

Abstract
Primary prevention of cardiovascular disease is governed at present by the risk factor model for cardiovascular events, a model which is widely accepted by physicians and professional associations, but which has important limitations: most critically, that effective treatment to reduce arterial damage is often delayed until the age at which cardiovascular events become common. This delay means that many of the early victims of vascular disease will not be identified in time. This delay also allows atherosclerosis to develop and progress unchecked within the arterial tree with the result that the absolute effectiveness of preventive therapy is limited by the time it is eventually initiated. The causal exposure model of vascular disease is an alternative to the risk factor model for cardiovascular events. Whereas the risk factor model aims to identify and treat those at markedly increased risk of vascular events within the next decade, the causal exposure model of vascular disease aims to prevent events by treating the causes of the disease when they are identified. In the risk factor model, age is an independent non-modifiable risk factor and the predictive power of age far outweighs that of the other risk factors. In the causal exposure model, age is the duration of time the arterial wall is exposed to the causes of atherosclerosis: apoB (apolipoprotein B) lipoproteins, hypertension, diabetes and smoking. Preventing the development of advanced atherosclerotic lesions by treating the causes of vascular disease is the simplest, surest and most effective way to prevent clinical events.
Keywords: age, atherosclerosis, lipoprotein, prevention, risk factor, vascular disease
Abbreviations: apo, apolipoprotein; BP, blood pressure; LDL, low-density lipoprotein; PCSK9, proprotein convertase subtilisin kexin 9
INTRODUCTION
Forty years ago, given the evidence available, it was appropriate to label LDL (low-density lipoprotein), BP (blood pressure) and smoking as risk factors rather than causes of vascular disease. That stance is no longer reasonable. All the links in the evidentiary chain of causality – temporality, strength, dose–response, specificity, consistency, biological plausibility and experimental confirmation – are in place. To pretend there is doubt is to disregard the masses of interlocking biological, pathophysiological, epidemiological and clinical trial results. But if LDL, BP and smoking cause vascular disease, why are they such weak risk factors for the likelihood of clinical events? Wald and Law, in particular, have enunciated and emphasized this paradox [1–3] and it is a fact that, except at the extremes, the level of LDL, however it is estimated, the level of BP and the extent of smoking only marginally influence the estimates of risk [4,5]. Indeed, Wald and Law [4] argue that, although LDL and BP cause vascular disease, they are of no practical value to identify those who would benefit from preventive therapy. That is the core of their argument that the polypill should be given to all those over 55 years of age.
In the conventional sense, this is correct: the causes of vascular disease are weak risk factors for vascular disease. However, it does not follow that we should not identify and treat the causes of vascular disease within individuals. On the contrary, we will try to demonstrate that the reason that the causes of vascular disease are weak risk factors for clinical events is primarily a function of how we treat age as a determinant of vascular disease.
AGE THE DOMINANT RISK FACTOR
Age is, by far, the dominant risk factor in any risk factor model for cardiovascular events. From 25 years of age, risk doubles every 8 years [5]. Indeed, once age and gender are taken into account, all of the accepted modifiable risk factors add only marginally to the predictive power of the risk factor engines such as Framingham [6,7]. But does that mean that age ‘causes’ vascular disease? Or does it point to the fact that the ‘causes’ of vascular disease act progressively over time?
The most important difference between the risk factor and causal exposure models is how they treat age. In the risk factor model, age is regarded as the simple, purely chronological, variable that it is thought to be in every day life, a variable, which is ‘known’ to be non-modifiable and which, operationally, is presumed to be independent of all the other risk factors. However, age is, in reality, a complex variable, pointing to, on the one hand, all the non-modifiable biological changes that occur within our arteries over time and, on the other, all the modifiable consequences of the cumulative injuries to our arteries over time due to LDL, BP, smoking and diabetes. Put simply, the injuries to arteries owing to age are due to exposure and to disintegration [8].
In the risk factor model of cardiovascular events:
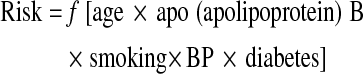 |
but actually age represents the duration of exposure of arteries to apoB, smoking and diabetes.
Therefore in the causal exposure model of vascular disease:
 |
STRENGTHS AND LIMITATIONS OF THE RISK FACTOR MODEL FOR CARDIOVASCULAR EVENTS
The strengths of the risk factor model are that it is well known, well accepted and that it works, that is it identifies risk within broad, but reasonable, limits for the next decade. To be sure, there are, in fact, multiple risk factor models: Framingham, SCORE, QRisk and Reynolds. Nevertheless, their predictions are quite similar because age is the principal determinant of risk in all.
Events early in life are much less common than events later in life. That simple reality is what makes age such an overwhelmingly powerful predictor. But the dominance of age is also the weakness of the risk factor model. Even numerically early events account for an important portion of the total toll of disease and early death is particularly tragic and costly. Thus, in Framingham, one in six events in men and one in ten in women occur before 55 years of age [9]. Moreover, the estimation of risk only holds in the short-term. Indeed, the majority of those at low short-term risk are at high long-term risk: in the Framingham Study, those in the lowest tertile of 10-year risk at 50 years of age face a lifetime risk of a coronary heart event almost as high as those in the highest tertile [10].
Most importantly, a low likelihood of a clinical event in the next decade does not equal a low likelihood that disease is not developing within our arterial walls during that decade. On the contrary, if the causes of vascular disease are present, there is a high likelihood that it is. The consequence is that prevention is too often delayed until disease is advanced and the potential for benefit from therapy correspondingly reduced. Moreover, there is a numbers mismatch at the core of the risk factor model which aims to identify the minority who are at greatest risk within the next decade, whereas the reality is the majority, two out of three in the case of the U.S.A., will suffer a significant cardiovascular event during their lifetimes [11].
STRENGTHS OF THE CAUSAL EXPOSURE MODEL: EARLY AND LATE PREVENTION
On the basis of ten large cohort studies in Western populations, Wald and Law [5] calculated that a reduction in LDL-cholesterol of 1.0 mmol/l at 50, 60 and 70 years of age would reduce vascular events by 56, 41 and 31% respectively, demonstrating that the same degree of LDL-lowering will produce much greater benefit earlier, rather than later, in life. In the BUPA (British United Provident Association) study, even though the absolute number of deaths due to ischaemic heart disease over a 12-year period was four times greater in those 55–64 years of age at entry compared with those 35–44 years of age at entry, reducing cholesterol by 0.6 mmol/l will prevent almost a two and a half times greater percentage of fatal events in the younger group compared with the older one [12]. This evidence supports the pathophysiological argument that the absolute gain from treating the younger rather than older subjects will be greater, arguing for earlier compared with later strategies of prevention.
Vascular disease is not inevitable. Lloyd-Jones and his co-workers [13] have shown a strikingly low lifetime incidence of vascular disease in those who do not manifest the major risk factors by 50 years of age. That the duration of the time arteries are exposed to apoB matters is illustrated by mutations in PCSK9 (proprotein convertase subtilisin kexin 9). Gain-of-function mutations in PCSK9 result in reduced numbers of LDL-receptors and higher plasma LDL levels, whereas loss-of-function mutations result in more LDL-receptor numbers and life-long lower plasma LDL levels. Strikingly, a gain-of-function mutation in PCSK9 that results in a reduction in LDL-cholesterol levels by 30% over a lifetime reduces clinical events by 88%. This decrease in events per mg of LDL-cholesterol is substantially more pronounced than the decrease in events per mg with LDL-lowering therapy [14]. This finding of augmented benefit from lifelong lower levels has been confirmed in other studies of mutations of this gene [15,16]. Finally, on the basis of the results of the AMORIS (Apolipoprotein-related MOrtality RISk) study, reducing the apoB/apoA-I ratio in all of those above the 80th percentile to the level of those in the 20th percentile would decrease deaths from acute myocardial infarction by 80% [17].
WHO SHOULD BE TREATED?
That a very high proportion of the population, approximately 75%, is at a high total lifetime risk of a significant cardiovascular event should not be surprising [18], since this percentage corresponds reasonably closely to the known total incidence of cardiovascular events in the population [13]. Indeed, this is the core of the polypill argument to treat all over 55 years of age with a combination of fixed low-dose pharmacological agents. But pharmacological therapy will produce clinical benefit only in those in whom that specific cause is present. Moreover, the degree of benefit will relate to the degree of abnormality, that is the absolute risk and the absolute benefit from pharmacological therapy depend on the absolute levels of the cause(s) of vascular disease. Thus patients with normal LDL levels but high BP do not require LDL-lowering therapy, whereas those with very high LDL levels require more intensive therapy than those with only moderately elevated LDL levels. Targeted therapy would be more cost-effective than population-based single dose therapy precisely because it is limited to those who need it most. The best choice of the biological marker of LDL will also improve outcome. For example, if the ATPIII (Adult Treatment Panel III) guidelines were applied to the U.S.A. population, substituting non-HDL (high-density lipoprotein)-cholesterol as the target of therapy for LDL-cholesterol would result in 300000 more events prevented over a 10-year period. However, if apoB were substituted for LDL-cholesterol, 800000 more events would be prevented over a 10-year period [19].
BARRIERS TO IMPLEMENTATION OF THE CAUSAL EXPOSURE MODEL
In the causal exposure model, therapeutic intervention would be based on detection of the causes of vascular disease, not the calculation of short-term risk. Age is the primary driver of decisions in both the risk factor and the polypill models. Therefore both discourage treatment of the causes of vascular disease in younger people. The causal exposure model would expand the numbers selected for early medical prevention of cardiovascular disease beyond the risk factor model and the polypill model. Treating more people for longer periods of time would necessarily change the usual estimates of cost and benefit.
However, the usual estimates are no longer valid because medication costs are plummeting. The patent periods of most of the major statins and antihypertensive agents have expired or are close to expiring. The price of generics, particularly in markets such as the U.S.A., has diminished dramatically with statins now costing less than $5 per month. Therefore cost is not the barrier to care it once was. Indeed, treatment of patients at intermediate risk for cardiovascular events has become cost-effective [20].
Safety must be a foremost concern. In the short- and medium-term, the safety of statins and antihypertensive agents has been established. Nevertheless, long-term safety cannot be taken for granted and, indeed, only recently has evidence emerged that the incidence of diabetes may be slightly increased with long-term ingestion of statins [21,22]. Naturally, this has been assumed to be a negative effect of statins, although it seems possible that this might represent a hazard of survival free of clinical events.
In any event, the balance of risk and benefit still strongly favour statin therapy, even when the impact of a potential increase in the risk of diabetes is taken into account [20]. That said, true assurance of long-term safety will only come with large numbers undergoing long-term therapy. The reality remains that cardiovascular disease is the major cause of death worldwide notwithstanding all the diagnostic tests and therapies that exist. Prevention is, therefore, the best strategy both from an individual and a societal perspective.
Ideally, the causal exposure model hypothesis should be put to the test in a randomized clinical trial. But this will never occur for many reasons, not least the several decades such a trial would require. However, it is possible to model the impact of different strategies to prevent vascular disease [19,20], and this evidence can be very helpful. Nevertheless, in the absence of unequivocal proof, guideline groups may find it impossible to recommend the causal exposure model and this alone may make widespread implementation impossible. Equally important, many patients are reluctant to take medication and would not accept the equation of potential long-term gain in exchange for early medical therapy. That, of course, would be their right. Our view is that patients should be aware of the risks that they face and the options available to avoid them. The choice is for the patient to make, a choice, which can be reviewed and revised as knowledge is gained.
Moreover, all the barriers that apply to early implementation of therapy to lower LDL also apply to hypertension and, in the case of hypertension, at least so far as the guideline groups are concerned and much of medical practice, they have been overcome. That is to say, virtually anyone with markedly increased BP will be offered the option of therapy even if their short- and medium-term risks of cardiovascular events are low. In this instance, the knowledge of the long-term hazards has overcome the barriers to therapy. The difference, we would contend, is that hypertension is recognized as a disease, whereas LDL, at least at present, is only a risk factor for disease.
Definition of when LDL-lowering therapy should be initiated needs to be defined for LDL. Early treatment is accepted when LDL levels are extreme, as in familial hypercholesterolaemia. Early treatment would not be accepted if only modest deviations from the norm are present. The challenge is to find the appropriate level at the appropriate age at which pharmacological therapy should be considered and this is where studies that systematically model outcome would be most helpful. With regard to LDL, statins will be most effective in those who are most abnormal. The younger the individual, the higher the threshold level for intervention that will be required. For those above 40 years of age, we suspect that treatment of those above the 70th percentile should be cost-effective, but this must be confirmed in quantitative modelling studies. It might also be helpful to couple the approach we suggest with appropriate non-invasive tests of vascular function. Identifying early abnormalities would greatly strengthen the case for earlier intervention. The difficulty is the lack of standardization and physiological validation of some of the most popular of these tests [23].
SUMMARY
The risk factor model for cardiovascular events is broadly known and broadly accepted by professionals and governments, and this facilitates the implementation of public health policy based on this model. The concept of appropriately weighting and integrating all of the relevant information is intuitively attractive and the method appears authoritative and precise. However, age, which is conventionally viewed as a non-modifiable risk factor, is the principal determinant of whether prevention will be initiated in the individual patient, not the modifiable causes of the disease process in the artery. Once we appreciate that age describes the length of time over which exposure to the causal factors of disease occurs, age becomes a modifiable risk factor for vascular disease. Once we couple this understanding with the recognition that the ability of therapy to prevent clinical events depends on the extent of disease within the arterial wall at the time therapy is initiated, the advantages of the causal exposure model for vascular disease over the risk factor model for cardiovascular events become apparent. After all, if disease in the wall is prevented, there will be no events to predict.
References
- 1.Law M. R., Wald N. J., Morris J. K. The performance of blood pressure and other cardiovascular risk factors as screening tests for ischaemic heart disease and stroke. J. Med. Screen. 2004;11:3–7. doi: 10.1177/096914130301100102. [DOI] [PubMed] [Google Scholar]
- 2.Wald N. J., Morris J. K. Assessing risk factors as potential screening tests: a simple assessment tool. Arch. Intern. Med. 2011;171:286–291. doi: 10.1001/archinternmed.2010.378. [DOI] [PubMed] [Google Scholar]
- 3.Wald N. J., Simmonds M., Morris J. K. Screening for future cardiovascular disease using age alone compared with multiple risk factors and age. PLoS ONE. 2011;6:e18742. doi: 10.1371/journal.pone.0018742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wald N. J., Law M. R. A strategy to reduce cardiovascular disease by more than 80% Br. Med. J. 2003;326:1419. doi: 10.1136/bmj.326.7404.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wald N. J., Law M. R. Screening for future coronary heart disease. In: Marmot M., Elliot P., editors. Coronary Heart Disease Epidemiology. Oxford: Oxford Medical Publications; 2005. p. 706. [Google Scholar]
- 6.Wang T. J., Gona P., Larson M. G., Tofler G. H., Levy D., Newton-Cheh C., Jacques P. F., Rifai N., Selhub J., Robins S. J. Markers for prediction of first major cardiovascular events and death. N. Engl. J. Med. 2006;355:2631–2639. doi: 10.1056/NEJMoa055373. [DOI] [PubMed] [Google Scholar]
- 7.Cook N. R., Buring J. E., Ridker P. M. The effect of including C-reactive protein in cardiovascular risk prediction models for women. Ann. Intern. Med. 2006;145:21–29. doi: 10.7326/0003-4819-145-1-200607040-00128. [DOI] [PubMed] [Google Scholar]
- 8.Sniderman A. D., Furberg C. D. Age: a modifiable risk factor for cardiovascular disease. Lancet. 2008;371:1547–1549. doi: 10.1016/S0140-6736(08)60313-X. [DOI] [PubMed] [Google Scholar]
- 9.Vasan R. S., Sullivan L. M., Wilson P. W., Sempos C. T., Sundstrom J., Kannel W. B., Levy D., D'Agostino R. B. Relative importance of borderline and elevated levels of coronary heart disease risk factors. Ann. Intern. Med. 2005;142:393–402. doi: 10.7326/0003-4819-142-6-200503150-00005. [DOI] [PubMed] [Google Scholar]
- 10.Lloyd-Jones D. M., Wilson P. W., Larson M. G., Beiser A., Leip E. P., D'Agostino R. B., Levy D. Framingham risk score and prediction of lifetime risk for coronary heart disease. Am. J. Cardiol. 2004;94:20–24. doi: 10.1016/j.amjcard.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Ingelsson E., Massaro J. M., Sutherland P., Jacques P. F., Levy D., D'Agostino R. B., Vasan R. S., Robins S. J. Contemporary trends in dyslipidemia in the Framingham Heart Study. Arch. Intern. Med. 2009;169:279–286. doi: 10.1001/archinternmed.2008.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law M. R., Wald N. J., Wu T., Hacksaw A., Bailey A. Systematic underestimation of association between serum cholesterol concentration and ischaemic heart disease in observational studies: data from the BUPA study. Br. Med. J. 1994;308:363–366. doi: 10.1136/bmj.308.6925.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lloyd-Jones D. M., Leip E. P., Larson M. G., D'Agostino R. B., Beiser A., Wilson P. W. F., Wolf P. A., Levy D. Prediction of lifetime risk for cardiovascular disease by risk factor burden at 50 years of age. Circulation. 2006;113:791–798. doi: 10.1161/CIRCULATIONAHA.105.548206. [DOI] [PubMed] [Google Scholar]
- 14.Cohen J. C., Boerwinkle E., Mosley T. H. J., Hobbs H. H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 15.Benn M., Nordestgaard B. G., Grande P., Schnohr P., Tybjaerg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J. Am. Coll. Cardiol. 2010;55:2833–2842. doi: 10.1016/j.jacc.2010.02.044. [DOI] [PubMed] [Google Scholar]
- 16.Davignon J., Dubue G., Seidah N. G. The influence of PCSK9 polymorphisms on serum low-density lipoprotein cholesterol and risk of atherosclerosis. Curr. Atheroscler. Rep. 2010;12:308–315. doi: 10.1007/s11883-010-0123-6. [DOI] [PubMed] [Google Scholar]
- 17.Sniderman A. D., Holme I., Aastveit A. A., Furberg C., Walldius G., Jungner I. Relation of age, the apoB/apoA-1 ratio and the risk of fatal myocardial infarction: Implications for primary prevention of cardiovascular disease. Am. J. Cardiol. 2007;100:217–221. doi: 10.1016/j.amjcard.2007.02.086. [DOI] [PubMed] [Google Scholar]
- 18.Marma A. K., Berry J. D., Ning H., Persell S. D., Lloyd-Jones D. M. Distribution of 10-year and lifetime predicted risks for cardiovascular disease in US adults: findings from the National Health and Nutrition Examination Survey 2003 to 2006. Circ. Cardiovasc. Qual. Outcomes. 2010;3:8–14. doi: 10.1161/CIRCOUTCOMES.109.869727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sniderman A. D., Williams K., Contois J. H., Monroe H. M., McQueen M. J., de Graaf J., Furberg C. D. A meta-analysis of LDL-C, non-HDL-C and apoB as markers of cardiovascular risk. Circ. Cardiovasc. Qual. Outcomes. 2011;4:337–345. doi: 10.1161/CIRCOUTCOMES.110.959247. [DOI] [PubMed] [Google Scholar]
- 20.Lazar L. D., Pletcher M. J., Coxson P. G., Bibbins-Domingo K., Goldman L. Cost-effectiveness of statin therapy for primary prevention in a low-cost statin era. Circulation. 2011;124:146–153. doi: 10.1161/CIRCULATIONAHA.110.986349. [DOI] [PubMed] [Google Scholar]
- 21.Catapano A. L., Reiner Z., De Backer G., Graham I., Taskinen M. R., Wiklund O., Agewall S., Alegraia E., Chapman M. J., Durrington P. ESC/EAS Guidelines for the management of dyslipidaemias The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) Atherosclerosis. 2011;217(Suppl. 1):1–44. doi: 10.1016/j.atherosclerosis.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Greenland P., Alpert J. S., Beller G. A., Benjamin E. J., Budoff M. J., Fayad Z. A., Foster E., Hlatky M. A., Hodgson J. M., Kushner F. G. ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2010;56:e50–e103. doi: 10.1016/j.jacc.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 23.de Graaf J., Holewijn S., Stalenhoef A. F., Sniderman A. D. Should preclinical vascular abnormalities be measured in asymptomatic adults to improve cardiovascular risk stratification? Curr. Opin. Lipidol. 2011;22:454–459. doi: 10.1097/MOL.0b013e32834c6245. [DOI] [PubMed] [Google Scholar]