithelium, T lymphocytes | Sensory neuronal ganglia | There are no known rodent α-herpesviruses. HSV1/2 can infect mice but do not recapitulate all aspects of human disease. |
| β | HCMV HHV6 HHV7 |
60–100% 90–100% 90–100% |
HCMV: Mononucleosis, congenital defects following transplacental infection. HHV6/7: infantile roseola | HCMV: mucosal epithelium HHV6/7: mucosal epithelium |
HCMV: myeloid lineage hematopoietic cells, smooth muscle cells, salivary and kidney epithelium HHV6/7: myeloid myeloid lineage hematopoietic cells, CD4+ T cells, salivary epithelium |
Rat and mouse CMV (MCMV) |
| γ | EBV, KSHV | 90–100% <5 to>50%d |
EBV: Mononucleosis, Burkitt’s and other lymphomas, nasopharyngeal carcinoma KSHV: Kaposi sarcoma, multicentric Castleman’s disease, peripheral effusion lymphoma |
EBV: oral epithelium KSHV: not known |
EBV: memory B cells KSHV: memory B cells, endothelial cells |
Murine gamma-herpesvirus 68 (MHV68, γHV68) |
Disease manifestations can vary quantitatively and qualitatively in the immune compromised individual
Frequency correlates strongly with number of sexual partners
Seroprevalence of VZV is estimated from studies occurring prior to the widespread adoption of live attenuated VZV vaccination
KSHV seroprevalence has a strong geographic bias
Infection with herpesviruses is conceptualized in three distinct phases corresponding to the clinical course of most patients: acute infection, latency, and reactivation. Acute infection with most herpesviruses, especially when it occurs in the first years of life, is asymptomatic or associated with only mild symptoms such as fever and rash. The acute phase is followed by a prolonged period called latency during which there is no overt evidence of disease. Clinically evident reactivation, often in the setting of immune compromise, can be associated with devastating morbidity and mortality ranging from painful genitourinary ulcers to deadly malignancies (2–5).
Many of the cellular and molecular events that correspond to these clinical stages of infection have been delineated. Lytic viral replication, which dominates acute infection, takes place initially at epithelial surfaces and is controlled by the host adaptive immune response. Herpesvirus latency at the molecular level is characterized by (i) the presence of the viral genome in the nucleus of the infected cell, generally as an episome, (ii) the absence of significant viral replication [as measured by classical, relatively insensitive methods including plaque assay, viral cellular transformation assays, and single-round polymerase chain reaction (PCR)], and (iii) minimal viral gene expression (6). Clinically silent, low-level viral reactivation events that stimulate an ongoing immune response are now known to persist into latency in spite of a lack of symptoms (7–9). A much more robust resurgence in viral gene expression and viral replication trigger the severe inflammatory response and tissue damage that characterize clinical reactivation.
Here we review data demonstrating that the lifelong interaction between latent herpesviruses and the host immune response not only keeps viral reactivation at bay but also modulates the host immune response to other antigens. For the purposes of this article, we define immune modulation as any effect of herpesvirus latency (including subclinical reactivation) that alters the host immune response to subsequent antigen exposure (infectious, non-infectious, self or altered self). Immune evasion by herpesviruses, including interference with viral antigen presentation, is essential for the acute and chronic phases of infection and has been expertly reviewed by others (10–12). Instead, we focus here on the evidence that herpesvirus infection alters the host immune response to non-herpesviral antigens. Specifically, a growing literature suggests that the anti-herpesvirus response triggers collateral damage. That is, herpesvirus latency may exacerbate seemingly unrelated diseases, including heterologous infections, autoimmunity, atherosclerosis, and cancer. But the consequences of latency are not always negative. Emerging data in both mice and humans suggests that herpesvirus latency may result in significant protection from secondary infections and allergic responses later in life. Lastly, since the vast majority of healthy people harbor multiple latent herpesviruses, we suggest that these viral passengers should be taken into account when modeling the ‘normal’ immune system. We propose that the study of mice intentionally infected with herpesvirus and other naturally occurring symbionts may lead to a better understanding of human immunology than can be accomplished with pathogen-free animals.
Immunologic hallmarks of herpesvirus latency
The cellular immunology of herpesvirus latency
For decades after the discovery of human herpesviruses, the lack of symptoms, absence of detectable viral replication, and paucity of viral gene expression during latency led virologists and clinicians to assume that this phase of infection was a period of viral quiescence. It followed naturally that the antiviral immune response should be resting—represented largely by memory T cells poised to respond to a reactivation event. It is now clear that neither the virus nor the host immune response is dormant during latency. The use of sensitive methods indicates that members of all three herpesvirus subfamilies produce infectious virions at mucosal and epithelial surfaces nearly continuously in apparently healthy individuals (7–9). Thus, clinically latent infection has a significant component of reactivation, during which some fraction of the pool of latently infected cells re-enters productive replication with high frequency.
Not surprisingly, such repetitive viral protein expression has profound effects on the adaptive antiviral response. The nature of the cellular immune response to three human herpesviruses, HSV1, EBV, and HCMV, has received considerable attention (13–15). Some features of these responses suggest they may have significant modulatory effects on the immune system in totum. First and foremost, the magnitude of the T-cell response during clinical latency is unusually high. Approximately 10–20% of circulating memory CD4+ and CD8+ T cells in healthy adults is specific for HCMV antigens (16). During acute symptomatic EBV infection presenting as infectious mononucleosis, these proportions are even higher, with up to 80% of all circulating CD8+ T cells specific for EBV antigens (14). This level decays rapidly with resolution of the acute infection, but even in healthy EBV carriers persists at an estimated 5–10% of CD8+ cells specific for EBV lytic and latent epitopes (17, 18). In the tonsils, a major site of latency and reactivation, the virus-specific fraction approaches 20%, indicating anatomic enrichment at the site of antigen expression (19). While the magnitude of the T-cell response against HSV1 and HSV2 is less dramatic systemically, there is significant enrichment of virus-specific cells both at the sites of latency and in epithelial tissues that support reactivation, even in the absence of clinically evident reactivation (20).
This expanded T-cell compartment appears to be highly functional. During stable latent infection, the majority of circulating CD8+ T cells specific for herpesvirus antigens have a resting memory phenotype, although late in HCMV infection a large proportion of CD8+ cells show signs of prolonged antigen-driven differentiation (15, 21). In both humans and mouse models, a subset of herpesvirus-specific T cells activate rapid cytokine production and cytolytic function following antigenic restimulation (22). Even in the absence of restimulation ex vivo, they display signs of recent antigen exposure including proliferation (23, 24). Interestingly, for most of the life of the human host, herpesvirus-specific lymphocytes do not develop signs of functional exhaustion seen in other chronic, highly-productive viral infections such as hepatitis C virus (HCV), human immunodeficiency virus (HIV), or persistent lymphocytic choriomeningitis virus (LCMV) (25). Far less is known about the CD4+ T-cell response to herpesviruses during latency, but this memory compartment also appears to be robustly and stably expanded, capable of rapidly exerting effector function (16, 23, 26). Thus, in spite of the absence of overt clinical disease, herpesvirus latency is a period of ongoing and intense interaction between the virus and the host, resulting in the production of a large pool of highly functional CD4+ and CD8+ T cells that is maintained for life.
This massive anti-herpesvirus T-cell compartment is essential for controlling viral reactivation, but it may also alter the outcome of heterologous infections, a phenomenon known as heterologous immunity (27, 28). Recent data in both human and mouse systems indicate that herpesvirus-specific CD8+ T cells are relatively unique in that they are activated in the setting of other infections, either via MHC-restricted cross-reactive peptides expressed by other pathogens or through antigen-independent bystander mechanisms. Evidence for immune modulation of this sort by herpesviruses in humans was recently provided by Sandalova et al. (29), who showed that EBV- and CMV-specific CD8+ T cells contribute to the expansion of the activated CD8+ T-cell compartment in response to natural infection with hepatitis B virus (HBV). In these subjects, who were acutely infected with HBV, nearly a quarter of the CD8+ T-cell pool was activated and this population included CD8+ T cells specific for herpesviruses. CMV- and EBV-specific CD8+ T cells showed a propensity to produce IFNγ during acute infection with HBV, indicating enhanced effector function. Bystander activation of herpesvirus-specific CD8+ T cells was also observed in patients acutely infected with Dengue virus, influenza A, adenovirus, HIV, and hantavirus infection (29–32).
Bystander activation of herpesvirus-specific T cells by a heterologous virus is not a universal phenomenon. Activation of CD8+ T cells specific for EBV and CMV was not observed during the CD8+ T-cell expansion that followed vaccination with attenuated yellow fever virus or vaccinia virus (33). In these experiments, the CD8+ T-cell response to the vaccines was robust, accounting for up to 40% of the peripheral CD8+ T-cell population. Therefore, the factors that allow for bystander activation do not seem to rely on the sheer magnitude of the CD8+ T-cell response. Rather, the nature of the acute secondary infection seems to play a role.
In situations where herpesvirus-specific CD8+ T cells are activated by acute secondary viral infection, activation of influenza-specific CD8+ T cells is absent or minimal (29). One key difference between CD8+ T cells specific for EBV or CMV versus those specific for influenza virus is the tonic exposure to antigen due to the near continuous reactivation of herpesviruses. Indeed, it has been postulated that herpesvirus reactivation (triggered by acute infection with another virus) is responsible for the activation of herpesvirus-specific T cells observed following infection with HIV and hantavirus. The transient detection of EBV DNA in the blood of a subset of hantavirus-infected individuals was offered as evidence in support of this hypothesis (32). However, the detection of EBV DNA did not correlate with the expansion of EBV-specific T cells. Furthermore, increased levels of EBV and CMV DNA were not detected in the sera of patients at any stage of infection with HBV, Dengue, influenza, and adenovirus (29), suggesting that reactivation of latent herpesviruses was not the cause of EBV- or CMV-specific T-cell activation. Sandalova et al. (29) addressed this discrepancy by testing the possibility that a different mechanism is at work. They were able to recapitulate the activated phenotype of EBV- and CMV-specific CD8+ T cells via the addition of exogenous IL-15. Interestingly, IL-15 did not activate CD8+ T cells specific for influenza, consistent with the dichotomy between herpesvirus- and influenza-specific T cells observed in vivo. The true mechanism driving bystander activation remains unknown, but IL-15 provides a plausible explanation whereby herpesvirus-specific CD8+ T cells (but not other antigen-experienced CD8+ T cells) become activated by heterologous infection. Alternatively, other studies have suggested that limited antigenic peptide similarity between herpesvirus antigens and proteins encoded by secondary pathogens may promote herpesvirus-specific T-cell activation via a major histocompatibility complex (MHC)- and T-cell receptor (TCR)-dependent mechanism (34).
Does bystander activation of herpesvirus-specific T cells impact host physiology? There is reason to believe, at least in the case of EBV and infectious mononucleosis, that heterologous immunity plays a role in the variable clinical course of acute herpesvirus infection at different ages. Acute infection with EBV triggers the activation of cross-reactive T cells specific for influenza A virus (35). Note that in this example of heterologous immunity, the cross-reactive T cells are specific for influenza A antigens but are activated by similar EBV peptides presented during acute EBV infection. It is possible that the exaggerated CD8+ T-cell response observed in patients with infectious mononucleosis, caused by cross reactivity with influenza-specific T cells, is responsible for the clinical severity of the disease in older patients (28, 35). Such cross reactivity, of course, is not expected in younger patients who generally do not experience mononucleosis during primary EBV infection, and who do not yet have a large population of heterologous memory cells. We revisit the importance of the age of the host in assessing the impact of herpesvirus infection below.
In summary, the developing picture of herpesvirus latency is one in which a substantial portion of the host T-cell compartment is specific for herpesvirus antigens, and these cells are periodically exposed to viral antigens expressed during frequent viral reactivation at mucosal sites. The capacity of these cells to surveil latency, and to curtail replication via either cytotoxic or noncytotoxic mechanisms, is central to maintaining a benign virus-host relationship. However, by virtue of herpesvirus-specific T-cell effector functions, the mucosal surfaces of humans are repeatedly exposed to inflammatory responses, with the potential to alter seemingly unrelated immune responses occurring at those sites. In addition, a significant proportion of herpesvirus-specific T cells become transiently activated in response to diverse secondary infections, and produce effector cytokines, including IFNγ, that can have profound immunomodulatory effects. Below, we discuss a growing body of circumstantial evidence that herpesvirus latency does in fact modulate the human immune response to other antigens in physiologically relevant ways.
The immunologic transcriptome of herpesvirus latency: insights from mouse models
To begin to dissect the mechanisms whereby latent herpesvirus infections might alter the host response to subsequent infection, we turned to the mouse. As there are no known mouse alphaherpesviruses, HSV1 infection of mice is commonly used as a model system (2). Mouse cytomegalovirus (MCMV) is a natural rodent virus closely related to HCMV (5), while murine gammaherpesvirus 68 (MHV68) is a pathogen of wild mice that is closely related to EBV and KSHV (36). As in EBV or HCMV infection in humans, murine herpesvirus latency is associated with robust expansion of virus-specific CD4+ and CD8+ T cells, and these cells display evidence of antigen exposure and proliferation during latent infection (37, 38). In the case of HSV1 infection in mice, there is an anatomically focused CD8+ T-cell response, recruited to latently infected trigeminal sensory neurons and capable of inhibiting viral reactivation via IFNγ secretion and noncytolytic granzyme-mediated degradation of viral transcription factors required for reactivation (39). In contrast, murine betaherpesvirus MCMV, and murine gammaherpesvirus MHV68, establish latency in hematopoietic cells and trigger a systemic lymphocyte response. MCMV infection is associated with memory inflation that resembles the expanding HCMV-specific CD8+ T cell response in humans (40).
By infecting mice with HSV1, MCMV, or MHV68, we were able to analyze the impact of latent infection with divergent herpesviruses on global host gene expression. Mice were infected intranasally to mimic the mucosal acquisition of the human herpesviruses. Since multiple herpesvirus infections are the rule among humans, we also co-infected mice with MCMV and MHV68 to determine whether dual infection would result in additive or synergistic effects on host transcription. One month after infection, at which point the acute infection with all three viruses has been cleared and latency established, microarray analysis was performed on total RNA harvested from the spleen, as an indicator of systemic immune modulatory effects of latent infection initiated at a mucosal site. We employed statistical techniques that allow the identification of patterns of gene expression unique to, or shared by, each herpesvirus tested (41, 42) (Fig. 1 and Supplementary Tables).
Fig. 1. Effects of herpesvirus infection on host transcription.
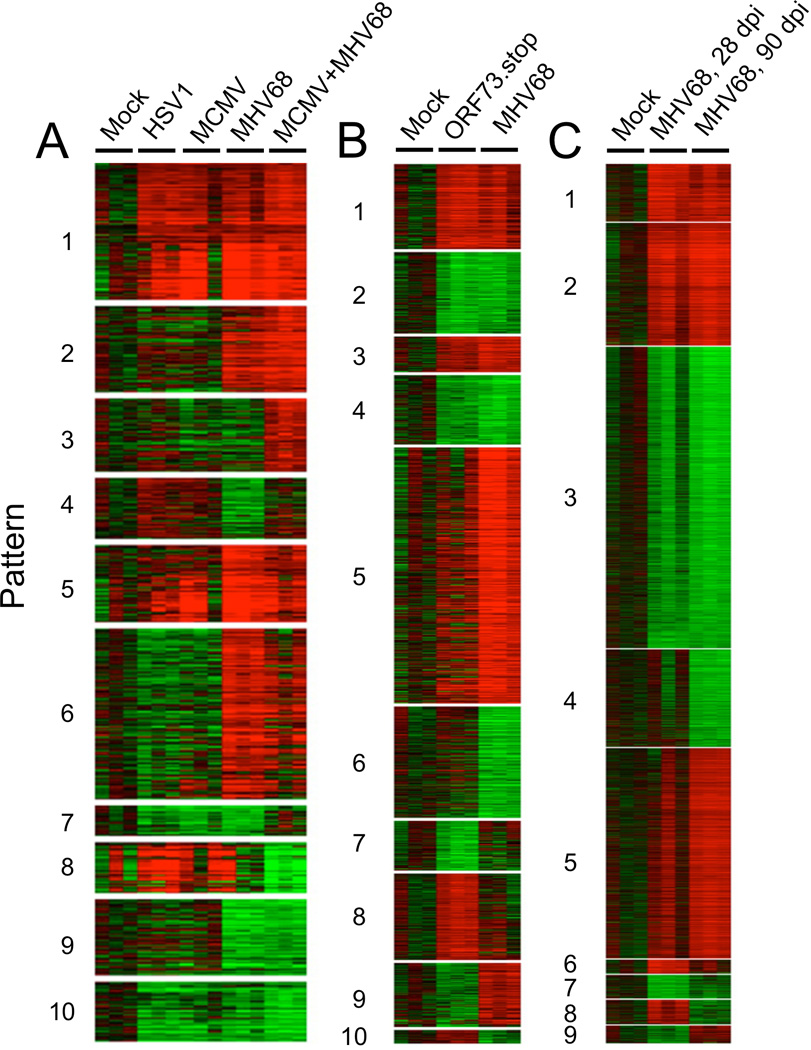
Wildtype (C57BL6/J) mice were infected with HSV1, MCMV, MHV68, latency-deficient MHV68 mutant (ORF73.stop), or both MCMV and MHV68 combined. All infections were performed with 1×105 plaque forming units of virus, administered intranasally. Twenty-eight or 90 days post infection (dpi, as indicated), total RNA was purified from mouse spleens and host transcript levels assessed using Illumina Mouse Gene 6 (v1.1) arrays and raw data pre-normalized using Beadstudio software. The extraction of gene expression patterns and identification of genes through EPIG was performed using the microarray data (41, 42). Briefly, intensity values from all of the probe sets on the arrays were log2-transformed and adjusted by systematic variation normalization. Distinct patterns of gene expression were extracted on the basis of the expression profile correlation values, the minimum cluster size for the patterns, and the cluster-partitioning resolution. From the patterns and with a signal-to-noise ratio of 3 (p < 0.001), magnitude of 0.5 (1.4-fold change), and a correlation r value of 0.64 (p < 0.001) of the gene profiles, probe sets were selected. In this data set, the mock-infected group (28 dpi) was used as a reference state. The average of the replicates of mock group arrays was aligned to 0 as a baseline, with all other treated samples adjusted by the same amount. Shown are heatmaps from biological triplicate experiments with one mouse per group that identify transcriptional signatures correlating with different single or multiple herpesvirus infections (A, 28 dpi), acute vs. latent MHV68 infection (B, 28 dpi), or early (28 dpi) vs. late (90 dpi) latent MHV68 infection (C). EPIG identified gene lists are available in Supplementary Tables linked from the online version of this manuscript.
Gene expression patterns between single or dual herpesvirus infected mice were complex, with multiple patterns of co- or divergently-regulated genes. By coupling pattern extraction with DAVID gene ontology analysis (43, 44), a number of useful concepts emerge (Fig. 1A). First, there were clusters of genes that were upregulated or downregulated in all herpesvirus-infected mice (patterns 1 and 5; and 10, respectively). Shared upregulated genes were highly enriched for those involved in cell division, with a minor enrichment for immune defense and inflammation, consistent with post-infection proliferation as a hallmark of all herpesvirus infections. Second, MHV68 infection induced more immune-response-associated gene expression than either HSV1 or MCMV (see the closely related patterns 2 and 6, which were highly enriched for these functional classes). The MHV68-driven immune response genes were dominated by an inflammatory cytokine component and included IFNγ, IL-18 receptor, signal transducer and activator of transcription 1 (STAT1), suppressor of cytokine signaling 3 (SOCS3), multiple chemokines, and a wide array of known IFNαβ-stimulated genes (Supplementary Tables).
It is clear that co-infection with two herpesviruses can have unexpected, emergent effects on host gene expression, since the transcription of some genes was altered predominantly in dual infection (patterns 3 and 8). Furthermore, some genes downregulated by MHV68 infection returned to baseline in MHV68/MCMV co-infected mice (pattern 4). Among those genes upregulated only in co-infection were several suggestive of heightened inflammation or enhanced lymphocyte response, including Nf-κB p50, Mapkbp1, Icos ligand, Pum1, and P-selectin ligand CD162 (Supplementary Tables).
To detect persistent or delayed gene expression patterns that are the result of acute infection, as opposed to transcriptional signatures specific for latent infection, we infected mice with a recombinant MHV68 mutant (ORF73.stop) that can replicate during acute infection but is incapable of establishing durable latency (45) (Fig. 1B). Comparison of ORF73.stop-induced gene expression to that induced by wildtype MHV68 identified transcripts that were similarly regulated following both acute-only (ORF73.stop) infection and acute + latent MHV68 infection (patterns 1–4). Genes upregulated or downregulated solely by acute infection were represented in patterns 7 and 8. In contrast, latency-specific differential gene signatures were evident in patterns 5 and 6. Gene ontology analysis indicates that the majority of the latency-specific host genes upregulated in pattern 5 were highly enriched for those related to the immune response. Patterns were also detected in which genes were inversely regulated by acute-only infection in comparison to acute + latent infection (patterns 9 and 10).
To determine whether transcriptional effects of latency evolve over time, or merely decay quantitatively without qualitative changes, we compared the transcriptional signatures in MHV68 infected mice at one and three months after infection (Fig, 1C). A number of genes were similarly upregulated or downregulated at both the early and later time points during latency (patterns 1 and 2; and 3, respectively). The consistently upregulated genes included IFNγ and multiple chemokines. However, it is also clear that the host response to latency matures with time, as a number of patterns emerged in which genes were only differentially expressed at one month (patterns 6, 7), three months (patterns 4, 5), or were inversely regulated at the two time points (patterns 8, 9).
These studies permit several conclusions about the impact of herpesvirus infection on host gene expression. First, the transcriptional effects of HSV1, MCMV, and MHV68 are unique. This is perhaps not surprising given the different tropism of these viruses, but it underscores the fact that the effects of latency will be difficult to generalize. Second, a significant component of the transcriptional signature during MHV68 infection can be attributed to immunomodulatory genes. In these experiments, MHV68 appeared to be the most immunomodulatory of the three viruses. This may be attributable to the anatomic restriction of HSV1 to the trigeminal ganglia (since we measured expression in the spleen) and the time-dependent inflation of the MCMV-specific T-cell response (which takes months to mature in the mouse system) or other factors (40). Third, co-infection with two viruses leads to emergent transcriptional signatures not predictable from individual herpesvirus infections. Fourth, latent infection confers a specific immunologic signature during MHV68 infection, since many immune response genes were not upregulated following infection with a latency-deficient viral mutant. Finally, the transcriptional signature of MHV68 latency evolves over time, with unique gene clusters evident at both one and three months. These observations highlight the ability of herpesvirus latency to profoundly and durably impact host immune function, and underscore the importance of defining these alterations triggered by latent herpesviruses (alone and in combined infections) over time. This data set provides an empirical foundation to begin such studies.
Herpesvirus latency modulates immunity to other pathogens
Given the capacity of herpesvirus latency to alter host gene expression and T-cell responses, it is reasonable to hypothesize that latent infection would alter the outcome of secondary infections. Indeed, this is the case. Data from multiple mouse models demonstrates the potential for latency to enhance resistance against secondary infection. In contrast, remarkably little is known about the potential benefits of herpesvirus latency in humans. Herpesvirus latency can also impact resistance to secondary infections in humans in a detrimental fashion, however. Among the risks identified so far is an increase in susceptibility to HIV infection, and a possible correlation between expanded HCMV memory T cells and immune senescence in the elderly.
Mutualistic symbiosis during murine betaherpesvirus and gammaherpesvirus latency
When heterologous immunity results in greater resistance to secondary infection, the outcome is termed cross protection. Early examples of this phenomenon were brought to light by Mackaness (46), who observed cross protection in mice infected with Mycobacterium tuberculosis, Listeria monocytogenes, and Brucella abortus. These cross protective effects are generally short-lived and are most robust when the secondary pathogen is administered during or shortly after the replication phase of the first pathogen. Given their lifelong persistence and continual low-level reactivation with resultant T-cell activation, we hypothesized that herpesviruses would trigger a durable cross protection. Indeed, increases in resistance against secondary heterologous infection have been observed in mice latently infected with the betaherpesvirus MCMV, which confers protection against vaccinia virus, lymphocytic choriomeningitis virus, and Pichinde virus (28, 47). The mechanism of MCMV-induced protection in these studies was not defined, and similarly durable cross protection was observed following infection with a non-persistent strain of LCMV. Therefore, it is unclear if cross-protection in this setting is conferred by latent infection or by prolonged consequences of acute infection.
We sought to determine whether durable cross-protection was a shared hallmark of herpesvirus latency by infecting mice with HSV1, MCMV, and MHV68 and challenging them one month later with heterologous bacterial and viral pathogens. We found that mice latently infected with either MHV68 or MCMV were highly resistant to challenge with L. monocytogenes, while mice infected with HSV1 were not (48). The flexibility of the murine system has allowed for several characteristics of this immune modulation by MHV68 to be delineated. Specifically, heterologous immunity against L. monocytogenes is not antigen-specific, in that it extends to protection from an unrelated bacterial pathogen, Yersinia pestis, and does not require CD4+ or CD8+ T cells at the time of secondary pathogen challenge (48). It is durable, lasting at least one fourth of the lifespan of the laboratory mouse even when the latent host is challenged with high doses of L. monocytogenes parenterally (49). Even more durable protection may be evident if physiologic doses and routes of secondary challenge are employed, but this hypothesis has not been tested. MHV68 latency is characterized by systemic activation of macrophages, which is also observed during MCMV latency, and elevated levels of TNFα, IFNγ, IL-6, and RANTES in the serum (Fig. 2). Both IFNγ and TNFα are genetically required for MHV68 latency to confer protection against L. monocytogenes, although the profound compromise in the ability of IFNγ−/− and TNFα−/− mice to resist L. monocytogenes challenge may mask latency-dependent cross protection in these backgrounds (48). The source of the systemic TNFα and IFNγ is unclear, since genetic ablation of the CD8+ T-cell compartment did not reduce the cross protection against L. monocytogenes (48). One possible source is natural killer (NK) cells, an important source of inflammatory cytokines during primary L. monocytogenes infection. MHV68 latency increases the cytotoxic capacity of NK cells via a process called arming (50). Increased granzyme B expression by murine NK cells, observed during latent MHV68 infection, is seen in multiple anatomic compartments and closely matches baseline granzyme B expression in peripheral NK cells from healthy adult humans (51) (Table 2).
Fig. 2. Latent MHV68 infection is associated with enhanced inflammatory cytokine production.

C57BL/6 mice were infected with MHV68 at 1×104 plaque forming units of virus, administered intranasally. Serum was harvested at the indicated time points (dpi) and assayed for cytokine concentrations via multiplex suspension array (RANTES, IL-6) or cytometric bead array (TNFα, IFNγ) according to the manufacturer's instructions (Bio Plex, Bio Rad Laboratories, Hercules, CA; BD Biosciences, San Diego, CA). Data are pooled from two to three independent experiments. Each dot represents one mouse. p values were calculated using a two-tailed Wilcoxon match pairs sign rank test, comparing mock to infected mice. *, p<0.05; **, p<0.01. Data for RANTES and IL-6, are unpublished results (Douglas W. White and Herbert W. Virgin). Data for TNFα and IFNγ are adapted from (48).
Table 2.
Changes in NK cell function induced by MHV68 latencya
| Granzyme B Expression By NK Cells | |
|---|---|
| Host | % NK cells that express granzyme B |
| healthy adult humans | ~40b |
| SPF mice | ~10 |
| MHV68 latently infected mice | ~40 |
| Protects Against RMA-S Lymphoma Transfer | |
| Condition | Lethality |
| Mock-infected | 80% |
| MHV68 latently infected | 10% |
| MHV68 latently infected, NK-depleted | 80% |
Heterologous immunity against L. monocytogenes is not observed during acute MHV68 infection, nor is it a delayed consequence of acute MHV68 infection. This was demonstrated using the MHV68 latency-defective ORF73.stop mutant, which neither activates macrophages nor confers cross protection against L. monocytogenes (48). In addition to our study, others have reported that mice latently infected with MHV68 are resistant to adenovirus (52) and plasmodium (53). Thus, latent infection with a gammaherpesvirus or a betaherpesvirus leads to significant protection from subsequent pathogen challenges in mouse models. These data highlight the possibility that herpesvirus latency has coevolved to promote host immunity to infectious disease, although no studies to date have addressed this potential in humans.
Synergistic co-pathogenesis between HIV and latent HSV2
In contrast to the benefits derived from betaherpesvirus or gammaherpesvirus latency in experimentally infected mice, alphaherpesvirus or betaherpesvirus latency can have profound negative consequences on HIV pathogenesis in humans. CMV, HHV6, HHV7, and HSV2 all affect the pathogenesis of simian immunodeficiency virus (SIV) and/or HIV (54–58). For instance, HIV/HSV2 co-infected patients shed more HIV than HSV2-negative controls, a difference that cannot be attributed simply to HIV RNA copy number in the plasma nor to peripheral CD4 counts (59). But perhaps the most dramatic effect of herpesviruses on HIV is the ability of HSV2 to increase the susceptibility of HIV-negative individuals to infection with HIV. One of the first hints that a herpesvirus could predispose to HIV infection came from the observation that genital ulcers are a risk factor for the acquisition of HIV (60). Indeed, the prevalence of HSV2 is as high as 90% in patients with HIV (61). This is not simply attributable to high-risk sexual behavior. A large number of observational studies indicate that HSV2 infection predisposes to an increase in the risk of acquiring HIV (62–64). The results of the Rakai Project study in particular, which was able to control for multiple confounders, are especially compelling in this regard. Whereas gonorrhea, Chlamydia, and trichomoniasis in HIV-negative partners of HIV-positive subjects had no detectable impact on susceptibility to HIV (65), the rate of transmission of HIV per sexual contact increased by fivefold if the HIV-susceptible individual was infected with HSV2 (61). Unfortunately, attempts to decrease HIV transmission by suppressing HSV2 with acyclovir have been disappointing (66–68), indicating that HSV2 replication may not be required for HSV2 to increase host susceptibility to HIV. One explanation for these results is the possibility that HSV2 latency and/or abortive reactivation (in the presence of acyclovir) are sufficient to modulate the host immune system in ways that enhance susceptibility to HIV.
The mechanisms by which HSV2 alters susceptibility to infection with HIV are unknown, but a number of observations suggest biologically plausible models. The degree to which these models call for immune modulation by HSV2, as we have defined it for this article, varies (57). First, HSV2 reactivation (even in the absence of clinically obvious lesions) results in a disruption of the mucosal barrier, which could allow for easier transmission of HIV. Furthermore, HSV2 lesions, even after healing, demonstrate an accumulation of dendritic cells and activated CD4+ T cells, which are potential targets for HIV infection (69). As a family, herpesviruses are well known for their ability to alter cytokine and interferon production by infected host cells. In theory, such alterations could increase susceptibility to HIV, but this area of investigation has not been explored in detail. The effect of HSV2 on HIV could be more direct. Indeed, HSV proteins can activate the HIV LTR (70). However, the relevance of this observation in vivo remains unknown. Such a mechanism would presumably require co-infection of HSV2 and HIV in the same cell. Interestingly, Moriuchi et al. (71) demonstrated that heat-inactivated HSV2 could trigger HIV replication in vitro in macrophages previously infected with HIV, indicating that HSV2 virions can alter the interaction between HIV and the host immune system without co-infecting the same cell.
While the synergy between HSV2 and HIV is clearly detrimental to humans exposed to HIV infection, this phenomenon also points to the possibility that infection with HSV2 (or biologically related HSV1) may alter the course of infection with other diseases acquired at mucosal sites and may even provide cross protection under some circumstances. To our knowledge, no epidemiologic studies have been performed with the aim of detecting putative protective effects conferred by alphaherpesvirus infection.
Inflation of HCMV-specific memory cells as a cofactor in immune senescence
The herpesvirus-specific lymphocyte response varies with the age of the host. An increasing portion of the CD8+ (and to a lesser degree CD4+) T-cell compartment in the elderly is directed against CMV such that a remarkable fraction (in some patients, >40%) of the T-cell repertoire is HCMV specific. This phenomenon is known as memory inflation. In most individuals, these CMV-specific T cells display signs of terminal differentiation, yet remain capable of proliferating in response to viral antigen, are cytotoxic, express IFNγ and TNFα, and control HCMV reactivation even in the very elderly (26, 40). However, immune senescence in some individuals may be attributable to the fact that these cells (CD28−CD57+) are by some measures hypofunctional or may crowd out naive T cells that might otherwise respond to new antigens (72–74). The presence of large numbers of these cells correlates with a diminished response to EBV (75) and a weak response to vaccination against influenza (76). In one study of aged, otherwise healthy individuals, HCMV infection correlated with inversion of the ratio of CD4+ to CD8+ T cells (<1) and conferred an increased risk of mortality (77). However, the expansion of HCMV memory is evident decades before other signs of frank immune senescence, and causal links between memory inflation and senescence are lacking. Despite the presence of large numbers of CD28−CD57+ CMV-specific T cells, CMV-positivity in infants is associated with a robust response against measles and staphylococcal enterotoxin B vaccines (78). Of note, a similar expansion of hypofunctional cells is not generally observed in response to EBV. The relationship between memory inflation and immune senescence, and the mechanisms that explain the differences between the young and aged, and between CMV and EBV, remain to be defined.
Herpesvirus latency as a cofactor in human inflammatory diseases
The potential for herpesvirus latency to affect human health goes beyond altering susceptibility to heterologous infections. Association studies and case reports have linked EBV (and other viruses) to a host of autoimmune diseases including multiple sclerosis (MS) and systemic lupus erythematosis (SLE). EBV is also postulated to play a role in the development of rheumatoid arthritis, perhaps by facilitating the generation of antibodies against citrullinated peptides (79). Hypotheses suggesting herpesviruses as etiologic agents in autoimmune disease are buoyed by the observation that viral syndromes sometimes precede the onset of autoimmune disease, as well as by experiments in which viruses have been shown to trigger or exacerbate autoimmune disease in animal models. Unfortunately, the interpretation of studies that show a link between herpesviruses and many human diseases is frequently hampered by the high prevalence of herpesviruses in healthy individuals. In the case of autoimmune diseases, this if further complicated by the potential for viral reactivation events which may occur more frequently in patients who are (iatrogenically or otherwise) immune suppressed due to their autoimmune disease. With these limitations in mind, a potential role for EBV in the pathogenesis of MS and SLE deserves careful consideration given the close epidemiologic links. We also briefly review a controversial association between HCMV and atherosclerosis. We next suggest that new immunologically informed models of cancer development suggest that latent herpesviruses have the potential to alter the progression or severity of human cancers. Finally, we describe compelling new data indicating that the news for herpesvirus-infected humans is not all bad, since a growing body of data indicate that early life infection with EBV and CMV may protect children from development of allergic disease.
EBV, systemic lupus erythematosis, and multiple sclerosis
Studies dating back four decades have found high titers of anti-EBV antibodies in SLE patients (80). Also, some SLE patients have elevated levels of EBV DNA in their blood (81, 82). In isolation, these findings could be explained simply by exaggerated EBV reactivation in SLE patients. But SLE patients seem to be more likely than the general population to be infected with EBV, even in studies of young patients where the seroprevalence of EBV in the control subjects is relatively low (83). Furthermore, mechanistic studies have suggested that molecular mimicry between EBV and self-antigens may play a role in the pathogenesis of SLE. For instance, Harley and James not only demonstrated homology between peptide epitopes in EBNA-1 (a latent protein produced by EBV) and the Smith antigen (a component of the spliceosome against which some SLE patients generate autoantibodies), but also demonstrated that immunization of mice and rabbits with EBNA-1 fragments leads to the development of anti-Smith antibodies and signs of SLE (84).
A relatively straightforward and popular hypothesis to explain these findings is that acute EBV infection triggers SLE in susceptible patients. However, analyses of multiple cohorts have found no evidence that infectious mononucleosis confers extra risk for the subsequent development of SLE (85–87). This, of course, does not rule out the possibility that immune modulation triggered by asymptomatic latent EBV could contribute to SLE. Still, the mechanisms by which EBV contributes to the pathogenesis of SLE, if any, do not appear to be as simple as an acute infection with EBV triggering autoimmune disease in a genetically susceptible host.
The association between EBV and MS is also compelling. Family studies, including data from monozygotic twins, and migration studies are largely consistent with the hypothesis that an environmental trigger, probably early in life, causes MS in a genetically susceptible host (88, 89). EBV is an attractive candidate for such a trigger. Infectious mononucleosis and MS are both observed more frequently in the West, in Caucasians, and in people living in northern latitudes. Nearly 100% of adults with MS are seropositive for EBV, while MS is rare in EBV-negative adults. As is the case with SLE, the association between MS and EBV is especially evident in pediatric patients where fewer of the control subjects are seropositive for EBV (90). Age at the time of infection with EBV may also play a role as infection in adolescence or early adulthood confers more risk than infection at an early age. Indeed, the risk of MS is two- to three-fold higher in people with a history of infectious mononucleosis than in the general population (89). Importantly, a history of infection with measles, mumps, rubella, or VZV confers no additional risk of MS. Finally, anti-EBNA-1 antibody titers not only correlate with an increased risk of MS, but an increase in the severity of the disease (91). Treatment with antivirals does not favorably alter the course of MS, but this finding has little bearing on the potential for EBV latency—which is minimally sensitive to antiviral drugs—to contribute to the pathogenesis of MS.
Evidence in animal models of the potential for gammaherpesvirus infection to exacerbate autoimmune disease in the central nervous system comes from studies of MHV68 in experimental allergic encephalitis-prone rats and mice (92). Interestingly, MHV68 latency also exacerbates other autoimmune conditions, including inflammatory bowel disease (93), perhaps by virtue of its ability to decrease levels of regulatory T cells (94).
While the links between EBV and certain autoimmune processes are strong, the mechanisms by which EBV might trigger or enhance susceptibility to these diseases remain unknown. Several models have been proposed by which a virus could alter the immune system resulting in an inappropriate activation and subsequent autoimmune disease (88, 95, 96). Molecular mimicry, that is cross-reactivity between viral and host epitopes, and epitope spread are among the oldest and most studied mechanisms proposed to be at the root of autoimmune diseases (not only with EBV and SLE as mentioned above, but also EBV and MS, (97–99). Unfortunately, examples in which this frequent phenomenon actually causes disease have been difficult to demonstrate (100, 101). And while this phenomenon is intriguing, the animal studies outlined above demonstrate that antigen cross reactivity at the level of the T-cell and B-cell receptor is not necessary for herpesvirus latency to result in profound alterations in the host immune response against unrelated antigens. Mechanisms that do not necessarily require antigen cross reactivity such as bystander activation of T cells (prominent in humans infected with EBV and CMV as outlined above), dysregulation of B-cell tolerance by viral proteins, adjuvant effects and virally encoded superantigens have also been discussed as potential culprits. A relatively new appreciation for the host cell machinery that alerts the innate immune system to the presence of microbes and coordinates the activation of the innate and adaptive immune responses (pattern recognition receptors and downstream signaling molecules) may bring new insights to the mechanisms by which viruses could contribute to autoimmunity (102).
Atherosclerosis and CMV
Atherosclerosis is a progressive inflammatory disease for which multiple pathogens have been proposed as cofactors, with significant focus on HCMV (103, 104). HCMV DNA is present in atherosclerotic and normal vascular tissue, and the anti-HCMV immune response may be increased in the context of severe disease (105–107). As mentioned previously, the interpretation of these results is clouded by the high prevalence of HCMV in most populations. However, animal studies bolster the assertion that CMV can drive atherosclerosis, since MCMV can promote disease in apoE−/− mice (genetically prone to accelerated atherosclerosis) (108). The mechanisms whereby CMV might promote atherosclerosis in vivo are unclear. Some studies suggest that chronic, unresolved inflammatory responses against latently infected cells distributed systemically or directly in the vessel wall may play a role (108, 109). Alternatively, immunomodulatory proteins secreted by CMV may alter smooth muscle cell motility or macrophage activation within atherosclerotic lesions (110, 111). Increased atherogenesis is also noted in latent MHV68-infected but not HSV1-infected apoE−/− mice (112), indicating this may be a shared capacity of beta- and gamma- but not alphaherpesviruses. In the MHV68 system, viral replication was detectable in aortic tissue, and antiviral drugs—which block reactivation but do not ablate latency—reduced atherogenesis (113), suggesting a requirement for viral replication. However, it is not clear whether replication needs to occur in the vessel wall or whether systemic effects of the virus can promote disease. In mice lacking the IFNγ receptor, MHV68 displays clear tropism for vascular smooth muscle. In these mice a persistently productive viral infection results in a robust but ineffective inflammatory infiltrate that ultimately causes a fatal vasculitis, confirming the capacity for an overactive—yet ineffective—antiviral immune response to promote vascular disease (114). Again, mouse models provide intriguing data that similar effects may occur in humans, especially those who experience herpesvirus infection later in life or are genetically predisposed to a prolonged anti-herpesvirus immune response.
Herpesviruses and cancer: a hypothesis
The human gammaherpesviruses EBV and KSHV are clearly associated with the development of multiple cancers arising from latently-infected cells, including Burkitt’s lymphoma, nasopharyngeal carcinoma, Kaposi sarcoma, and multicentric Castleman’s disease (3, 4). Both of these viruses encode putative oncogenes, and in the case of EBV, these genes are sufficient to immortalize human B cells in vitro. In a genocentric model of cancer progression, herpesvirus infection therefore functions simply as a mutagen, delivering exogenous oncogenes to the cell and thereby promoting primary or secondary steps of cancer progression (Fig. 3A).
Fig. 3. Hypothetical relationships between herpesvirus immune modulation during latency and cancer development.
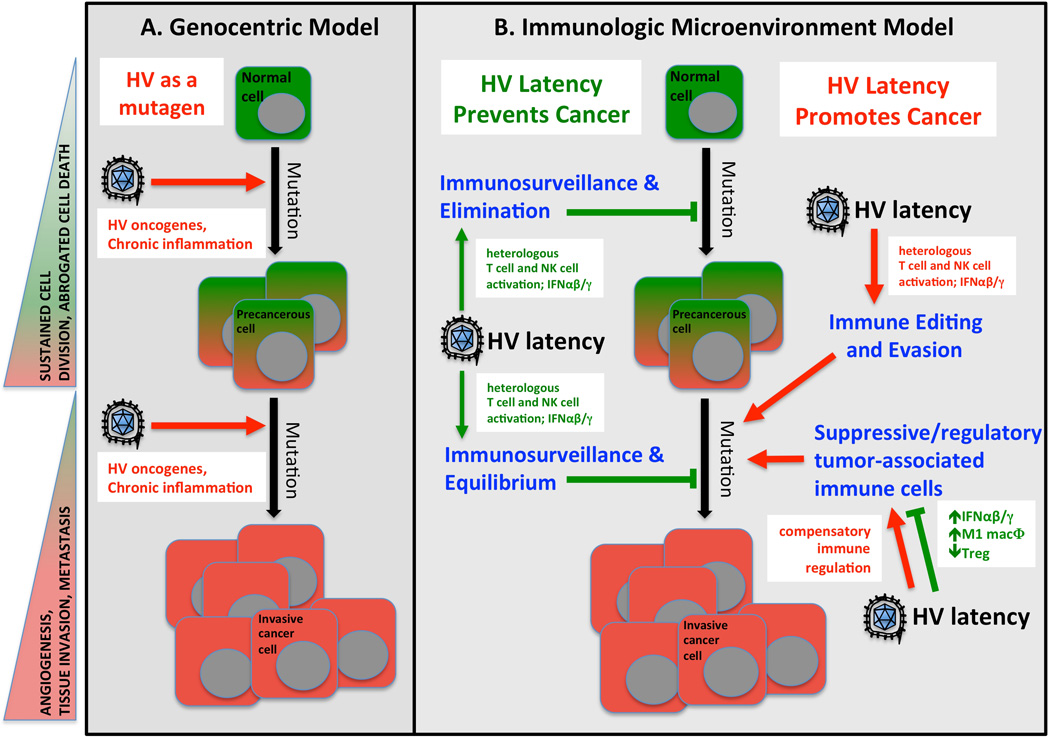
Shown are two models of cancer development that emphasize either genetic changes in the cancerous cell (Genocentric Model, A) or the immunologic microenvironment of the developing tumor (B) as driving forces in cancer progression. In genocentric models of cancer (A), the predominant view of herpesviruses (HV) is one of mutagenesis, either directly via introduction of viral oncogenes or indirectly. In immunologic models of cancer progression, HV may have more diverse mechanisms of interaction with the nascent cancer cell, including enhanced immunosurveillance, tumor elimination, or maintenance of pre-cancerous equilibrium triggered by latency-driven immune modulation. In contrast, the immune modulatory environment triggered by HV latency may promote more rapid tumor immunoediting and selection of immune evasion or suppressive tumor cells. In addition, compensatory regulatory events triggered by HV latency may enhance the development or recruitment of immunosuppressive cells into the tumor microenvironment. Hallmarks of cancer progression are shown the left in shaded triangles. Potential negative (tumor-promoting) mechanisms triggered by herpesvirus (HV) latency are indicated in red text with red arrows. Potential beneficial (tumor-inhibiting) mechanisms triggered by HV latency are indicated in green text with green arrows or blocked bars. The predominant modes of interaction between the immune system and cancerous cells are shown in blue text and based on (118).
However, the mechanisms by which gammaherpesvirus mediate transformation in vivo are not as simple as this model would predict. Although EBV infection is nearly universal, lymphoma attributable to EBV is rare unless the host is immune deficient (as in the case of post-transplant lymphoproliferative disease or AIDS-associated EBV lymphoma) or co-infected with HIV or malaria (as in the case of Burkitt’s lymphoma) (3). Similarly, KSHV infection is generally asymptomatic in the HIV-negative host (4), but HIV-positive patients routinely succumb to KS with lesions that are characterized by an inflammatory infiltrate and cytokine production. This inflammatory component seems to be an essential cofactor in tumor progression (115). Thus, EBV and KSHV infection alone are not sufficient to induce malignancy in most hosts but require either secondary genetic changes (often translocations of cellular oncogenes including c-myc in the case of Burkitt’s lymphoma) or immune suppression permitting unrestrained expression of viral oncogenes (116).
One potential mechanism whereby gammaherpesviruses could promote oncogenesis is genotoxic stress, secondary to unresolved inflammation, in a host that is predisposed to an unusually vigorous antiherpesvirus immune response (117). This model holds that it is not viral oncogenes alone that drive tumorigenesis, but the host response to the persistent virus that drives mutation of host genes. Subsequent virus-driven transcriptional changes promoting cellular proliferation, or blocking cell death, may stimulate the growth and survival of the mutated host cell. In this respect, our data showing that multiple herpesvirus infections in mice upregulate genes associated with the cell cycle is of particular interest (Fig. 1A).
It is also important to note that latency-induced immune modulation in humans and mice might enhance anti-tumor immunosurveillance. Indeed, mice that are latently infected with MHV68 are resistant to lethal lymphoma challenge, a phenotype mediated by latency-induced arming of host NK cells (50) (Table 2). Current theories suggest that the immune response against tumor cells can detect newly transformed cells and eliminate them before clinically evident tumors develop (Fig. 3B). Even if elimination is not possible, the anti-tumor immune response can hold pre-cancerous cells in a dormant state known as equilibrium (118). NK cells, CD8+ T cells, IFNγ, classically activated inflammatory (M1) macrophages (119) and IFNαβ are known mediators of both elimination and equilibrium (118). Given the upregulation of each of these anti-tumor effectors in latently infected humans and mice, it is plausible that herpesvirus latency may confer a state of enhanced tumor resistance. To our knowledge, no studies have addressed this possibility in humans.
Heightened anti-tumor immunity during latent infection may represent a double-edged sword. Enhanced surveillance of tumors also leads to increased selective pressure on tumor cells, which may result in latency-driven immune editing of tumors and selection of less immunogenic, more aggressive cancers that can evade tumor immunity (118). In addition, progression and metastasis of many tumors is spurred by the recruitment of a suppressive immune cell component, including alternatively activated anti-inflammatory (M2) macrophages, regulatory T cells, and myeloid-derived suppressor cells (118). The inflammatory state present during MHV68 latency seems unlikely to promote such an immune suppressive tumor microenvironment. However, negative-feedback compensatory processes may be triggered subsequent to latency-driven inflammation, and these homeostatic mechanisms may foster the development and recruitment of suppressive tumor-infiltrating leukocytes. Along these lines, it was recently reported that the same stimuli which drive M1 macrophage activation also result in the compensatory expansion of anti-inflammatory regulatory macrophages that express the transcription factor FoxP3 (120).
Given these variables, it is difficult to predict the effects of herpesvirus latency on cancer progression, and any effects may be virus-, time- and tumor-specific. The evidence that herpesviruses modulate the immune system in humans, coupled with an understanding of the importance of the anti-tumor immune response in cancer progression, indicate that studies addressing the role of herpesvirus infection in human cancer are merited. A few studies have suggested roles for EBV in cancers not classically associated with EBV, including breast cancer (121, 122). Several cancers have been linked to HCMV, which is not known to be oncogenic, but the links are tenuous so far (123, 124). A weakness of many studies correlating herpesviruses with cancer is the assumption that any herpesvirus-driven mechanism must be detrimental and must involve the presence of the virus in tumor cells, or within the tumor microenvironment (125, 126). We propose that the function of the host immune system is skewed systemically by herpesvirus latency, and the fundamental nature of the interaction between the tumor and the immune system—a critical determinant of tumor progression—is thereby altered. According to this mechanism, the amount of time that has passed since the patient was infected and the patient-specific response to latency, rather than viral tropism, may be the predominant variables that determine whether herpesviruses will impact tumorigenesis. Small animal studies of tumor incidence, progression, and metastasis in the presence of herpesvirus latency may enable identification of mechanisms linking latency to tumorigenesis, thereby permitting more refined human studies to determine whether herpesvirus are significant cofactors in cancer outcomes.
Does herpesvirus latency protect humans from atopy and allergy?
Among the many studies suggesting that herpesvirus-driven inflammation may exacerbate human disease, there is a suggestion that these same modulatory events—if delivered at the appropriate time—may provide a beneficial developmental cue to the human immune system. Some of the most compelling data that early herpesvirus infection alters the immune response against heterologous antigens in humans in potentially beneficial ways comes from studies in which researchers observed an inverse correlation between EBV seropositivity and the risk of atopy (127, 128). For example, Nilsson et al. (127) tested for antibodies against a variety of viruses (including HCMV, EBV, HSV, HHV6, and VZV) in two-year-old children. In the same cohort, they measured serum IgE levels and performed skin testing against a range of food and inhalant allergens to detect IgE sensitization. IgE sensitization was significantly reduced in children who were seropositive for EBV. Prior infection with HCMV alone did not confer protection against IgE sensitization, but co-infection with EBV and HCMV reduced the risk of IgE sensitization to a greater degree than did EBV infection alone. In a follow-up study, early infection with EBV (before two years of age) correlated with a decreased risk of IgE sensitization while late infection with EBV (after age two but before age five) conferred an increased risk (129). That infection with HCMV does not have the same effect as EBV, as well as the finding that the protective effects are observed in younger children but not in older cohorts, have been observed by others as well (128, 130, 131). Saghafian-Hedengren et al. (132) also examined lymphocyte function and serum cytokines in children infected with EBV or co-infected with EBV and HCMV and found that the presence of the viruses correlated with fewer IFNγ-producing NK cells and reduced serum IFNγ in the periphery. While some earlier studies did not observe a potential protective role for EBV in atopy (133, 134), the bulk of the available data to date demonstrate clear alterations in the human immune system attributable to herpesvirus latency and justify a closer examination of EBV infection before and after two years of age with respect to the risk of atopy, as well as cytokine production and lymphocyte function at anatomic sites where herpesvirus reactivation is common and allergic sensitization is initiated..
Since atopy is associated with asthma, and the incidence of asthma is increasing contemporaneously with an increase in the age of seroconversion to EBV and CMV (135–139), it is reasonable to ask whether early infection with EBV and/or CMV might protect against asthma. A recent study from Wu et al. (140) adds another dimension to this interesting question. These authors studied over 95,000 infants and found that their risk of developing asthma varied with their age at the time of the peak of winter virus infections. Infants born four months before the peak of the winter virus season were more likely to develop asthma than infants born twelve months prior to the peak. We speculate that one explanation for this finding has to do with the timing of the infants' acquisition of herpesviruses relative to their exposure to winter (asthmogenic) viruses. That is, herpesvirus infection, acquired before the winter virus season, might protect against asthma by altering the immune response against winter viruses such as respiratory syncytial virus. In contrast, we hypothesize that winter viruses might be more likely to cause asthma in infants not previously exposed to herpesvirus infection.
Future directions
Beyond the war metaphor: toward an ecological understanding of herpesvirus-host interactions
Virologists and clinicians alike have historically addressed the herpesvirus-host interaction using the so-called ‘war metaphor’: the virus invades and attacks the host, the host mounts a multi-pronged defense, and the virus uses of an array of sophisticated means to subvert or evade host defenses. The war metaphor seems especially apt in light of the AIDS epidemic, which has illustrated the destructive capacity of herpesviruses in patients with reduced defenses. However, many of the concepts we have discussed here are clearer when the war perspective is substituted with an evolutionary-ecological perspective (141–143). From this perspective, herpesviruses represent co-evolved symbionts that are remarkably well suited to persist, with little cost to the host. In addition, there is a growing appreciation that the viruses we carry constitute part of our ‘normal’ makeup and have profound, often positive, effects both in health and disease (144, 145).
In light of the fact that humans have been co-evolving with herpesviruses since the mammalian radiation (146), our finding that MHV68 induces cross protection led us to speculate that the relationship between herpesviruses and their hosts might be more appropriately termed one of mutualistic symbiosis. The term symbiosis is often used to connote a relationship of mutual benefit. But, in the strictest sense, symbiosis refers to any intimate relationship between two species. Some symbiotic relationships provide benefit for both members and are therefore termed mutualistic. Importantly, most symbiotic relationships are not purely parasitic or mutualistic. They often contain elements of mutualism and parasitism that may vary with developmental and environmental conditions. This form of symbiosis seems to most appropriately describe the relationship between humans and herpesviruses. We propose that the long evolutionary history between herpesviruses and mammals has selected for hosts with the ability to harbor latent herpesviruses because they gain a significant benefit in the form of cross protection from secondary infections early in life (Fig. 4A). In our studies, the mechanism of cross protection does not involve the introduction of a novel viral-induced mechanism of immunity to the host, but rather an upregulated set point of host innate immunity, likely triggered in a bystander manner by lymphocyte-derived cytokines that activate anti-bacterial phagocytes. However, other mechanisms may be required to explain the broad cross protection against multiple viral, bacterial and parasitic pathogens observed during MHV68 latency (48, 52, 53).
Fig. 4. Age-dependent outcomes in herpesvirus immunomodulation.

(A). Herpesvirus latency as a mutualistic symbiosis to promote cross protection from secondary infection. This hypothesis postulates that all organisms are born with a basal level of innate resistance to infection. Specific pathogen-free mice represent this basal level. As humans acquire multiple herpesviruses during their early years of life, latency-driven immune responses trigger increased resistance to secondary infections. This cross protection may be transient or durable as described in the text. (B). If herpesviruses are acquired during early life, beneficial immune modulation including cross protection from infections and immune skewing away from Th2-driven allergic inflammation may result. In contrast, following herpesvirus infection later in life, latency-driven inflammation may exacerbate host inflammatory diseases. If infections are acquired predominantly in late childhood through adulthood, this model predicts that the primary outcomes for the host will be pathologic.
Assuming mouse models of cross protection are predictive of the state in herpesvirus-infected humans, we predict that benefits of latency would be maximal during the several months following acquisition of each herpesvirus in early childhood and would then wane with time. However, given the evidence that herpesvirus reactivation is nearly constitutive in most individuals, we predict that mucosal resistance to infection would be increased above baseline for a prolonged period (7–9). Interestingly, the selective advantage conferred by latent infection need not be large in order to profoundly alter virus-host coevolution: a survival advantage conferred to less than one percent of latently infected individuals would ensure that latency became the predominant form of herpesvirus-host interaction within a few thousand generations (147, 148).
From this perspective, a disturbing trend among developed, higher socio-economic demographics is the increasing age of seroconversion to multiple herpesviruses. Multiple societal trends could contribute to delayed herpesvirus infection, which we hypothesize could have at least two opposing negative effects on human immunologic function: (i) decreased cross protection from other pathogens and allergic disease in early childhood, and (ii) exacerbation of inflammatory diseases that typically strike later in life, including some autoimmune diseases, cancers, and cardiovascular disease (Fig. 4B). Longitudinal studies designed to address the impact of early or delayed herpesvirus infection on a variety of human disease outcomes are needed to address these possibilities. These studies should be designed to identify potentially beneficial consequences of herpesvirus infection, including resistance to subsequent infection.
Implications for herpesvirus vaccine development
An evolutionary and ecological understanding has significant implications for plans to develop herpesvirus vaccines. Currently, vaccines are in development against HSV1, HSV2, HCMV, and EBV, and show varying efficacy at preventing disease (149–151). One trend arising from these efforts is the realization that sterilizing herpesvirus vaccines—those that prevent natural infection—may be impossible to design, perhaps due to the capacity of herpesviruses to establish latency with minimal replication (152, 153).
Diseases caused by just one herpesvirus are preventable using an FDA-approved vaccine (marketed as Varivax and Zostavax) that contains a live attenuated strain of VZV. Varivax is highly effective at preventing chicken pox when administered to children, but immunity wanes and periodic boosters may be required throughout adulthood. In an increased dose, the same attenuated vaccine strain (Zostavax) provides significant protection from VZV reactivation manifest as herpes zoster (HZ), or shingles (154). Once shown to be safe and effective, the VZV vaccine quickly became mandatory for most school age children in the USA.
Only a very small fraction of children experience severe symptoms following natural VZV infection. The risk of hospitalization and death is much higher in adults but is still quite rare (~0.4 deaths per year per million individuals in the United States, prior to widespread vaccination) (154). The deployment of Varivax has dramatically altered the natural ecology of VZV circulation, making natural wildtype infection much less likely. This may place unvaccinated children at an increased risk of severe VZV infection since it could delay their first exposure until adulthood. Perhaps due to waning vaccine-induced immunity, even prior vaccinees are at increased risk of moderate to severe VZV disease if re-exposed to natural VZV later in life but prior to vaccine boost (155). In addition, early reports suggest that near universal childhood VZV vaccination has increased the incidence of adult HZ (156, 157), perhaps because community acquired re-exposure to VZV boosts adult immunity (158). These and other concerns are cited as rationale for current European policies of administering Varivax only to high risk patients, rather than universally (159). Widespread adoption of VZV vaccination was predicted to reduce lost workforce productivity resulting from parents that miss work to care for children with chickenpox (160, 161). It is not clear whether the putative economic benefits of VZV vaccination can be realized in light of a potential increase in the rate of HZ, or if routine boosters are needed for all vaccine recipients (160, 161).
Further herpesvirus vaccine deployment and subsequent reduction of natural infection rates may be of concern if co-evolved human-herpesvirus relationships confer mutualistic advantages, as we hypothesize in this review. How then should vaccine design proceed? First, careful longitudinal studies following naturally infected and uninfected cohorts for a variety of clinical outcomes should be performed, similar to the studies described above correlating atopy with EBV and HCMV seroconversion. If benefits of natural HV infection in humans become clear, then we should strive to develop live, attenuated, latency-competent vaccines that confer these benefits while still protecting from the risks of natural infection. Conversely, if life-long inflammation triggered by viral latency is found to exacerbate of other inflammatory states, then we should be careful to ensure that live herpesvirus-based vaccine vectors do not worsen these effects, especially given prospects to use HCMV as a vaccine vector (162). In short, caution and vigilance are advisable prior to the disruption of poorly understood virus-host equilibria that have adapted over hundreds of millions of years. Accordingly, vaccination of those at highest risk for severe herpesvirus-induced disease (e.g., HCMV naïve pregnant women, EBV-naive adolescents or organ-transplant candidates, or VZV-seronegative adolescents and adults), as opposed to universal childhood vaccination, might be the most reasonable approach until the impact of herpesviruses on human immune physiology is more carefully studied.
Is it time to intentionally re-introduce the virobiota into laboratory mice?
The development of inbred, genetically manipulable, pathogen-free mice has brought an undeniable wealth of insight into the genetic and cellular basis of immune function and the pathogenesis of infectious disease. However, this reductionist approach may not reveal the insights necessary to unravel some of the most vexing human immunologic and inflammatory diseases. There are a variety of durable immunologic phenotypes that arise as a result of herpesvirus infection. This suggests that a more complete understanding of normal immune function must take these effects into account.
How do we render the mouse a more predictive model of human immune physiology? The path forward would seem to start with a careful definition of natural persistent flora of feral mice, including but not limited to herpesviruses. Indeed, initial evidence suggests that immune function in feral mice is quite distinct from that in pathogen free laboratory mice (163). The ultimate goal may be the creation of a new immune standard: the specific-pathogen-colonized mouse. We predict that such models will allow a renaissance of immunologic studies with the potential to more accurately reflect human immunology as it actually exists. This new approach may provide essential insight into the genetic mechanisms that matter most against the noisy, variable background of immune stimulation with which human evolution has had to cope—and upon which normal immune function may in fact rely.
Supplementary Material
Acknowledgments
Support for this work came from the NIH (K08AI079011 to D.W.W.; R01AI080775 to E.S.B) an Abbott Scholar Award (to D.W.W.), the Gundersen Lutheran Medical Foundation, and a Homeland Security STEM fellowship (R.S.B). We thank Cherie Ochsenfeld and Rebecca Doerge, Purdue University Department of Statistics, for preliminary analyses of microarray data. We are grateful for the expert assistance of Jeff W. Chou, Wake Forest University School of Medicine, Division of Public Health Sciences, in final analysis of microarray data. We wish to thank Herbert W. Virgin for his committed and inspiring mentorship and for numerous critical conversations that have shaped our thinking on these topics.
Footnotes
All authors affirm that no conflicts of interest exist.
References
- 1.Pellett PE, Roizman B. The Family Herpesviridae: A Brief Introduction. In: Knipe DM, Howely PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2479–2500. [Google Scholar]
- 2.Roizman B, Knipe DM, Whitley RJ. Herpes Simplex Viruses. In: Knipe DM, Howely PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2501–2602. [Google Scholar]
- 3.Rickinson AB, Kieff E. Epstein-Barr Virus. In: Knipe DM, Howely PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2655–2700. [Google Scholar]
- 4.Ganem D. Kapsoi's Sarcoma-Associated Herpesvirus. In: Knipe DM, Howely PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2847–2888. [Google Scholar]
- 5.Mocarski ES, Shenk T, Pass RF. Cytomegaloviruses. In: Knipe DM, Howely PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2701–2772. [Google Scholar]
- 6.Virgin HW, Speck SH. Unraveling immunity to gamma-herpesviruses: a new model for understanding the role of immunity in chronic virus infection. Curr Opin Immunol. 1999;11:371–379. doi: 10.1016/s0952-7915(99)80063-6. [DOI] [PubMed] [Google Scholar]
- 7.Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009;5 doi: 10.1371/journal.ppat.1000496. e1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tronstein E, et al. Genital shedding of herpes simplex virus among symptomatic and asymptomatic persons with HSV-2 infection. J Am Med Assoc. 2011;305:1441–1449. doi: 10.1001/jama.2011.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ling PD, et al. The dynamics of herpesvirus and polyomavirus reactivation and shedding in healthy adults: a 14-month longitudinal study. J Infect Dis. 2003;187:1571–1580. doi: 10.1086/374739. [DOI] [PubMed] [Google Scholar]
- 10.Means RE, Lang SM, Jung JU. Human gammaherpesvirus immune evasion strategies. In: Arvin A, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. [PubMed] [Google Scholar]
- 11.Gewurz BE, Vyas JM, Ploegh HL. Herpesvirus evasion of T-cell immunity. In: Arvin A, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007. [PubMed] [Google Scholar]
- 12.Vandevenne P, Sadzot-Delvaux C, Piette J. Innate immune response and viral interference strategies developed by human herpesviruses. Biochem Pharmacol. 2010;80:1955–1972. doi: 10.1016/j.bcp.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Decman V, Freeman ML, Kinchington PR, Hendricks RL. Immune control of HSV-1 latency. Viral Immunol. 2005;18:466–473. doi: 10.1089/vim.2005.18.466. [DOI] [PubMed] [Google Scholar]
- 14.Callan MF. The immune response to Epstein-Barr virus. Microbes Infect. 2004;6:937–945. doi: 10.1016/j.micinf.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Moss P, Khan N. CD8(+) T-cell immunity to cytomegalovirus. Hum Immunol. 2004;65:456–464. doi: 10.1016/j.humimm.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Sylwester AW, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hislop AD, Annels NE, Gudgeon NH, Leese AM, Rickinson AB. Epitope-specific evolution of human CD8(+) T cell responses from primary to persistent phases of Epstein-Barr virus infection. J Exp Med. 2002;195:893–905. doi: 10.1084/jem.20011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan LC, et al. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J Immunol. 1999;162:1827–1835. [PubMed] [Google Scholar]
- 19.Hislop AD, et al. Tonsillar homing of Epstein-Barr virus-specific CD8+ T cells and the virus-host balance. J Clin Invest. 2005;115:2546–2555. doi: 10.1172/JCI24810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheridan BS, Knickelbein JE, Hendricks RL. CD8 T cells and latent herpes simplex virus type 1: keeping the peace in sensory ganglia. Expert Opin Biol Ther. 2007;7:1323–1331. doi: 10.1517/14712598.7.9.1323. [DOI] [PubMed] [Google Scholar]
- 21.van de Berg PJ, et al. Cytomegalovirus infection reduces telomere length of the circulating T cell pool. J Immunol. 2010;184:3417–3423. doi: 10.4049/jimmunol.0903442. [DOI] [PubMed] [Google Scholar]
- 22.Hislop AD, et al. EBV-specific CD8+ T cell memory: relationships between epitope specificity, cell phenotype, and immediate effector function. J Immunol. 2001;167:2019–2029. doi: 10.4049/jimmunol.167.4.2019. [DOI] [PubMed] [Google Scholar]
- 23.Heller KN, Upshaw J, Seyoum B, Zebroski H, Munz C. Distinct memory CD4+ T-cell subsets mediate immune recognition of Epstein Barr virus nuclear antigen 1 in healthy virus carriers. Blood. 2007;109:1138–1146. doi: 10.1182/blood-2006-05-023663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belz GT, Doherty PC. Virus-specific and bystander CD8+ T-cell proliferation in the acute and persistent phases of a gammaherpesvirus infection. J Virol. 2001;75:4435–4438. doi: 10.1128/JVI.75.9.4435-4438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim PS, Ahmed R. Features of responding T cells in cancer and chronic infection. Curr Opin Immunol. 2010;22:223–230. doi: 10.1016/j.coi.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Libri V, et al. Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+ CD45RA+ CD27 T cells: the potential involvement of interleukin-7 in this process. Immunology. 2011;132:326–339. doi: 10.1111/j.1365-2567.2010.03386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol. 2002;2:417–426. doi: 10.1038/nri820. [DOI] [PubMed] [Google Scholar]
- 28.Welsh RM, Che JW, Brehm MA, Selin LK. Heterologous immunity between viruses. Immunol Rev. 2010;235:244–266. doi: 10.1111/j.0105-2896.2010.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandalova E, et al. Contribution of herpesvirus specific CD8 T cells to anti-viral T cell response in humans. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001051. e1001051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doisne JM, et al. CD8+ T cells specific for EBV, cytomegalovirus, and influenza virus are activated during primary HIV infection. J Immunol. 2004;173:2410–2418. doi: 10.4049/jimmunol.173.4.2410. [DOI] [PubMed] [Google Scholar]
- 31.Papagno L, et al. Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PLoS Biol. 2004;2:E20. doi: 10.1371/journal.pbio.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuuminen T, et al. Human CD8+ T cell memory generation in Puumala hantavirus infection occurs after the acute phase and is associated with boosting of EBV-specific CD8+ memory T cells. J Immunol. 2007;179:1988–1995. doi: 10.4049/jimmunol.179.3.1988. [DOI] [PubMed] [Google Scholar]
- 33.Miller JD, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 34.Cornberg M, et al. CD8 T cell cross-reactivity networks mediate heterologous immunity in human EBV and murine vaccinia virus infections. J Immunol. 2010;184:2825–2838. doi: 10.4049/jimmunol.0902168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clute SC, et al. Cross-reactive influenza virus-specific CD8+ T cells contribute to lymphoproliferation in Epstein-Barr virus-associated infectious mononucleosis. J Clin Invest. 2005;115:3602–3612. doi: 10.1172/JCI25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Virgin HW, et al. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71:5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton E, Mandal P, Speck SH. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol. 2011;29:351–397. doi: 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
- 38.Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol. 2006;177:450–458. doi: 10.4049/jimmunol.177.1.450. [DOI] [PubMed] [Google Scholar]
- 39.Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snyder CM, Cho KS, Bonnett EL, van Dommelen S, Shellam GR, Hill AB. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29:650–659. doi: 10.1016/j.immuni.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou JW, Zhou T, Kaufmann WK, Paules RS, Bushel PR. Extracting gene expression patterns and identifying co-expressed genes from microarray data reveals biologically responsive processes. BMC Bioinformatics. 2007;8:427. doi: 10.1186/1471-2105-8-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chou JW, Paules RS, Bushel PR. Systematic variation normalization in microarray data to get gene expression comparison unbiased. J Bioinform Comput Biol. 2005;3:225–241. doi: 10.1142/s0219720005001028. [DOI] [PubMed] [Google Scholar]
- 43.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 44.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moorman NJ, Willer DO, Speck SH. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J Virol. 2003;77:10295–10303. doi: 10.1128/JVI.77.19.10295-10303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mackaness GB. The immunological basis of acquired cellular resistance. J Exp Med. 1964;120:105–120. [PubMed] [Google Scholar]
- 47.Selin LK, Varga SM, Wong IC, Welsh RM. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J Exp Med. 1998;188:1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barton ES, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 49.Yager EJ, et al. gamma-Herpesvirus-induced protection against bacterial infection is transient. Viral Immunol. 2009;22:67–72. doi: 10.1089/vim.2008.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White DW, et al. Latent herpesvirus infection arms NK cells. Blood. 2010;115:4377–4383. doi: 10.1182/blood-2009-09-245464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104:2840–2848. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen Y, McGuffie BA, Anderson VE, Weinberg JB. Gammaherpesvirus modulation of mouse adenovirus type 1 pathogenesis. Virology. 2008;380:182–190. doi: 10.1016/j.virol.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haque A, Rachinel N, Quddus MR, Haque S, Kasper LH, Usherwood E. Co-infection of malaria and gamma-herpesvirus: exacerbated lung inflammation or cross-protection depends on the stage of viral infection. Clin Exp Immunol. 2004;138:396–404. doi: 10.1111/j.1365-2249.2004.02652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biancotto A, et al. Evolution of SIV toward RANTES resistance in macaques rapidly progressing to AIDS upon coinfection with HHV-6A. Retrovirology. 2009;6:61. doi: 10.1186/1742-4690-6-S2-I3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lisco A, et al. Viral interactions in human lymphoid tissue: Human herpesvirus 7 suppresses the replication of CCR5-tropic human immunodeficiency virus type 1 via CD4 modulation. J Virol. 2007;81:708–717. doi: 10.1128/JVI.01367-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lisco A, Vanpouille C, Margolis L. War and peace between microbes: HIV-1 interactions with coinfecting viruses. Cell Host Microbe. 2009;6:403–408. doi: 10.1016/j.chom.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 57.Van de Perre P, et al. Herpes simplex virus and HIV-1: deciphering viral synergy. Lancet Infect Dis. 2008;8:490–497. doi: 10.1016/S1473-3099(08)70181-6. [DOI] [PubMed] [Google Scholar]
- 58.Webster A, et al. Cytomegalovirus infection and progression towards AIDS in haemophiliacs with human immunodeficiency virus infection. Lancet. 1989;2:63–66. doi: 10.1016/s0140-6736(89)90312-7. [DOI] [PubMed] [Google Scholar]
- 59.LeGoff J, et al. Cervicovaginal HIV-1 and herpes simplex virus type 2 shedding during genital ulcer disease episodes. AIDS. 2007;21:1569–1578. doi: 10.1097/QAD.0b013e32825a69bd. [DOI] [PubMed] [Google Scholar]
- 60.Greenblatt RM, et al. Genital ulceration as a risk factor for human immunodeficiency virus infection. AIDS. 1988;2:47–50. doi: 10.1097/00002030-198802000-00008. [DOI] [PubMed] [Google Scholar]
- 61.Corey L, Wald A, Celum AL, Quinn TC. The effects of herpes simplex virus-2 on HIV-1 acquisition and transmission: a review of two overlapping epidemics. J Acquir Immune Defic Syndr. 2004;35:435–445. doi: 10.1097/00126334-200404150-00001. [DOI] [PubMed] [Google Scholar]
- 62.Wald A, Link K. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J Infect Dis. 2002;185:45–52. doi: 10.1086/338231. [DOI] [PubMed] [Google Scholar]
- 63.Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS. 2006;20:73–83. doi: 10.1097/01.aids.0000198081.09337.a7. [DOI] [PubMed] [Google Scholar]
- 64.Brown JM, et al. Incident and prevalent herpes simplex virus type 2 infection increases risk of HIV acquisition among women in Uganda and Zimbabwe. AIDS. 2007;21:1515–1523. doi: 10.1097/QAD.0b013e3282004929. [DOI] [PubMed] [Google Scholar]
- 65.Gray RH, et al. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet. 2001;357:1149–1153. doi: 10.1016/S0140-6736(00)04331-2. [DOI] [PubMed] [Google Scholar]
- 66.Watson-Jones D, et al. Effect of herpes simplex suppression on incidence of HIV among women in Tanzania. N Engl J Med. 2008;358:1560–1571. doi: 10.1056/NEJMoa0800260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Celum C, et al. Effect of aciclovir on HIV-1 acquisition in herpes simplex virus 2 seropositive women and men who have sex with men: a randomised, double-blind, placebo-controlled trial. Lancet. 2008;371:2109–2119. doi: 10.1016/S0140-6736(08)60920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Celum C, et al. Acyclovir and transmission of HIV-1 from persons infected with HIV-1 and HSV-2. N Engl J Med. 2010;362:427–439. doi: 10.1056/NEJMoa0904849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rebbapragada A, et al. Negative mucosal synergy between Herpes simplex type 2 and HIV in the female genital tract. AIDS. 2007;21:589–598. doi: 10.1097/QAD.0b013e328012b896. [DOI] [PubMed] [Google Scholar]
- 70.Mosca JD, et al. Activation of human immunodeficiency virus by herpesvirus infection: identification of a region within the long terminal repeat that responds to a trans-acting factor encoded by herpes simplex virus 1. Proc Natl Acad Sci USA. 1987;84:7408–7412. doi: 10.1073/pnas.84.21.7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moriuchi M, Moriuchi H, Williams R, Straus SE. Herpes simplex virus infection induces replication of human immunodeficiency virus type 1. Virology. 2000;278:534–540. doi: 10.1006/viro.2000.0667. [DOI] [PubMed] [Google Scholar]
- 72.Khan N. The immunological burden of human cytomegalovirus infection. Arch Immunol Ther Exp (Warsz) 2007;55:299–308. doi: 10.1007/s00005-007-0037-3. [DOI] [PubMed] [Google Scholar]
- 73.Pawelec G, Larbi A, Derhovanessian E. Senescence of the human immune system. J Comp Pathol. 2010;142 Suppl:S39–S44. doi: 10.1016/j.jcpa.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 74.Gress RE, Deeks SG. Reduced thymus activity and infection prematurely age the immune system. J Clin Invest. 2009;119:2884–2887. doi: 10.1172/JCI40855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khan N, et al. Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J Immunol. 2004;173:7481–7489. doi: 10.4049/jimmunol.173.12.7481. [DOI] [PubMed] [Google Scholar]
- 76.Trzonkowski P, et al. Association between cytomegalovirus infection, enhanced proinflammatory response and low level of anti-hemagglutinins during the anti-influenza vaccination--an impact of immunosenescence. Vaccine. 2003;21:3826–3836. doi: 10.1016/s0264-410x(03)00309-8. [DOI] [PubMed] [Google Scholar]
- 77.Wikby A, Johansson B, Olsson J, Lofgren S, Nilsson BO, Ferguson F. Expansions of peripheral blood CD8 T-lymphocyte subpopulations and an association with cytomegalovirus seropositivity in the elderly: the Swedish NONA immune study. Exp Gerontol. 2002;37:445–453. doi: 10.1016/s0531-5565(01)00212-1. [DOI] [PubMed] [Google Scholar]
- 78.Miles DJ, et al. Cytomegalovirus infection induces T-cell differentiation without impairing antigen-specific responses in Gambian infants. Immunology. 2008;124:388–400. doi: 10.1111/j.1365-2567.2007.02787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pratesi F, Tommasi C, Anzilotti C, Chimenti D, Migliorini P. Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2006;54:733–741. doi: 10.1002/art.21629. [DOI] [PubMed] [Google Scholar]
- 80.Evans AS, Rothfield NF, Niederman JC. Raised antibody titres to E.B. virus in systemic lupus erythematosus. Lancet. 1971;1:167–168. doi: 10.1016/s0140-6736(71)91937-4. [DOI] [PubMed] [Google Scholar]
- 81.Kang I, et al. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J Immunol. 2004;172:1287–1294. doi: 10.4049/jimmunol.172.2.1287. [DOI] [PubMed] [Google Scholar]
- 82.Moon UY, et al. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Res Ther. 2004;6:R295–R302. doi: 10.1186/ar1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J Clin Invest. 1997;100:3019–3026. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poole BD, Gross T, Maier S, Harley JB, James JA. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J Autoimmun. 2008;31:362–371. doi: 10.1016/j.jaut.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ulff-Moller CJ, Nielsen NM, Rostgaard K, Hjalgrim H, Frisch M. Epstein-Barr virus-associated infectious mononucleosis and risk of systemic lupus erythematosus. Rheumatology. 2010;49:1706–1712. doi: 10.1093/rheumatology/keq148. [DOI] [PubMed] [Google Scholar]
- 86.Strom BL, Reidenberg MM, West S, Snyder ES, Freundlich B, Stolley PD. Shingles, allergies, family medical history, oral contraceptives, and other potential risk factors for systemic lupus erythematosus. Am J Epidemiol. 1994;140:632–642. doi: 10.1093/oxfordjournals.aje.a117302. [DOI] [PubMed] [Google Scholar]
- 87.Cooper GS, Dooley MA, Treadwell EL, St Clair EW, Gilkeson GS. Risk factors for development of systemic lupus erythematosus: allergies, infections, and family history. J Clin Epidemiol. 2002;55:982–989. doi: 10.1016/s0895-4356(02)00429-8. [DOI] [PubMed] [Google Scholar]
- 88.Kakalacheva K, Munz C, Lunemann JD. Viral triggers of multiple sclerosis. Biochim Biophys Acta. 2011;1812:132–140. doi: 10.1016/j.bbadis.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ascherio A, Munger KL. Epstein-barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol. 2010;5:271–277. doi: 10.1007/s11481-010-9201-3. [DOI] [PubMed] [Google Scholar]
- 90.Alotaibi S, Kennedy J, Tellier R, Stephens D, Banwell B. Epstein-Barr virus in pediatric multiple sclerosis. J Am Med Assoc. 2004;291:1875–1879. doi: 10.1001/jama.291.15.1875. [DOI] [PubMed] [Google Scholar]
- 91.Farrell RA, et al. Humoral immune response to EBV in multiple sclerosis is associated with disease activity on MRI. Neurology. 2009;73:32–38. doi: 10.1212/WNL.0b013e3181aa29fe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Peacock JW, Elsawa SF, Petty CC, Hickey WF, Bost KL. Exacerbation of experimental autoimmune encephalomyelitis in rodents infected with murine gammaherpesvirus-68. Eur J Immunol. 2003;33:1849–1858. doi: 10.1002/eji.200323148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nelson DA, Petty CC, Bost KL. Infection with murine gammaherpesvirus 68 exacerbates inflammatory bowel disease in IL-10-deficient mice. Inflamm Res. 2009;58:881–889. doi: 10.1007/s00011-009-0059-x. [DOI] [PubMed] [Google Scholar]
- 94.Gasper-Smith N, Marriott I, Bost KL. Murine gamma-herpesvirus 68 limits naturally occurring CD4+CD25+ T regulatory cell activity following infection. J Immunol. 2006;177:4670–4678. doi: 10.4049/jimmunol.177.7.4670. [DOI] [PubMed] [Google Scholar]
- 95.Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. 2009;9:246–258. doi: 10.1038/nri2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Niller HH, Wolf N, Ay E, Minarovits J. Epigenetic dysregulation of epstein-barr virus latency and development of autoimmune disease. Adv Exp Med Biol. 2011;711:82–102. doi: 10.1007/978-1-4419-8216-2_7. [DOI] [PubMed] [Google Scholar]
- 97.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lunemann JD, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med. 2008;205:1763–1773. doi: 10.1084/jem.20072397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jilek S, et al. Strong EBV-specific CD8+ T-cell response in patients with early multiple sclerosis. Brain. 2008;131:1712–1721. doi: 10.1093/brain/awn108. [DOI] [PubMed] [Google Scholar]
- 100.Rose NR, Mackay IR. Molecular mimicry: a critical look at exemplary instances in human diseases. Cell Mol Life Sci. 2000;57:542–551. doi: 10.1007/PL00000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801. doi: 10.1038/ni0901-797. [DOI] [PubMed] [Google Scholar]
- 102.Chervonsky AV. Influence of microbial environment on autoimmunity. Nat Immunol. 2010;11:28–35. doi: 10.1038/ni.1801. [DOI] [PubMed] [Google Scholar]
- 103.Mahmoudi M, Curzen N, Gallagher PJ. Atherogenesis: the role of inflammation and infection. Histopathology. 2007;50:535–546. doi: 10.1111/j.1365-2559.2006.02503.x. [DOI] [PubMed] [Google Scholar]
- 104.Stassen FR, Vainas T, Bruggeman CA. Infection and atherosclerosis. An alternative view on an outdated hypothesis. Pharmacol Rep. 2008;60:85–92. [PubMed] [Google Scholar]
- 105.Nieto FJ, et al. Cohort study of cytomegalovirus infection as a risk factor for carotid intimal-medial thickening, a measure of subclinical atherosclerosis. Circulation. 1996;94:922–927. doi: 10.1161/01.cir.94.5.922. [DOI] [PubMed] [Google Scholar]
- 106.Sorlie PD, et al. Cytomegalovirus/herpesvirus and carotid atherosclerosis: the ARIC Study. J Med Virol. 1994;42:33–37. doi: 10.1002/jmv.1890420107. [DOI] [PubMed] [Google Scholar]
- 107.Hendrix MG, Dormans PH, Kitslaar P, Bosman F, Bruggeman CA. The presence of cytomegalovirus nucleic acids in arterial walls of atherosclerotic and nonatherosclerotic patients. Am J Pathol. 1989;134:1151–1157. [PMC free article] [PubMed] [Google Scholar]
- 108.Vliegen I, Duijvestijn A, Grauls G, Herngreen S, Bruggeman C, Stassen F. Cytomegalovirus infection aggravates atherogenesis in apoE knockout mice by both local and systemic immune activation. Microbes Infect. 2004;6:17–24. doi: 10.1016/j.micinf.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 109.Krebs P, Scandella E, Bolinger E, Engeler D, Miller S, Ludewig B. Chronic immune reactivity against persisting microbial antigen in the vasculature exacerbates atherosclerotic lesion formation. Arterioscler Thromb Vasc Biol. 2007;27:2206–2213. doi: 10.1161/ATVBAHA.107.141846. [DOI] [PubMed] [Google Scholar]
- 110.Melnychuk RM, et al. Mouse cytomegalovirus M33 is necessary and sufficient in virus-induced vascular smooth muscle cell migration. J Virol. 2005;79:10788–10795. doi: 10.1128/JVI.79.16.10788-10795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vliegen I, Duijvestijn A, Stassen F, Bruggeman C. Murine cytomegalovirus infection directs macrophage differentiation into a pro-inflammatory immune phenotype: implications for atherogenesis. Microbes Infect. 2004;6:1056–1062. doi: 10.1016/j.micinf.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 112.Alber DG, Vallance P, Powell KL. Enhanced atherogenesis is not an obligatory response to systemic herpesvirus infection in the apoE-deficient mouse: comparison of murine gamma-herpesvirus-68 and herpes simplex virus-1. Arterioscler Thromb Vasc Biol. 2002;22:793–798. doi: 10.1161/01.atv.0000016046.94521.68. [DOI] [PubMed] [Google Scholar]
- 113.Alber DG, Powell KL, Vallance P, Goodwin DA, Grahame-Clarke C. Herpesvirus infection accelerates atherosclerosis in the apolipoprotein E-deficient mouse. Circulation. 2000;102:779–785. doi: 10.1161/01.cir.102.7.779. [DOI] [PubMed] [Google Scholar]
- 114.Weck KE, et al. Murine gamma-herpesvirus 68 causes severe large-vessel arteritis in mice lacking interferon-gamma responsiveness: a new model for virus-induced vascular disease. Nat Med. 1997;3:1346–1353. doi: 10.1038/nm1297-1346. [DOI] [PubMed] [Google Scholar]
- 115.Martellotta F, Berretta M, Vaccher E, Schioppa O, Zanet E, Tirelli U. AIDS-related Kaposi's sarcoma: state of the art and therapeutic strategies. Curr HIV Res. 2009;7:634–638. doi: 10.2174/157016209789973619. [DOI] [PubMed] [Google Scholar]
- 116.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 117.Hold GL, El-Omar EM. Genetic aspects of inflammation and cancer. Biochem J. 2008;410:225–235. doi: 10.1042/BJ20071341. [DOI] [PubMed] [Google Scholar]
- 118.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 119.Lamagna C, Aurrand-Lions M, Imhof BA. Dual role of macrophages in tumor growth and angiogenesis. J Leukoc Biol. 2006;80:705–713. doi: 10.1189/jlb.1105656. [DOI] [PubMed] [Google Scholar]
- 120.Manrique SZ, et al. Foxp3-positive macrophages display immunosuppressive properties and promote tumor growth. J Exp Med. 2011;208:1485–1499. doi: 10.1084/jem.20100730. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 121.Hippocrate A, Oussaief L, Joab I. Possible role of EBV in breast cancer and other unusually EBV-associated cancers. Cancer Lett. 2011;305:144–149. doi: 10.1016/j.canlet.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 122.Mazouni C, et al. Epstein-Barr virus as a marker of biological aggressiveness in breast cancer. Br J Cancer. 2011;104:332–337. doi: 10.1038/sj.bjc.6606048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Michaelis M, Baumgarten P, Mittelbronn M, Driever PH, Doerr HW, Cinatl J., Jr Oncomodulation by human cytomegalovirus: novel clinical findings open new roads. Med Microbiol Immunol. 2011;200:1–5. doi: 10.1007/s00430-010-0177-7. [DOI] [PubMed] [Google Scholar]
- 124.Soroceanu L, Cobbs CS. Is HCMV a tumor promoter? Virus Res. 2011;157:193–203. doi: 10.1016/j.virusres.2010.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Khan G, Philip PS, Al Ashari M, Houcinat Y, Daoud S. Localization of Epstein-Barr virus to infiltrating lymphocytes in breast carcinomas and not malignant cells. Exp Mol Pathol. 2011;91:466–470. doi: 10.1016/j.yexmp.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 126.Joshi D, Quadri M, Gangane N, Joshi R. Association of Epstein Barr virus infection (EBV) with breast cancer in rural Indian women. PLoS One. 2009;4:e8180. doi: 10.1371/journal.pone.0008180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nilsson C, et al. Does early EBV infection protect against IgE sensitization? J Allergy Clin Immunol. 2005;116:438–444. doi: 10.1016/j.jaci.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Calvani M, Alessandri C, Paolone G, Rosengard L, Di Caro A, De Franco D. Correlation between Epstein Barr virus antibodies, serum IgE and atopic disease. Pediatr Allergy Immunol. 1997;8:91–96. doi: 10.1111/j.1399-3038.1997.tb00150.x. [DOI] [PubMed] [Google Scholar]
- 129.Saghafian-Hedengren S, Sverremark-Ekstrom E, Linde A, Lilja G, Nilsson C. Early-life EBV infection protects against persistent IgE sensitization. J Allergy Clin Immunol. 2010;125:433–438. doi: 10.1016/j.jaci.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 130.Sidorchuk A, Lagarde F, Pershagen G, Wickman M, Linde A. Epstein-Barr virus infection is not associated with development of allergy in children. Pediatr Infect Dis J. 2003;22:642–647. doi: 10.1097/01.inf.0000076510.41038.a8. [DOI] [PubMed] [Google Scholar]
- 131.Sidorchuk A, Wickman M, Pershagen G, Lagarde F, Linde A. Cytomegalovirus infection and development of allergic diseases in early childhood: interaction with EBV infection? J Allergy Clin Immunol. 2004;114:1434–1440. doi: 10.1016/j.jaci.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 132.Saghafian-Hedengren S, et al. Herpesvirus seropositivity in childhood associates with decreased monocyte-induced NK cell IFN-gamma production. J Immunol. 2009;182:2511–2517. doi: 10.4049/jimmunol.0801699. [DOI] [PubMed] [Google Scholar]
- 133.Laske N, et al. Infantile natural immunization to herpes group viruses is unrelated to the development of asthma and atopic phenotypes in childhood. J Allergy Clin Immunol. 2002;110:811–813. doi: 10.1067/mai.2002.128593. [DOI] [PubMed] [Google Scholar]
- 134.Strannegard IL, Strannegard O. Epstein-Barr virus antibodies in children with atopic disease. Int Arch Allergy Appl Immunol. 1981;64:314–319. doi: 10.1159/000232709. [DOI] [PubMed] [Google Scholar]
- 135.Akinbami LJ, Schoendorf KC. Trends in childhood asthma: prevalence, health care utilization, and mortality. Pediatrics. 2002;110:315–322. doi: 10.1542/peds.110.2.315. [DOI] [PubMed] [Google Scholar]
- 136.Takeuchi K, et al. Prevalence of Epstein-Barr virus in Japan: trends and future prediction. Pathol Int. 2006;56:112–116. doi: 10.1111/j.1440-1827.2006.01936.x. [DOI] [PubMed] [Google Scholar]
- 137.Figueira-Silva CM, Pereira FE. Prevalence of Epstein-Barr virus antibodies in healthy children and adolescents in Vitoria, State of Espirito Santo, Brazil. Rev Soc Bras Med Trop. 2004;37:409–412. doi: 10.1590/s0037-86822004000500008. [DOI] [PubMed] [Google Scholar]
- 138.Staras SA, Flanders WD, Dollard SC, Pass RF, McGowan JE, Jr, Cannon MJ. Cytomegalovirus seroprevalence and childhood sources of infection: A population-based study among pre-adolescents in the United States. J Clin Virol. 2008;43:266–271. doi: 10.1016/j.jcv.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 139.Chandler SH, Alexander ER, Holmes KK. Epidemiology of cytomegaloviral infection in a heterogeneous population of pregnant women. J Infect Dis. 1985;152:249–256. doi: 10.1093/infdis/152.2.249. [DOI] [PubMed] [Google Scholar]
- 140.Wu P, et al. Evidence of a causal role of winter virus infection during infancy in early childhood asthma. Am J Respir Crit Care Med. 2008;178:1123–1129. doi: 10.1164/rccm.200804-579OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Haine ER. Symbiont-mediated protection. Proc Biol Sci. 2008;275:353–361. doi: 10.1098/rspb.2007.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gonzalez JP, et al. Pathocenosis: a holistic approach to disease ecology. Ecohealth. 2010;7:237–241. doi: 10.1007/s10393-010-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.in Ending the War Metaphor: The Changing Agenda for Unraveling the Host-Microbe Relationship: Workshop Summary2006. Washington (DC): [PubMed] [Google Scholar]
- 144.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 145.Roossinck MJ. The good viruses: viral mutualistic symbioses. Nat Rev Microbiol. 2011;9:99–108. doi: 10.1038/nrmicro2491. [DOI] [PubMed] [Google Scholar]
- 146.McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EA. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol. 1995;247:443–458. doi: 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- 147.Barton ES, White DW, Virgin HW. Herpesvirus latency and symbiotic protection from bacterial infection. Viral Immunol. 2009;22:3–4. doi: 10.1089/vim.2008.0100. author reply 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Felsenstein J, et al. PopG v3.3. University of Washington: p. A one-locus, two-allele genetic simulation program; 2008. [Google Scholar]
- 149.Sung H, Schleiss MR. Update on the current status of cytomegalovirus vaccines. Expert Rev Vaccines. 2010;9:1303–1314. doi: 10.1586/erv.10.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schiller JT, Lowy DR. Vaccines to prevent infections by oncoviruses. Annu Rev Microbiol. 2010;64:23–41. doi: 10.1146/annurev.micro.112408.134019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dasgupta G, Chentoufi AA, Nesburn AB, Wechsler SL, BenMohamed L. New concepts in herpes simplex virus vaccine development: notes from the battlefield. Expert Rev Vaccines. 2009;8:1023–1035. doi: 10.1586/erv.09.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sokal EM, et al. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis. 2007;196:1749–1753. doi: 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- 153.Li H, Ikuta K, Sixbey JW, Tibbetts SA. A replication-defective gammaherpesvirus efficiently establishes long-term latency in macrophages but not in B cells in vivo. J Virol. 2008;82:8500–8508. doi: 10.1128/JVI.00186-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Marin M, Zhang JX, Seward JF. Near elimination of varicella deaths in the US after implementation of the vaccination program. Pediatrics. 2011;128:214–220. doi: 10.1542/peds.2010-3385. [DOI] [PubMed] [Google Scholar]
- 155.Chaves SS, et al. Loss of vaccine-induced immunity to varicella over time. N Engl J Med. 2007;356:1121–1129. doi: 10.1056/NEJMoa064040. [DOI] [PubMed] [Google Scholar]
- 156.Goldman GS. Universal varicella vaccination: efficacy trends and effect on herpes zoster. Int J Toxicol. 2005;24:205–213. doi: 10.1080/10915810591000659. [DOI] [PubMed] [Google Scholar]
- 157.Civen R, et al. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954–959. doi: 10.1097/INF.0b013e3181a90b16. [DOI] [PubMed] [Google Scholar]
- 158.Thomas SL, Wheeler JG, Hall AJ. Contacts with varicella or with children and protection against herpes zoster in adults: a case-control study. Lancet. 2002;360:678–682. doi: 10.1016/S0140-6736(02)09837-9. [DOI] [PubMed] [Google Scholar]
- 159.Sengupta N, et al. Varicella vaccination in Europe: are we ready for a universal childhood programme? Eur J Pediatr. 2008;167:47–55. doi: 10.1007/s00431-007-0424-0. [DOI] [PubMed] [Google Scholar]
- 160.Goldman GS. Cost-benefit analysis of universal varicella vaccination in the U.S. taking into account the closely related herpes-zoster epidemiology. Vaccine. 2005;23:3349–3355. doi: 10.1016/j.vaccine.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 161.Rozenbaum MH, van Hoek AJ, Vegter S, Postma MJ. Cost-effectiveness of varicella vaccination programs: an update of the literature. Expert Rev Vaccines. 2008;7:753–782. doi: 10.1586/14760584.7.6.753. [DOI] [PubMed] [Google Scholar]
- 162.Hansen SG, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473:523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Boysen P, Eide DM, Storset SK. Natural killer cells in free-living Mus musculus have a primed phenotype. Molec Ecology. 2011 doi: 10.1111/j.1365-294X.2011.05269.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.