

Abstract
Ubiquitous calpains (calpain I & II) are generally recognized as cytosolic proteins. Recently, mitochondrial localized calpain I (μ-calpain) has been identified. Activation of mito-u-calpain cleaves apoptosis inducing factor (AIF), a flavoprotein located within the mitochondrial intermembrane space, in liver mitochondria, but not in brain mitochondria. We first tested if activation of mito-u-calpain cleaves AIF in isolated heart mitochondria. A decrease in AIF content within mitochondria increases cardiac injury during ischemia-reperfusion by augmenting oxidative stress. We hypothesize that the activation of mito-u-calpain by calcium overload during ischemia-reperfusion results in decreased AIF content within mitochondria by cleaving AIF. The u-calpain was present within mouse heart mitochondria, mostly in the intermembrane space. Exogenous calcium treatment induced a calpain-dependent decrease of mitochondrial AIF content in isolated mouse heart mitochondria. This process was blocked by a calpain inhibitor (MDL-28170). The Mitochondrial u-calpain activity was increased by 160% ± 15% during ischemia-reperfusion compared to time control. In contrast, the mitochondrial AIF content was decreased by 52% ± 7% during reperfusion vs. time control in the buffer perfused mouse heart. Inhibition of mito-u-calpain using MDL-28170 decreased cardiac injury by preserving AIF content within mitochondria during ischemia-reperfusion. Thus, activation of mito-u-calpain is required to release AIF from cardiac mitochondria. Inhibition of calpains using MDL-28170 decreases cardiac injury by inhibiting both cytosolic calpains and mito-u-calpain during ischemia-reperfusion.
Keywords: mitochondria, calpastatin, calpain, ischemia-reperfusion, calcium
Calpains are a family of Ca2+-dependent cysteine proteases including 15 ubiquitous isoforms and additional tissue-specific isoforms [1]. There are two ubiquitous calpains: calpain I (μ-calpain) and calpain II (m-calpain). μ-Calpain is activated in the presence of micro-molar (μM) concentrations of calcium, whereas activation of m-calpain requires milli-molar (mM) calcium [1, 2]. Calpain activity is regulated in vivo by the endogenous inhibitor calpastatin [2, 3]. μ-calpain and m-calpain are generally considered to be localized in the cytoplasm [1, 3, 4]. Calpain 10 is a mitochondrial calpain located within the mitochondrial matrix [5]. Recently another mitochondrial localized calpain, mitochondrial μ-calpain (mito-μ-calpain), has been identified in liver, brain, and heart mitochondria [4, 6].
Apoptosis-inducing factor (AIF) is a mitochondrial flavoprotein that functions as an antioxidant when it is located within the mitochondrial intermembrane space [4, 7]. In contrast, a release of AIF from mitochondria triggers caspase-independent apoptotic cell death [7]. AIF is reported to be anchored on the mitochondrial inner membrane and requires cleavage before release from mitochondria [4, 8]. Incubation of calcium with isolated and purified liver mitochondria cleaves AIF to its truncated form (t-AIF) that is released from mitochondria into cytosol, whereas the formation of t-AIF is inhibited by a calpain inhibitor [4]. These results indicate that calcium-mediated mito-u-calpain activation increases t-AIF formation in isolated liver mitochondria. In isolated brain mitochondria, however, inhibition of mito-u-calpain does not prevent calcium-mediated t-AIF formation [6], suggesting that activation of mito-u-calpain is not required to cleave AIF in the isolated brain mitochondria. Thus, the link between mito-u-calpain activation and AIF cleavage may be tissue dependent. Mito-u-calpain is present in heart mitochondria [4], but its location within heart mitochondria is unclear. It is unknown if activation of mito-u-calpain is required to cleave AIF to tAIF in the isolated heart mitochondria.
AIF can be released from mitochondria into cytosol and relocate to the nucleus during cell stress conditions [4, 7, 9]. Cardiac ischemia-reperfusion results in a decrease in the content of AIF within mitochondria [10]. However, it remains unclear if a release of AIF from mitochondria results from activation of mito-u-calpain during ischemia-reperfusion. In the present study, we first investigated the distribution of mito-u-calpain within cardiac mitochondria and tested if activation of mito-u-calpain is required to cleave AIF in isolated heart mitochondria. Next, we studied if ischemia-reperfusion led to mito-u-calpain activation and AIF cleavage in cardiac mitochondria. Lastly, we tested if inhibition of mito-u-calpain using a calpain inhibitor can prevent AIF cleavage during ischemia-reperfusion.
METHODS
The experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of Virginia Commonwealth University (VCU) and the McGuire Department of Veterans Affairs Medical Center.
Isolation and purification of cardiac mitochondria from the mouse heart
The mouse heart was harvested and cardiac mitochondria were isolated based on our published protocol (see details in Supplemental Material) [11]. Trypsin (5 mg/g tissue) was used to isolate mouse heart mitochondria for this study because trypsin treatment effectively removed any adherent cytosolic proteins from the outer mitochondrial membrane [4, 12].
Separation of mitochondrial components
To further localize mito-μ-calpain, mitochondrial components were separated by digitonin (0.05 mg/ml) treatment and differential centrifugation [13]. Digitonin was used to permeabilize the outer mitochondrial membrane. Mouse heart mitochondria were incubated with 0.05 mg/ml digitonin for 30 min. followed by centrifugation at 5000×g for 10 min. The pellet consisted of mitochondria with a permeabilized outer membrane [digitonin pellet (Dig)]. Digitonin (0.05 mg/ml) was selected since this concentration of digitonin only permeabilizes the outer mitochondrial membrane [14]. The supernatant was further centrifuged at 100,000×g for 30 min. The subsequent pellet was the mitochondrial outer membrane fraction (MF), and corresponding supernatant was the soluble fraction (SF) that included proteins from the mitochondrial intermembrane space [14]. Western blotting was performed to identify proteins in each component[11].
Incubation of isolated and purified mitochondria in vitro
Isolated and trypsin-purified mouse heart mitochondria (1 mg/ml) were incubated in buffer (composition, in mM: 100 KCl, 100 Tris·HCl, and 0.1% 2-mercaptoethanol) for 20 min at 30 °C. Calcium (400 uM) was used to activate mito-u-calpain, and MDL-28170 (MDL, 10 uM) was used to inhibit mito-u-calpain. At the end of incubation, sample buffer was directly added into the incubation medium and heated at 95 °C for 5 min to stop the reaction. The contents of AIF and t-AIF were detected using immunoblotting (see below).
Preparation of mouse heart for perfusion
Male C57BL/6 mice [2–3 months of age (22 – 28 g)] were anesthetized [14] and hearts were excised and retrograde perfused via the aorta in the Langendorff mode (Supplemental Material). In untreated hearts, the heart was buffer-perfused for 15 min followed by 30 min global ischemia at 37 °C and 30 min reperfusion. In the group treated with calpain inhibitor, hearts underwent the same perfusion protocol except that MDL-28170 (MDL) was included in identical K-H buffer for the entire perfusion period [15]. MDL was first dissolved in DMSO and then diluted in Krebs-Henseleit buffer. The final DMSO concentration was less than 0.01% and had no effect on myocardial function based upon DMSO vehicle experiments (data not shown). Hearts in the time control group were buffer-perfused without ischemia. All hearts were paced at 420 beats per min [16] during the 15 min equilibration period and after 15 min reperfusion. Coronary effluent was collected during the entire reperfusion period in order to determine LDH activity [17].
Measurement of mitochondrial calpain activity
Mito-μ-calpain activity was determined using a commercially available kit: (QIA 120, Calbiochem, San Diego, CA) [4, 12]. Succ-Leu-Tyr-AMC (7-Amino-4-methylcoumarin) was cleaved by calpain to form a fluorescent product that is quantified (excitation: 380 nm; emission:460 nm) [18]. Mitochondria (0.5 mg/ml) were incubated in buffer containing 100 mM Tris-HCl, pH 7.5, 10 mM 2-mercaptoethanol, 100 mM KCl, 5 mM CaCl2, and 20 uM Succ-Leu-Tyr-AMC for 30 min at 30 °C. The intensity of fluorescence was determined (Victor 2, PerkinElmer, Waltham MA) [4]. A standard curve was generated with known concentrations of AMC to quantify the calpain activity [4, 12].
Statistical Analysis
Data were expressed as the mean ± standard error of the mean. Differences among groups were compared by one-way analysis of variance with post hoc comparisons performed using the Student-Newman-Keuls test of multiple comparisons (Sigmastat 3.5, ProgramPaketet, Gothenburg, Sweden). A difference of p<0.05 was considered significant.
RESULTS
Localization of mito-u-calpain within mitochondria
Isolated mouse heart mitochondria were purified by trypsin treatment to remove potential contamination by cytosolic calpain [4]. Tubulin (a cytosolic marker) was used to assess the purity of the isolated mitochondria [19]. Tubulin was detected in cytosol but not in the isolated mitochondria (Figure 1A), indicating that trypsin treatment effectively removed potential cytosolic contamination. Trypsin treatment did not alter the content of VDAC (voltage dependent ion channel), a mitochondrial outer membrane protein (Figure 1A). Lack of VDAC in cytosol excluded significant mitochondrial contamination of the cytosolic fraction (Figure 1A). μ-calpain was detected both in purified mitochondria and cytosol, whereas m-calpain was only detected in cytosol (Figure 1B), indicating that u-calpain is located in both cytosol and in mitochondria.
Fig. 1. The purity of isolated mitochondria and localization of mito-u-calpain.
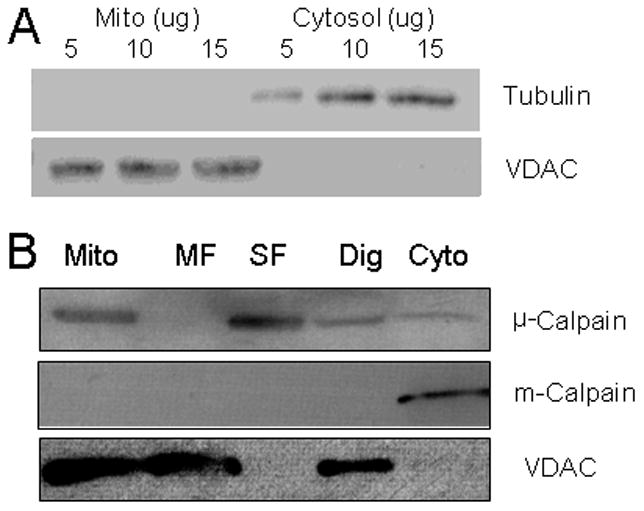
Panel A: demonstration of the purity of the mitochondria used for study. Tubulin, a cytosolic marker, is only detected in cytosol but not in mitochondria. In contrast, VDAC, a mitochondrial marker, is only detected in mitochondria. Thus, trypsin treatment effectively removed cytosolic contamination from mitochondria, and cytosol is also devoid of mitochondrial contamination.
Panel B: the localization of mito-μ-calpain. Mito-u-calpain is detected in both cytosolic and mitochondrial fractions, whereas m-calpain is only detected in the cytosolic fraction. The permeabilization of the outer mitochondrial membrane by digitonin decreased mito-μ-calpain content compared to intact mitochondria (Dig lane), suggesting that mito-u-calpain is located in the outer mitochondrial membrane or within the inter-membrane space. The abundant mito-μ-calpain present in the soluble fraction (SF) but not in the membrane fraction (MF) indicates that mito-μ-calpain is mainly located within the intermembrane space. VDAC was detected in mitochondria, Dig, MF, but not SF, consistent with the outer membrane localization of VDAC.
To further localize mitochondrial μ-calpain, submitochondrial components were separated using digitonin treatment and differential centrifugation [13]. Permeabilization of the outer mitochondrial membrane by digitonin decreased mito-μ-calpain content compared to intact mitochondria (Figure 1B, Dig lane), suggesting that the mito-μ-calpain was located either in the outer membrane or within the intermembrane space. The abundant mito-μ-calpain present in the soluble fraction (SF) but not in the membrane fraction (MF) indicated that mito-μ-calpain was mainly located within the intermembrane space. VDAC was detected in mitochondria, Dig, MF, but not SF, consistent with localization of VDAC to the outer membrane in contrast to mito-u-calpain (Figure 1B).
Activation of mito-u-calpain decreased AIF content in the isolated mitochondria
In order to study the link between mito-u-calpain activation and AIF cleavage, the total amount of AIF and tAIF in the entire incubation media were quantified after incubation. Incubation of mouse heart mitochondria in the presence of high dose exogenous calcium (400 uM) decreased AIF content compared to mitochondria without calcium treatment (0 uM calcium). Inhibition of mito-u-calpain using MDL preserved AIF content in calcium treated mitochondria (Figure 2A and B). Interestingly, calcium treatment also decreased t-AIF content compared to mitochondria that were not treated with calcium. MDL treatment also attenuated calcium-mediated t-AIF degradation in isolated mitochondria (Figure 2A and B).
Fig. 2. Activation of mito-u-calpain decreased AIF content within mitochondria.

Panel A: calcium-mediated AIF cleavage. Isolated mouse heart mitochondria were exposed to 0 or 400 uM calcium in the presence or absence of MDL (10 uM). Calcium stimulation significantly decreased AIF content compared to mitochondria without calcium treatment, whereas the content of AIF was preserved in MDL-treated mitochondria compared to mitochondria without MDL treatment (Panel B). Calcium stimulation also decreased t-AIF content compared to untreated (no calcium) mitochondria. Inhibition of mito-u-calpain using MDL attenuated the t-AIF degradation compared to untreated (no MDL) treated mitochondria (Panel C). Data are expressed as mean ± SEM; * p<0.05 vs. 0 μM calcium treated mitochondria.
Panel B: Quantification of total AIF content in the incubation medium.
Panel C: Quantification of total t-AIF content in the incubation medium.
Mito-u-calpain was activated in cardiac mitochondria during ischemia-reperfusion
Ischemia and reperfusion substantially increased mito-μ-calpain activity in isolated mouse heart mitochondria compared to the time control group (Figure 3A). However, the protein content of mito-μ-calpain following ISC-REP was not different from time controls (Figure 3B), indicating that the increased mito-μ-calpain activity during REP is not due to the alteration of mito-u-calpain protein content. Calpastatin was also identified within trypsin-purified cardiac mitochondria (Figure 3B). Ischemia-reperfusion did not alter the content of calpastatin in mouse heart mitochondria, suggesting that the increased mito-μ-calpain activity during ischemia-reperfusion was not due to the loss of the endogenous inhibitor. ISC-REP did not alter the contents of cytosolic μ-calpain and m-calpain in the mouse heart (data not shown).
Fig. 3. The activities of mito-μ-calpain during ischemia-reperfusion.

Panel A: the activity of mito-μ-calpain is significantly increased in mouse heart mitochondria following ischemia-reperfusion compared to time control. The calpain inhibitor MDL effectively prevents mito-μ-calpain activation during ischemia-reperfusion.
Panel B: immunoblotting shows that ischemia and reperfusion does not alter the protein content of mito-μ-calpain compared to time control [Mean ± SEM: time control 1.12 ± 0.18 (Arbitrary unit, n=4); ISC-REP, 0.90 ± 0.03 (n=4); p=NS vs. IR]. Calpastatin is an endogenous inhibitor of calpain. Ischemia-reperfusion also does not alter the content of calpastatin localized within mitochondria [Mean ± SEM: time control 1.07 ± 0.33 (Arbitrary unit, n=4); IR, 0.96 ± 0.08 (n=4); p=NS vs. IR], suggesting that the increased mito-μ-calpain activity is not due to the loss of its endogenous inhibitor during ischemia-reperfusion. Subunit 4 of cytochrome oxidase (COX) is used as the loading control. Data are expressed as mean ± SEM; * p<0.05 vs. time control, † p<0.05 vs. untreated hearts.
Calpain inhibition prevented the loss of AIF from mitochondria
Activation of mito-u-calpain cleaves AIF to its truncated form- tAIF [4]. Mitochondrial AIF content was decreased during reperfusion compared to time control, supporting that activation of mito-u-calpain results in a cleavage of AIF to tAIF in the buffer perfused heart during ischemia-reperfusion. MDL treatment preserved AIF content in mitochondria following ischemia-reperfusion (Figure 4). The decreased AIF content observed in mitochondria following reperfusion should indicate the formation of tAIF. In contrast, tAIF content was also decreased in mitochondria following reperfusion vs. Time control. MDL treatment maintained the contents of AIF and t-AIF compared to untreated IR hearts, indicating that activation of mito-μ-calpain contributed to decreased AIF and t-AIF content within mitochondria (Figure 4).
Fig. 4. Inhibition of mito-u-calpain decreased the formation of truncated AIF (t-AIF) and attenuated t-AIF loss from mitochondria during ischemia-reperfusion.

Ischemia-reperfusion decreased the content of full length AIF compared to time control, supporting that activation of mito-u-calpain cleaved AIF. MDL treatment tended to protect AIF during reperfusion. Ischemia-reperfusion also decreased t-AIF content compared to time control, suggesting that t-AIF was released from mitochondria into cytosol during reperfusion. MDL treatment prevented the loss of t-AIF from mitochondria, supporting that calpain inhibition decreased the permeability of the mitochondrial outer membrane. Subunit 4 of cytochrome oxidase was used as a loading control. Data are expressed as mean ± SEM; * p<0.05 vs. time control. † p<0.05 vs. MDL treatment.
MDL treatment decreased myocardial injury during ischemia-reperfusion
MDL decreases cardiac injury in buffer perfused rat [20] and rabbit hearts [12, 18]. The release of LDH into coronary effluent was increased in hearts following ischemia-reperfusion compared to time control, whereas MDL treatment decreased LDH release compared to untreated hearts [Mean ± SEM: Time control, 77 ± 39 (mU/g tissue, n=7); ischemia-reperfusion, 764 ± 78 (n=8)*; MDL + ischemia-reperfusion, 476 ± 54 (n=5)*#; *p<0.05 vs. time control, #p<0.05 vs. ischemia-reperfusion]. These results support that inhibition of calpains decreased cardiac injury during ischemia-reperfusion in the isolated mouse heart.
DISCUSSION
In the present study, we first identified that μ-calpain is present in mouse heart mitochondria. We further localized mito-u-calpain to the mitochondrial intermembrane space by separation of submitochondrial components. Next, we found that activation of mito-μ-calpain was required to cleave AIF to tAIF in isolated cardiac mitochondria. Third, we found that ischemia-reperfusion activated mito-μ-calpain and decreased AIF content within mitochondria. MDL prevented mito-μ-calpain activation during ischemia-reperfusion accompanied by preservation of AIF content within mitochondria. Thus, activation of mito-u-calpain is required to cleave AIF to t-AIF in cardiac mitochondria. Calpain inhibitors protect the heart during ischemia-reperfusion not only by affecting cytosolic calpain activity, but also through regulating mito-μ-calpain activity.
In order to understand the relationship between mito-μ-calpain activation and the cleavage of AIF in heart mitochondria, the submitochondrial location of mito-μ-calpain was determined. Mito-μ-calpain is located in the intermembrane space and thus can accesses AIF. Activation of mito-u-calpain increases AIF cleavage in isolated liver mitochondria [4] and rat retinal mitochondria [21]. However, calcium overload induced AIF cleavage may be independent of mito-u-calpain activation in isolated brain mitochondria [6]. In the present study, purified mitochondria were used for incubation. The potential contamination from cytosolic calpains was excluded. Incubation of isolated mouse heart mitochondria with exogenous calcium increased AIF cleavage, and this process was sensitive to a calpain inhibitor. These results indicate that activation of mito-u-calpain cleaves and decreases AIF content in heart mitochondria. Thus, the link between mito-u-calpain activation and AIF cleavage was clearly established for the heart.
Thus, AIF is a target for mito-u-calpain cleavage in heart mitochondria. AIF has a pro-survival role when located within mitochondria and a pro-apoptotic role following release from mitochondria and translocation to the nucleus [7, 22]. Genomic down regulation of mitochondrial AIF content in the Harlequin mouse increases myocardial infarct size by augmenting oxidative stress during ischemia-reperfusion, supporting that AIF contributes an antioxidant role within mitochondria [22]. AIF released from mitochondria relocates to the nucleus and increases caspase-independent apoptosis by activating poly(ADP-ribose) polymerase-1 [7]. Ischemia-reperfusion results in the increased generation of reactive oxygen species [17, 23, 24] and calcium overload within mitochondria [25]. The increased oxidative stress during ischemia-reperfusion may lead to oxidative modification of AIF that facilitates calpain-mediated AIF cleavage and subsequent release from mitochondria [8]. Ischemia-reperfusion decreases mitochondrial AIF in rat heart mitochondria [10]. In the present study, we found that ischemia-reperfusion resulted in decreased mitochondrial AIF content accompanied by increased mito-u-calpain activity. Prevention of mito-u-calpain activation using MDL treatment maintained mitochondrial AIF content during ischemia-reperfusion. These results support that activation of mito-u-calpain contributes to decreased AIF content within mitochondria in the buffer perfused mouse heart during reperfusion [22].
MDL treatment decreases myocardial injury in buffer perfused rat and rabbit hearts, and the mechanism of protection has been proposed to be inhibition of cytosolic calpains [12, 18, 20]. In the present study, MDL decreased LDH release in the isolated mouse heart during REP, supporting that inhibition of calpain also decreases cardiac injury in buffer perfused mouse hearts as well. MDL treatment decreased mito-u-calpain activity in the isolated control mitochondria and mitochondria following ischemia-reperfusion. Our results expand the mechanism by which MDL treatment decreases cardiac injury during ischemia-reperfusion to include inhibition of mito-u-calpain in addition to the previously described cytosolic calpain [12, 18].
In addition to mito-μ-calpain, mitochondria contain other isoforms of calpain including calpain 10 [5]. Activation of calpain 10 by calcium overload decreases complex I activity via the cleavage of complex I subunits [5]. Inhibition of calpain 10 using selective peptides protects complex I by preventing Ca2+ induced cleavage of complex I subunits (NDUFV2) [26]. MDL is primarily an inhibitor of μ-calpain and m-calpain [12, 18, 20]. In the present study, MDL treatment did not dramatically improve oxidative phosphorylation compared to untreated hearts during ischemia-reperfusion (Supplemental Data). These results suggest that MDL has a minimal effect on calpain 10 activity in the mouse heart following ischemia-reperfusion. Since specific inhibitors of cytosolic calpains, mito-u-calpain, and calpain 10 are lacking, the current pharmacological approach cannot differentiate the roles of individual calpain in ischemia-reperfusion-mediated mitochondrial damage. This forms of a major limitation of the current study, and requires further investigation in the future using combined pharmacological and genomic approaches to up- and down- regulate individual calpain activities during ischemia-reperfusion.
Supplementary Material
HIGHLIGHTS.
In cardiac mitochondria, μ-calpain is present and is mainly located in the intermembrane space.
Activated mitochondrial calpain cleaves AIF and decreases its content within heart mitochondria
Mitochondrial calpain is activated during cardiac ischemia and reperfusion.
Mitochondrial calpain activation contributes to a release of AIF from mitochondria during ischemia-reperfusion
The calpain inhibitor MDL-28170 prevents the activation of mitochondrial μ-calpain, the loss of AIF from mitochondria and decreases injury in ischemia-reperfused hearts
Acknowledgments
This work was supported by a Scientist Development Grant (11SDG5120011) from the American Heart Association (QC) and the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (EJL), Program Project 2PO1AG15885 from the National Institutes of Health (EJL), and the Pauley Heart Center, Virginia Commonwealth University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 2.Thompson VF, Goll DE. Purification of mu-calpain, m-calpain, and calpastatin from animal tissues. Methods Mol Biol. 2000;144:3–16. [PubMed] [Google Scholar]
- 3.Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozaki T, Tomita H, Tamai M, Ishiguro S. Characteristics of mitochondrial calpains. J Biochem. 2007;142:365–376. doi: 10.1093/jb/mvm143. [DOI] [PubMed] [Google Scholar]
- 5.Arrington DD, Van Vleet TR, Schnellmann RG. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol Cell Physiol. 2006;291:C1159–1171. doi: 10.1152/ajpcell.00207.2006. [DOI] [PubMed] [Google Scholar]
- 6.Joshi A, Bondada V, Geddes JW. Mitochondrial micro-calpain is not involved in the processing of apoptosis-inducing factor. Exp Neurol. 2009;218:221–227. doi: 10.1016/j.expneurol.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 8.Norberg E, Gogvadze V, Vakifahmetoglu H, Orrenius S, Zhivotovsky B. Oxidative modification sensitizes mitochondrial apoptosis-inducing factor to calpain-mediated processing. Free Radic Biol Med. 2010;48:791–797. doi: 10.1016/j.freeradbiomed.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 9.Kar P, Samanta K, Shaikh S, Chowdhury A, Chakraborti T, Chakraborti S. Mitochondrial calpain system: an overview. Arch Biochem Biophys. 2010;495:1–7. doi: 10.1016/j.abb.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 10.Javadov S, Choi A, Rajapurohitam V, Zeidan A, Basnakian AG, Karmazyn M. NHE-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc Res. 2008;77:416–424. doi: 10.1093/cvr/cvm039. [DOI] [PubMed] [Google Scholar]
- 11.Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC. Mitochondrial-targeted Signal Transducer and Activator of Transcription 3 (STAT3) Protects against Ischemia-induced Changes in the Electron Transport Chain and the Generation of Reactive Oxygen Species. J Biol Chem. 2011;286:29610–29620. doi: 10.1074/jbc.M111.226209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–29186. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- 13.Matas J, Young NT, Bourcier-Lucas C, Ascah A, Marcil M, Deschepper CF, Burelle Y. Increased expression and intramitochondrial translocation of cyclophilin-D associates with increased vulnerability of the permeability transition pore to stress-induced opening during compensated ventricular hypertrophy. J Mol Cell Cardiol. 2009;46:420–430. doi: 10.1016/j.yjmcc.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 14.Badugu R, Garcia M, Bondada V, Joshi A, Geddes JW. N terminus of calpain 1 is a mitochondrial targeting sequence. J Biol Chem. 2008;283:3409–3417. doi: 10.1074/jbc.M706851200. [DOI] [PubMed] [Google Scholar]
- 15.Chen Q, Hoppel CL, Lesnefsky EJ. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J Pharmacol Exp Ther. 2006;316:200–207. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 16.Reichelt ME, Willems L, Hack BA, Peart JN, Headrick JP. Cardiac and coronary function in the Langendorff-perfused mouse heart model. Exp Physiol. 2009;94:54–70. doi: 10.1113/expphysiol.2008.043554. [DOI] [PubMed] [Google Scholar]
- 17.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 18.Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- 19.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hernando V, Inserte J, Sartorio CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J Mol Cell Cardiol. 2010;49:271–279. doi: 10.1016/j.yjmcc.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Mizukoshi S, Nakazawa M, Sato K, Ozaki T, Metoki T, Ishiguro S. Activation of mitochondrial calpain and release of apoptosis-inducing factor from mitochondria in RCS rat retinal degeneration. Exp Eye Res. 2010;91:353–361. doi: 10.1016/j.exer.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 22.van Empel VP, Bertrand AT, van der Nagel R, Kostin S, Doevendans PA, Crijns HJ, de Wit E, Sluiter W, Ackerman SL, De Windt LJ. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ Res. 2005;96:e92–e101. doi: 10.1161/01.RES.0000172081.30327.28. [DOI] [PubMed] [Google Scholar]
- 23.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem. 1993;268:18532–18541. [PubMed] [Google Scholar]
- 24.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Cherednichenko G, Hernandez L, Halow J, Camacho SA, Figueredo V, Schaefer S. Preconditioning limits mitochondrial Ca2+ during ischemia in rat hearts: role of KATP channels. Am J Physiol Heart Circ Physiol. 2001;280:H2321–2328. doi: 10.1152/ajpheart.2001.280.5.H2321. [DOI] [PubMed] [Google Scholar]
- 26.Rasbach KA, Arrington DD, Odejinmi S, Giguere C, Beeson CC, Schnellmann RG. Identification and optimization of a novel inhibitor of mitochondrial calpain 10. J Med Chem. 2009;52:181–188. doi: 10.1021/jm800735d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.