

Abstract
Lethal toxin, a key virulence factor produced by Bacillus anthracis, induces cell death, in part by disrupting numerous signaling pathways, in mouse macrophages. However, exposure to sublethal doses of lethal toxin allows some cells to survive. Because these pro-survival signaling events occur within a few hours after exposure to sublethal doses, we hypothesized that acute phase proteins might influence macrophage survival. Our data show that serum amyloid A (SAA) is produced in response to lethal toxin treatment. Moreover, pre-treatment of macrophages with exogenous SAA protected macrophages from lethal toxin-mediated death. Exogenous SAA activated the p38 mitogen activated protein kinase (MAP) kinase pathway, while lethal toxin mutants incapable of p38 activation were incapable of causing cell death. Chemical inhibition of the p38 activation pathway abrogated the protective effects of SAA. These data show that SAA affords protection against lethal toxin in mouse macrophages and link this response to the p38 pathway.
Keywords: anthrax lethal toxin, acute phase proteins, serum amyloid A
1. Introduction
Pulmonary anthrax is a multi-stage infectious disease characterized by an initial mild illness followed by a rapidly progressing systemic stage that is generally fatal. The causative pathogen, Bacillus anthracis, secretes high levels of anthrax toxin, which inactivates critical components of the host immune response and thus facilitates disease progression. Anthrax toxin is a secreted, tripartite toxin that functions in binary combinations of protective antigen (PA) plus edema factor (EF) or lethal factor (LF), yielding edema toxin (ET) and lethal toxin (LT) respectively. PA is not itself a toxin, but is required for entry of both EF and LF peptides into host cells. Recent studies on the effects of anthrax toxin have identified multiple host immune cells that are inactivated by anthrax toxins (reviewed in [1;2]). However, it is also now apparent that cells of the innate immune response, macrophages in particular, are not simply passive targets of intoxication, but instead mount adaptive responses that provide resistance to one of the toxins, lethal toxin (LT). Understanding the mechanisms by which macrophages respond to LT will provide new insights that could be helpful for future treatment regimens.
Intoxication by LT results in LF-mediated cleavage of mitogen activated protein kinase kinases (MAPKKs), leading to disruption in downstream signaling by c-Jun N-terminal kinase, p38, and extracellular response kinases (ERK) 1 and 2 [3-6]. This loss in MAPKK signaling leads to prominent transcriptional changes and altered cell physiology. Although many cell types remain viable after exposure to LT despite proteolytic cleavage of MAPKKs, other cell types are susceptible to LT-induced cytotoxicity. Murine macrophages are especially susceptible to LT-induced cytotoxicity [5-9].
One of the first steps in early innate immune responses is the rapid production of acute phase proteins, such as C-reactive protein (CRP), alpha-1-trypsinogen, transferrin, serum amyloid P (SAP) and serum amyloid A (SAA), with concomitant increases in serum levels of these proteins by as much as 2,000-fold (reviewed in [10]). Although synthesis of acute phase proteins was initially thought to be limited to hepatocytes, it is now clear that many cell types at infection sites, including macrophages, can also produce them [11;12]. The precise functions of many of these proteins remain obscure, but accumulating evidence suggests that several of these proteins, including SAA, participate directly in protective responses against microbes [13-16].
SAA is produced as at least four distinct active isoforms in mice and humans [11]. There are notable differences in regulatory elements in the promoters of the different isoforms and these may vary among different strains of mice [17;18]. Several independent studies confirmed production of SAA by macrophages within inflammation sites [19-21]. Upon inflammation, these small peptides (~12 kDa) increase by more than 1,000-fold, reaching serum concentrations as high as 1 mg/ml in as short a time as 20 minutes [11]. Most circulating SAA is bound to high density lipoprotein, but SAA also binds to formyl peptide receptor-like 1 where it may affect angiogenesis [22]. SAA has been shown to modulate cellular physiology by binding to cell surface receptors. For example, SAA activates MAPKK pathways and regulates lipid uptake and cholesterol efflux by binding to scavenger receptor class B type I receptors [23-25]. By binding to Toll-like receptor 2, SAA also activates p38, JNK and ERK signaling [26]. Together, these data suggest that SAA may modulate very early events after macrophage exposure to LT, and that these events could influence cell survival.
In the current study, we examined the effects of SAA on macrophage sensitivity to LT. Our results suggest that early SAA production confers resistance to LT-induced death in mouse macrophages, revealing for the first time an early innate immune response that alters the outcome of LT-intoxication.
2. Materials and Methods
2.1.Lethal Toxin
Recombinant PA and LF were prepared from E. coli as previously described [7]. Mutated LF containing a His to Cys mutation at amino acid 719 was prepared similarly. Lethal toxin (LT) is defined as 1μg/ml PA plus 500 ng/ml LF unless otherwise indicated. LPS was removed from recombinant PA and LF using polymyxin B chromatography (Sigma) according to the manufacturer’s directions and was tested for remaining endotoxin using the Limulus amoebocyte assay (Biowhitaker). Protein concentrations were determined by colorimetric assay (Protein Assay Concentrate, Bio-Rad) at 595nm.
2.2.Macrophage Preparation and Cell Culture
Balb/c and C57BL/6 mice were maintained and used according to institutional regulatory guide lines. Bone marrow was extracted from male 14 to 20 week-old femurs and tibias. Cells were filtered through 40μm cell strainers (BD Falcon) and cultured at 1×105 to 1×106 cells/ ml at 37°C in RPMI 1640 supplemented with 10% FCS, 2% pen/strep, and 15% CMG supernatant (kindly provided by Dr. M. Coggeshall, this institution) as a source of macrophage colony stimulating factor [27]. After five days, cultures were treated with PA, LF, SAA or LPS (E. coli 0111:B4, Sigma) at the doses and times indicated. Macrophages were harvested on day 5 using cell dissociation buffer (Sigma) for 10 minutes at 37°C and then washed with PBS containing 3% FCS. Macrophage viability was assessed by three methods, with similar results: visual assessment of trypan blue exclusion (500 cells per sample from triplicate cultures), flow cytometry analysis of propidium iodide uptake, and MTT (thiazolyl blue tetrazolium bromide) assay (Sigma) according to the manufacturer’s directions. Briefly, 5mg/ml MTT in PBS containing 3% FCS was added to each well and incubated overnight at 37°C without CO2 followed by 100 μL DMSO as an extraction buffer. The absorbence at 560nm was read using a Synergy HT Multi-Detection Microplate Reader (Biotek) spectrophotometer. Background values were determined from wells without cells. A Student’s t test was used to determine p-values and statistical significance. Recombinant human apo-SAA (rSAA) (PeproTech, Inc.), a composite of SAA1 and 2 reflecting the acute phase isoforms of SAA rather than constitutively expressed isoforms was used, and contained < 0.1ng/μg endotoxin. The p38 inhibitor SB 202190 was obtained from Sigma.
2.3.RT-PCR
Following treatment with LT (500 ng/ml LF) for 3 hr, the macrophages (1×105) were transferred to TRI-reagent (Molecular Research Center, Inc.) and total RNA was extracted following the manufacturer’s protocol. cDNA was generated from mRNA with M-MLV reverse transcriptase (Promega) and amplified with primers for actin [28], SAA1, SAA2, SAA3, SAA4, C reactive protein, and serum amyloid P using primers indicated in Table I. RT-PCR conditions consisted of 40 cycles of 94°C for 1 min, the indicated annealing temperature (Table I) for 1 min, and 72°C for 1 min, with an initial cycle of 94°C for 5 min. C57BL/6 liver extracts were used as positive control samples for RT-PCR of each transcript. The resulting bands were quantified relative to actin using the LumiAnalyst software, Ver.3.0 (Media Cybernetics).
Table 1.
List of Primers and Annealing Temperatures for RT-PCR.
| Gene | 5′-3′ Sequence | Temp. | PCR Product Size |
|---|---|---|---|
| SAA1-F | GACATGTGGCGAGCCTACACTG | 66.2°C | 426 bp |
| SAA1-R | CTGCAAGAGTAACTCAGTTCTG | ||
| SAA2-F | CAGGATGAAGCTACTCACCAG | 64.3°C | 352bp |
| SAA2-R | CAGGAGGTCTGTAGTAATTGG | ||
| SAA3-F | GGAGTTGACAGCCAAAGATG | 60.0°C | 309bp |
| SAA3-R | GCCAGCAGGTCGGAAGTGG | ||
| SAA4-F | CATATAGAGTCGCATCATGAGCG | 65.5°C | 483bp |
| SAA4-R | CAAGCTGAGTGACTATCACAG | ||
| CRP-F | GTATGGCGGTGACTTTGATG | 60.0°C | 525bp |
| CRP-R | CTCATCTATGTGAGGGAGAAG | ||
| SAP-F | GACCAAGCATGGACAAGCTACTG | 57.2°C | 780bp |
| SAP-R | CGGCCATCTGATGTCCATGAG |
2.4.Flow Cytometry
For each treated sample, 1×106 cells were added to a 96-well v-bottom plate and spun down at 1500 rpm for 3 min. The cells were stained with fluorescently labeled antibodies in the dark at 4°C for 15 minutes in PBS containing 3% FCS, and then analyzed using FACS LSR II. Data were analyzed using the Cellquest software (BD Bioscience). Dead cells were excluded by propidium iodide staining. Antibodies used were α-CD36 (BD Pharmingen) with secondary antibody of α-Ig κ PE (BD Pharmingen) and MAC-1 APC (eBioscience).
2.5.Western Blotting and ELISAs
Macrophages were harvested and resuspended in sample buffer at 50,000 cells per 20 μL sample buffer (0.5M Tris-HCl pH 6.8, glycerol, 10% SDS, 2-β-mercaptoethanol, bromophenol blue). Samples were boiled, separated via SDS-PAGE (12%), transferred to nitrocellulose membranes and blocked for 1 hr in 5% powdered milk in TBST (2M Tris-HCl pH 8.0, NaCl, Tween 20). Membranes were incubated overnight with anti-phospho-p38, stripped and then incubated with p38 antibodies (both from Cell Signaling Technology) overnight. Proteins were detected using the SuperSignal West Dura Extended Duration Substrate chemiluminescence system (Pierce). SAA concentrations in macrophage supernatants were measured by ELISA (Immunoassay Kit, Biosource International), according to the manufacturer’s protocol and used antibodies against human SAA1 and 2 that are broadly cross reactive with mouse SAAs.
3. RESULTS
3.1. LT induces SAA production in murine macrophages
Macrophages from Balb/c mice appear to be more sensitive to LT-induced cytotoxicity than macrophages from C57Bl/6 mice [7-9]. Therefore, we analyzed the responses to LT of macrophages from both of these strains. After three hours of treatment with LT or with PA alone, we assessed viability by trypan blue exclusion, MTT assay, and propidium iodide exclusion (Figures 1A and B, and not shown). As expected, PA alone affected viability only marginally by MTT assay (Figure 1B). Both Balb/c and C57Bl/6 macrophages showed a dose-dependent decrease in viability after treatment with LT, although the Balb/c macrophages were more sensitive than those from the C57Bl/6 strain (Figure 1A and B). All three viability methods gave similar results, so only the MTT assays are shown for the following experiments in this study.
Figure 1.

Lethal toxin induces serum amyloid A in murine macrophages. Bone marrow Balb/c and C57Bl/6 macrophages were untreated, treated with 1 μg/ml PA and increasing doses of LF (LT) for 3 hours, or were treated with 1 μg/ml PA alone (open symbols). A. Trypan blue-exclusion was used to assess viability. Data represent cells from triplicate cultures. B. Viability was also assessed by MTT production in triplicate cultures. Background absorbance was < 0.100. Data are representative of 3-5 experiments. C. Balb/c macrophages were treated with media alone (Unt), or 25 μg/ml LPS for 15 minutes, or with LT (500 ng/ml LF) for the designated times, and mRNA levels of SAA 1, 2, 3 and 4 were measured by RT-PCR and are expressed as units normalized to actin levels. Data are representative of 3-5 experiments. D. Supernatants were harvested from the macrophage cultures assessed in (B), and SAA levels were measured by ELISA. Data are representative of 2 experiments. Error bars indicate one standard deviation in A, B and D.
To determine if LT treatment induced acute phase proteins, macrophages were stimulated with maximum doses of LT (500 ng LF plus 1 μg/ml PA) for 15 minutes and RNA levels of acute phase proteins were assessed by RT-PCR. Because high doses of endotoxin, or LPS, effectively enhance production of all acute phase proteins, cells were treated with LPS (25 μg/ml) for three hours as a positive control. RNA isolated from cells at time 0, prior to treatment, was used to measure basal expression levels. RNA levels of C reactive protein and serum amyloid P were not altered by LT treatment (data not shown). There are five serum amyloid A (SAA) proteins in the mouse, one of which, SAA5, is ubiquitously and constitutively expressed [18] and therefore was not measured in these experiments. For SAA1-4, Balb/c and C57Bl/6 macrophages responded similarly; results from Balb/c macrophages are shown in Figure 1C. SAA1 mRNA was low at all time points, while SAA3 mRNA was constitutively expressed at high levels (Figure 1C and not shown). However, new SAA4 transcripts were induced by LT treatment at the earliest measured time points, but decreased to negligible amounts by thirty minutes. SAA2 transcripts were also induced, but at later time points, and decreased to undetectable levels within an hour of induction.
Supernatants from LT-treated macrophages were assessed for SAA protein levels to determine if transcription correlated with secreted SAA protein. Supernatants from untreated control, and LPS- (25 μg/ml) and LT-treated macrophages were harvested after three hours and examined for the presence of SAA protein by ELISA. Under each condition, SAA protein levels were similar in cultures of Balb/c and C57Bl6 macrophages. Untreated control cultures for both strains of macrophages showed low levels of SAA, consistent with constitutive SAA3 production. LT treatment increased secreted SAA levels by two-fold, similar to the LT positive control (Figure 1D). Although the protein assays available cannot distinguish among the four isotypes of SAA, these data indicate that LT induces production of the acute phase reactant SAA within three hours.
3.2. Recombinant SAA protects macrophages from LT-induced cytotoxicity
Earlier studies showed that low dose treatment of macrophage cell lines for 6 hours resulted in cells which were then resistant to LT-induced cytotoxicity [7], and we hypothesized that resistance might be due to an early acute phase response, and the production of SAA. Therefore, we tested whether exogenous SAA conferred any protection against LT-mediated death. Macrophages were pretreated for 30 minutes with recombinant SAA (rSAA) (1μg/ml) and then treated with LT at various concentrations (1μg/ml PA + 0, 1, 25, 50, 100, or 500 ng/ml LF) in the continued presence of rSAA. Pretreatment of macrophages with SAA showed statistically significant (p < 0.05) prevention of LT-induced death of Balb/c and C57Bl/6 macrophages, from 25 ng/ml LF through the highest dose used (Figure 2A). Although the LT tests were carefully prepared to remove contaminating endotoxin, we were concerned that the commercially prepared SAA may have contained low levels of endotoxin (< 0.1 ng/ml) that could have contributed to the increased viability of the macrophages. Indeed, cells treated with 25 μg/ml LPS exhibited as much SAA in culture supernatants as did cells treated with LT (Figure 1D). Therefore, we tested whether various concentrations of LPS alone, starting at 0.1 ng/ml, could rescue cells from LT-induced death. Even at concentrations as high as 500 ng/ml, LPS pretreatment did not protect macrophages from LT-induced cytotoxicity (Figure 2B). We did find that doses above 5 μg/ml were effective in rescuing viability when used as a pretreatment, consistent with acute phase SAA production as shown in Figure 1D. However, the levels of endotoxin present in the rSAA were below levels that could have induced endogenous SAA from the cells. These data indicate that rSAA pretreatment can protect murine macrophages from the cytotoxic effects of LT.
Figure 2.

Pretreatment with exogenous SAA protects macrophages from LT induced death. A. Macrophages were pre-treated with 1μg/ml rSAA for 30 minutes prior to addition of media with 1μg/ml PA alone (CON) or 1μg/ml PA with increasing doses of LF (LT). Viability was assessed for triplicate cultures 3 hours later. Data are representative of 3 experiments. B. Macrophages were treated with increasing doses of LPS for 30 minutes before addition of LT (1μg/ml PA plus 500 ng/ml LF) and viability was measured for triplicate cultures 3 hours after addition of LT. Average absorbances for cultures with no LT were 0.846 for Balb/c and 0.925 for C57Bl/6 macrophages. Background absorbances were <0.100. Data are representative of 2 experiments. Background absorbance was < 0.100 for A and B. Error bars indicate one standard deviation.
3.3. Mutated LT that fails to disrupt p38 activation does not induce cytotoxicity of macrophages
Several earlier studies indicated that LT mediates cytotoxicity by cleaving MAPKKs leading to disruption of downstream signaling pathways including p38 activation [2-6]. Therefore, we tested whether a mutant form of LF (LFH719C) that cannot cleave MAPKKs and does not lead to p38 inactivation [7] induced cytotoxicity of macrophages under these conditions. Balb/c and C57Bl/6 macrophages were treated with wild type or mutated lethal toxin (PA plus LFH719C), and viability was assessed by MTT assay. Although wild type LT caused does-dependent cell death of the macrophages of both strains, mutant LT did not significantly alter macrophage viability for either strain at any dose tested (Figure 3). These data confirmed that the ability of LF to cleave MAPKKs is required for LT-induced death of mouse macrophages under the tested conditions.
Figure 3.

Mutant LF that cannot cleave MAPKKs does not kill macrophages. Wild type or mutant LF was added to macrophages with 1 μg/ml PA at the indicated doses, and viabilities of triplicate cultures were measured after 3 hours. Error bars indicate one standard deviation. Data are representative of 2 experiments.
3.4. SAA and LT activate common MAPKK pathways in mouse macrophages
We hypothesized that pretreatment with rSAA might confer protection from LT-induced death by activating downstream pro-survival mediators prior to the LT-mediated disruption of upstream signaling pathways. We queried if activation of downstream mediators occurred in response to LT treatments that resulted in SAA transcription in Figure 1C, or in response to exogenous SAA. Therefore, we assessed whether, and at what time points, treatment with LT or with rSAA resulted in activation of p38 in our culture system. C57Bl/6 and Balb/c macrophages were treated with maximum doses of LT (500 ng LF plus 1 μg/ml PA) or with rSAA (1 μg/ml) and western blotted for p38 and phosphorylated p38 at various time points. Both C57Bl/6 and Balb/c cells macrophages showed weak and transient increases in phospho-p38 after LT treatment, starting after ~5 minutes in C57Bl/6 macrophages and after ~15 minutes in Balb/c macrophages (Figure 4A). In contrast, treatment of Balb/c and C57Bl/6 macrophages with rSAA caused robust activation of p38 after only two minutes and activation was maintained for up to 15 minutes in Balb/c cells (Figure 4B). Next we assessed how the secreted SAA protein might activate intracellular p38 signaling pathways. The scavenger receptor CD36 is a receptor for SAA [23;29]. Therefore, we tested whether stimulation of the CD36 receptor with an anti-CD36 antibody also resulted in p38 activation in C57Bl/6 and Balb/c macrophages. Similar results were observed for C57Bl/6 and Balb/c macrophages (Figure 4C and not shown). As expected, an isotype specific control antibody did not activate p38, and treatment with LPS (25 μg/ml) or LT (500 ng LF plus 1 μg/ml PA) did result in p38 activation. Treatment with anti-CD36 induced phospho-p38 levels similar to those observed with rSAA treatment, suggesting that rSAA may signal through the CD36 receptor. These data suggest that exogenous SAA and LT can activate common intracellular signaling pathways, such as the p38 pathway.
Figure 4.

LT and SAA activate p38 in C57Bl/6 and Balb/c macrophages. A. Macrophages were treated with LT (500 ng/ml LF) for the indicated times and cells were western blotted for p38 and phospho-p38. Levels of phospho-p38 relative to total p38 were quantified and time 0’ was arbitrarily set as 1.0. Data are averaged from three individual experiments and standard error bars are shown. B. Macrophages from Balb/c and C57Bl/6 mice were treated with 1 μg/ml rSAA and harvested directly into sample buffer at the times indicated. Levels of p38 and phospho-p38 were detected by western blotting and quantified by densitometry. Levels of phospho-p38 relative to total p38 from three individual experiments are presented as average values with standard error bars. Values for time 0’ were arbitrarily given a value of 1.0. C. C57Bl/6 macrophages were untreated (CON), or treated with LPS (25 μg/ml), LT, rSAA, anti-CD36, or an unrelated isotype control antibody (CON-Ab) for 30 minutes prior to harvesting and assessing p38 and phospho-p38 levels by western blot. Data are representative of 2 experiments.
3.5. Inhibition of p38 signaling interferes with SAA-mediated protection from LT-induced cytotoxicity
We used an inhibitor of the p38 pathway, SB 202190, to directly test whether p38 activation was required for SAA-mediated protection against LT-induced cytotoxicity. To ensure that cell death caused by high doses of SB 202190 did not affect our results, we tested the viability of macrophages with drug treatment at varying doses. Although viability of both macrophage strains was somewhat reduced by even low doses of SB 202190, as determined by the sensitive MTT assay, the majority of cells survived after four hours of treatment with 0.5 ng/ml to 10 μg/ml SB202190 (Figure 5A). These doses were sufficient to completely abolish p38 activation within 30 minutes, as assessed by western blotting (Figure 5B). Therefore, Balb/c and C57Bl/6 macrophages were pretreated for 30 minutes with 0.5 ng/ml SB 202190 or with DMSO carrier control, prior to treatment with exogenous SAA (for 30 minutes) and/or LT (500 ng/ml LF) for three additional hours. Results for Balb/c and C57Bl/6 macrophages are shown in Figure 5C and Figure 5D, respectively. Consistent with our original findings, LT treatment reduced cell viability (p < 0.002), and pretreatment with rSAA prevented LT-induced cytotoxicity (p < 0.008, Figure 5C, 5D, left bars). Treatment with DMSO alone or SB 202190 only modestly affected viability (p > 0.05). Pre-treatment with SB 202190 did not affect LT-induced cytotoxicity. Therefore, the p38 inhibitor SB 202190 neither directly induced cytotoxicity nor contributed to the cytotoxic effects of LT. However, pretreatment with SB202190 dramatically reduced the viability of macrophages treated with LT and exogenous rSAA (p < 0.003), essentially abrogating the protective effects of rSAA (Figure 5C, 5D). These data suggest that SAA-mediated protection against LT-induced cell death requires the p38 MAP kinase pathway.
Figure 5.
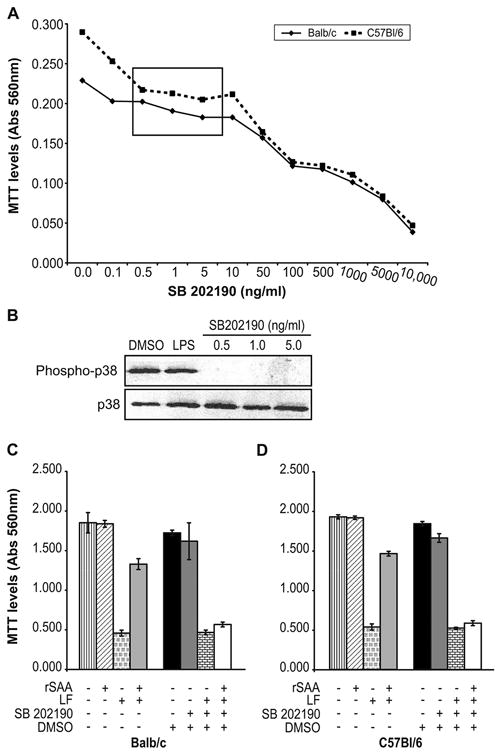
Activation of p38 MAP kinase pathways is required for macrophage survival after LT exposure. A. Macrophages were treated for 4 hours with the indicated doses of the p38 inhibitor SB 202190, and viability was measured for triplicate cultures. Background absorbance levels were less than 0.080. The box indicates doses of SB 202190 that inactivated p38 but had minimal effects on cell viability. Standard deviations were < 0.04. B. C57BL/6 macrophages were treated for 30 minutes with DMSO solvent control, LPS (25 μg/ml), or the indicated doses of SB 202190, were lysed and western blotted for p38 and phospho-p38. Macrophages from Balb/c (C) or C57Bl/6 mice (D) were pretreated for 30 minutes with 0.5 ng/ml SB 202190 or DMSO, then treated with 1 μg/ml rSAA or media for an additional 30 minutes prior to LT addition. Viability was assessed for triplicate cultures four hours after pretreatment began. Background absorbance levels were < 0.080. Error bars indicate one standard deviation. Data are representative of 2 experiments.
4. Discussion
The long term presence of lethal toxin in host cells contributes to the pathology of anthrax infection. Therefore, understanding the cellular response to LT has important implications for the prevention and treatment of anthrax. The major findings of these studies show that macrophages produce SAA in response to LT and that treatment with SAA protects macrophages from LT. Furthermore, the ability of SAA to activate p38 MAP kinase appears to play a direct role in the protective effects of SAA. Therefore, this work indicates that SAA is a critical contributor to inducible resistance to LT, and for the first time suggests a role for this acute phase protein in protection against anthrax.
The four functional forms of murine SAA are differentially expressed in various extrahepatic cell types where they are differentially transcribed in response to pro-inflammatory cytokines [18]. Each of the SAA isoforms in the mouse are >74% identical at the nucleotide level indicating that the proteins are highly conserved. Therefore, they may serve equivalent or overlapping functions, particularly at the high concentrations that are induced physiologically in acute phase responses [10]. Consistent with this idea, a composite form of recombinant human SAA-1,2, both acute response SAA isoforms in man [10], prevented LT-induced cytotoxicity of Balb/c and C57Bl/6 macrophages, although only SAA-4 was rapidly and specifically induced in response to LT. SAA2 levels also increased with LT treatment, albeit more slowly and for a shorter duration compared to SAA-4. Future studies may demonstrate that specific isoforms of SAA provide enhanced protection against LT or other pathogens.
The mechanisms that influence a cell’s susceptibility to LT are not certain, but several observations suggest that both inherent and inducible factors contribute to differential susceptibility to LT. For example, polymorphisms in the gene encoding Nalp1b influence sensitivity to LT by Nalp1 activation of caspase-1 [30]. In addition, some cell types, such as macrophages are particularly sensitive to the effects of LT. Furthermore, sensitive macrophages can be rendered resistant to LT through a mechanism termed toxin-induced resistance (TIR) [7]. It was recently suggested that TIR involves the differential induction of apoptosis related proteins, Bnip and BnipL, as well as the Akt-dependent regulation of glycogen synthase kinase 3 beta activity [31]. Yet, whether macrophage sensitivity to LT is modulated under more dynamic conditions, such as those encountered during early stages of the host immune response, is not known.
Macrophages from both mouse strains that we examined were equally capable of responding to exogenous SAA, despite the fact that Balb/c macrophages were slightly more sensitive to the effects of LT than C57Bl/6 macrophages. Although alternative schemes might be imagined, and SAA may be of secondary importance to those processes, we propose the following hypothetical model to explain how toxin-induced resistance of cells might occur. Initial exposure of macrophages to low doses of LT causes the immediate release of SAA protein. SAA then activates p38 in either the same cell or nearby cells, perhaps via the CD36 receptor. After p38 activation, (which can clearly occur within five minutes after exposure to LT), and induction of downstream regulatory pathways, further activation of p38 is unnecessary for cell survival. Thus, the complete cleavage of MEK kinases by LT, which occurs within 6 hours in vitro and prevents further p38 activation [6], would be irrelevant because downstream mediators have already been activated. It is premature to speculate much on the identity of those downstream mediators, although it seems likely that they may include transcription factors and anti-apoptotic proteins. The identification of these downstream survival factors is of key importance for understanding how SAA protects macrophages from LT-induced cytotoxicity and is the subject of future studies.
Others have shown that SAA activates p38, JNK1 and 2 and ERK proteins in human intestinal epithelial cell lines resulting in NF-κB activation and IL-8 secretion [32]. Although we could not demonstrate activation of ERK or JNK pathways (data not shown), in our system SAA-induced p38 phosphorylation was necessary to protect murine macrophages from LT-induced cytotoxicity. It is unclear whether SAA might abrogate some of the effects of LT in human cells. However, small doses of SAA have recently been shown to extend human neutrophil viability in culture by delaying constitutive apoptosis [33], suggesting that SAA protect human as well as murine macrophages from LT-induced cell death. Thus, our data have important implications for understanding how acute phase responses may contribute to protection from the pathology of anthrax.
Acknowledgments
The authors wish to thank A. Windell for technical assistance, S. Wasson and B. Hurt for manuscript preparation, and Drs. M. Coggeshall and D. Capra for critical reading of the manuscript.
Footnotes
This work was supported by AI062629 (CFW and JDB) from the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xu L, Frucht DM. Bacillus anthracis: A multi-faceted role for anthrax lethal toxin in thwarting host immune defenses. Int J Biochem Cell Biol. 2006 doi: 10.1016/j.biocel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Baldari CT, Tonello F, Paccani SR, Montecucco C. Anthrax toxins: a paradigm of bacterial immune suppression. Trends Immunol. 2006;27:434–440. doi: 10.1016/j.it.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Moayeri M, Leppla SH. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med. 2009;30:439–455. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tonello F, Montecucco C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol Aspects Med. 2009;30:431–438. doi: 10.1016/j.mam.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Turk BE. Manipulation of host signalling pathways by anthrax toxins. Biochem J. 2007;402:405–417. doi: 10.1042/BJ20061891. [DOI] [PubMed] [Google Scholar]
- 6.Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–2051. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 7.Salles II, Tucker AE, Voth DE, Ballard JD. Toxin-induced resistance in Bacillus anthracis lethal toxin-treated macrophages. Proc Natl Acad Sci U S A. 2003;100:12426–12431. doi: 10.1073/pnas.2134042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moayeri M, Martinez NW, Wiggins J, Young HA, Leppla SH. Mouse susceptibility to anthrax lethal toxin is influenced by genetic factors in addition to those controlling macrophage sensitivity. Infect Immun. 2004;72:4439–4447. doi: 10.1128/IAI.72.8.4439-4447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha SD, Ng D, Lamothe J, Valvano MA, Han J, Kim SO. Mitochondrial proteins Bnip3 and Bnip3L are involved in anthrax lethal toxin-induced macrophage cell death. J Biol Chem. 2007;282:26275–26283. doi: 10.1074/jbc.M703668200. [DOI] [PubMed] [Google Scholar]
- 10.Cray C, Zaias J, Altman NH. Acute phase response in animals: a review. Comp Med. 2009;59:517–526. [PMC free article] [PubMed] [Google Scholar]
- 11.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 12.Upragarin N, Landman WJ, Gaastra W, Gruys E. Extrahepatic production of acute phase serum amyloid A. Histol Histopathol. 2005;20:1295–1307. doi: 10.14670/HH-20.1295. [DOI] [PubMed] [Google Scholar]
- 13.Kaur S, Singh PP. Serum amyloid P-component-mediated inhibition of the uptake of Mycobacterium tuberculosis by macrophages, in vitro. Scand J Immunol. 2004;59:425–431. doi: 10.1111/j.0300-9475.2004.01412.x. [DOI] [PubMed] [Google Scholar]
- 14.Badolato R, Wang JM, Stornello SL, Ponzi AN, Duse M, Musso T. Serum amyloid A is an activator of PMN antimicrobial functions: induction of degranulation, phagocytosis, and enhancement of anti-Candida activity. J Leukoc Biol. 2000;67:381–386. doi: 10.1002/jlb.67.3.381. [DOI] [PubMed] [Google Scholar]
- 15.Hari-Dass R, Shah C, Meyer DJ, Raynes JG. Serum amyloid A protein binds to outer membrane protein A of gram-negative bacteria. J Biol Chem. 2005;280:18562–18567. doi: 10.1074/jbc.M500490200. [DOI] [PubMed] [Google Scholar]
- 16.Mold C, Du Clos TW. C-reactive protein increases cytokine responses to Streptococcus pneumoniae through interactions with Fc gamma receptors. J Immunol. 2006;176:7598–7604. doi: 10.4049/jimmunol.176.12.7598. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto K-I, Shiroo M, Migita S. Diverse gene expression for isotypes of murine serum amyloid A protein during acute phase reaction. Science. 1986;232:227–229. doi: 10.1126/science.3456645. [DOI] [PubMed] [Google Scholar]
- 18.Thorn CF, Whitehead AS. Differential transcription of the mouse acute phase serum amyloid A genes in response to pro-inflammatory cytokines. Amyloid. 2002;9:229–236. doi: 10.3109/13506120209114098. [DOI] [PubMed] [Google Scholar]
- 19.O’Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Local expression of the serum amyloid A and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum. 2004;50:1788–1799. doi: 10.1002/art.20301. [DOI] [PubMed] [Google Scholar]
- 20.Yamada T, Wada A, Itoh K, Igari J. Serum amyloid A secretion from monocytic leukaemia cell line THP-1 and cultured human peripheral monocytes. Scand J Immunol. 2000;52:7–12. doi: 10.1046/j.1365-3083.2000.00734.x. [DOI] [PubMed] [Google Scholar]
- 21.Meek RL, Benditt EP. Amyloid A gene family expression in different mouse tissues. J Exp Med. 1986;164:2006–2017. doi: 10.1084/jem.164.6.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH. Serum amyloid A binding to formyl peptide receptor-like 1 induces synovial hyperplasia and angiogenesis. J Immunol. 2006;177:5585–5594. doi: 10.4049/jimmunol.177.8.5585. [DOI] [PubMed] [Google Scholar]
- 23.Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem. 2005;280:2954–2961. doi: 10.1074/jbc.M411555200. [DOI] [PubMed] [Google Scholar]
- 24.Munteanu A, Taddei M, Tamburini I, Bergamini E, Azzi A, Zingg JM. Antagonistic effects of oxidized low density lipoprotein and alpha-tocopherol on CD36 scavenger receptor expression in monocytes: involvement of protein kinase B and peroxisome proliferator-activated receptor-gamma. J Biol Chem. 2006;281:6489–6497. doi: 10.1074/jbc.M508799200. [DOI] [PubMed] [Google Scholar]
- 25.van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J Biol Chem. 2005;280:35890–35895. doi: 10.1074/jbc.M505685200. [DOI] [PubMed] [Google Scholar]
- 26.Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting Edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. 2008;181:22–26. doi: 10.4049/jimmunol.181.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riches DWH, Underwood GA. Expression of Intereron-beta during the triggering phase of macrophage cytocidal activation. J Biol Chem. 1991;36:24785–24792. [PubMed] [Google Scholar]
- 28.Webb CF, Dou S, Buchanan KL, Resta R, Smithson G, Smith EA. Reassessment of germline heavy chain transcripts from two murine VH families. Mol Immunol. 1997;34:743–750. doi: 10.1016/s0161-5890(97)00084-9. [DOI] [PubMed] [Google Scholar]
- 29.Baranova IN, Vishnyakova TG, Bocharov AV, Kurlander R, Chen Z, Kimelman ML, Remaley AT, Csako G, Thomas F, Eggerman TL, Patterson AP. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem. 2005;280:8031–8040. doi: 10.1074/jbc.M405009200. [DOI] [PubMed] [Google Scholar]
- 30.Boyden ED, Dietrich WF. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nature Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 31.Ha S-D, Ng D, Pelech SL, Kim SO. Critical role of the phosphatidylinositol 3-kinase/AKT/glycogen synthase kinase-3beta signaling pathway in recovery from anthrax lethal toxin-induced cell cycle arrest and MEK cleavage in macrophages. J Biol Chem. 2007;282:36230–36239. doi: 10.1074/jbc.M707622200. [DOI] [PubMed] [Google Scholar]
- 32.Jijon HB, Madsen KL, Walker JW, Allard B, Jobin C. Serum amyloid A activates NF-kappaB and proinflammatory gene expression in human and murine intestinal epithelial cells. Eur J Immunol. 2005;35:718–726. doi: 10.1002/eji.200425688. [DOI] [PubMed] [Google Scholar]
- 33.Kebir DE, Jozsef L, Khreiss T, Pan W, Petasis NA, Serhan CN, Filep JG. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: a novel mechanism for resolution of inflammation. J Immunol. 2007;179:616–622. doi: 10.4049/jimmunol.179.1.616. [DOI] [PubMed] [Google Scholar]