

Abstract
Purpose
To report, for the first time, that X-linked incomplete congenital stationary night blindness (CSNB2A) and Åland island eye disease (AIED) phenotypes coexist in a molecularly confirmed pedigree and to present novel phenotypic characteristics of calcium channel alpha-1F subunit gene (CACNA1F)-related disease.
Methods
Two affected subjects (the proband and his maternal grandfather) and an unaffected obligate carrier (the proband’s mother) underwent detailed ophthalmological evaluation, fundus autofluorescence imaging, and spectral-domain optical coherence tomography. Goldmann visual field assessment and full-field electroretinogram (ERG) were performed in the two affected subjects, and multichannel flash visual evoked potential was performed on the proband. Scotopic 15 Hz flicker ERG series were performed in both affected subjects to evaluate the function of the slow and fast rod pathways. Haplotype analysis using polymorphic microsatellite markers flanking CACNA1F was performed in all three family members. The proband’s DNA was sequenced for mutations in the coding sequence of CACNA1F and nyctalopin (NYX) genes. Segregation analysis was performed in the family.
Results
Both affected subjects had symptoms of nonprogressive nyctalopia since childhood, while the proband also had photophobia. Both cases had a distance visual acuity of 20/50 or better in each eye, normal contrast sensitivity, and an incomplete type of Schubert-Bornschein ERGs. The proband also had high myopia, a mild red-green color deficit, hypopigmented fundus, and foveal hypoplasia with no evidence of chiasmal misrouting. Spectral-domain optical coherence tomography confirmed the presence of foveal hypoplasia in the proband. The clinical phenotype of the proband and his maternal grandfather fit the clinical description of AIED and CSNB2A, respectively. The fundus autofluorescence and the visual fields were normal in both cases; the scotopic 15 Hz flicker ERG demonstrated only fast rod pathway activity in both. Both affected cases shared the same haplotype across CACNA1F. The proband carried a novel hemizygous c.1807G>C mutation (p.G603R) in the CACNA1F gene. The change segregated with the disease phenotypes and was not identified in 360 control chromosomes. No mutations were identified in NYX.
Conclusions
This report of a missense mutation in CACNA1F causing AIED and CSNB2A phenotypes in a family confirms that both diseases are allelic and that other genetic or environmental modifiers influence the expression of CACNA1F. This is the first report to suggest that in CACNA1F-related disease, the rod system activity is predominantly from the fast rod pathways.
Introduction
Congenital stationary night blindness (CSNB) is a clinically and genetically heterogenous disorder and may follow autosomal dominant, autosomal recessive, or X-linked recessive patterns of inheritance. The clinical features may vary between subtypes, but affected individuals often complain of nyctalopia since childhood and commonly present with nystagmus, reduced visual acuity, strabismus, and myopic refractive error [1]. Although commonly referred to as a stationary disease with a normal-looking retina, except for myopic changes, there are reports of progressive vision loss [2–4], optic atrophy [2], and retinal pigment epithelial abnormalities [2,4] in CSNB. Electroretinography (ERG) aids in diagnosing and classifying CSNB. Broadly, two distinct electrophysiological patterns are recognized; the Riggs type of ERG [5] is observed when the deficit involves phototransduction, and the Schubert-Bornschein type of ERG [6] is observed when the deficit is post-phototransduction. The cardinal feature of Schubert-Bornschein ERG is an electronegative response to standard flash (normal a-wave, reduced b-wave and b:a ratio <1.0) under scotopic conditions. Miyake et al. [1] further classified patients with the Schubert-Bornschein type of ERG into complete and incomplete phenotypes based on the absence or presence of rod function, respectively, to dim white light stimulation and noted that involvement of the cone system also differed between the groups. Mutations in the α1 subunit of the L-type voltage gated Ca2+ channel gene (CACNA1F; OMIM *300110) is associated with X-linked incomplete CSNB (CSNB2A; OMIM #300071) [7,8]. Expressed predominantly in the outer and inner nuclear layers of the retina [7], these channels support Ca2+ influx under relatively depolarized conditions, which is necessary for tonic glutamate release from rod and cone photoreceptors [9,10]. Mouse and rat models of Cacna1f also confirm the deficit to be in the transmission of the signal from photoreceptors to second-order neurons [7,11].
Åland island eye disease (AIED; OMIM #300600) or Forsius-Eriksson syndrome is an X-linked disorder first described in Norwegian descendants on the Åland Islands in the Sea of Bothnia [12]. Affected males have nystagmus, myopia, reduced visual acuity, red-green color vision deficits, iris trans-illumination defects, foveal hypoplasia, and a blonde fundus without evidence of chiasmal misrouting [12,13]. Although an incomplete type of Schubert-Bornschein ERG was not observed originally in AIED, this has subsequently been described [14]. Two reports of AIED pedigrees with mutations identified in CACNA1F have been published [15,16]; hence, CSNB2A and AIED are considered to be allelic variants.
The present study describes, for the first time, two affected members of the same pedigree with distinct features of AIED in one and of incomplete CSNB (CSNB2A) in the other, both associated with a novel p.G603R missense mutation in the CACNA1F gene. This study also presents novel electrophysiology data describing the slow and fast rod pathway activity in CACNA1F- related disease.
Methods
Clinical evaluation
The study protocol was approved by the Research Ethics Board at The Hospital for Sick Children and adhered to the Tenets of the Declaration of Helsinki. Three members (including two affected males) of a Caucasian family were evaluated (Figure 1A).
Figure 1.

The family pedigree, chromatogram, and amino-acid conservation map are shown. A: Pedigree of the family studied demonstrating the X-linked inheritance pattern. The haplotypes for the polymorphic DNA markers flanking CACNA1F (DXS1003 and DXS7132; proximal and distal, respectively) in the three family members are also included. B: Chromatogram of the portion of CACNA1F exon 14 showing the c.1807G>C hemizygous change in cases II 1 and IV 1. Case III 1 is a female carrier and had no manifestations. C: Amino acid conservation map across species demonstrating that glycine at position 603 is conserved lower down to sticklebacks and zebrafish.
All three subjects underwent ophthalmologic evaluation that included best corrected visual acuity (BCVA) and color vision assessment (Hardy-Rand-Rittler plates), contrast sensitivity measurement (Pelli-Robson charts), slit-lamp evaluation, dilated retinal examination, fundus autofluorescence imaging (Visucam; Carl Zeiss Meditec, Dublin, CA), and spectral domain optical coherence tomography (SD-OCT, Cirrus; Carl Zeiss Meditec). Goldmann visual field analysis was performed on the two affected subjects.
Electrophysiology
Full-field ERG incorporating the International Society for Clinical Electrophysiology of Vision (ISCEV) Standards [17] was performed on the two affected individuals. The four ERG responses evaluated include dim light scotopic response [flash intensity, 0.01 cd-seconds per square meter (cd.s.m−2)], combined rod-cone response to standard flash (2.29 cd.s.m−2), single flash photopic response (2.29 cd.s.m−2), and 30 Hz flicker response (2.29 cd.s.m−2). Multichannel flash visual evoked potential with active electrodes placed at scalp positions Oz, O1, O2, PO7, and PO8 was performed on the proband (case IV 1) [18,19]. Additionally, a series of 15 Hz flicker ERGs was performed under scotopic conditions to study the slow (rod → rod On bipolar cell → AII amacrine) and fast (rod → rod-cone gap junction → cone On bipolar cell) rod pathways [20]. Serial 15 Hz stimuli incrementing in intensity from −3.40 log scotopic troland-second (scot td-sec) to +0.35 log scot td-sec (steps of 0.1–0.3 log units) were presented using a Ganzfield bowl (Espion, Diagnosys LLC, Boston, MA) using equivalent techniques as reported previously [21]. Stimuli were created by combining colored light-emitting diodes to generate white flashes of 4 ms duration. At lower light intensities (<–1.0 log scot td-sec), 100 trials in length (each trial lasted 200 ms and was composed of 3 flashes) were recorded and averaged; this was reduced to 30 trials at higher flash intensities to minimize patient discomfort. In adults with normal retinal function, slow rod pathway activity predominates at lower intensities (<–1.97 log scot td-sec), and fast rod pathway activity predominates at higher intensities (>–1.37 log scot td-sec) [21,22]; however, inter-laboratory variations have been reported [23,24].
Genetic analysis
Haplotype analysis was performed on all three members of the family using polymorphic microsatellite DNA markers flanking CACNA1F (DXS1003 and DXS7132; PCR conditions for both: 1.5 mM MgCl2; annealing temperature: 57 °C; cycles: 35). All exons and exon/intron boundaries of CACNA1F and nyctalopin (NYX) genes were sequenced in the proband (case IV 1) at the University of Colorado Denver DNA diagnostic laboratory. The CACNA1F exon 14 harboring the variant of interest in all three subjects was amplified and sequenced at the Héon laboratory (PCR conditions: 1.5 mM MgCl2, 1× Q Solution (Qiagen GmbH, Hilden, Germany); annealing temperature: 62.3 °C; cycles: 36; forward primer: 5′-GCC TGA ATA CCG AGC ACA TT-3′; reverse primer: 5′-TGT TGA GGC TGT TTG AGG-3′). The allele frequency of the novel change (c.1807G>C) was evaluated in 360 ethnically matched control chromosomes with amplification-refractory mutation system assay designed for the mutant C allele (PCR conditions: 1.0 mM MgCl2, 1× Q solution; annealing temperature: 60.7 °C; cycles: 38; forward primer: 5′-TGA CTG CTT TGT GGT CAG TC-3′; reverse primer: same as above).
Results
The inheritance pattern of the phenotype was determined to be X-linked (Figure 1A). The detailed phenotypic and genotypic characteristics are described below.
Phenotypic characteristics
Proband (case IV 1)
A six-year-old male child presented with a history suggestive of nyctalopia and photophobia from childhood. Parents have not noted any nystagmus. The symptoms have remained stationary since birth. The family history is significant for similar symptoms in the maternal grandfather’s generation (Figure 1A). The child was born blond but now tans easily. On examination, he was orthophoric and had minimal nystagmus in the right eye. He had high myopic refraction, and his BCVA was 20/50 and 20/40 in the right and left eyes, respectively. Color vision testing showed a mild deficit in the red-green axis. His log contrast sensitivity was 1.65 in each eye. The iris did not show any trans-illumination defects. The fundus was markedly hypopigmented and showed foveal hypoplasia (Figure 2A,B). The SD-OCT demonstrated a shallow foveal pit, the presence of inner plexiform and inner nuclear layers at the fovea, and a widened outer nuclear layer consistent with Grade 1 foveal hypoplasia (Figure 2C). The autofluorescence was normal in the posterior pole (Figure 2D).The Goldman visual fields showed normal boundaries to I4e and III4e stimulus targets.
Figure 2.
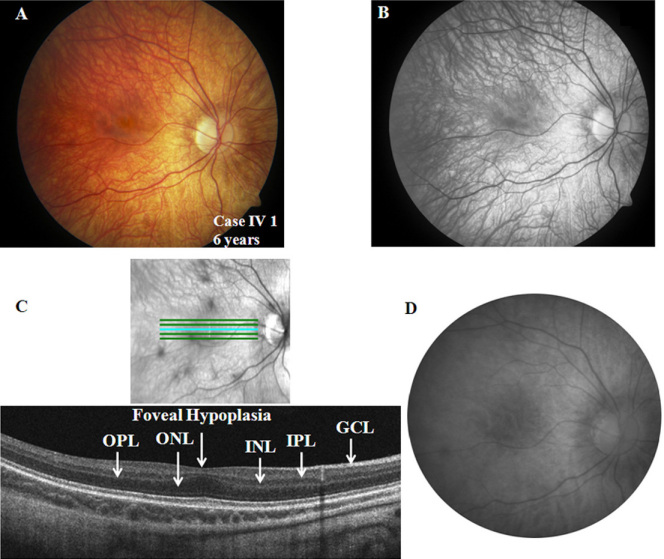
Ocular phenotypic characteristics of the proband (case IV 1; only right eye details shown). A, B: Fundus photograph and red free image showing hypopigmented fundus and foveal hypoplasia, respectively. C: Spectral domain optical coherence tomography demonstrating widening of the outer nuclear layer (ONL) at the fovea. The layers inner to ONL, including the outer plexiform layer (OPL), the inner nuclear layer (INL), and the inner plexiform layer (IPL), are all present at the fovea consistent with foveal hypoplasia. The ganglion cell layer (GCL) is also labeled. D: Fundus autofluorescence image of the posterior pole revealed normal autofluorescence.
Full-field ERG showed severely reduced dim light scotopic response, electronegative configuration of combined rod-cone response, severely reduced and delayed single flash photopic response, and markedly reduced 30 Hz flicker response (Figure 3A; compare with Figure 3C traces of a control subject). The scotopic 15 Hz flicker showed no discernable rod activity between intensities of −3.40 log scot td-sec and −0.46 log scot td-sec (Figure 4A). The rod activity was demonstrable first at an intensity of +0.19 log scot td-sec and was noted at higher intensities, suggesting residual activity in the fast rod pathways (Figure 4A; compare with Figure 4C traces of a control subject). Multichannel visual evoked potential showed no inter-hemispheric difference to monocular stimulation (Pearson’s correlate=0.8), thus ruling out the presence of chiasmal misrouting. The phenotypic characteristics of the proband were consistent with AIED with an incomplete Schubert-Bornschein type of ERG.
Figure 3.

Full-field electroretinogram (ERG) characteristics in case IV 1 (A), case II 1 (B), and a control subject (C) are presented. Dim light scotopic response (dark adapted [DA] ERG to 0.01 cd-seconds per square meter–DA 0.01 ERG) showed severe b-wave reduction in case IV 1 (3A) and was at the lower limit of normal in case II 1 (B). Both cases showed electronegative configuration to combined rod-cone response (DA ERG to 2.29 cd-seconds per square meter – DA 2.29 ERG; A and B), and case IV 1 showed additional a-wave involvement. Single flash photopic response (light adapted (LA) ERG to 2.29 cd-seconds per square meter–LA 2.29 ERG) showed severe reduction and delay of the a- and b-waves with reduced b/a ratio in both subjects (A and B). The 30 Hz flicker response (LA 30 Hz flicker) showed severely reduced and delayed b-waves in both cases (A and B). Please note that the scale on the y-axis is different for the cases and the control.
Figure 4.

Scotopic 15 Hz flicker electroretinogram characteristics in the proband (A), case II 1 (B), and a control subject (C) are presented. Various intensities between −3.4 log scot td-sec and +0.35 log scot td-sec were performed. Dashed lines show the response filtered using a simple Fourier domain, bandpass filter (13–18 Hz). In the control panel (C), the rod system activity observed between −3.40 log scot td-sec and −2.0 log scot td-sec intensities originates from the slow rod pathway. At an intensity of −1.52 log scot td-sec, there is no discernable rod activity denoting neutralization of activity between the slow and fast rod pathways (C). At intensities of −1.30 log scot td-sec, the rod system activity reappears (which is out of phase to the response seen at −2.0 log scot td-sec and originates in the fast rod pathways), which progressively increases in amplitude and shows progressive advancement in phase (C). In case IV 1 and case II 1, the rod activity is discernable only at intensities higher than +0.19 log scot td-sec and −0.62 log scot td-sec, respectively (figures A and B, respectively). This suggests that only residual fast rod pathway activity is seen in CACNA1F-related disease.
Case II 1
The 56-year-old grandfather of the proband had complaints of non-progressive nyctalopia since childhood but had no symptoms of photophobia. On examination, he was orthophoric with a BCVA of 20/50 in each eye, and he had no significant refractive error. His color vision and contrast sensitivity (1.65 log units) were normal in each eye. The anterior segment evaluation was normal. The fundus showed normal pigmentation; however, the foveal reflex was dull (Figure 5A). The fundus autofluorescence was normal (Figure 5B), and Goldmann visual field testing showed normal boundaries to I4e and III4e stimulus targets. The SD-OCT showed a normal foveal pit and central retinal thickness (Figure 5C).
Figure 5.

Ocular phenotypic characteristics of case II 1 (only left eye details are shown) A: Fundus photograph showing normal pigmentation and dull foveal reflex. B: Fundus autofluorescence image of the posterior pole revealed normal autofluorescence. C: Spectral domain optical coherence tomography of the left eye revealed normal foveal contour and central retinal thickness. All relevant outer and inner retinal layers have been labeled: the outer nuclear layer (ONL), the outer plexiform layer (OPL), the inner nuclear layer (INL), the inner plexiform layer (IPL), and the ganglion cell layer (GCL).
Full-field ERG showed borderline normal dim light scotopic response, electronegative configuration of combined rod-cone response, severely reduced and delayed single flash photopic response, and markedly reduced 30 Hz flicker response (Figure 3B; compare with Figure 3C traces of a control subject). The scotopic 15 Hz flicker showed no discernable rod activity between intensities of −3.40 log scot td-sec and −0.72 log scot td-sec (Figure 4B). Rod activity was first noted at −0.62 log scot td-sec and was subsequently present at all higher flash intensities, suggesting residual activity in the fast rod pathways (Figure 4B; compare with Figure 4C traces of a control subject). The phenotypic characteristics of the subject were consistent with incomplete CSNB (CSNB2A).
Case III 1
The 36-year-old mother of the proband was asymptomatic. On examination, she had exophoria, and her BCVA was 20/20 in each eye. Her color vision and contrast sensitivity were normal. Fundus evaluation, autofluorescence, and SD-OCT were unremarkable.
Genetic results
Haplotype analysis of the region adjoining CACNA1F supported the role of the gene in this family (Figure 1A). A novel hemizygous missense mutation c.1807G>C (NM_005183.2; p.G603R) was identified in the proband and the change segregated with the disease phenotype (Figure 1B). The mutation is located on the S3 helical trans-membrane domain repeat II of the CACNA1F gene (CACNA1F encodes for the α1 subunit of a L-type Ca2+ channel that contains four repeat domains, each of which has six transmembrane domains (S1–S6); NP_005174.2). The novel c.1807G>C change was not seen in 360 control chromosomes, and the glycine at position 603 is conserved in lower vertebrates such as stickleback and zebrafish (UCSC Conservation track; Figure 1C). The p.G603R was predicted to be possibly damaging by Polyphen v. 2.09 with a PSIC score difference of 1.525. The mutation was predicted also to affect protein function by SIFT with a score of 0.00. No mutations were identified in NYX.
Discussion
This is the first report in a molecularly confirmed CACNA1F pedigree with two affected members showing distinct classical phenotypes of AIED and CSNB2A, respectively. Since the association of CSNB2A with CACNA1F [7,8], few instances of deletions, either in CACNA1F (two pedigrees) or in the regions of Xp21 (one case), were reported with the classical AIED phenotype [15,16,25], suggesting AIED and CSNB2A are allelic with specific mutations causing each particular phenotype. The novel p.G603R mutation identified in this family confirms AIED and CSNB2A are allelic. Furthermore, the phenotypic variability observed in this family suggests the presence of other modifiers (probably genetic) yet to be identified in CACNA1F- related disease. This finding implies that the phenotypic variability observed between CSNB2A and AIED is not a consequence of intragenic allelic heterogeneity alone. Before the CACNA1F gene was discovered, a pedigree of X-linked incomplete CSNB was reported with some cases showing hypopigmented fundus (with no foveal hypoplasia), supporting this concept [26]. Further, Boycott et al. [27] showed the absence of one of the three major features of CSNB (nyctalopia, myopia, and nystagmus) in approximately three quarters of a large cohort of patients carrying the same CACNA1F mutation, which also suggests the presence of modifying factors.
Although AIED and CSNB2A have many overlapping clinical features, the former has additional features such as progressive myopia, dyschromatopsia, iris trans-illumination defects, and foveal hypoplasia [1,12]. The AIED phenotype in this study demonstrated all the features except for iris trans-illumination deficits. Animal models demonstrate abnormal ribbon synapses in photoreceptors and abnormal synaptogenesis in that a portion of second-order neurons and horizontal cells synapse in the outer nuclear layer or retinal pigment epithelium rather than in the outer plexiform layer [9–11,28–30]. This may affect centrifugal migration of the inner retinal layers at the fovea, which happens in human eyes during development [from 24 weeks gestation to 15 months postpartum [31]] and may result in foveal hypoplasia seen in the AIED phenotype. The SD-OCT demonstrated a shallow foveal pit, persistence of all the inner retinal layers, and widening of the outer nuclear layer in the AIED phenotype, consistent with Grade 1 foveal hypoplasia [32]. The autofluorescence in the posterior pole was normal in the two affected cases, suggesting a normal lipofuscin level at the retinal pigment epithelium in CACNA1F-related disease. There was no evidence of visual field constriction in either affected individual as observed previously [33]. Rare progressive phenotypes with central field scotomas also have been reported [2].
The AIED and CSNB2A phenotypes in this study revealed an incomplete Schubert-Bornschein type of ERG, as previously reported [2,14]. Interestingly, the individual with the CSNB2A phenotype, who was much older (the grandfather of the proband), showed milder scotopic ERG abnormalities. Both individuals showed severe and identical photopic ERG abnormalities. It is tempting to relate the severe abnormality in ERG directly to the AIED phenotype, but the proband also had high myopia, which is known to affect ERG amplitude [34,35], thus precluding an accurate conclusion. This is the first report to assess scotopic 15 Hz flicker ERGs in CACNA1F phenotypes. There was no rod activity from the slow rod pathways in either affected individual. The rod activity appeared at intensities of +0.19 and −0.62 log scot td-sec in the AIED and CSNB2A phenotypes, respectively. This reflects residual activity originating in the fast rod pathways in CACNA1F-related disease. The appearance of fast rod activity at a higher intensity in the AIED phenotype is probably a consequence of a more severe scotopic ERG abnormality observed in this subject (see above). Further, cone activity under scotopic conditions is known not to appear until intensities of +0.75 log scot td-sec [36], thus ruling out a cone contribution to the observed scotopic 15 Hz flicker responses in this study. Previously, two genes associated with complete CSNB (NYX and transient receptor potential cation channel, subfamily M, member 1 [TRPM1]) [21,24] have been associated with identical scotopic 15 Hz ERG findings as those now found in CACNA1F-related disease. On the contrary, calcium binding protein 4 (CABP4) related autosomal recessive incomplete CSNB shows preservation of slow rod pathways and severely attenuated or absent fast rod pathways [23]. Hence, the scotopic 15 Hz flicker ERG could be useful in differentiating between CACNA1F- and CABP4-related incomplete CSNB if the inheritance pattern is unknown.
To conclude, this pedigree with a novel p.G603R mutation in CACNA1F confirms that CSNB2A and AIED are indeed allelic variants, but also illustrates that the two are not mutation specific. An incomplete Schubert-Bornschein type of ERG was seen in both phenotypes, and the residual rod activity demonstrable in CACNA1F-related disease arises predominantly from the fast rod pathways. The underlying modifying factor responsible for the phenotypic variability remains to be elucidated.
Acknowledgments
We thank the family members for active participation in the study. We thank Cynthia VandenHoven (Ophthalmic Photographer), Joanne Sutherland (Genetic Counselor), Catherine Deveault (Research Coordinator) and Melissa Cotesta (Orthoptist) for appropriate assistance. We thank Carole Panton (Electrophysiology Department) for editorial assistance.
References
- 1.Miyake Y, Yagasaki K, Horiguchi M, Kawase Y, Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch Ophthalmol. 1986;104:1013–20. doi: 10.1001/archopht.1986.01050190071042. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura M, Ito S, Piao CH, Terasaki H, Miyake Y. Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family. Arch Ophthalmol. 2003;121:1028–33. doi: 10.1001/archopht.121.7.1028. [DOI] [PubMed] [Google Scholar]
- 3.Zeitz C, Kloeckener-Gruissem B, Forster U, Kohl S, Magyar I, Wissinger B, Mátyás G, Borruat FX, Schorderet DF, Zrenner E, Munier FL, Berger W. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet. 2006;79:657–67. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wycisk KA, Zeitz C, Feil S, Wittmer M, Forster U, Neidhardt J, Wissinger B, Zrenner E, Wilke R, Kohl S, Berger W. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet. 2006;79:973–7. doi: 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riggs LA. Electroretinography in cases of night blindness. Am J Ophthalmol. 1954;38:70–8. doi: 10.1016/0002-9394(54)90011-2. [DOI] [PubMed] [Google Scholar]
- 6.Schubert G, Bornschein H. Analysis of the human electroretinogram. Ophthalmologica. 1952;123:396–413. doi: 10.1159/000301211. [DOI] [PubMed] [Google Scholar]
- 7.Strom TM, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber BH, Wutz K, Gutwillinger N, Rüther K, Drescher B, Sauer C, Zrenner E, Meitinger T, Rosenthal A, Meindl A. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:260–3. doi: 10.1038/940. [DOI] [PubMed] [Google Scholar]
- 8.Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:264–7. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 9.Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE, Bech-Hansen NT. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet. 2005;14:3035–46. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- 10.Chang B, Heckenlively JR, Bayley PR, Brecha NC, Davisson MT, Hawes NL, Hirano AA, Hurd RE, Ikeda A, Johnson BA, McCall MA, Morgans CW, Nusinowitz S, Peachey NS, Rice DS, Vessey KA, Gregg RG. The nob2 mouse, a null mutation in Cacna1f: anatomical and functional abnormalities in the outer retina and their consequences on ganglion cell visual responses. Vis Neurosci. 2006;23:11–24. doi: 10.1017/S095252380623102X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu Y, Wang L, Zhou J, Guo Q, Liu N, Ding Z, Li L, Liu X, An J, Yan G, Yao L, Zhang Z. A naturally-occurring mutation in Cacna1f in a rat model of congenital stationary night blindness. Mol Vis. 2008;14:20–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Forsius H, Eriksson AW. A new eye syndrome with x–chromosomal transmission. a family clan with fundus albinism, fovea hypoplasia, nystagmus, myopia, astigmatism and dyschromatopsia. Klin Monatsbl Augenheilkd. 1964;144:447–57. [PubMed] [Google Scholar]
- 13.van Dorp DB, Eriksson AW, Delleman JW, van Vliet AG, Collewijn H, van Balen AT, Forsius HR. Aland eye disease: no albino misrouting. Clin Genet. 1985;28:526–31. doi: 10.1111/j.1399-0004.1985.tb00421.x. [DOI] [PubMed] [Google Scholar]
- 14.François J, International Society for Clinical Electroretinography. The Clinical value of electroretinography. Symposium in Ghent, August 1–4, 1966, under the sponsorship of the ISCERG. Edited by J. François. Basel: Karger; 1968. [Google Scholar]
- 15.Wutz K, Sauer C, Zrenner E, Lorenz B, Alitalo T, Broghammer M, Hergersberg M, de la Chapelle A, Weber BH, Wissinger B, Meindl A, Pusch CM. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur J Hum Genet. 2002;10:449–56. doi: 10.1038/sj.ejhg.5200828. [DOI] [PubMed] [Google Scholar]
- 16.Jalkanen R, Bech-Hansen NT, Tobias R, Sankila EM, Mäntyjärvi M, Forsius H, de la Chapelle A, Alitalo T. A novel CACNA1F gene mutation causes Aland Island eye disease. Invest Ophthalmol Vis Sci. 2007;48:2498–502. doi: 10.1167/iovs.06-1103. [DOI] [PubMed] [Google Scholar]
- 17.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 18.American Clinical Neurophysiology Society Guideline 5: Guidelines for standard electrode position nomenclature. J Clin Neurophysiol. 2006;23:107–10. doi: 10.1097/00004691-200604000-00006. [DOI] [PubMed] [Google Scholar]
- 19.American Clinical Neurophysiology Society Guideline 9B: Guidelines on visual evoked potentials. J Clin Neurophysiol. 2006;23:138–56. doi: 10.1097/00004691-200604000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Sharpe LT, Fach CC, Stockman A. The spectral properties of the two rod pathways. Vision Res. 1993;33:2705–20. doi: 10.1016/0042-6989(93)90230-t. [DOI] [PubMed] [Google Scholar]
- 21.Scholl HP, Langrova H, Pusch CM, Wissinger B, Zrenner E, Apfelstedt-Sylla E. Slow and fast rod ERG pathways in patients with X-linked complete stationary night blindness carrying mutations in the NYX gene. Invest Ophthalmol Vis Sci. 2001;42:2728–36. [PubMed] [Google Scholar]
- 22.Scholl HP, Langrova H, Weber BH, Zrenner E, Apfelstedt-Sylla E. Clinical electrophysiology of two rod pathways: normative values and clinical application. Graefes Arch Clin Exp Ophthalmol. 2001;239:71–80. doi: 10.1007/s004170000232. [DOI] [PubMed] [Google Scholar]
- 23.Littink KW, van Genderen MM, Collin RW, Roosing S, de Brouwer AP, Riemslag FC, Venselaar H, Thiadens AA, Hoyng CB, Rohrschneider K, den Hollander AI, Cremers FP, van den Born LI. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest Ophthalmol Vis Sci. 2009;50:2344–50. doi: 10.1167/iovs.08-2553. [DOI] [PubMed] [Google Scholar]
- 24.van Genderen MM, Bijveld MM, Claassen YB, Florijn RJ, Pearring JN, Meire FM, McCall MA, Riemslag FC, Gregg RG, Bergen AA, Kamermans M. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009;85:730–6. doi: 10.1016/j.ajhg.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weleber RG, Pillers DA, Powell BR, Hanna CE, Magenis RE, Buist NR. Aland Island eye disease (Forsius-Eriksson syndrome) associated with contiguous deletion syndrome at Xp21. Similarity to incomplete congenital stationary night blindness. Arch Ophthalmol. 1989;107:1170–9. doi: 10.1001/archopht.1989.01070020236032. [DOI] [PubMed] [Google Scholar]
- 26.Hawksworth NR, Headland S, Good P, Thomas NS, Clarke A. Aland island eye disease: clinical and electrophysiological studies of a Welsh family. Br J Ophthalmol. 1995;79:424–30. doi: 10.1136/bjo.79.5.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boycott KM, Pearce WG, Bech-Hansen NT. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can J Ophthalmol. 2000;35:204–13. doi: 10.1016/s0008-4182(00)80031-9. [DOI] [PubMed] [Google Scholar]
- 28.Lodha N, Bonfield S, Orton NC, Doering CJ, McRory JE, Mema SC, Rehak R, Sauvé Y, Tobias R, Stell WK, Bech-Hansen NT. Congenital stationary night blindness in mice - a tale of two cacna1f mutants. Adv Exp Med Biol. 2010;664:549–58. doi: 10.1007/978-1-4419-1399-9_63. [DOI] [PubMed] [Google Scholar]
- 29.Specht D, Wu SB, Turner P, Dearden P, Koentgen F, Wolfrum U, Maw M, Brandstätter JH, tom Dieck S. Effects of presynaptic mutations on a postsynaptic Cacna1s calcium channel colocalized with mGluR6 at mouse photoreceptor ribbon synapses. Invest Ophthalmol Vis Sci. 2009;50:505–15. doi: 10.1167/iovs.08-2758. [DOI] [PubMed] [Google Scholar]
- 30.Raven MA, Orton NC, Nassar H, Williams GA, Stell WK, Jacobs GH, Bech-Hansen NT, Reese BE. Early afferent signaling in the outer plexiform layer regulates development of horizontal cell morphology. J Comp Neurol. 2008;506:745–58. doi: 10.1002/cne.21526. [DOI] [PubMed] [Google Scholar]
- 31.Yuodelis C, Hendrickson A. A qualitative and quantitative analysis of the human fovea during development. Vision Res. 1986;26:847–55. doi: 10.1016/0042-6989(86)90143-4. [DOI] [PubMed] [Google Scholar]
- 32.Thomas MG, Kumar A, Mohammad S, Proudlock FA, Engle EC, Andrews C, Chan WM, Thomas S, Gottlob I. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography a predictor of visual acuity? Ophthalmology. 2011;118:1653–60. doi: 10.1016/j.ophtha.2011.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allen LE, Zito I, Bradshaw K, Patel RJ, Bird AC, Fitzke F, Yates JR, Trump D, Hardcastle AJ, Moore AT. Genotype-phenotype correlation in British families with X linked congenital stationary night blindness. Br J Ophthalmol. 2003;87:1413–20. doi: 10.1136/bjo.87.11.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westall CA, Dhaliwal HS, Panton CM, Sigesmun D, Levin AV, Nischal KK, Héon E. Values of electroretinogram responses according to axial length. Doc Ophthalmol. 2001;102:115–30. doi: 10.1023/a:1017535207481. [DOI] [PubMed] [Google Scholar]
- 35.Flitcroft DI, Adams GG, Robson AG, Holder GE. Retinal dysfunction and refractive errors: an electrophysiological study of children. Br J Ophthalmol. 2005;89:484–8. doi: 10.1136/bjo.2004.045328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Birch DG, Fish GE. Rod ERGs in retinitis pigmentosa and cone-rod degeneration. Invest Ophthalmol Vis Sci. 1987;28:140–50. [PubMed] [Google Scholar]