

Abstract
The structure and dynamics of the hepatitis delta virus ribozyme (HDVr) are studies using molecular dynamics simulations at several stages along its catalytic reaction path, including reactant, activated precursor, transition state mimic and product states, departing from an initial structure based on the C75U mutant crystal structure (PDB: 1VC7). Results of five 350 ns molecular dynamics simulations reveal a spontaneous rotation of U-1 that leads to an in-line conformation and support the role of protonated C75 as the general acid in the transition state. Our results provide rationale for the interpretation of several important experimental results, and make experimentally testable predictions regarding the roles of key active site residues that are not obvious from any available crystal structures.
Keywords: Ribozyme, HDV, MD simulation, in-line conformation, general acid
The hepatitis delta virus (HDV) ribozyme is a small catalytic RNA motif that is essential for viral replication during the HDV life cycle.1–4 Recently, HDV-like ribozymes have been found to be widely distributed in Nature, including in human genes, where they likely play a variety of important biological roles.5 The HDV ribozyme (HDVr) catalysis reaction starts with an inline nucleophilic attack of the U-1:O2′ to the adjacent scissile phosphate, followed by cleavage of the P-O5′ bond of G1 to produce a 2′,3′-cyclic phosphate and a 5′ hydroxy-terminus. Extensive structural and biochemical evidence suggest a catalytic mechanism involving acid-base catalysis; however the detailed catalytic reaction mechanism of HDV ribozyme is still not resolved.
The first crystal structure of HDVr was reported in the product form6 with an overall fold consistent with mutagenesis and chemical probing studies of the solution conformation.7,8 In this product structure, C75:N3 is in a position to form a hydrogen bond with G1:O5′ (the leaving group); hence, it is reasonable to suggest that C75 acts as the general acid in phosphodiester bond cleavage reaction,9,10 a hypothesis that is well supported by inactivation of the ribozyme by C75 mutation and a variety of experimental approaches.9,11–13 Importantly, pre-activation of the 5′O leaving group by substitution with a 5′S bridging phosphorothioate renders the ribozyme insensitive to C75 mutation.10 Nevertheless, in the structures of the precleavage HDVr inactivated by C75U mutation and in the absence of Mg2+ ions, the active site has different arrangement in that U75 is posed to serve as the general base for cleavage reaction,14 an interpretation that has been supported in certain molecular dynamics studies. 15–17
Although not absolutely required for catalysis,18 the presence of divalent metal ions at millimolar levels significantly enhances the HDVr reactivity2,3,7,19–21 It is believed that there is a hydrated Mg2+ ion near the active site.9,14,22,23 This Mg2+ ion is likely to be involved in the HDVr catalytic reaction as it has been shown that can compete with Mg2+ binding and inhibit HDVr activity9,24 The active site Mg2+ ion has been shown to directly interact with critical active site residues,25,26 and modification of the linkage at the scissile phosphate can alter metal ion preference.20,27
Recently, Raman crystallographic experiments have determined that the pKa value of C75 is shifted toward neutrality in a Mg2+-dependent fashion,12 and further, that protonation of C75 may be coupled with changes in inner-sphere coordination of a divalent metal ion binding.24,25 Subsequent crystallographic28 and molecular dynamics29,30 studies have provided new information about the HDVr active site, and in particular, the nature of metal ion binding at a site involving a G·U wobble at the cleavage site and a rare reverse G·U wobble located near the active site. Nonetheless, the conformational events that lead to a catalytically active state where U-1 is poised for in-line attack are not well understood, as this residue was not resolved in the recent crystallographic study, but rather modeled based on the conformation of the inhibitor strand of the hammerhead ribozyme. Further, there has been relatively little reported work on the HDVr structure and dynamics at different key stages along the catalytic reaction path.
The present study examines results from a series of molecular dynamics simulations of HDVr at different stages along the reaction path. While previously simulation studies on HDVr focused on only the reactant states or the product state, 15,29–33 here we report results from a series of five 350-ns molecular dynamics simulations of HDV ribozyme (summarized in Table 1), the first focusing on different points along the reaction coordinates: 1) the reactant state (RT) with neutral C75 (C750), 2) the activated precursor state with the nucleophile (U-1:O2′) deprotonated (dRT) and C75 protonated (C75+), 3) the early transition state mimic (ETS), 4) the late transition state mimic (LTS), and 5) the product state (Prod). The ETS and LTS transition state mimics are formed by defining new chemical bonds between the nucleophile O2′ and the active center P atom, and different bond lengths derived from high-level ab initio quantum calculations. The same protocol has been previously utilized in various simulation work of hammerhead ribozymes, where significant differences were observed in the ETS and LTS simulation results.34,35 The unrefined starting structures were based on the 2.45 Å crystal structure of the C75U mutant (PDB:1VC7),14 which contains positions for the U-1 residue, but that differs significantly from crystallographic data for the product structure6 and from recent crystallographic data of HDVr bound to an inhibitor RNA containing a deoxynucleotide at the cleavage site.28 In this structure, a metal ion is at the position between U75 and G25 and is directly coordinated to U75:O4, which cannot occur in the wild-type. Hence in our simulations, the active Mg2+ ion was initially placed to be bound to G1:N7, in accord with the position suggested by Chen et al.26 In the first 10 ns of simulation, the Mg2+–G1:N7 distance was restrained to 2.0 Å. Initial simulations with the active site metal ion placed at the original position of the C75U crystal structure indicated that, with the native C75, the metal ion does not bind stably at this position. Recent crystallographic structure of the inhibited reactant28 could be an alternative choice of the starting structure, however, the key nucleophile residue U-1 is missing in this structure hence it was not selected here. The full details about the simulation protocol are provided in the supporting information.
Table 1.
Summary of simulations reported in the present work with their abbreviations used in the text. In the table “TS” refers to “transition state”, and “0” and “+” refer to neutral and protonated (at the N3 position) C75. Systems were solvated in a 60×60×120 Å3 box of TIP3P waters43 and an ion atmosphere corresponding to a 0.14 M bulk NaCl concentration, with residue C41 protonated. All simulations were carried out in the NPT ensemble at 1 atm and 298 K under periodic boundary conditions and using the smooth particle-mesh Ewald method44,45 for calculation of electrostatic interactions. For each system, 10 ns of water/ion equilibration followed by additional 10 ns of solute equilibration were performed, followed by 350 ns of production simulation, the last 300 ns of which was used for analysis. Simulations were performed with the NAMD simulation package (version 2.7b3)46 using the AMBER47,48 parm99 force field with the α/γ corrections for nucleic acids.49
| Abbrev. | State | C75 |
|---|---|---|
| RT-C750-Mg | reactant | 0 |
| dRT-C75+-Mg | activated precursor | + |
| ETS | early TS mimic | + |
| LTS | late TS mimic | + |
| Prod | product | 0 |
The focus of this work is to present structure models of different stages of the HDVr reaction path. These simulation-derived models outline critical residues and their interactions as shown in Figure 1. Different stages are shown: Figure 1(A): the neutral reactant state (RT-C750-Mg); Figure 1(B): the precursor state with the nucleophile deprotonated and C75 protonated (dRT-C75+-Mg); Figure 1(C): the early transition state mimic (ETS); Figure 1(D): the late transition state mimic (LTS); Figure 1(E): the product state (Prod). Statistical analyses were performed for the key geometry indexes, including bond distances, angles, dihedrals, and hydrogen bonds (H-bond). All indexes reported in the following sections were calculated from the last 300 ns out of the total 350 ns of trajectory for each simulation with a sampling rate of 100 ps. The hydrogen bonds are defined as formed when the distance between the donor and the acceptor is less than 3.5 Å and the angle is greater than 150 degrees. The H-bond is reported in terms of percentage of the number of snapshots with formed hydrongen bonds compared to the total number of snapshots of each trajectory. The time series of mentioned H-bond distances are shown in Figure S1.
Figure 1.
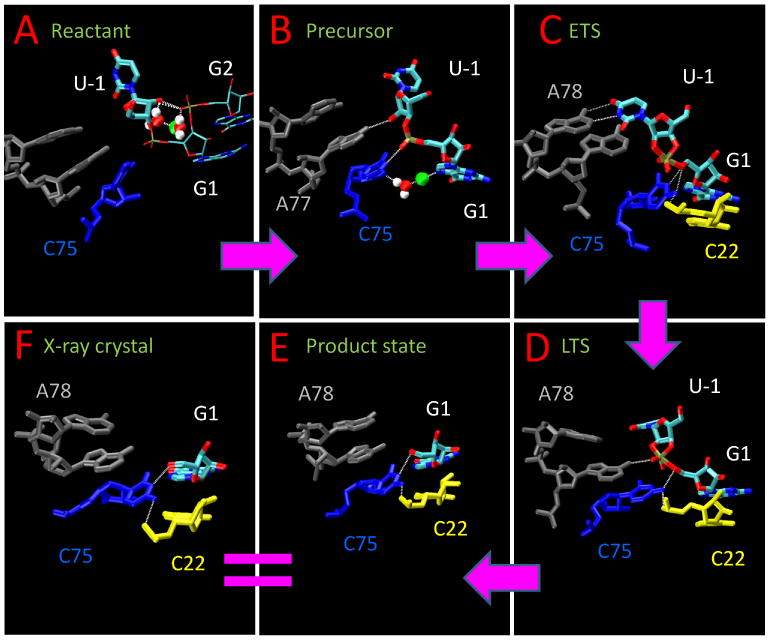
Graphic summary of the simulation results. Shown are representative snapshots from simulations listed in Table 1: A: the neutral reactant state; B: the precursor state with the nucleophile deprotonated and C75 protonated; C: the early transition state mimic; D: the late transition state mimic; E: the product state; F: the crystal product structure (PDBID: 1C×0).
In the neutral reactant state, G2:O2P positions U-1:O2′ for general base activation by the active site Mg2+ ion
Substitution of sulfur at the G2:O2P position has a significant effect on HDVr activity that cannot be rescued by thiophilic metals,36 and as yet, the origin of this effect remains unclear. In the RT-C750-Mg simulation (Figure 1(A)), G2:O2P forms a hydrogen bond with the U-1:O2′ nucleophile (72% in a 300 ns trajectory). The interaction between G2:O2P and U-1:O2′ is further facilitated by a Mg2+-mediated water bridge involving two inner sphere water molecules (Figure Figure 1(A)).
The C75 position is held near the active site by the H-bond between C75:N4 and the scissile phosphate G1:O2P (45%). Therefore, C75 is not in a position near the leaving group to act as a general acid, which is consistent with the precleavage crystal structure14 and other simulation results.15 However, the mechanisms cannot be unambiguously defined based on structural evidence corresponding to a single point along the reaction coordinate alone.37 In this case, it is particularly precarious to assume that C75 does not act as the general acid, given the growing body of contrasting experimental evidence.28 Furthermore, based on its position in the precleavage state and the results from the RT-C750-Mg simulation, rearrangement of active site interactions are needed for C75 to participate in this catalytic mode.
In the activated precursor state, A77, the active site Mg2+, and C75, collectively hold the in-line conformation formed after a rigid rotation of U-1
A representative snapshot from the simulation of the activated precursor state with the nucleophile deprotonated and C75 protonated (dRT-C75+-Mg) is shown in Figure Figure 1(B). During the simulations, the U-1 residue spontaneously undergoes a rigid rotation and reaches a near in-line conformation around 20 ns, with the 2′O-P-5′O angle around 140 degrees, rotates back to around 90 degrees around 130 ns, and then rotates back to above 140 degrees at around 200 ns. The in-line angle is kept around 140 degrees after 200 ns (average 138 degrees, with maximum 162 degrees). This observation shows that U-1 is able to adopt multiple conformations, consistent with recent crystallographic data of an inhibited precleavage structure where the electron density of U-1 was observed to be disordered.28 After 200 ns the in-line conformation of U-1 is stabilized by a new H-bond between A77:N6 and the nucleophile (U-1:O2′) (38%), between C75:N3 and G1:O2P (30%), and between between C75:N4 and G1:O2P (56%). The A77:N6/U-1:O2′ H-bond interaction is intriguing in that it provides a rationale for the hitherto unexplained importance of the exocyclic NH2 group of A77 identified through mutagenesis experiments.38–40 The active site Mg2+ is directly bound to G1:N7 (2.25 Å) and also bound to C75:O2′ through a water molecule (5.20 Å). These interactions are consistent with experimental evidence suggesting that a previously unobserved hydrated magnesium ion interacts with G1:N7,26 as well as the observation that the reactivity of the HDVr is reduced by 28 fold when C75 is mutated to deoxy-C75.40
In the early transition state mimic simulation, U-1 forms a canonical WCpair with A78, and C75+ forms strong H-bond with the leaving group
A dramatic change in the base pair hydrogen bonding occurs in the early transition state mimic simulation, whereby a WC base pair forms between A78 and U-1, and is shown in Figure 1(C). The occupancy of the H-bond between A78:N6 and U-1:O4 is 86% while between A78:N1 and U-1:N3 is 76%. This WC-pair provides a rational for the importance of the identity of A78,38–40 as well as the nucleobase preference of the −1 position, 32,41 and suggests that experiments involving correlated mutations in the 78 and −1 positions may provide further insight into the importance of this interaction, as it has been suggested that the U-1 preference can be altered under different conditions.41
In the ETS, the protonated C75+ moves to a position where it is available to act as the general acid, and is held in place by a strong H-bond between C75:N4 (the exocyclic amine) and G1:OO5′ (84%). At the same time, the exocyclic amine of C75 forms an additional hydrogen bonding interaction with C22:O2P (57%) which helps to hold its position. The H-bond between C75:N4 and C22:O2P was observed crystallographically in the post-cleavage structure6 and in a recent precleavage structure28 but not in the precleavage structure.14 The exocyclic amine of C75 (N4) thus appears to play an important role in maintaining the proper active site fold near the transition state, in agreement with conclusions from experiments that investigated the incorporation of 6-azauridine into the genomic HDVr active site.42 The Mg2+ loses its direct coordination with G1:N7 and moves slightly away from the active site center when C75+ moves towards G1:OO5′, which is consistent with reported anti-cooperative binding of the Mg and C75 protonation.26
In the late transition state, the position of the general acid is maintained by H-bond interactions between A77:N6 and G1:O2P, and C75:N4 and C22:O2P
In the late transition state mimic, cleavage of the P-O5′ bond has progressed, and there is an accumulation of negative charge at the G1:O5′ position, leading to slight changes in the active site hydrogen bonding (Figure 1(D). In the LTS, C75 has a similar position compared to the ETS and the strong H-bond between C75:N4 and G1:OO5′ is maintained (65%). However, the WC pair between A78 and U-1, formed in the ETS, is broken. Instead, due to the shift in the position the reactive phosphate group, the exocyclic amine group of A77:N6 now forms a strong H-bond with G1:O2P (40%). The H-bond bond between C75:N4 and C22:O2P is more pronounced in LTS (86%) than in ETS (57%). The Mg2+ further loses both direct and indirect coordination interactions with G1:N7 and C75:O2′ and exits from the active site, as the reaction proceeds to a late stage. This ejection of a metal from the HDVr active site in the late stages of the reaction has been inferred previously from crystallographic data.14
In the product state, the Mg2+ exits the active site and the simulation converges toward the crystal structure of the product complex
In our simulation of the product state, A77 and A78 lose all H-bond interactions with the subtract as U-1 no longer exists in the active site as shown in Figure 1(E). The H-bond between C75:N3 and G1:O5′, reported in the post-cleavage structure, is formed (56%), as well as the H-bond between C75:N4 and C22:O2P (43%). The strong H-bond between C75:N4 and G1:O5′ observed in other stages no longer exists as C75 is now in its neutral state, having donated its proton to the O5′ leaving group. Our simulation of the product state converge reasonably closely to that of the crystallographic data of the product structure,6 with the exception of formation of a rare reverse G·U wobble, also observed in a recent crystallographic structure of HDVr bound to an inhibitor RNA containing a deoxynucleotide at the cleavage site.28 This reverse wobble may be further stabilized by divalent metal ion binding.
A key point is that our simulations of the product state were not initiated from the product crystallographic structure6 but, rather, a precleavage structure of the C75U mutant.14 Thus, the observation that despite beginning with a distinct geometry along the reaction coordinate our simulations converge in both structure and hydrogen bonding pattern very closely to that of the post-cleavage structure,6 shown in Figure 1(E, F) and Figure 2, provides an important internal check on reliability of our simulations.
Figure 2.
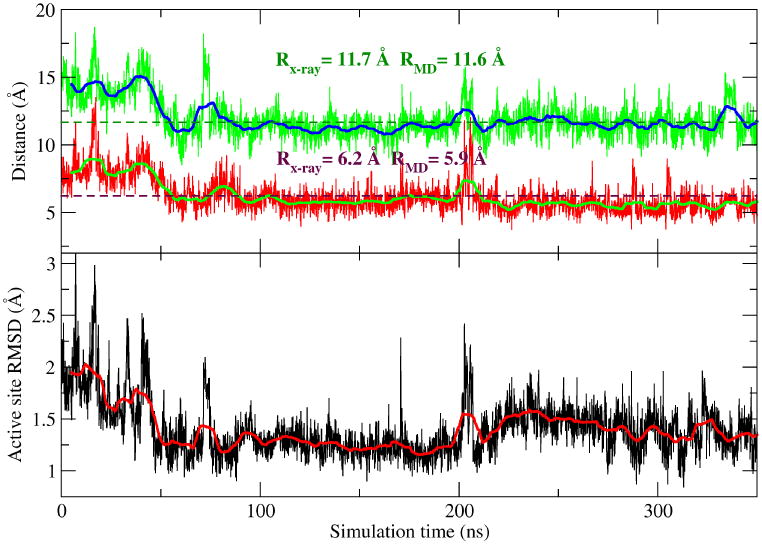
The product simulation converges towards the product (post-cleavage) structure.14 The upper plot shows two distances in the simulations vs the crystal distance: the first is the distance (in green) between A78:N6 and G3:O2P while the second one between A77:N6 and G2:O2P. RMD refers to the average value from the simulations (from 50 ns to 350 ns). Rx–ray refers to the distance in the crystal structure (PDBID: 1C×0). The crystal structure distances are also plotted as dotted lines. The lower plot shows the active site RMSD of the product simulation respect to the product crystal structure. The active site is defined as the collection of G1, G2, G3, C75, A77, and A78. Data are shown every 100 ps and the smooth solid lines along the data curves are the window-averaged results with window size=10.
The details of the catalytic mechanism of HDVr have been the topic of considerable discussion and debate, originating from varying mechanistic interpretations derived from crystallographic data and biochemical experiments. Of particular focus was the crystallographic structure of the C75U mutant in the precleavage state14 that suggested the role of C75 as a general base rather than general acid, as was inferred by previous crystallographic data of the native product.6 It was for this reason that we used this structure of the former as a departure point for our simulations of the native HDV at several stages along the reaction coordinate.
Our results suggest that the position of C75, which is initially close to the U-1:O2′ in the reactant state, prefers to adopt a hydrogen bonding interaction with the G1:O2P, whereas the nucleophile interacts with G2:O2P and a hydrated Mg2+ ion through a metal-mediated water bridge. These results do not support the role of C75 as that of a general base. Although the crystallographic structure of the C75U mutant was not in an in-line conformation required for nucleophilic attack, in the activated precursor simulation, U-1 reorients so as to form an in-line conformation that is stabilized by hydrogen bond interactions with A77.
The simulations of the transition state mimics, on the other hand, indicate that protonated C75, adopts an orientation where it is poised to act as a general acid, acquiring interactions with the scissile phosphate and G1:O5′ leaving group, and being held in place, in part, by a hydrogen bond between the exocyclic amine of C75 and a non-bridge phosphoryl oxygen of C22 that supports a role for this functional group in maintaining the active site fold.42 These changes in hydrogen bonding are accompanied by weakening and ejection of a divalent metal ion from the active site, consistent with crystallographic data.14
In the product state, the MD simulation results are observed to relax to within 1.5 Å RMSD from the product crystal structure, despite departing from a structure derived from the C75U pre-cleavage structure. The only notable difference is that the simulations did not form a reverse G25·U20 wobble as observed somewhat weakly in the product crystal structure6 and more pronounced in a recent crystal structure of an inhibited reactant,28 where G25 is in a syn conformation. This is not unexpected since the time scales of the simulations are likely not sufficient for G25 to flip from a anti conformation, seen in our simulations, to syn conformation to form a reverse wobble pair with U20.
Furthermore, based on this inhibited reactant structure and MD simulations, an alternate metal binding site near the G25/U20 pair has been proposed,28,29 where the G25·U20 reverse wobble base pair provides an environment for Mg2+ to bind to G25:N7 and nearby active site residues. On the other hand, in our simulations, G25 is in the anti conformation and its N7 is not facing the active site hence G25:N7 cannot provide such binding environment.
In all our simulations reported here (total more than 1.5 microsecond), no G25:U20 reverse wobble base pair has been observed, consistent with a previously reported MD studies. 15,31 Therefore, simulations with different G25 conformations may be needed to further explore the Mg2+ binding site near G25 and its relationship with the binding site we proposed in the present work. These results provide impetus for experiments focused on examination of HDVr activity with chemical modifications at the G25 position, including an N7 deaza modification to eliminate binding of Mg2+ to N7, or an 8-bromo substitution to favor the syn conformation required by the reverse wobble pair.
In conclusion, we present a set of extended molecular dynamics simulations for HDVr at different stages along the reaction path, and characterize a conformational transition of U-1 into an in-line active conformation in the activated precursor state. Our simulations support the role of C75 as the general acid, and identify several key residue interactions at different stages of the reaction. Our results provide explanations for the observed importance of several active site residues, and suggest specific hypotheses that can be experimentally tested. Although simulations starting with other crystal structures may further explore different possible active site conformations, this work provides a departure point for further investigations into the catalytic chemical steps of the HDVr mechanism.
Supplementary Material
Acknowledgments
The authors are grateful for financial support provided by the National Institutes of Health (GM62248 to DY). Computational resources from The Minnesota Supercomputing Institute for Advanced Computational Research (MSI) were utilized in this work. This research was supported in part by the National Science Foundation through TeraGrid resources provided by Ranger at TACC and Kraken at NICS under grant number TG-CHE100072.
Footnotes
Supporting Information Available: Additional computational details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kuo MY, Sharmeen L, Dinter-Gottlieb G, Taylor J. Characterization of self-cleaving RNA sequences on the genome and antigenome of human hepatitis delta virus. J Virol. 1988;62:4439–4444. doi: 10.1128/jvi.62.12.4439-4444.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharmeen L, Kuo MY, Dinter-Gottlieb G, Taylor J. Antigenomic RNA of human hepatitis delta virus can undergo self-cleavage. J Virol. 1988;62:2674–2679. doi: 10.1128/jvi.62.8.2674-2679.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu HN, Lin YJ, Lin FP, Makino S, Chang MF. Human hepatitis δ virus RNA subfragments contain an autocleavage activity. Proc Natl Acad Sci USA. 1989;86:1831–1835. doi: 10.1073/pnas.86.6.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai MMC. The molecular biology of hepatitis delta virus. Annu Rev Biochem. 1995;64:259–286. doi: 10.1146/annurev.bi.64.070195.001355. [DOI] [PubMed] [Google Scholar]
- 5.Webb CHT, Riccitelli NJ, Ruminski DJ, Luptak A. Widespread Occurrence of Self-Cleaving Ribozymes. Science. 2009;326:953. doi: 10.1126/science.1178084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferré-D'Amaré Adrian R, Zhou K, Doudna JA. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 7.Perrotta AT, Been MD. The self-cleaving domain from the genomic RNA of hepatitis delta virus: sequence requirements and the effects of denaturant. Nucleic Acids Res. 1990;18:6821–6827. doi: 10.1093/nar/18.23.6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wadkins TS, Perrotta AT, Ferre-D'Amare AR, Doudna JA, Been MD. A nested double pseudoknot is required for self-cleavage activity of both the genomic and antigenomic hepatitis delta virus ribozymes. RNA. 1999;5:720–727. doi: 10.1017/s1355838299990209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakano S, Chadalavada DM, Bevilacqua PC. General Acid-Base Catalysis in the Mechanism of a Hepatitis Delta Virus Ribozyme. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 10.Das S, Piccirilli J. General acid catalysis by the hepatitis delta virus ribozyme. Nature Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 11.Oyelere AK, Kardon JR, Strodel SA. pKa perturbation in genomic hepatitis delta virus ribozyme catalysis evidenced by nucleotide analogue interference mapping. Biochemistry. 2002;41:3667–3675. doi: 10.1021/bi011816v. [DOI] [PubMed] [Google Scholar]
- 12.Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. Direct Measurement of a pKa near Neutrality for the Catalytic Cytosine in the Genomic HDV Ribozyme Using Raman Crystallography. J Am Chem Soc. 2007;129:13335–13342. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- 13.Cerrone-Szakal AL, Siegfried NA, Bevilacqua PC. Mechanistic Characterization of the HDV Genomic Ribozyme: Solvent Isotope Effects and Proton Inventories in the Absence of Divalent Metal Ions Support C75 as the General Acid. J Am Chem Soc. 2008;130:14504–14520. doi: 10.1021/ja801816k. [DOI] [PubMed] [Google Scholar]
- 14.Ke A, Zhou K, Ding F, Cate JHD, Doudna JA. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature. 2004;429:201–205. doi: 10.1038/nature02522. [DOI] [PubMed] [Google Scholar]
- 15.Krasovska MV, Sefcikova J, Špačková Nad's, Šponer Jiří, Walter NG. Structural dynamics of precursor and product of the RNA enzyme from the hepatitis delta virus as revealed by molecular dynamics simulations. J Mol Biol. 2005;351:731–748. doi: 10.1016/j.jmb.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 16.Banáš P, Rulíšek L, Hánošová V, Svozil D, Walter NG, Šponer J, Otyepka M. General Base Catalysis for Cleavage by the Active-Site Cytosine of the Hepatitis Delta Virus Ribozyme: QM/MM Calculations Establish Chemical Feasibility. J Phys Chem B. 2008;112:11177–11187. doi: 10.1021/jp802592z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banás P, Jurecka P, Walter NG, Sponer J, Otyepka M. Theoretical studies of RNA catalysis: hybrid QM/MM methods and their comparison with MD and QM. Methods. 2009;49:202–216. doi: 10.1016/j.ymeth.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fedoruk-Wyszomirska A, Giel-Pietraszuk M, Wyszko E, Szymański M, Ciesiołka J, Barciszewska MZ, Barciszewski J. The mechanism of acidic hydrolysis of esters explains the HDV ribozyme activity. Mol Biol Rep. 2009;36:1647–1650. doi: 10.1007/s11033-008-9364-7. [DOI] [PubMed] [Google Scholar]
- 19.Suh YA, Kumar PK, Taira K, Nishikawa S. Self-cleavage activity of the genomic HDV ribozyme in the presence of various divalent metal ions. Nucleic Acids Res. 1993;21:3277–3280. doi: 10.1093/nar/21.14.3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shih Ih, Been MD. Ribozyme cleavage of a 2′,5′-phosphodiester linkage: Mechanism and a restricted divalent metal-ion requirement. RNA. 1999;5:1140–1148. doi: 10.1017/s1355838299990763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shih Ih, Been MD. Catalytic strategies of the hepatitis delta virus ribozymes. Annu Rev Biochem. 2002;71:887–917. doi: 10.1146/annurev.biochem.71.110601.135349. [DOI] [PubMed] [Google Scholar]
- 22.Bevilacqua PC, Yajima R. Nucleobase catalysis in ribozyme mechanism. Curr Opin Chem Biol. 2006;10:455–464. doi: 10.1016/j.cbpa.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 23.Nakano Si, Bevilacqua PC. Mechanistic Characterization of the HDV Genomic Ribozyme: A Mutant of the C41 Motif Provides Insight into the Positioning and Thermodynamic Linkage of Metal Ions and Protons. Biochemistry. 2007;46:3001–3012. doi: 10.1021/bi061732s. [DOI] [PubMed] [Google Scholar]
- 24.Gong B, Chen JH, Bevilacqua PC, Golden BL, Carey PR. Competition Between Co(NH3)63+ and Inner Sphere Mg2+ Ions in the HDV Ribozyme. Biochemistry. 2009;48:11961–11970. doi: 10.1021/bi901091v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong B, Chen Y, Christian EL, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. Detection of Innersphere Interactions between Magnesium Hydrate and the Phosphate Backbone of the HDV Ribozyme Using Raman Crystallography. J Am Chem Soc. 2008;130:9670–9672. doi: 10.1021/ja801861s. [DOI] [PubMed] [Google Scholar]
- 26.Chen JH, Gong B, Bevilacqua PC, Carey PR, Golden BL. A Catalytic Metal Ion Interacts with the Cleavage Site GU Wobble in the HDV Ribozyme. Biochemistry. 2009;48:1498–1507. doi: 10.1021/bi8020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shih Ih, Been M. Kinetic scheme for intermolecular RNA cleavage by a ribozyme derived from hepatitis delta virus RNA. Biochemistry. 2000;39:9055–9066. doi: 10.1021/bi000499+. [DOI] [PubMed] [Google Scholar]
- 28.Chen JH, Yajima R, Chadalavada DM, Chase E, Bevilacqua PC, Golden BL. A 1.9 A Crystal Structure of the HDV Ribozyme Precleavage Suggests both Lewis Acid and General Acid Mechanisms Contribute to Phosphodiester Cleavage. Biochemistry. 2010;49:6508–6518. doi: 10.1021/bi100670p. [DOI] [PubMed] [Google Scholar]
- 29.Veeraraghavan N, Ganguly A, Chen JH, Bevilacqua PC, Hammes-Schiffer S, Golden BL. Metal binding motif in the active site of the HDV ribozyme binds divalent and monovalent ions. Biochemistry. 2011;50:2672–2682. doi: 10.1021/bi2000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veeraraghavan N, Ganguly A, Golden BL, Bevilacqua PC, Hammes-Schiffer S. Mechanistic strategies in the HDV ribozyme: chelated and diffuse metal ion interactions and active site protonation. J Phys Chem B. 2011;115:8346–8357. doi: 10.1021/jp203202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krasovska MV, Sefcikova J, Réblová K, Schneider B, Walter Nils G, S J. Cations and Hydration in Catalytic RNA: Molecular Dynamics of the Hepatitis Delta Virus Ribozyme. Biophys J. 2006;91:626–638. doi: 10.1529/biophysj.105.079368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sefcikova J, Krasovska MV, Šponer J, Walter NG. The genomic HDV ribozyme utilizes a previously unnoticed U-turn motif to accomplish fast site-specific catalysis. Nucleic Acids Res. 2007;35:1933–1946. doi: 10.1093/nar/gkl1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veeraraghavan N, Bevilacqua PC, Hammes-Schiffer S. Long-Distance Communication in the HDV Ribozyme: Insights from Molecular Dynamics and Experiments. J Mol Biol. 2010;402:278–291. doi: 10.1016/j.jmb.2010.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee TS, Silva-Lopez C, Martick M, Scott WG, York DM. Insight into the role of Mg2+ in hammerhead ribozyme catalysis from x-ray crystallography and molecular dynamics simulation. J Chem Theory Comput. 2007;3:325–327. doi: 10.1021/ct6003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee TS, Silva Lopez C, Giambaşu GM, Martick M, Scott WG, York DM. Role of Mg2+ in Hammerhead ribozyme catalysis from molecular simulation. J Am Chem Soc. 2008;130:3053–3064. doi: 10.1021/ja076529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeoung YH, Kumar PK, Suh YA, Taira K, Nishikawa S. Identification of phosphate oxygens that are important for self-cleavage activity of the HDV ribozyme by phosphorothioate substitution interference analysis. Nucleic Acids Res. 1994;22:3722–3727. doi: 10.1093/nar/22.18.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee TS, York DM. Computational mutagenesis studies of hammerhead ribozyme catalysis. J Am Chem Soc. 2010;132:13505–13518. doi: 10.1021/ja105956u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar PK, Suh YA, Miyashiro H, Nishikawa F, Kawakami J, Taira K, Nishikawa S. Random mutations to evaluate the role of bases at two important single-stranded regions of genomic HDV ribozyme. Nucleic Acids Res. 1992;20:3919–3924. doi: 10.1093/nar/20.15.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu HN, Huang ZS. Mutagenesis analysis of the self-cleavage domain of hepatitis delta virus antigenomic RNA. Nucleic Acids Res. 1992;20:5937–5941. doi: 10.1093/nar/20.22.5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishikawa F, Shirai M, Nishikawa S. Site-specific modification of functional groups in genomic hepatitis delta virus (HDV) ribozyme. Eur J Biochem. 2002;269:5792–5803. doi: 10.1046/j.1432-1033.2002.03280.x. [DOI] [PubMed] [Google Scholar]
- 41.Shih Ih, Been MD. Energetic contribution of non-essential 5′ sequence to catalysis in a hepatitis delta virus ribozyme. EMBO J. 2001;20:4884–4891. doi: 10.1093/emboj/20.17.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oyelere AK, Strobel SA. Site specific incorporation of 6-azauridine into the genomic HDV ribozyme active site. Nucleosides Nucleotides Nucleic Acids. 2001;20:1851–1858. doi: 10.1081/NCN-100107196. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 44.Essmann U, Perera L, Berkowitz ML, Darden T, Hsing L, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 45.Sagui C, Darden TA. Molecular Dynamics Simulations of Biomolecules: Long-Range Electrostatic Effects. Annu Rev Biophys Biomol Struct. 1999;28:155–179. doi: 10.1146/annurev.biophys.28.1.155. [DOI] [PubMed] [Google Scholar]
- 46.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kaleé L, Schulten K. Scalable Molecular Dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Case DA, et al. AMBER 7. University of California San Francisco; San Francisco: 2002. [Google Scholar]
- 48.Pearlman DA, Case DA, Caldwell JW, Ross WR, Cheatham T, III, DeBolt S, Ferguson D, Seibel G, Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structure and energetic properties of molecules. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- 49.Pérez Alberto, Marchán Iván, Svozil D, Sponer J, C TE, III, Laughton CA, Orozco M. Refinement of the AMBER Force Field for Nucleic Acids:Improving the Description of α/γ Conformers. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.