

Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease in developed countries. The core motor symptoms are attributable to the degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc). Why these neurons succumb in PD is not clear. One potential clue has come from the observation that the engagement of L-type Ca2+ channels during autonomous pacemaking elevates the sensitivity of SNc DA neurons to mitochondrial toxins used to create animal models of PD, suggesting that Ca2+ entry is a factor in their selective vulnerability. Recent work has shown that this Ca2+ entry also elevates mitochondrial oxidant stress and that this stress is exacerbated by deletion of DJ-1, a gene associated with an early onset, recessive form of PD. Epidemiological data also supports a linkage between L-type Ca2+ channels and the risk of developing PD. This review examines the hypothesis that the primary factor driving neurodegenerative changes in PD is the metabolic stress created by Ca2+ entry, particularly in the face of genetic or environmental factors that compromise oxidative defenses or proteostatic competence.
Keywords: Parkinson’s disease, pacemaking, calcium, endoplasmic reticulum, mitochondrial oxidant stress, L-type channels
PD is a disabling neurodegenerative disorder that is strongly associated with aging, increasing exponentially in incidence above the age of 65 (de Rijk et al., 1997, de Lau et al., 2004). The incidence of PD is expected to rise dramatically worldwide with increased life expectancy (Dorsey et al., 2007). Although there are signs of distributed neuropathology (as judged by Lewy body formation) (Braak et al., 2004), the motor symptoms of PD, including bradykinesia, rigidity, and resting tremor, are the first to appear clinically and are clearly linked to the degeneration and death of SNc DA neurons (Hornykiewicz, 1966, Riederer and Wuketich, 1976). The efficacy of the clinical gold-standard treatment of L-DOPA - a DA precursor - is testament to the centrality of DA neurons in the motor symptoms of PD.
Theories of selective vulnerability
The mechanisms responsible for the preferential loss of DA neurons in PD have been debated for decades. A widely held theory implicates DA itself, suggesting that oxidation of cytosolic DA (and its metabolites) leads to the production of cytotoxic free radicals (Greenamyre and Hastings, 2004). However, there are reasons to doubt this type of cellular stress alone is responsible for the loss of DA neurons in PD. For example, there is considerable regional variability in the vulnerability of DA neurons in PD, with some areas being devoid of pathological markers (Matzuk and Saper, 1985, Kish et al., 1988, Saper et al., 1991, Ito et al., 1992, Damier et al., 1999). Moreover, L-DOPA administration (which relieves symptoms by elevating DA levels in PD patients) does not appear to accelerate disease progression (Fahn, 2005), suggesting that DA itself is not a significant source of reactive oxidative stress, at least in the short term. Sulzer and colleagues have recently reported that Ca2+ entry through L-type channels stimulates DA metabolism in SNc DA neurons, pushing cytosolic DA concentrations into a toxic range with L-DOPA loading(Mosharov et al., 2009). As noted below, the oxidant stress created by DA could interact with other factors to promote cell loss. However, the frank death or phenotypic decline of a variety of non-DA neurons in PD argues that DA itself is not likely to be the principal culprit in the disease.
Mitochondrial dysfunction clearly plays a role in PD (Henchcliffe and Beal, 2008, Schapira, 2008b, Vila et al., 2008). Toxins that nominally target mitochondria create a parkinsonian phenotype (Przedborski et al., 2004). Moreover, many of the genes implicated in familial and idiopathic forms of PD code for mitochondrially-linked proteins (Moore et al., 2005a, Abou-Sleiman et al., 2006, Schapira, 2008a). Additional evidence for mitochondrial involvement in PD comes from the study of human PD patients. In postmortem tissue samples of the SNc from sporadic PD patients, there is a substantial decrease in the activity of mitochondrial complex I (Mann et al., 1994); this deficit is specific to PD patients (Gu et al., 1997) and appears to reflect oxidative damage to complex I (Keeney et al., 2006). On the other hand, experimental stresses such as 1-methyl-4-phenylpyridinium (MPP+) that selectively target mitochondria in dopaminergic cells do not necessarily implicate these organelles in clinical disease, as any toxin that acts on specifically on SNc cells will mimic the signs of PD. Nevertheless, oxidative damage to other cellular components such as lipids, proteins and DNA has been found in the SNc of PD brains (Zhang et al., 1999) and the source of this oxidative stress appears to be mitochondrial. Superoxide and reactive oxygen species (ROS) are generated by inefficiencies in the ETC, which is responsible for creating the electrochemical gradient across the inner mitochondrial membrane that drives ATP synthase and the conversion of adenosine diphosphate to ATP (Nicholls, 2002). Elevated superoxide production could be responsible for the high level of somatic DNA mutations in SNc DA neurons (Soong et al., 1992). The physical proximity of mitochondrial DNA (mtDNA) to the site of superoxide generation makes it an even more vulnerable target. The mitochondrial genome encodes 13 proteins involved in the mitochondria respiratory chain, 7 of which are involved in the formation of complex I (Schapira, 2008a). The number of mtDNA mutations present in clonally-expanded clusters of SNc mitochondria is positively correlated with age and negatively correlated with cytochrome oxidase activity (a marker for functional respiratory activity) (Kraytsberg et al., 2006). Their clonal nature indicates that these mutations are due to the expansion of a somatic mutation, not a genetic mutation present at birth.
Because many of these mtDNA mutations impair ETC function (Bender et al., 2006), they could contribute to a bioenergetic impairment in SNc DA neurons, proteostatic dysfunction (e.g., Lewy body formation) and death. In fact, disruption of the gene for mitochondrial transcription factor A (Tfam) reduces mtDNA copy number and respiratory chain function in DA neurons in a way that mimics accumulating mtDNA mutations and produces an adult onset form of PD that is accompanied by intraneuronal inclusions and cell death (Ekstrand et al., 2007). Recently, a novel regulator of PGC-1alpha and mitochondrial biogenesis has been identified that interacts with the PD-associated protein parkin (so-called parkin interacting substrate or PARIS) (Shin et al., 2011). Removing parkin elevates the activity of PARIS, leading to the suppression of PGC-1alpha and the selective loss of SNc dopaminergic neurons. This exciting discovery clearly suggests that mitochondrial stress in SNc DA neurons is a factor in PD, but it does not elucidate why mitochondrial stress and the need to generate new mitochondria is any greater in SNc DA neurons than in other types of neuron (see below).
Another organelle that has been widely linked to pathogenesis in PD is the endoplasmic reticulum (ER) (Wang and Takahashi, 2007, Bandopadhyay and de Belleroche, 2010, Gleichmann and Mattson, 2011). The ER is an integral component of the cellular machinery responsible for the production, delivery and degradation of proteins, a process referred to as proteostasis (Balch et al., 2008). One of the hallmarks of PD is the formation of Lewy bodies (LBs), an abnormal protein aggregate found in SNc DA neurons and elsewhere in the brain (Del Tredici and Braak, 2004). These depositions reflect a deficiency in proteostasis that may arise from protein misfolding or defects in the ubiquitin-proteasome degradation systems, and are accompanied by signs of ER stress and an attempt to sequester cytotoxic proteins (Ryu et al., 2002).
The other major function of the ER is Ca2+ homeostasis (Berridge, 2002). The ER forms a continuous, intracellular network, allowing it to regulate both local and global Ca2+ signals. As in other neurons, the ER network in SNc DA neurons extends throughout the somatodendritic tree (Schwyn and Fox, 1974, Mogami et al., 1997, Park et al., 2000, Choi et al., 2006). High affinity ATP-dependent transporters move Ca2+ from the cytoplasm into the ER lumen. The absence of high-affinity, anchored intraluminal Ca2+ buffers and the physical continuity of the lumen within the cell (Mogami et al., 1997, Park et al., 2000) allows the ER to rapidly (~30 μm/s) redistribute Ca2+ between intracellular compartments, thus avoiding pro-apoptotic accumulations in the cytosol (Choi et al., 2006). Ca2+ sequestered in the ER is released at sites where it can be pumped back across the plasma membrane or where it can be used to modulate cellular function (Rose and Konnerth, 2001, Cui et al., 2004, Verkhratsky, 2005, Bardo et al., 2006, Park et al., 2008). However, the storage capacity of the ER is limited. BCL-2 family proteins, which are implicated in apoptosis, control the ER Ca2+ concentration and are capable of adjusting it in response to stress (Hetz, 2007).
Movement of the ER away from its Ca2+ set point can compromise proteostasis. In part, this is a consequence of the fact that Ca2+ is an allosteric regulator of protein processing and folding (Paschen and Mengesdorf, 2005, Toescu, 2005). Depleting ER Ca2+ stores induces signs of ER stress and the unfolded-protein-response (Paschen and Mengesdorf, 2005, Toescu, 2005). Conversely, proteostatic deficits in Alzheimer’s disease have been associated with high ER Ca2+ concentrations and large changes in cytosolic Ca2+ concentration upon ER release (LaFerla, 2002, Stutzmann et al., 2006). The endoplasmic stress sensor, ATF6a, has been implicated in the death of DA neurons induced by neurotoxins (Egawa et al., 2011). Moreover, ATF4, a transcriptional factor controlled by the unfolded protein response, regulates the expression of parkin – an E3 ubiquitin ligase involved in protein degradation and mitophagy (Bouman et al., 2011).
In neurons, mitochondria partner with the ER in Ca2+ homeostasis. Mitochondria are commonly found physically coupled to the ER by mitochondria-associated membrane (MAM) (Hayashi et al., 2009). These regions of juxtaposition create multifunctional signaling microdomains (Csordas et al., 2006, Rizzuto and Pozzan, 2006, de Brito and Scorrano, 2008). For example, within these microdomains, Ca2+ released by the ER through ryanodine (RYR) and inositol trisphosphate (IP3) receptors runs down the steep potential gradient (~-200 mV) across the inner mitochondrial membrane into the matrix through a pore called the Ca2+ uniporter (Kirichok et al., 2004). Matrix Ca2+ stimulates enzymes of the tricarboxylic acid (TCA) cycle that produce reducing equivalents for oxidative phosphorylation (OXPHOS) (McCormack et al., 1990). This Ca2+ shuttling creates a means by which the ER can regulate mitochondrial ATP production to match the needs of both Ca2+ homeostasis and proteostasis.
Another interesting linkage between mitochondria and proteostasis involves an interaction between PTEN-induced putative kinase 1 (PINK1) and parkin. Like parkin, mutations in the gene coding for PINK1 are associated with rare, recessive forms of PD (Moore et al., 2005a, Abou-Sleiman et al., 2006, Schapira, 2008a). Work in flies has provided evidence that PINK1 and parkin interact in regulating mitochondrial turnover (Guo, 2010). Recent work has put this model on firm footing in mammalian cells. PINK1 is a mitochondrial kinase that is proteolytically processed by healthy, polarized mitochondria, but accumulates when the inner mitochondrial membrane becomes significantly depolarized, indicative of damage (Narendra et al., 2010). PINK1 promotes recruitment of parkin to mitochondria, which triggers their mitoautophagy. By this mechanism, depolarization of the inner membrane can alert the cell to the presence of a damaged organelle and trigger its removal. Mutations in either PINK1 or parkin that are associated with PD disrupt this process (Narendra et al., 2010).
What is missing from this discussion to this point is why SNc DA neurons should be any more vulnerable to develop dysfunction in mitochondria or the ER than any other neuron. There is no evidence for heterogeneity in these organelles across cell types. Furthermore, there is no evidence of selective regional expression of genes associated with familial forms of PD that would be predictive of disease progression (Moore et al., 2005b). One possible explanation is that the intracellular environment in which these organelles find themselves brings out the pathological potential of genetic mutations, environmental toxins or age. A corollary is that a unique phenotypic function of these cells renders them more vulnerable to disruptions in another pathway compared with other neurons.
Ca2+ handling in SNc DA neurons – a unifying mechanism?
One of the common elements in scenarios discussed thus far is Ca2+. Ca2+ stimulates DA synthesis and the toxic impact of L-DOPA loading (Mosharov et al., 2009). Ca2+ is a principal modulator of the ER and mitochondria. Recent work has shown that a distinctive feature of SNc DA neurons is the way they handle Ca2+. Unlike the vast majority of neurons in the brain, adult SNc DA neurons are autonomously active, generating broad, slow action potentials regularly (2-4 Hz) in the absence of synaptic input (Grace and Bunney, 1983)(Figure 1). This pacemaking activity is believed to be important in maintaining ambient DA levels in regions that are innervated by these neurons, particularly the striatum (Romo and Schultz, 1990). While most neurons rely exclusively on monovalent cation channels to drive pacemaking, SNc DA neurons also engage ion channels that allow extracellular Ca2+ to enter the cytoplasm (Ping and Shepard, 1996, Bonci et al., 1998, Puopolo et al., 2007) leading to elevated intracellular Ca2+ concentrations (Wilson and Callaway, 2000, Chan et al., 2007).
Figure 1. Pacemaking firing in SNc DA neurons is associated with Ca2+ influx via L-type channels.
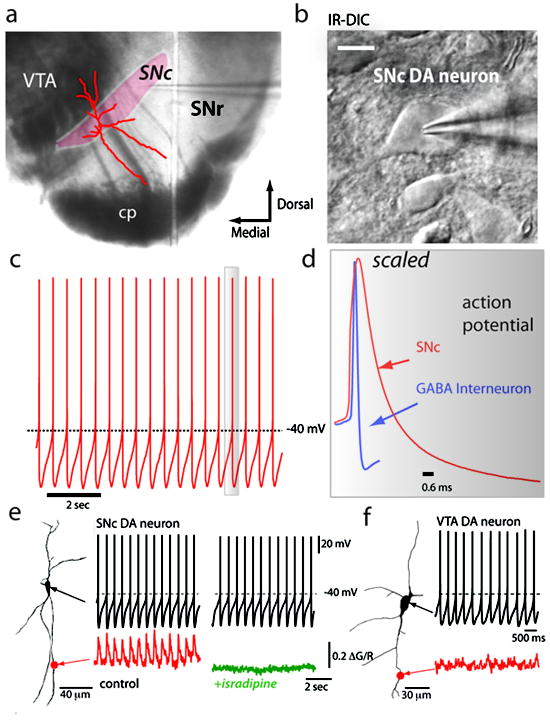
a) Macroscopic view of a coronal midbrain slice section illustrating the SNc region as the site of electrophysiological recording and optical imaging. b) SNc DA neurons illustrated by infrared digital interference contrast (IR-DIC) optics from coronal mibrain slices. c) Representative pacemaking firing trace in whole-cell patch current clamp configuration from an SNc DA neuron. d) From panel ‘c’, one scaled action potential spike showing that SNc DA neurons display broad action potentials compared to a GABAergic interneuron recorded from the substantia nigra pars reticulata (SNr). e) Ca2+ imaging of dendrites using two-photon laser scanning microscopy (2PLSM) in an SNc DA neuron displaying pacemaking firing synchronized to Ca2+ transients per spike in a distal dendrite (marked by the red dot). Experiments were performed as described previously. Blocking L-type channels with 5 μM isradipine attenuates dendritic Ca2+ transients without affecting pacemaking rate. f) Recording from a ventral tegmental area (VTA) DA neuron showing pacemaking firing but this population of cells lack dendritic Ca2+ oscillations. Figures e-f represents data adapted from previous work (Guzman et al., 2010).
The L-type Ca2+ channels used by SNc DA neurons in pacemaking have a distinctive Cav1.3 pore-forming subunit encoded by Cacna1d (Striessnig et al., 2006, Chan et al., 2007). Cav1.3 Ca2+ channels are relatively rare, constituting only about 10% of the all the L-type Ca2+ channels found in the brain (Sinnegger-Brauns et al., 2009). Channels with this subunit differ from other L-type Ca2+ channels in that they open at relatively hyperpolarized potentials, allowing them to contribute to the mechanisms driving the membrane potential to spike threshold underlying autonomous pacemaking (Chan et al., 2007, Puopolo et al., 2007, Guzman et al., 2009). Until recently, it was thought that L-type Ca2+ channels were essential for pacemaking in SNc DA neurons making them less than ideal drug targets if pacemaking was necessary to maintain physiologically important levels of DA in target structures (Nedergaard et al., 1993, Mercuri et al., 1994). This inference was based upon the ability of L-type channel antagonists (dihydropyridines) to halt pacemaking. However, we know now that at the concentrations of dihydropyridine (DHP) necessary to stop pacemaking, other ion channels are being antagonized, complicating the interpretation of previous studies; at lower, channel-specific DHP concentrations, pacemaking continues, unaltered in rate and regularity, in spite of near complete antagonism of L-type channels (Guzman et al., 2009). The ability of SNc DA neurons to continue pacemaking under these circumstances reflects the robustness of the multi-channel pacemaking mechanism and that Cav1.3 Ca2+ channels play a supportive, but not necessary role.
Why Cav1.3 channels are found at significantly higher density in SNc DA neurons than in neighboring VTA DA neurons is not entirely clear (Puopolo et al., 2007, Guzman et al., 2009, Khaliq and Bean, 2010). Cav1.3 Ca2+ channels do participate in the postsynaptic response to activation of glutamatergic synapses and burst spiking, but their role is modest in comparison with NMDA receptors (Blythe et al., 2009, Deister et al., 2009). It is also possible that, as noted above, Ca2+ entry through L-type channels helps maintain an adequate level of DA synthesis (Mosharov et al., 2009). However, as with burst spiking, this modulation appears not to be of major physiological significance.
The sustained engagement of Cav1.3 Ca2+ channels during pacemaking comes at an obvious metabolic cost to SNc DA neurons. Because of its involvement in cellular processes ranging from the regulation of enzyme activity to programmed cell death, Ca2+ is under very tight homeostatic control, with a cytosolic set point near 100 nM – 10,000 times lower than the concentration of Ca2+ in the extracellular space (Berridge et al., 2000, Orrenius et al., 2003) Ca2+ entering neurons is rapidly sequestered or pumped back across the steep plasma membrane concentration gradient; this process requires energy stored in ATP or in ion gradients that are maintained with ATP-dependent pumps. This makes use of Ca2+ in cellular processes several times more expensive energetically on a per charge basis than monovalent ions, like Na+, which has a transmembrane concentration ratio of near 10. In most neurons this isn’t a significant issue as Ca2+ channel opening is a rare event, occurring primarily during very brief action potentials. This makes the task and the metabolic cost to the cell readily manageable. But in SNc DA neurons, where Cav1.3 Ca2+ channels are open much of the time, the magnitude and the spatial extent of Ca2+ influx are much larger (Wilson and Callaway, 2000).
Recent work by our group has shown that maintained opening of L-type Ca2+ channels in SNc DA neurons creates a basal mitochondrial oxidant stress (Guzman et al., 2010). These studies utilized a transgenic mouse that expressed a mitochondrially-targeted redox-sensitive variant of green-fluorescent protein (mito-roGFP) (Hanson et al., 2004, Guzman et al., 2010) expressed under control of the tyrosine hydroxylase (TH) promoter (Fig. 2). The use of roGFP allowed mitochondrial matrix redox state to be quantitatively estimated, something not possible with conventional redox probes. Using two photon laser scanning microscopy to monitor mito-roGFP in brain slices from young adult mice, we found that the engagement of plasma membrane Cav1.3 L-type calcium channels during normal autonomous pacemaking created an oxidant stress in the mitochondria that was specific to the vulnerable SNc DA neurons and not apparent in neighboring VTA DA neurons. The oxidant stress engaged defenses that induced transient, mild mitochondrial depolarization or uncoupling. The mild uncoupling was not affected by deletion of cyclophilin D, which is a component of the permeability transition pore, but was attenuated by genipin and purine nucleotides, which are antagonists of cloned uncoupling proteins. Knocking out DJ-1 (also known as PARK7 in humans and Park7 in mice), which is a gene associated with an early-onset form of Parkinson’s disease, downregulated the expression of two uncoupling proteins (UCP4 (SLC25A27) and UCP5 (SLC25A14)), compromised calcium-induced uncoupling and increased oxidation of matrix proteins specifically in SNc dopaminergic neurons. The results with the DJ-1 knockout – showing that the impact of DJ-1 deletion depend upon a physiological phenotype that engages mitochondrial oxidant defenses – provides an example of how mutations in a widely expressed gene can affect a select subpopulation of neurons.
Figure 2. SNc DA neurons display elevated oxidant stress attenuated by blockade of L-type Ca2+ channels.
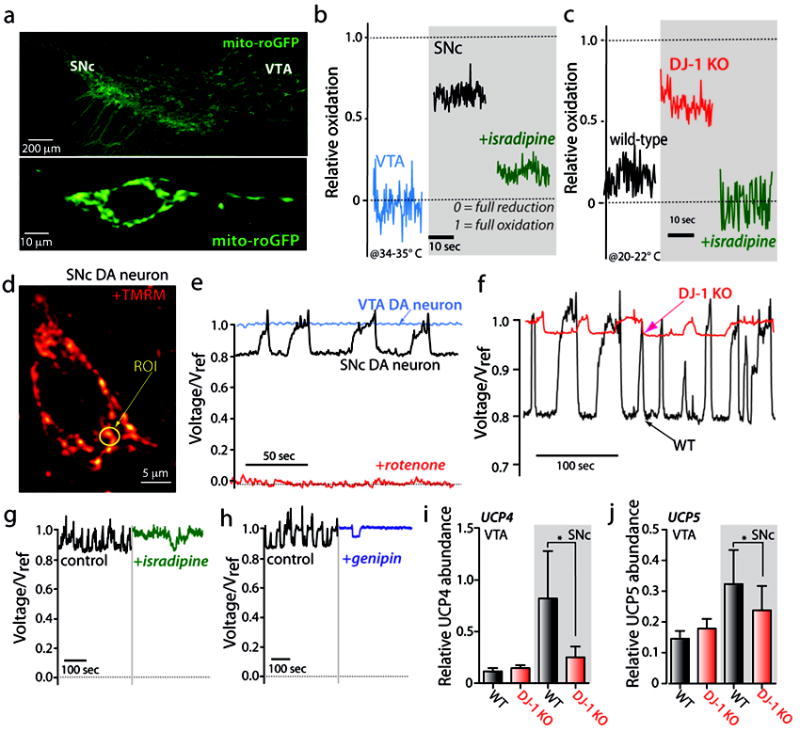
a) Representative confocal microscopy images from coronal midbrain slices of a transgenic mouse that expresses a mitochondrial redox-sensitive form of green fluorescent protein (mito-roGFP) under the regulation of the tyrosine hydroxylase promoter DNA sequence. b) Relative oxidation plots showing that SNc DA neurons (black trace) displays increased oxidation compared to VTA. Acute application of 5 μM isradipine (green trace) decrease oxidant stress with relative oxidation values closer to VTA neurons (blue trace). Relative oxidation plots are calculated by calibrating the mito-roGFP signal with dithiothreitol (DTT) and aldrithiol to fully reduce and oxidize the mito-roGFP signal respectively. Fluorescence values of the mito-roGFP under control conditions (F), DTT (FDTT), and aldrithiol (Fald) are taken from the same neuron, and relative oxidation is calculated by the following A value of 0 represents full equation: 1-[(F-Fald)/(FDTT-Fald)]. A value of 0 represents full reduction whereas a value of 1 represents full oxidation. c) Loss of function of DJ-1 protein (pink trace) exacerbated oxidant stress in SNc DA neurons compared to wild-type mice. Pretreatment of SNc DA neurons in DJ-1 knock-out mice with 200 nM isradipine decreased oxidant stress with relative oxidation values similar to naïve SNc DA neurons from wild-type mice. d) Representative 2PLSM image from an SNc DA neuron labeled with tetramethyl rhodamine methyl ester dyes to monitor mitochondrial membrane potential (MMP). e) SNc DA neurons have flickering changes of MMP that are absent in VTA DA neurons. The change in MMP in SNc DA neurons represents a 20% amplitude change in voltage. f) Gene-targeted deletion of DJ-1 reduced the amplitude and frequency of flickering mitochondrial activity. g) Blockade of L-type channels with isradipine (5 μM) decreased frequency of MMP flickering activity, suggesting that Ca2+ impacts the mild mitochondrial depolarization or uncoupling phenomena in SNc DA neurons. h) Antagonism of uncoupling proteins (UCPs) with 100 μM genipin decreased MMP flickering activity. i-j) SNc DA neurons containing mild depolarization of the MMP showed increased relative mRNA abundance of the uncoupling proteins UCP4 and UCP5 (gray bars). DJ-1 knock-out mice however, display decreased levels of UCP4 and UCP5 compared to WT, suggesting that the decreased amplitude and frequency changes in MMP in DJ-1 knock-out mice are related to a decrement in the proteins that induce uncoupling. This figure summarizes data adapted from previous work (Guzman et al., 2010).
Aging related cell loss and dysfunction is widely viewed to be a direct consequence of accumulated mtDNA and organelle damage produced by ROS and related reactive molecules generated by the ETC in the course of oxidative phosphorylation (Harman, 1972, Wallace, 2005). Our work suggests that this decline should be accelerated in SNc DA neurons, as they have a high basal metabolic rate, as judged by mitochondrial redox state. Studies of the aging primate brain provide strong support for this inference (Collier et al., 2007). DA neurons in the ventral tier of the primate SNc – neuron homologous to those in the rodent SNc examined in our study – show clear phenotypic downregulation with advanced age as well as signs of oxidant damage, whereas as neurons in the dorsal tier of the SNc or in the ventral tegmental area show little or no decline. More importantly from the standpoint of PD, this distribution matches that seen in PD patients and in animal models following 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine administration (Collier et al., 2011), suggesting that the mechanisms underlying the aging related cell loss and those underlying PD are similar. Age is undoubtedly the single strongest risk factor for PD (Calne and Langston, 1983, Gibb and Lees, 1991).
Is PD an inevitable consequence of aging? It would appear the answer would be yes if humans lived forever. But, we don’t. The question then is whether the trajectory of cell loss in the SNc leads to crossing of the symptomatic threshold (~75% loss) before some other system malfunction causes death. Clearly, there is considerable variation between individuals on this point. Genetic factors certainly could account for a large part of this variation (Calne and Langston, 1983, Moore et al., 2005a, Sulzer, 2007). These factors could increase (or decrease) the rate at which vulnerable neurons age by compromising (or enhancing) ER or mitochondrial function. Environmental factors, like toxin exposure (Betarbet et al., 2006), could alter the trajectory of cell loss. Other factors, like brain trauma or elevated inflammatory responses could also alter the trajectory of cell loss (Klegeris et al., 2007). Unfortunately, at present, there are not viable strategies for altering the negative impact of genetic polymorphisms, toxin exposure or head injury on the degeneration of SNc DA neurons.
Do other vulnerable neurons share the SNc phenotype?
Can the Ca2+-mediated cellular aging hypothesis account for the vulnerability of other cell types in PD? Other regions of the brain that have cell loss paralleling that of the SNc are the locus ceruleus (LC) and hypothalamic tuberomamillary nucleus (German et al., 1992b, Del Tredici and Braak, 2004). The neurons of the LC and the tuberomamillary neurons are similar to SNc DA neurons in several respects. Like SNc DA neurons, both LC and tuberomamillary neurons are autonomous pacemakers that engage L-type Ca2+ channels (Williams et al., 1984, Stevens and Haas, 1996, Taddese and Bean, 2002). Another population of neurons that show LB pathology is found in the dorsal motor nucleus of the vagus (DMV) (Del Tredici and Braak, 2004). Although these neurons have not been characterized in depth, they are autonomous pacemakers (McCann and Rogers, 1990, Travagli et al., 1991). DA neurons in the olfactory bulb also are autonomous pacemakers and rely upon Ca2+ channels (although not L-type channels) (Pignatelli et al., 2005). While olfactory deficits have been associated with PD (Postuma et al., 2006), there is no obvious loss of olfactory bulb DA neurons (Huisman et al., 2008). Although this would seem to run counter to the Ca2+ hypothesis, this could simply be a consequence of the capacity of this region for adult neurogenesis (Pignatelli et al., 2009).
It is also worth considering DA neurons in the VTA. These neurons are slow pacemakers, but do not have a significant sub-threshold L-type Ca2+ channel current during pacemaking (Chan et al., 2007, Khaliq and Bean, 2010). These neurons are relatively intact in PD patients and in animal models of PD (Kish et al., 1988, German et al., 1992a, Pignatelli et al., 2005, Belzunegui et al., 2007).
Are L-type Ca2+ channels a viable therapeutic target?
Although there are several factors governing the loss of SNc DA neurons, most of them cannot be manipulated. The exception is the engagement of L-type Ca2+ channels. These channels are antagonized by orally-deliverable dihydropyridines with good brain bioavailability that have a long record of safe use in humans (Yamada et al., 1990, German et al., 1992a, Chan et al., 2007, Guzman et al., 2010, Ilijic et al., 2011). SNc DA neurons that express the Ca2+ binding protein calbindin have a diminished sensitivity to toxins and to PD (Yamada et al., 1990, German et al., 1992a).
Is there evidence that DHP use might work in humans to prevent or slow PD? Calcium channel antagonists (CCAs), including the DHPs used in animal studies, are commonly used in clinical practice to treat hypertension, creating a potential database to be mined. A case-control study of hypertensive patients found a significant reduction in the observed risk of PD with CCA use, but not with medications that reduce blood pressure in other ways (Becker et al., 2008). More recently, a large Danish data set has been examined (Ritz et al., 2010). The authors agreed with the main conclusions of the Becker et al. study but extended their findings by showing that only DHPs that cross the blood-brain barrier (BBB) are associated with reduced PD risk (~30%). Given the short period of treatment in many cases (~2 years), variable dosing, and low relative affinity of DHPs for Cav1.3 Ca2+ channels (compared to Cav1.2 channels (Mannhold et al., 1995, Kupsch et al., 1996, Eisenberg et al., 2004), this is a surprisingly strong association and lends further credence to the proposition that a BBB permeable and potent Cav1.3 antagonist could be a very effective neuroprotective agent.
That said, these studies are not a substitute for a controlled clinical trial. In the absence of a selective Cav1.3 Ca2+ channel antagonist, the DHP isradipine is the most attractive drug for such a trial. Isradipine has a relatively higher affinity for Cav1.3 Ca2+ channels than the other known DHP and has good brain bioavailability (Koschak et al., 2001, Scholze et al., 2001). At the doses used to treat hypertension, isradipine has relatively minor side effects (Fitton and Benfield, 1990). The question is whether it will prove neuroprotective at doses tolerated by the general population. Pharmacokinetic studies by our group have found that plasma concentrations of isradipine achieved in mice that are protected against systemic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration are very close to those achieved in humans with a very well tolerated daily dose of isradipine (~2-4 ng/ml in mice; 1-2 ng/ml in humans @ 10 mg/day, Dynacirc CR), suggesting that neuroprotection is achievable. A more recent study using an intrastriatal 6-hydroxydopamine model has shown that systemic administration of isradipine produces a dose-dependent protection of both SNc DA axon terminals and cell bodies at plasma concentrations in a similar range (IC50~4-8 ng/ml) (Ilijic et al., 2011). The plasma concentrations required for significant protection against this acute challenge were greater than those needed in the chronic MPTP model, but were are still near the range achievable in humans. It is important to note that these studies suggest that protection is afforded by partial antagonism of Cav1.3 channels, minimizing any complications that might attend near complete disruption of these channels. Using a modulated receptor model derived from an early paper by Bean (Bean, 1984), Ilijic et al. estimated that roughly 40-60% of the Cav1.3 channels were antagonized at the half-maximal dose for protection from intrastriatal 6-OHDA injection. Given the slow progression of PD, it is highly likely that a more modest antagonism would suffice in humans.
The ideal candidate for DHP therapy would be in the very early stages of SNc loss, prior to the onset of symptoms. Unfortunately, there are no biomarkers that would allow the identification of presymptomatic PD patients. As a consequence, the most likely subjects for a clinical trial are is those that have been recently diagnosed with PD. In these early stage patients, SNc DA cell loss is substantial (>60%) and the remaining neurons might be compromised in ways not seen in healthy tissue. Nevertheless, disease progression could be tracked in a longitudinal study to determine whether treated patients exhibit a slower rate of motor deficit progression. There is growing evidence that inflammation could have an important impact on disease progression at this stage (Hirsch and Hunot, 2009). Antagonism of L-type channels might be helpful but not enough to significantly slow progression if this is the case.
It is also worth considering how DHPs compare with other drugs that are being tested in clinical neuroprotection trials for PD. Although early trials with creatine, coenzyme Q10, and the mitochondrial antioxidant compound MitoQ have been disappointing (Shults et al., 1997), this is not sufficient evidence to reject the idea that oxidative stress contributes to PD. Coenzyme Q10 is an electron acceptor for complexes I and II that appears compromised in PD patients (Shults et al., 1997) and is neuroprotective in animal models of PD (Beal et al., 1998). MitoQ is a mitochondria-targeted version of coenzyme Q10 that functions as a scavenger of mitochondrial oxidants (Smith and Murphy, 2011). Creatine is a substrate for ATP production that can both improve mitochondrial efficiency and reduce oxidative stress by buffering fluctuations in cellular energy production (Matthews et al., 1999). One possibility is that antioxidant treatment initiated after the point of clinical diagnosis is ineffective because the cells have already accumulated a critical level of damage. By analogy, myocardial damage in ischemia-reperfusion is mediated by excessive oxidant stress, yet antioxidants given in the days after myocardial infarction are ineffective at rescuing cell function because the oxidant damage is already done. It is also important to consider that the effect of antioxidants will depend on where the oxidant stress is generated and where the critical targets of these oxidants reside. Clearly an antioxidant distributed for example in membranes could be minimally effective in scavenging oxidants in the mitochondrial matrix. If antioxidant therapy by itself is ineffective, then approaches aimed at improving mitochondrial function rather than attacking the source of stress on mitochondria could be more effective. For example, deprenyl could prove to have neuroprotective effects by virtue not of its ability to inhibit the degradation of DA, but by its ability to induce the expression of antioxidant defenses (Magyar and Szende, 2004). Because their sites of action differ within the chain of events leading to oxidative stress and mitochondrial dysfunction, a combination therapy could prove more effective than any one alone.
Conclusions
Ca2+-mediated cellular stress has long been thought to be important in neurodegeneration, but it usually is envisioned as a late stage consequence of organelle damage inflicted by some other challenge. The unusual reliance of SNc DA neurons on voltage-dependent L-type Ca2+ channels in autonomous pacemaking suggests that the mitochondrial and ER stress created by sustained Ca2+ entry could be responsible for their selective vulnerability, rather than simply a late stage consequence. This hypothesis is consistent with the centrality of the ER and mitochondria – key organelles in Ca2+ homeostasis – in prevailing models of pathogenesis in PD. Genetic mutations and environmental challenges could easily synergize with this basal stress, hastening cellular aging and eventual death. Recent work has provided new evidence for this hypothesis, showing that Ca2+ entry through L-type channels elevates mitochondrial oxidant stress in SNc DA neurons and this stress is elevated in at least one genetic model of PD. The scientific data, together with the long safety record of DHPs and epidemiological evidence suggesting brain penetrating DHPs reduce the risk of PD, provide a strong motivation for moving ahead with human trials.
Highlights.
The motor symptoms of Parkinson’s disease largely are attributable to the death of dopaminergic neurons in the substantia nigra pars compacta (SNc)
The selective vulnerability of these neurons might be due to their engagement of low threshold L-type Ca2+ channels during pacemaking. This engagement elevates mitochondrial oxidant stress.
These channels are antagonized by dihydropyridines that are approved for human use and have been linked by epidemiological studies to reduced risk of PD.
Acknowledgments
This work was supported by grants from the Hartman Foundation, USAMRMC and NIH (NS047085, RR025355 and HL35440).
Footnotes
Disclosure: The authors have no competing financial interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Bandopadhyay R, de Belleroche J. Pathogenesis of Parkinson’s disease: emerging role of molecular chaperones. Trends Mol Med. 2010;16:27–36. doi: 10.1016/j.molmed.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Bardo S, Cavazzini MG, Emptage N. The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol Sci. 2006;27:78–84. doi: 10.1016/j.tips.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Beal MF, Matthews RT, Tieleman A, Shults CW. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998;783:109–114. doi: 10.1016/s0006-8993(97)01192-x. [DOI] [PubMed] [Google Scholar]
- Bean BP. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc Natl Acad Sci U S A. 1984;81:6388–6392. doi: 10.1073/pnas.81.20.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C, Jick SS, Meier CR. Use of antihypertensives and the risk of Parkinson disease. Neurology. 2008;70:1438–1444. doi: 10.1212/01.wnl.0000303818.38960.44. [DOI] [PubMed] [Google Scholar]
- Belzunegui S, San Sebastian W, Garrido-Gil P, Izal-Azcarate A, Vazquez-Claverie M, Lopez B, Marcilla I, Lanciego JL, Luquin MR. The number of dopaminergic cells is increased in the olfactory bulb of monkeys chronically exposed to MPTP. Synapse. 2007;61:1006–1012. doi: 10.1002/syn.20451. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH, Lund S, Na HM, Taylor G, Bence NF, Kopito R, Seo BB, Yagi T, Yagi A, Klinefelter G, Cookson MR, Greenamyre JT. Intersecting pathways to neurodegeneration in Parkinson’s disease: Effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis. 2006;22:404–420. doi: 10.1016/j.nbd.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Blythe SN, Wokosin D, Atherton JF, Bevan MD. Cellular Mechanisms Underlying Burst Firing in Substantia Nigra Dopamine Neurons. J Neurosci. 2009;29:15531–15541. doi: 10.1523/JNEUROSCI.2961-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Grillner P, Mercuri NB, Bernardi G. L-Type calcium channels mediate a slow excitatory synaptic transmission in rat midbrain dopaminergic neurons. J Neurosci. 1998;18:6693–6703. doi: 10.1523/JNEUROSCI.18-17-06693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, Gasser T, Augustin R, Trumbach D, Irrcher I, Park DS, Wurst W, Kilberg MS, Tatzelt J, Winklhofer KF. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011;18:769–782. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- Calne DB, Langston JW. Aetiology of Parkinson’s disease. Lancet. 1983;2:1457–1459. doi: 10.1016/s0140-6736(83)90802-4. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Choi YM, Kim SH, Chung S, Uhm DY, Park MK. Regional interaction of endoplasmic reticulum Ca2+ signals between soma and dendrites through rapid luminal Ca2+ diffusion. J Neurosci. 2006;26:12127–12136. doi: 10.1523/JNEUROSCI.3158-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12:359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Lipton J, Daley BF, Palfi S, Chu Y, Sortwell C, Bakay RA, Sladek JR, Jr, Kordower JH. Aging-related changes in the nigrostriatal dopamine system and the response to MPTP in nonhuman primates: diminished compensatory mechanisms as a prelude to parkinsonism. Neurobiol Dis. 2007;26:56–65. doi: 10.1016/j.nbd.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Okamoto T, Morikawa H. Spontaneous opening of T-type Ca2+ channels contributes to the irregular firing of dopamine neurons in neonatal rats. J Neurosci. 2004;24:11079–11087. doi: 10.1523/JNEUROSCI.2713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D (28K) immunohistochemistry. Brain. 1999;122(Pt 8):1421–1436. doi: 10.1093/brain/122.8.1421. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Giesbergen PC, de Rijk MC, Hofman A, Koudstaal PJ, Breteler MM. Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology. 2004;63:1240–1244. doi: 10.1212/01.wnl.0000140706.52798.be. [DOI] [PubMed] [Google Scholar]
- de Rijk MC, Tzourio C, Breteler MM, Dartigues JF, Amaducci L, Lopez-Pousa S, Manubens-Bertran JM, Alperovitch A, Rocca WA. Prevalence of parkinsonism and Parkinson’s disease in Europe: the EUROPARKINSON Collaborative Study European Community Concerted Action on the Epidemiology of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;62:10–15. doi: 10.1136/jnnp.62.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deister CA, Teagarden MA, Wilson CJ, Paladini CA. An Intrinsic Neuronal Oscillator Underlies Dopaminergic Neuron Bursting. J Neurosci. 2009;29:15888–15897. doi: 10.1523/JNEUROSCI.4053-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Tredici K, Braak H. Idiopathic Parkinson’s disease: staging an alpha-synucleinopathy with a predictable pathoanatomy. In: Kahle PJ, Haas C, editors. Molecular Mechanisms of Parkinson’s Disease. Georgetown, TX.: Landes Bioscience; 2004. pp. 1–32. [Google Scholar]
- Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. The endoplasmic reticulum stress sensor, ATF6alpha, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem. 2011;286:7947–7957. doi: 10.1074/jbc.M110.156430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg MJ, Brox A, Bestawros AN. Calcium channel blockers: an update. Am J Med. 2004;116:35–43. doi: 10.1016/j.amjmed.2003.08.027. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. Does levodopa slow or hasten the rate of progression of Parkinson’s disease? J Neurol. 2005;252(Suppl 4):IV37–IV42. doi: 10.1007/s00415-005-4008-5. [DOI] [PubMed] [Google Scholar]
- Fitton A, Benfield P. Isradipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cardiovascular disease. Drugs. 1990;40:31–74. doi: 10.2165/00003495-199040010-00004. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye KF, Sonsalla PK, Brooks BA. Midbrain dopaminergic cell loss in Parkinson’s disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells. Ann N Y Acad Sci. 1992a;648:42–62. doi: 10.1111/j.1749-6632.1992.tb24523.x. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye KF, White CL, 3rd, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. 1992b;32:667–676. doi: 10.1002/ana.410320510. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1991;54:388–396. doi: 10.1136/jnnp.54.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleichmann M, Mattson MP. Neuronal calcium homeostasis and dysregulation. Antioxid Redox Signal. 2011;14:1261–1273. doi: 10.1089/ars.2010.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--2. Action potential generating mechanisms and morphological correlates. Neuroscience. 1983;10:317–331. doi: 10.1016/0306-4522(83)90136-7. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Hastings TG. Biomedicine Parkinson’s--divergent causes, convergent mechanisms. Science. 2004;304:1120–1122. doi: 10.1126/science.1098966. [DOI] [PubMed] [Google Scholar]
- Gu M, Gash MT, Cooper JM, Wenning GK, Daniel SE, Quinn NP, Marsden CD, Schapira AH. Mitochondrial respiratory chain function in multiple system atrophy. Mov Disord. 1997;12:418–422. doi: 10.1002/mds.870120323. [DOI] [PubMed] [Google Scholar]
- Guo M. What have we learned from Drosophila models of Parkinson’s disease? Prog Brain Res. 2010;184:3–16. doi: 10.1016/S0079-6123(10)84001-4. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends Cell Biol. 2009;19:81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- Hetz CA. ER stress signaling and the BCL-2 family of proteins: from adaptation to irreversible cellular damage. Antioxid Redox Signal. 2007;9:2345–2355. doi: 10.1089/ars.2007.1793. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:925–964. [PubMed] [Google Scholar]
- Huisman E, Uylings HB, Hoogland PV. Gender-related changes in increase of dopaminergic neurons in the olfactory bulb of Parkinson’s disease patients. Mov Disord. 2008;23:1407–1413. doi: 10.1002/mds.22009. [DOI] [PubMed] [Google Scholar]
- Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol Dis. 2011 doi: 10.1016/j.nbd.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Goto S, Sakamoto S, Hirano A. Calbindin-D28k in the basal ganglia of patients with parkinsonism. Ann Neurol. 1992;32:543–550. doi: 10.1002/ana.410320410. [DOI] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci. 2010;30:7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease Pathophysiologic and clinical implications. N Engl J Med. 1988;318:876–880. doi: 10.1056/NEJM198804073181402. [DOI] [PubMed] [Google Scholar]
- Klegeris A, McGeer EG, McGeer PL. Therapeutic approaches to inflammation in neurodegenerative disease. Curr Opin Neurol. 2007;20:351–357. doi: 10.1097/WCO.0b013e3280adc943. [DOI] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Kupsch A, Sautter J, Schwarz J, Riederer P, Gerlach M, Oertel WH. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res. 1996;741:185–196. doi: 10.1016/s0006-8993(96)00917-1. [DOI] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Magyar K, Szende B. (-)-Deprenyl, a selective MAO-B inhibitor, with apoptotic and anti-apoptotic properties. Neurotoxicology. 2004;25:233–242. doi: 10.1016/S0161-813X(03)00102-5. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, Schapira AH. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol. 1994;36:876–881. doi: 10.1002/ana.410360612. [DOI] [PubMed] [Google Scholar]
- Mannhold R, Rekker RF, Sonntag C, ter Laak AM, Dross K, Polymeropoulos EE. Comparative evaluation of the predictive power of calculation procedures for molecular lipophilicity. J Pharm Sci. 1995;84:1410–1419. doi: 10.1002/jps.2600841206. [DOI] [PubMed] [Google Scholar]
- Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, Kaddurah-Daouk R, Beal MF. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 1999;157:142–149. doi: 10.1006/exnr.1999.7049. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Saper CB. Preservation of hypothalamic dopaminergic neurons in Parkinson’s disease. Ann Neurol. 1985;18:552–555. doi: 10.1002/ana.410180507. [DOI] [PubMed] [Google Scholar]
- McCann MJ, Rogers RC. Oxytocin excites gastric-related neurones in rat dorsal vagal complex. The Journal of physiology. 1990;428:95–108. doi: 10.1113/jphysiol.1990.sp018202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Bonci A, Calabresi P, Stratta F, Stefani A, Bernardi G. Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br J Pharmacol. 1994;113:831–838. doi: 10.1111/j.1476-5381.1994.tb17068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogami H, Nakano K, Tepikin AV, Petersen OH. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane patch. Cell. 1997;88:49–55. doi: 10.1016/s0092-8674(00)81857-7. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005a;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Moore DJ, Zhang L, Troncoso J, Lee MK, Hattori N, Mizuno Y, Dawson TM, Dawson VL. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum Mol Genet. 2005b;14:71–84. doi: 10.1093/hmg/ddi007. [DOI] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard S, Flatman JA, Engberg I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J Physiol. 1993;466:727–747. [PMC free article] [PubMed] [Google Scholar]
- Nicholls D. Mitochondrial bioenergetics, aging, and aging-related disease. Sci Aging Knowledge Environ. 2002;2002:pe12. doi: 10.1126/sageke.2002.31.pe12. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Park MK, Choi YM, Kang YK, Petersen OH. The endoplasmic reticulum as an integrator of multiple dendritic events. Neuroscientist. 2008;14:68–77. doi: 10.1177/1073858407305691. [DOI] [PubMed] [Google Scholar]
- Park MK, Petersen OH, Tepikin AV. The endoplasmic reticulum as one continuous Ca(2+) pool: visualization of rapid Ca(2+) movements and equilibration. Embo J. 2000;19:5729–5739. doi: 10.1093/emboj/19.21.5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W, Mengesdorf T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium. 2005;38:409–415. doi: 10.1016/j.ceca.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Pignatelli A, Ackman JB, Vigetti D, Beltrami AP, Zucchini S, Belluzzi O. A potential reservoir of immature dopaminergic replacement neurons in the adult mammalian olfactory bulb. Pflugers Arch. 2009;457:899–915. doi: 10.1007/s00424-008-0535-0. [DOI] [PubMed] [Google Scholar]
- Pignatelli A, Kobayashi K, Okano H, Belluzzi O. Functional properties of dopaminergic neurones in the mouse olfactory bulb. J Physiol. 2005;564:501–514. doi: 10.1113/jphysiol.2005.084632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping HX, Shepard PD. Apamin-sensitive Ca(2+)-activated K+ channels regulate pacemaker activity in nigral dopamine neurons. Neuroreport. 1996;7:809–814. doi: 10.1097/00001756-199602290-00031. [DOI] [PubMed] [Google Scholar]
- Postuma RB, Lang AE, Massicotte-Marquez J, Montplaisir J. Potential early markers of Parkinson disease in idiopathic REM sleep behavior disorder. Neurology. 2006;66:845–851. doi: 10.1212/01.wnl.0000203648.80727.5b. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C, Vila M. MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J Bioenerg Biomembr. 2004;36:375–379. doi: 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–656. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer P, Wuketich S. Time course of nigrostriatal degeneration in parkinson’s disease A detailed study of influential factors in human brain amine analysis. J Neural Transm. 1976;38:277–301. doi: 10.1007/BF01249445. [DOI] [PubMed] [Google Scholar]
- Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol. 2010;67:600–606. doi: 10.1002/ana.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- Romo R, Schultz W. Dopamine neurons of the monkey midbrain: contingencies of responses to active touch during self-initiated arm movements. J Neurophysiol. 1990;63:592–606. doi: 10.1152/jn.1990.63.3.592. [DOI] [PubMed] [Google Scholar]
- Rose CR, Konnerth A. Stores not just for storage intracellular calcium release and synaptic plasticity. Neuron. 2001;31:519–522. doi: 10.1016/s0896-6273(01)00402-0. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci. 2002;22:10690–10698. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Sorrentino DM, German DC, de Lacalle S. Medullary catecholaminergic neurons in the normal human brain and in Parkinson’s disease. Ann Neurol. 1991;29:577–584. doi: 10.1002/ana.410290602. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008a;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Progress in neuroprotection in Parkinson’s disease. Eur J Neurol. 2008b;151(Suppl):5–13. doi: 10.1111/j.1468-1331.2008.02055.x. [DOI] [PubMed] [Google Scholar]
- Scholze A, Plant TD, Dolphin AC, Nurnberg B. Functional expression and characterization of a voltage-gated CaV1.3 (alpha1D) calcium channel subunit from an insulin-secreting cell line. Mol Endocrinol. 2001;15:1211–1221. doi: 10.1210/mend.15.7.0666. [DOI] [PubMed] [Google Scholar]
- Schwyn RC, Fox CA. The primate substantia nigra: a Golgi and electron microscopic study. J Hirnforsch. 1974;15:95–126. [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shults CW, Haas RH, Passov D, Beal MF. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from parkinsonian and nonparkinsonian subjects. Ann Neurol. 1997;42:261–264. doi: 10.1002/ana.410420221. [DOI] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol. 2009;75:407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- Smith RA, Murphy MP. Mitochondria-targeted antioxidants as therapies. Discov Med. 2011;11:106–114. [PubMed] [Google Scholar]
- Soong NW, Hinton DR, Cortopassi G, Arnheim N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat Genet. 1992;2:318–323. doi: 10.1038/ng1292-318. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Haas HL. Calcium-dependent prepotentials contribute to spontaneous activity in rat tuberomammillary neurons. J Physiol. 1996;493(Pt 3):747–754. doi: 10.1113/jphysiol.1996.sp021419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, Pelster G, Singewald N. Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans. 2006;34:903–909. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Toescu EC. Normal brain ageing: models and mechanisms. Philos Trans R Soc Lond B Biol Sci. 2005;360:2347–2354. doi: 10.1098/rstb.2005.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA, Rossiter CD, Vicini S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. The American journal of physiology. 1991;260:G531–536. doi: 10.1152/ajpgi.1991.260.3.G531. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- Vila M, Ramonet D, Perier C. Mitochondrial alterations in Parkinson’s disease: new clues. J Neurochem. 2008;107:317–328. doi: 10.1111/j.1471-4159.2008.05604.x. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HQ, Takahashi R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson’s disease. Antioxid Redox Signal. 2007;9:553–561. doi: 10.1089/ars.2006.1524. [DOI] [PubMed] [Google Scholar]
- Williams JT, North RA, Shefner SA, Nishi S, Egan TM. Membrane properties of rat locus coeruleus neurones. Neuroscience. 1984;13:137–156. doi: 10.1016/0306-4522(84)90265-3. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Callaway JC. Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J Neurophysiol. 2000;83:3084–3100. doi: 10.1152/jn.2000.83.5.3084. [DOI] [PubMed] [Google Scholar]
- Yamada T, McGeer PL, Baimbridge KG, McGeer EG. Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 1990;526:303–307. doi: 10.1016/0006-8993(90)91236-a. [DOI] [PubMed] [Google Scholar]
- Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154:1423–1429. doi: 10.1016/S0002-9440(10)65396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]