

Abstract
Mast cells (MCs) are well known effectors of allergic reactions and are considered sentinels in the skin and mucosa. In addition, through their production of cathelicidin, mast cells have the capacity to oppose invading pathogens. We therefore hypothesized that mast cells could act as sentinels in the skin against viral infections using antimicrobial peptides. Here, we demonstrate that mast cells react to Vaccinia virus (VV) and degranulate using a membrane-activated pathway that leads to antimicrobial peptide discharge and virus inactivation. This finding was supported using a mouse model of viral infection. Mast cell-deficient (Kitwsh−/−) mice were more susceptible to skin VV infection than the wild-type animals, while Kitwsh−/− mice reconstituted with mast cells in the skin showed a normal response to VV. Using mast cells derived from mice deficient in cathelicidin antimicrobial peptide, we showed that antimicrobial peptides are one important antiviral granule component in vivo skin infections.
In conclusion, our paper demonstrates that: MC presence protects mice from VV skin infection. MC degranulation is required for protecting mice from VV. Neutralizing antibody to the L1 fusion entry protein of VV inhibits degranulation apparently by preventing S1PR2 activation by viral membrane lipids. Antimicrobial peptide release from mast cell granules is necessary to inactivate VV infectivity.
Introduction
Only recently, the multifunctional nature of mast cells (MCs) has been revealed through their involvement in both innate and adaptive immune responses. Recent insight into the various functions of MCs has shown that these innate immune effectors possess the dual ability to kill microbes and to modify classical adaptive immune responses (1). MCs recognize bacteria with the aid of opsonins and through the expression of Toll-like receptors (TLRs) (2, 3) and the FimH receptor CD48 (4). MC activation subsequently leads to the expression of receptors and production of mediators that influence the activity of neutrophils, T cells and dendritic cells (5).
We have recently shown that MCs have the capacity to release molecules like antimicrobials peptides that are able to mediate an antiviral response (6). We therefore predicted that MCs could directly sense circulating viruses and use their granule contents to limit viral invasion. Viral recognition via the innate immune system is more challenging than recognition of other pathogen classes because of the relative lack of conserved and specific features (7). Hence, many cells of the innate immune system use nucleic acids as a means of detecting viral infection. In mammals, TLR3 is the sensor of double-stranded RNA. Single-stranded viral RNA is sensed by TLR7 and TLR8, while viral DNA is detected by TLR9. RIG-I, MDA5, and LGP2 play a major role in pathogen sensing of RNA virus infection to initiate and modulate antiviral immunity. VV can be sensed by MDA5 (8). MCs possess TLRs and they are functional (9–12). It is known that MCs use interferon to respond to virus infection when stimulated through TLR3 (9, 10). MCs capacity to produce interferon and TNF alpha has already been recognized as a potential to modulate T cell response to viral infections (13). However, it is not known if MCs are able to recognize the surface components of the virus and degranulate using a membrane-activated pathway rather than the TLR 3, 7, 8 or 9 signaling pathways.
The granules released by MCs are diverse in composition and contain the important antimicrobial peptide, cathelicidin. Antimicrobial peptides are ancient and potent host defense molecules that serve as natural and endogenous antibiotics (14) (15–19). In mammals, defensins and cathelicidin are the two major classes of antimicrobial peptides(20). Cathelicidin has been shown to have activity against viruses in vitro (21). Here, we have investigated, using MCs derived from mice deficient in cathelicidin, the possible involvement of cathelicidin as a mast cell anti-viral granular component in vivo and in vitro.
VV, the laboratory prototype for the poxvirus family, has a 200-year history of intentional human infection via the skin for the purpose of smallpox prophylaxis (22). Major complications of vaccination such as eczema vaccinatum are more common in individuals with abnormal skin immunity (23, 24). VV is a member of the Orthopoxvirus genus of the Poxviridae family, which includes variola (smallpox) virus, monkeypox virus, cowpox virus and ectromelia virus. VV is enveloped, contains double-stranded DNA, and has a 200-kb genome that encodes most of the proteins required for its cytoplasmic replication. VV infects skin and can cause skin lesions or rashes (25, 26). VV infection is a well-established in vivo model for study of skin infection (23, 24) (27–29). We therefore chose this mouse model to study the interaction of skin MCs and VV.
Early reports indicated VV enters cells through different routes including endocytosis (30, 31) and plasma membrane fusion (32–36). Recently VV has been shown to enter cells both by fusion with the plasma membrane and endocytic vacuoles depending to some extent on the virus strain and cell type (37, 38). The endocytic pathway involves macropinocytosis (39) or fluid phase uptake (40). In our study we will provide evidence that fusion of the mature virion (MV) is required to start the VV-MC interaction and response. The cell-derived lipid membranes of both the MV and enveloped (EV) virions contain many lipids including sphingomyelin (41). Sphingomyelin in the cell membrane can be converted to sphingosine-1-phosphate (S1P) which can activate the S1P2 G-coupled receptor (S1PR2) in an autocrine manner to stimulate MC degranulation (42–44). We will present data that demonstrate that this pathway is activated upon VV encounter and leads to mast cell degranulation.
There have been a few reports of mast cell involvement in viral infections through the initiation of a chemokine-dependent host response (45–50), and of histamine release in response to viral contact (45, 51, 52); however, the direct capacity of MCs to kill VV through antimicrobial peptides has not been reported before. Here, we show that MCs sense VV, degranulate, and can subsequently kill VV using their antimicrobial peptides. Using MCs derived from mice deficient in cathelicidin, we demonstrate that cathelicidin is a critical anti-viral granular component in vivo and in vitro. This study provides a novel insight into the role of MCs in antiviral response, the interaction of viruses with MCs, and suggests unexplored therapeutic avenues to resist viral infections.
MATERIALS AND METHODS
Mouse
Mast cell-deficient (C57BL/6-Kitwsh−/−) mice were a donation from Dr. Besmer’s laboratory (Developmental Biology Program, Memorial-Sloan Kettering Cancer Center at Cornell University, NY). The animals were bred at our facility. The Veteran Affairs and Institutional Animal Care and Use Committee approved all animal experiments. These mice have been extensively studied since they were generated (53, 54). Kitwsh−/− mice bearing the W-sash (Wsh) inversion mutation have mast cell deficiency but lack anemia and sterility. Adult Kitwsh−/− mice had a profound deficiency in MCs in all tissues examined but normal levels of major classes of other differentiated lymphoid cells. In adulthood, these mice may develop myeloid and megakaryocyte dysplasia in the spleen (55, 56). In our case 20–30 % mice exhibit splenomegaly. Hematopoietic abnormalities extend to the bone marrow and are reflected by neutrophilia and thrombocytosis. Kitwsh−/− mice can accept transplantation of genetically compatible bone marrow-derived cultured MCs with normal c-kit gene expression. The reconstitution of MCs can be done by adoptive transfer of these cells via intraperitoneal, intradermal or intravenous injection, without the development of other donor-derived hematopoietic cells (57, 58). The levels of lymphoid cells, including TCR gamma delta, are normal in adult Kitwsh−/− mice, and these animals do not exhibit a high incidence of spontaneous pathology affecting the skin, stomach or duodenum (59–61). Another mast cell-deficient WBB6F1/J-KitW/KitW-v mice (The Jackson Laboratory) were also used in this study. WBB6F1/J-KitW/KitW-v double heterozygotes are viable but sterile because of germ cell deficiency. They are also mast cell deficient. WBB6F1/J-KitW/KitW-v double heterozygotes lack intermediate cells, derived from melanoblasts, in the stria vascularis resulting in endocochlear degeneration, loss of endocochlear potential, and hearing impairment.
Cnlp−/− C57BL/6 mice were generated in Dr Gallo’s laboratory as previously described (62). Cnlp−/− has been backcrossed in C57BL/6 and used with C57BL/6 background for the last 5 years. Tlr2−/− were acquired and bred in our facility. Sex-matched wild type C57BL/6 littermate mice were used as wild-type controls throughout the study.
Cells
Primary MCs were generated by extracting bone marrow cells from the femurs of 5- to 8-week-old mice and culturing cells in RPMI 1640 medium (Invitrogen) supplemented with 10% inactivated FBS (Thermo Fisher Scientific), 25 mM HEPES (pH 7.4), 4 mM L-glutamine, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 50 µM 2-mercaptoethanol, 100 IU/ml penicillin and 100 µg/ml streptomycin. Recombinant murine IL-3 (1 ng/ml, R&D Systems) and recombinant murine stem cell factor (20 ng/ml, R&D Systems), both shown to support the in vitro growth and differentiation of the mast cell precursor, were also included (58, 63). Mouse mast cells derived from bone marrow cells with SCF and IL-3 become mature after 4 weeks of culture in vitro. We use SCF and IL-3, not WEHI supernatant (supernatant from a murine myelomonocytic leukemia cell line [WEHI-3 cells], rich in interleukin 3) as a source of mast cell growth factors to differentiate mast cells.
After 4 weeks, MCs were consistently generated as confirmed by the expression of CD117 (c-Kit) and FcεRI; cell maturation was confirmed by metachromatic staining with toluidine blue. The purity of MCs was greater than 98%. MCs were derived from bone marrow cells of Cnlp−/−, Tlr2−/− C57BL/6 mice, S1PR2−/− Balb/C ByJ mice and their wild-type littermate mice. Bone marrow cells of S1PR2−/− Balb/C ByJ mice and their wild-type littermate mice were a kind donation of Dr. Jerold Chun from The Scripps Research Institute.
MC adoptive transfer into MC-deficient mice
After culturing a sufficient number of donor MCs from mouse bone marrow cells, we counted cells and resuspended them in an appropriate volume of cold PBS, then kept on ice until intradermal cell injection(use 27-gauge needle and 1 ml syringe), within the back skin of anesthetized Kitwsh−/− mice. The number of total transferred MCs for one recipient mouse was 6×106 in 400 µl PBS (8×50 µl injections in two rows, down the length of shaved back) (63). Cell maturation was confirmed by metachromatic staining with toluidine blue prior to the transfer. A skin biopsy of recipient mouse was performed to demonstrate that MCs were resident in the skin at the time the experiment was performed after transfer (Supplementary Fig. 1G–H). The interval between mast cell transfer and the infection with VV is 2 weeks. The 2-week time interval is a modification of the original protocol that uses the mice 4–6 weeks from the adoptive transfer of the cells.
Viable cell counts
We used 0.4% Trypan Blue Stain (Invitrogen), a well-accepted method, for staining dead cells. Trypan blue is a dye which can enter cells with compromised membranes (64)
Virus
The purified VV (strain Western Reserve), and recombinant VV vB5R-GFP (65) and anti-L1 7D11 neutralizing monoclonal antibody for VV (66) have been previously described. BS-C-1 monkey kidney cells were grown to confluence in DMEM medium (Invitrogen) supplemented with 10% FBS for propagation of VV, and at 48–72 h post infection of BS-C-1 cells with VV, viruses were isolated and purified by sedimentation through two 36% (wt/vol) sucrose cushions followed by one sedimentation on a 25–40% (wt/vol) continuous sucrose gradient; the visible virus band was collected, and virus was pelleted and stored at −80 °C. On thawing for experiments, virus was sonicated on ice for 1 min before use (67). Virus plaque assays were performed on human keratinocyte cell line HaCaT or BS-C-1 monkey kidney cells as previously described with supernatant of tissue homogenate or cell culture media (68). Anti-L1 7D11 monoclonal antibody neutralization assay was performed as previously described (66). Briefly, viruses were incubated with 100% plaque reduction titer of 0.01 mg/ml anti-L1 7D11 monoclonal antibody in DMEM medium (Invitrogen) without serum for 30 min at room temperature before added to mast cells. UVC-inactivated VV was obtained by inactivating VV with UVC (254 nm) radiation dose of 7.6 mWs/cm2 at room temperature as previously described (69) (70).
Murine challenge with VV
UCSD IACUC System subcommittee on animal studies approved procedures. The backs of sex-matched adult littermates were shaved and hair was removed by chemical depilation (Nair; Church & Dwight). Mice were inoculated with 106 PFU of vB5R-GFP VV by scarification with 15 pricks into the skin (29) (27). At 3 days after infection, mice were euthanized with CO2 and the lesion areas were pictured alongside with a scale bar. Lesion size was measured using ImageJ (NIH) (71). And skin lesions and spleens were collected and homogenized with a Mini-Beadbeater (Biospec Products) for assessing cytokines concentrations by ELISA, VV titer by plaque assay and virus gene expression by quantitative real-time PCR. VV DNA expression was assessed using the same primers that we used for quantifying VV mRNA, but the substrate was the extracted DNA. For cromolyn experiments, wild type C57BL/6 littermate mice were injected intraperitoneally with 10 mg/kg body weight of cromolyn sodium salt (Sigma) once everyday for 4 days before experiments, and VV was inoculated on the fourth day after cromolyn injection, then cromolyn was still injected intraperitoneally once every day until the mice were sacrificed (72).
The mouse experiments were performed on groups of 4–9 mice for each treatment and repeated 3 times.
Histology
Animals were sacrificed on the third day of the experiment, and a skin sample of tissue adjacent to the infection wound was collected, fixed with buffered formalin, and embedded in paraffin for hematoxylin and eosin staining. Some sections were stained with toluidine blue to identify MCs and mast cell degranulation (73). If any immature mast cells were present in the section they were not stained by formalin fixation.
Real-time quantitative RT-PCR
Trizol Reagent (Invitrogen) was used to isolate total RNA. One µg of total RNA for cDNA synthesis by the iSCRIPT cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions was applied for each reaction; it was amplified by real-time RT-PCR in an ABI 7300 Real-Time PCR system (Applied Biosystems). The primers and probes used for real-time RT-PCR were from Applied Biosystems; RNA analysis reagents (TaqMan Master Mix reagents kit) were from Applied Biosystems. We used the comparative ΔΔCT method to determine the quantification of gene expression, normalized the target gene expression in the test samples to the endogenous reference GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) level and reported them as the fold difference relative to GAPDH gene expression in untreated baseline control (74). We performed all the assays in triplicate and repeated the experiments at least 3 times.
Early gene expression of VV
VV early gene expression was evaluated by using quantitative real-time PCR normalized to housekeeping gene GAPDH. Briefly, MCs were cultured in 48-well plates at a concentration of 106 cells/well. VV (106 PFU) was added to corresponding wells and incubated for 1 hour in a total volume of 100 µl, then 500 µl of mast cell media was added for additional 24 hour. RNA was isolated from cultured cells or homogenized tissues using TRIzol (Invitrogen) according to the manufacturer’s instructions. After reverse transcription with the iScript cDNA synthesis kit (Bio-Rad Laboratories), real-time PCR was performed by using an ABI 7300 Real-Time PCR system (Applied Biosystems). The following primer sequences were used to assay for the vaccinia gene transcripts: forward, 5'-GCCAATGAGGGTTCGAGTTC-3' and reverse, 5'-AACAACATCCCGTCGTTCATC-3'. This region of the genome encodes a subunit of a DNA-directed RNA polymerase expressed within 2 hour of viral entry (75). The TaqMan probe, 6FAM-ATTGAATTCTCTTCCCGCGGATGCTG, was purchased from Applied Biosystems. Samples were amplified in MicroAmp Optical 96-well reaction plates (Applied Biosystems) in a 25-µl volume containing 2X TaqMan Master Mix (Applied Biosystems), 200 nM forward primer, 200 nM reverse primer, 80 nM probe and template RNA. Thermal cycling conditions were 50°C for 2 min and 95°C for 10 min for the first cycle. Subsequently, samples were amplified for 50 cycles at 95°C for 15 s and 60°C for 1 min.
Flow Cytometry (FACS)
After treatment with vB5R-GFP VV, MCs were counter-stained with 1 µg/ml of propidium iodide (PI, Sigma) on ice according to the manufacturer’s instructions. Cells were analyzed with the Guava EasyCyte 8HT two laser, 6 color microcapillary-based benchtop flow cytometer (Millipore) for assessing infection rate and cell viability.
MC degranulation assay
Degranulation was assessed by measuring the activity of β-hexosaminidase in the supernatants (76–78) of 1×105 MCs in 200 µl Tyrode's buffer (0.1% bovine serum albumin, 0.1% glucose, 2 mmol/L MgCl2, 137.5 mmol/L NaCl, 12 mmol/L NaHCO3, 2.6 mmol/L KCl, pH 7.4) incubated for 1 hr with 1×105 PFU VV, HSV-1, adenovirus, or UVC-inactivated VV. For each sample assayed, supernatant aliquots (20 µl) were mixed with substrate solution (100 µl), which consisted of 1 mM 4-methylumbelliferyl-2-acetamide-2-deoxy-β-D-glucopyranoside (Calbiochem) in 0.1 M sodium citrate buffer (pH 4.5), and were incubated for 30 min at 37°C. The reaction was then stopped by the addition of 12 µl of 0.2 M glycine (pH 10.7). The reaction mixtures were excited at 365 nm and measured at 460 nm in a fluorescence plate reader (Gemini EM microplate spectrofluorometer, Molecular Devices). To determine the total cellular content of this enzyme, an equivalent number of cells were lysed with 1 % triton-X-100 (Sigma). Release of β-hexosaminidase was calculated as the percentage of the total enzyme content. MC stabilizer, cromolyn (Sigma, 1 µg/ml, 5 min at 37°C), was used to block mast cell degranulation.
MC granule isolation
To isolate the MC granules and the granule remnants we followed a classic protocol (79). Briefly, MC granule release, from Cnlp−/− MCs and wild type MCs, was induced with compound 48/80 (1 µg/ml, 5 min at 37°C), then MCs were separated by centrifugation and the supernatant containing granules and granule remnants were used and treated with VV. For each condition, 1×106 MCs were used in 1 ml Tyrode's buffer. Baseline control was the supernatant from MCs treated with PBS. VV stock solution 10 µl (5×105PFU) was added to 1 ml solution containing mast cell granules for 24 hour at 37°C. The outcome was measured as VV titer in the supernatant by the plaque assay performed on BS-C-1 cells.
ELISA
We used ELISA kits (eBioscience) to determine mouse interleukins in all supernatants of tissue homogenate or cell culture media according to the manufacturers' instructions. For tissue samples, we normalized ELISA measurements to total protein content of the sample (using BCA Protein Assay kit from Pierce) (80).
Fluorescence images
MCs were stained with 1 µg/ml of propidium iodide (PI, Sigma) on ice, then attached to a glass slide by using Shandon Cytospin 2 cytocentrifuge (Thermo Fisher Scientific). Prior to Cytospin Cells were resuspended at 106/ml. Each slide had the same amount of spun cells. Slides were mounted in ProLong Anti-Fade reagent with DAPI (Molecular Probes). For autophagy assay, we used LC3B antibody Kit for autophagy (Invitrogen) according to the manufacturers' instructions. We imaged the MCs using the Bx51 research microscope (Olympus) and X-Cite 120 fluorescence illumination systems (EXFO Photonic Solutions).
Golgi membrane separation
The Golgi membrane separation was done by gradient ultracentrifugation as previously described (81). Briefly, the whole homogenate of HaCaT cells (human keratinocyte line) is adjusted to 1.4 M sucrose (Sigma) and layered under a discontinuous sucrose density gradient of 1.2 M and 0.8 M sucrose. After centrifugation for 2 hour in an ultracentrifuge at 110,000 × g, 4°C., the Golgi membranes, which have a much lower density than those of the other membrane particles, float upwards to band at the 1.2 M/0.8 M sucrose interface.
Autophagy assay for mast cell
We used LC3B Antibody Kit for Autophagy (Invitrogen) according to manufacturer's instructions.
Sphingosine 1-phosphate (S1P) signal blocking assay
A S1P receptor 2 (S1PR2) specific antagonist, JTE-013 (10 µM, Tocris Bioscience), was added to MCs 30 min before virus infection (MOI=1) S1P (20 µM, Cayman Chemical) was added to MCs immediately after virus inoculation. The IC50=22 nM of JTE013 is used for human and rat receptors. However, in mouse mast cells, most research used JTE013=10 µM (82). In some experiments, desipramine (10 µM, Sigma), an inhibitor of acidic sphingomyelinases, and N,N-dimethylsphingosine (DMS, 10 µM, Cayman Chemical), a competitive inhibitor of sphingosine kinase, were added to MCs 30 min before virus infection.
Dot blot analysis
Supernatants from in vitro MCs culture were collected 24 hours after VV infection (MOI=1), and 100 µl was applied to a nitrocellulose membrane (Whatman) using S&S Minifold I Dot-Blot System (Schleicher & Schuell). The membrane was blocked in Odyssey Blocking Buffer (LI-COR) and then incubated with a rabbit polyclonal anti-CRAMP antibody (0.25 µg/ml) (Antibodies against the CRAMP peptide were prepared by Quality Controlled Biochemicals, Hopkinton, MA.) in Odyssey Blocking Buffer for 1 hour. Incubation with the secondary antibody, IRDye 680 Goat anti-rabbit secondary antibody (LI-COR) diluted 1:15,000 in Odyssey Blocking Buffer, was performed for 1 hour. Immunoreactivity was visualized using an Odyssey Infrared Imaging System (LI-COR) according to the manufacturer's instructions. All incubations were performed at room temperature.
Statistical analyses
All data are presented as mean ± SD. At least 3 independent experiments were performed to assess the reproducibility of the different experiments. The two-tailed t-test and one-way or two-way ANOVA with Bonferroni’s post-test of GraphPad Prism Version 4 were used to determine significance between two groups or multiple groups. For all statistical tests, P values <0.05 were considered statistically significant (*P<0.05, **P<0.01, ***P<0.001).
RESULTS
Mast cell deficient mice are more susceptible to VV infections
To study the role of dermal MCs in combating skin initiated viral infections, we infected mast cell-deficient mice (Kitwsh−/−) and wild-type littermates (C57BL/6) with VV. Mice were scarified and 106 plaque forming units (PFU) of VV was applied on the lower back. The Kitwsh−/− mice developed lesions at 24 hours, and the lesions increased in size over time while the wild-type littermates developed only a few pinpoint lesions at 3 days post-infection (Fig. 1A–B). A histology section of the wild-type skin at the inoculation site showed a significant increase in the number of MCs (Supplementary Fig. 1A), thereby implicating MCs in the viral response.
Figure 1. VV infection in mast cell-deficient Kit wsh−/− mice.
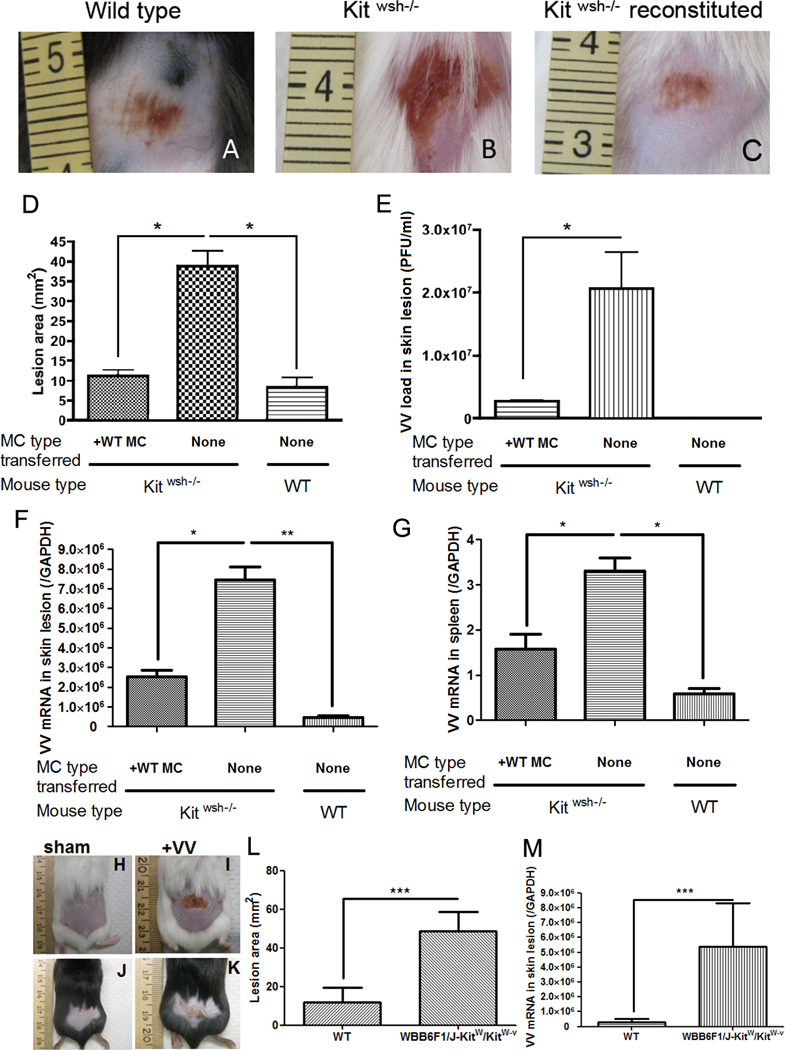
106 PFU of VV was applied on the lower backs of Kitwsh−/− mice and their wild-type littermate control mice. (A) Left image is representative of the reaction seen at 72 hours in wild type mice. (B) The center image is representative of the skin lesion in Kitwsh−/− mice. (C) The right image is Kitwsh−/− mice reconstituted with mast cells. (D) Quantification of lesion size (mm2) at 72 h post-infection. (E) Quantification of viral load (in PFU/ml) in the skin lesions at 72 h post-infection. (F) Expression of VV early gene at the skin lesions was quantified by real-time quantitative RT-PCR. (G) Expression of VV early gene in the spleen was quantified by real-time quantitative RT-PCR. *P<0.05. VV infection in mast cell-deficient WBB6F1/J-KitW/KitW-v double heterozygotes mice (H–M) 106 PFU of VV was applied on the lower backs of WBB6F1/J-KitW/KitW-v double heterozygotes mice and their wild-type littermate control mice. (H) Sham control of wild type mice scarified without VV application at 72 hours (I) Upper right image is representative of the skin lesion in WBB6F1/J-KitW/KitW-v double heterozygotes mice. (J) Sham control of WBB6F1/J-KitW/KitW-v double heterozygotes mice scarified without VV application at 72 hours (K) Lower right image is representative of the reaction seen at 72 hours in wild type mice. The right image is Kitwsh−/− mice reconstituted with mast cells. (L) Quantification of lesion size (mm2) at 72 h post-infection. (M) Expression of VV early gene at the skin lesions was quantified by real-time quantitative RT-PCR at 72 h post-infection.
Three independent experiments with 5 mice per group were performed for each experiment
Our evaluations were based on the wound development after viral scarification. Since it was previously reported (83) that abnormal wound repair occurs in of Kitwsh−/− mice we also ruled out the possibility of an abnormality in the scarification healing of Kitwsh−/− mice, by performing in parallel, the scarification of Kitwsh−/− mice without virus application. In our scarification model, Kitwsh−/− mice displayed normal scarification repair (Supplementary Fig. 1B).
To ensure that our observation was not related to the particular strain of mice that we were using, we also repeated the experiment using of a second mast cell deficient model such as the W/W-v. As you can see in Fig. 1 H–M, we obtained overlapping results, clinically (Fig. 1H–L) and biologically (Fig. 1M).
Reconstituted mast cell deficient mice have normal susceptibility to VV infections
To confirm that the susceptibility of Kitwsh−/− mice to VV was directly related to mast cell activity and not to other unknown defects present in the mice, we used an adoptive transfer mouse model. Mast cell-deficient mice were used as recipients of MCs derived from wild-type littermates. MCs derived from the bone marrow of wild-type littermates were differentiated in culture and injected intra-dermally into the Kitwsh−/− mice upon maturation. A skin biopsy was performed to demonstrate that MCs were resident in the skin at least 2 weeks after transfer (Supplementary Fig. 1 F–J). After 2 weeks, the reconstituted Kitwsh−/− mice and two control groups, including non-reconstituted mast cell-deficient mice and wild-type littermates, were scarified in the presence of 106 PFU of VV. The mast cell-deficient mice reconstituted with wild-type MCs had significantly smaller lesions than the non-reconstituted mice (Fig. 1C). The lesion size was quantified with NIH ImageJ software (71, 84) and expressed in mm2 (Fig.1D). After 3 days, the lesioned skin and spleens were excised and processed in TRIzol for RNA extraction to determine viral replication. We quantified the early expression of VV (DNA-directed RNA polymerase mRNA which encodes a gene subunit expressed within 2 hour of viral entry) in the wound site and spleen by real-time quantitative polymerase chain reaction (qPCR). Kitwsh−/− mice had a significantly higher expression of viral RNA compared to wild-type littermates (Fig. 1F–G). To determine whether early gene expression was followed by virus replication, we harvested and homogenized the tissue and seeded the supernatant on BS-C-1 cells to count plaque formation (PFU) (Fig. 1E). Kitwsh−/− mice had a significantly higher level of infectious virus than wild-type littermates. Mast cell reconstituted Kitwsh−/− mice showed similar levels of infectious virus as compared to the wild-type littermates that correlated with the wound clinical presentation (Fig. 1E). The reconstituted mice showed similar viral levels to the wild-type littermates, as the mast cell reconstitution of Kitwsh−/− mice prevented the spreading of the virus to the circulation and the spleen (Fig. 1G). We studied the number of MCs at the site of inoculation (supplementary Fig. 1J) and their capacity to degranulate in wild-type and Kitwsh−/− reconstituted mice (supplementary Fig. 2 A–B). The results showed no differences between reconstituted and wild-type littermates. A panel of pro-inflammatory and anti-inflammatory cytokines was tested and TNFα, IL-6 and IL-10 showed significant changes. The reconstituted mice showed a higher level of TNF-α (columns 1, 2 and 3 vs 4 Fig. S1C) and a lower level of IL-10 (columns 1, 2 and 3 vs 4 Fig. S1E) as expected. Levels of IL-6, a pro-inflammatory cytokine, increased in all the infected groups (columns 1, 2 and 3 vs 4 Fig S1D) when compared to non-infected groups, but were much higher in the Kitwsh−/− non-reconstituted infected mice (Supplementary Fig. 1 C–E). Kitwsh−/− mice have been reported to have myeloid and megakaryocyte hyperplasia which could have explained the defective immunity noticed in the mice after viral challenge(60); however, since reconstitution with MCs promoted normal viral response, we concluded that the defect observed was primarily due to the absence of MCs. The absence of MCs impairs the capacity of the skin to respond to VV infections and can be corrected by the reconstitution of MCs in the skin.
MC degranulation reduces the severity of VV infection
On the basis of the in vivo data, we next sought to determine the mechanism of MC antiviral action. We hypothesized that MCs can act against pathogens through their ability to degranulate and release antimicrobial molecules. We demonstrate that MCs are resistant to VV infection up to a MOI of 0.1 (Fig. 2A–C) and they begin to degranulate on contact with VV as evidenced by β-hexosaminidase release (1 hour observation in Fig. 2D). Addition of cromolyn, a well-described inhibitor of mast cell degranulation (5) (Fig. 2D), increased viral susceptibility and resulted in an increased VV-induced cell death (Fig. 2E). Cromolyn itself has no inhibitory activity on VV (Supplementary Fig 2 C).
Figure 2. Mast cells degranulate upon contact with VV.
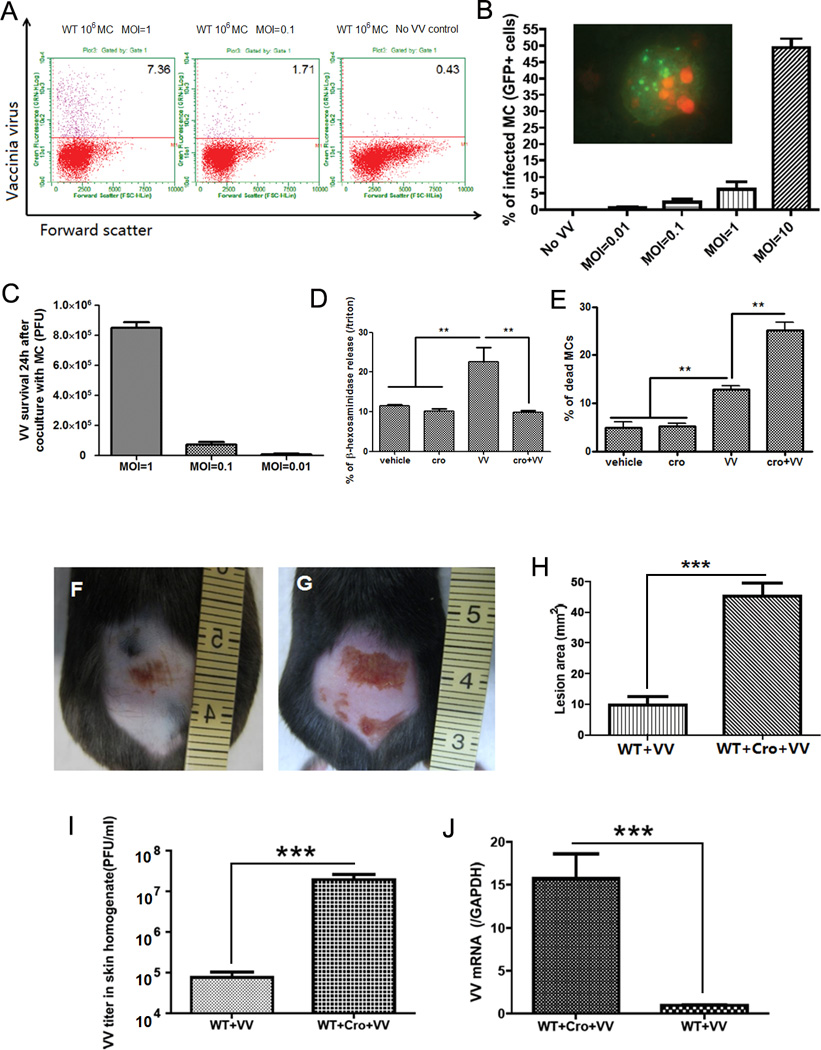
(A) Mast cells were infected with VV at different MOI for 24 h and analyzed by FACS. The right plot shows resting mast cells before virus interaction; the two other plots show cells after contact with VV. The upper quadrant number is the percentage of infected mast cells. (B) FACS analysis of VV expression in mast cells infected with VV (GFP+ cells) at different MOI. The insert shows green fluorescence in mast cells infected with VV. The nucleus was stained in red with propidium iodide (PI). (C) VV titer 24 h after co-culture VV with mast cells at different MOI. VV titer in the supernatant was expressed as PFU by plaque assay in BS-C-1 cells. (D) Mast cell β-hexosaminidase release (as index of granule content release) 1 h after contact with VV. The granule release was blocked by adding cromolyn (cro) to the medium. **P<0.01 compared to other groups. (E) Mast cell death was measured with trypan blue 1 h after contact with VV. The number of dead cells was increased by cromolyn in the medium. *P<0.05. All the in vitro experiments have been performed in triplicate (F) Wild-type C57BL/6 mice at 72 h following VV inoculation. (G) Cromolyn pretreated wild-type C57BL/6 mice at 72 h following VV inoculation. (H) Quantification of skin lesion size (mm2) in non-pretreated and cromolyn-pretreated mice at 72 h post infection. (I) Quantification of viral load in skin lesions expressed as PFU/ml. (J) Quantification of VV early gene expression in skin lesions by real-time quantitative RT-PCR. ***P<0.001. Three independent experiments with 4 mice per group were performed for each experiment
To provide better insight into the role of mast cell degranulation in viral immunity, we investigated the antiviral activity of MC granules in vivo. We pretreated wild-type littermates with cromolyn sodium for 4 days before VV application and every 24 hours for the duration of the experiment. Mice pretreated with cromolyn developed a lesion that was compatible in size to the Kitwsh−/− mice lesion and significantly larger than lesions on the wild-type littermates (Fig. 2F–H). Viral replication was also higher in the cromolyn pretreated when compared to untreated (Fig. 2I–J), suggesting that MC degranulation attenuates VV infection.
VV envelope fusion induces MC degranulation independent of TLR2
Since these observations demonstrate that MCs respond to VV infection, we next wanted to determine how MCs degranulate upon encountering VV. We hypothesized that the VV envelope contains the signal for mast cell degranulation. Because the envelops contains many different components partly from cells and partly specific to the virus, we tested our hypothesis in different ways. First to rule out that cell contaminants were responsible of inducing degranulation, we used purified VV (Fig. 3A) in comparison with unpurified VV and we showed that there are no differences in degranulation. Second we used short wavelength UVC (254 nm) inactivated VV (the virions lack infectivity but not fusion capacity with the cells (85, 86) (Fig. 3A). We demonstrated that UVC inactivated VV still induced cell degranulation (Fig. 3A). Third, we blocked one viral membrane protein L1 essential for viral fusion with a specific neutralizing antibody to L1 (Fig 3A). In the presence of the neutralizing antibody the cells did not degranulate suggesting that the function of the L1 protein in mediating virus entry is required for mast cell degranulation.
Figure 3. Mast cells degranulate on contact with viral envelopes.
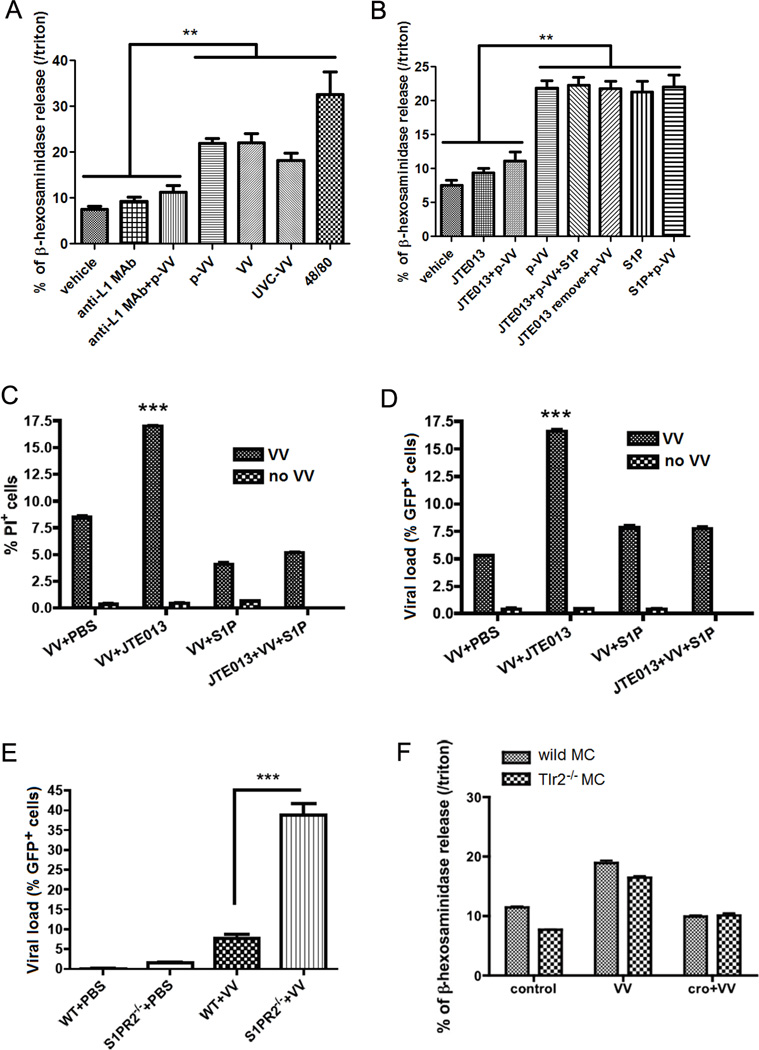
(A) β-hexosaminidase release of mast cells upon contact with live purified VV (p-VV), UVC-inactivated VV (UVC-VV), live unpurified VV at MOI=1 for 1h. VV, p-VV and UVC-VV induced mast cell degranulation. Anti-L1 MAb control (Anti-L1 MAb) and Anti-L1 MAb pretreated virus (Anti-L1 MAb +VV) did not induced degranulation. 48/80 is the positive control for degranulation. NS: no significant difference between VV and p-VV treatment. **P<0.01 compared to other treatments. (B) Mast cell β-hexosaminidase release (as index of granule content release) on contact with purified VV (p-VV) at MOI=1 for 1h. The granule release was blocked by JTE013, the S1PR2 specific antagonists (JTE013+p-VV). When JTE013 was removed from the medium, p-VV inoculation (JTE013 remove+p-VV) can induce mast cell degranulation, which showed that mast cells were not damaged by JTE013 addition. ***P<0.001. (C) FACS analysis of mast cells stained with PI as index of cell damage 24 h after VV infection at MOI=1. The cells pretreated with JTE013 had the most damage, but addition of S1P with JTE013 prevented the cell damage. ***P<0.001 compared to all the other groups. (D) FACS analysis of mast cells inoculated with GFP+ VV at MOI=1 for 24 h (GFP+ cells as viral load marker). The cells pretreated with JTE013 (JTE013+VV) had the highest viral load, and addition of S1P with JTE013 (JTE013+VV+S1P) decreased mast cell viral load. ***P<0.001 compared to all the other columns. (E) FACS analysis of mast cell from S1PR2−/− Balb/C ByJ mice and their wild type littermate control mice. Mast cells were inoculated with GFP+ VV at MOI=1 for 24 h. The S1PR2−/− cells (S1PR2−/− +VV) had the highest viral load (GFP+ cells). ***P<0.001. (F) β-hexosaminidase release of mast cells from Tlr2−/− mice and their wild-type littermates on contact with VV at MOI=1 for 1 h with or without cromolyn (cro) pretreatment. All the in vitro experiments have been performed in triplicate
When VV entering cells by membrane fusion, sphingomyelin and other PS lipids in VV envelopes can be discharged in MC membrane; the discharge will lead to a chain of events that includes the formation of sphingosine-1-phosphate (S1P) and activation of the S1P2 G-coupled receptor (S1PR2). This final event stimulates MC degranulation (42–44). To verify that this is the exact pathway involved in mast cell-VV degranulation, we added S1PR2 receptor-specific antagonist, JTE013 (87) to MCs before VV inoculation. In other cell types JTE013 also blocks S1PR4, but MCs lack this receptor (44). We show that MCs treated with JTE013 failed to degranulate in response to VV (Fig. 3B); moreover, they were more susceptible to virus-induced cell death (Fig. 3C) and exhibited a higher viral load (Fig. 3D). The exogenous addition of S1P rescued the effect of the JTE013 block (Fig. 3B–D). And the removal of JTE013 from the medium reverted the capacity to respond to VV, demonstrating that MCs were specifically blocked but not damaged. Because the use of JTE013 at the required blocking concentration may raise the question of the specificity of its effects, we also challenged S1PR2 knockout MCs (S1PR2−/−) with VV (Fig. 3E) and demonstrated that these cells were more sensitive to viral infection. To support previously discussed results, the infection (Supplementary Fig. 3C) was proportional to the amount of degranulation (Supplementary Fig. 3D). In Supplementary Fig. 3E, we demonstrated that as well as blocking S1PR2 with JTE013, the block of sphingosine kinase (the enzyme that generates S1P) with N,N-dimethylsphingosine (DMS) significantly increased the percentage of infected MCs, which can be rescued by addition of S1P. Whereas inhibiting acidic sphingomyelinases, a non-membrane enzyme involved in sphingomyelin metabolism, with desipramine does not increase VV infectivity in MCs. Because sphingomyelin is present in both viral membranes and Golgi membranes, we isolated Golgi membranes and demonstrated that they were able to induce degranulation in a dose dependent manner (Fig. 3A and Supplementary Fig 3B); third, we also showed that HSV-1 enveloped virus induced mast cell degranulation (Supplementary Fig. 3A); On the other hand, adenoviruses, which are non-enveloped viruses, failed to induce a significant amount of degranulation (Supplementary Fig. 3A).
TLR2, one of the pattern recognition receptors in the skin, has been previously implicated in VV infectivity and cytokine responses in different cell types (88, 89). To investigate if TLR2 was important for VV-induced degranulation in MCs, we utilized Tlr2−/− MCs. MCs were derived from the bone marrow of wild-type and Tlr2−/− mice. Wild-type and Tlr2−/− MCs were efficiently infected by VV and their capacity to degranulate was equivalent to the wild-type cells (Fig. 3F). These data suggest that TLR2 is not necessary for VV-induced degranulation in MCs.
Expression of Cathelicidin (mCamp) is critical in MC defense against VV in vivo
We have demonstrated that MCs function through degranulation to attenuate VV infection. We next wanted to investigate how degranulation influences the MC antiviral capacity. Cathelicidin antimicrobial peptides, previously shown to have antiviral activity in vitro and in vivo (90, 91), are expressed by MCs in vivo (6). We demonstrated that mCamp is released during VV-induced degranulation (Fig. 4A). Notably, in the cromolyn-pretreated mice, the mCamp levels at the site of the infection were lower than in the non-pretreated mice (Fig. 4B). A similar low level of mCamp was found in non-reconstituted Kitwsh−/− mice at the site of VV infection in skin when compared to wild type littermates (Fig. 4C). To verify the biological relevance of these findings, we reconstituted Kitwsh−/− mice with Cnlp−/− MCs (deficient in cathelicidin antimicrobial peptides) or wild-type MCs. Wound development and viral load were compared after viral challenge. The Kitwsh−/− mice reconstituted with wild-type MCs had a reduced viral load and reduced wound development when compared to Kitwsh−/− mice reconstituted with Cnlp−/− MCs as assessed by the wound size measurements (Fig. 4D–H). This correlated with VV titer from the wound sites (Fig. 4I). Moreover, Kitwsh−/− mice reconstituted with Cnlp−/− MCs also showed an increased VV expression in the skin (Fig. 4I), spleen (Fig. 4J) and elevated levels of skin IL-6 (Fig. 4K). Only reconstitution with wild-type MCs maintained skin IL-6 levels comparable to wild-type littermates (Fig. 4K). The observed differences were not due to a different level of cell infiltration in the lesions (Supplementary Fig. 4A–C)
Figure 4. Antimicrobial peptide release is important for mast cell antiviral function.
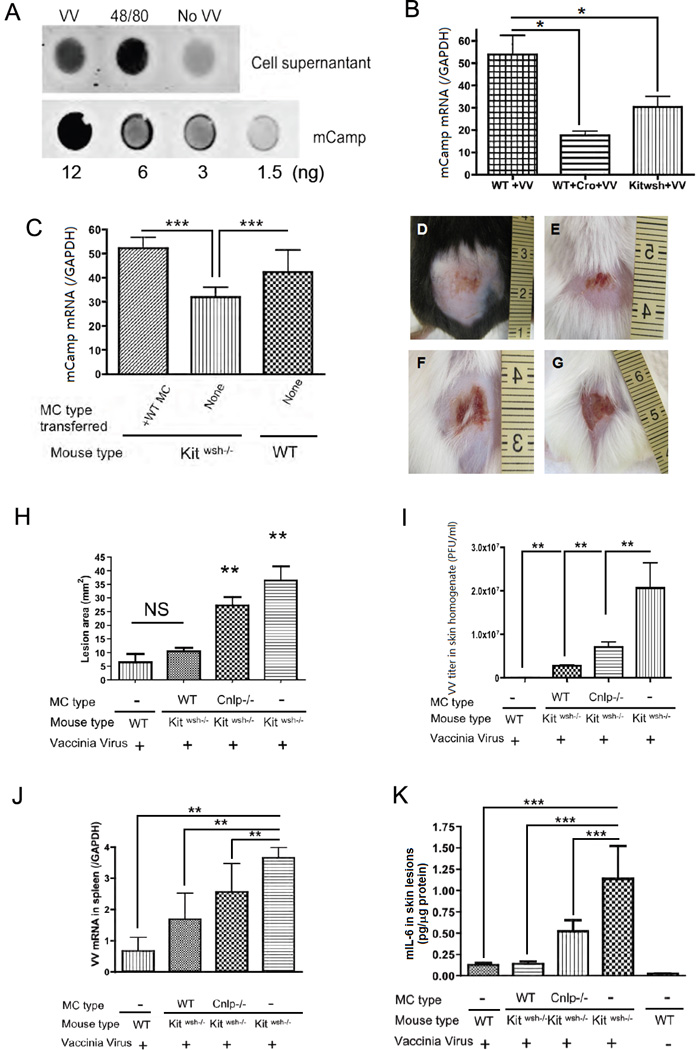
(A) Dot blot of the mCamp released in the supernatant when mast cells are exposed to VV at MOI=1 for 1 h and to compound 48/80 as positive control; the lower lane shows the standard blot of mCamp synthetic peptide. Experiments have been performed in triplicate (B) mCamp expression by real-time quantitative RT-PCR in VV induced skin lesions of Kitwsh−/− mice, their wild type littermates and cromolyn (Cro) pretreated wild type littermates 3 days post infection. *P<0.05. (C) mCamp expression by real-time quantitative RT-PCR in VV induced skin lesions of Kitwsh−/− mice, their wild type littermates, and Kitwsh−/− mice reconstituted with wild type mast cells 3 days post infection. ***P<0.001 compared to all the other groups. (D–K) Kitwsh−/− mice reconstituted with mast cells from Cnlp−/− mice and their wild type littermates. Representative lesions from wild-type littermates (D), Kitwsh−/− mice reconstituted with wild type mast cells (E) and Kitwsh−/− mice reconstituted with Cnlp−/− mast cells (F) and Kitwsh−/− mice (G) at 72 h following VV scarification. (H) Quantification of lesion size (mm2) in wild-type littermates, Kitwsh−/− mice and mast cell-reconstituted Kitwsh−/− mice at 72 h post infection. **P<0.01 compared to other groups. NS: no significant difference. (I) Quantification of VV load (PFU/ml) in skin lesions. **P<0.01. (J) Quantification of VV early gene expression in the spleen by real-time quantitative RT-PCR. **P<0.01. (K) Mouse IL-6 ELISA quantification in skin lesions homogenate expressed as pg/µg tissue protein. ***P<0.001. Three independent experiments with 5 mice per group were performed for each experiment
To investigate whether cathelicidin expression in MCs is directly coupled with their antiviral response, we compared the infectivity of Cnlp−/− MCs to wild-type MCs. We derived MCs from the bone marrow of wild-type and Cnlp−/− mice and infected with VV. Cnlp−/− MCs were more sensitive to virus compared to wild-type MCs. They demonstrated more viral RNA and DNA (Fig. 5A–B), a higher VV titer in the supernatant and more VV infected GFP+ cells when compared to wild type (Fig. 5C–E). To further confirm that the Cnlp−/− MCs impaired capacity to kill VV was linked to their granule content and release, MCs were pretreated with or without cromolyn sodium. As illustrated in Fig. 5F, Cnlp−/− cells have no advantage from the release of their granules upon viral encounter. Additionally, purified Cnlp−/− mast cell granules fail to kill VV, further demonstrating that the effects observed are not due to differences in granule release (Fig. 5G). The Wild type granules are directly killing VV, a dose-dependent effect that had not been observable with granules from Cnlp−/− MCs (Fig. 5G). Cnlp−/− MCs in vitro characterization is presented in the Supplementary Fig. 4D–H and shows that granule composition in TNF-α and metachromasia is similar between Cnlp−/− and wild-type cells and that there were no changes in the number of degranulating cells or in autophagy formation on viral contact. When examining at 72 hours post VV infection in vivo, the presence of Cnlp−/− MCs is still an advantage over having no MCs at all (Fig. 4I–J).
Figure 5. Cathelicidin expression is important in mast cell defense toward VV.
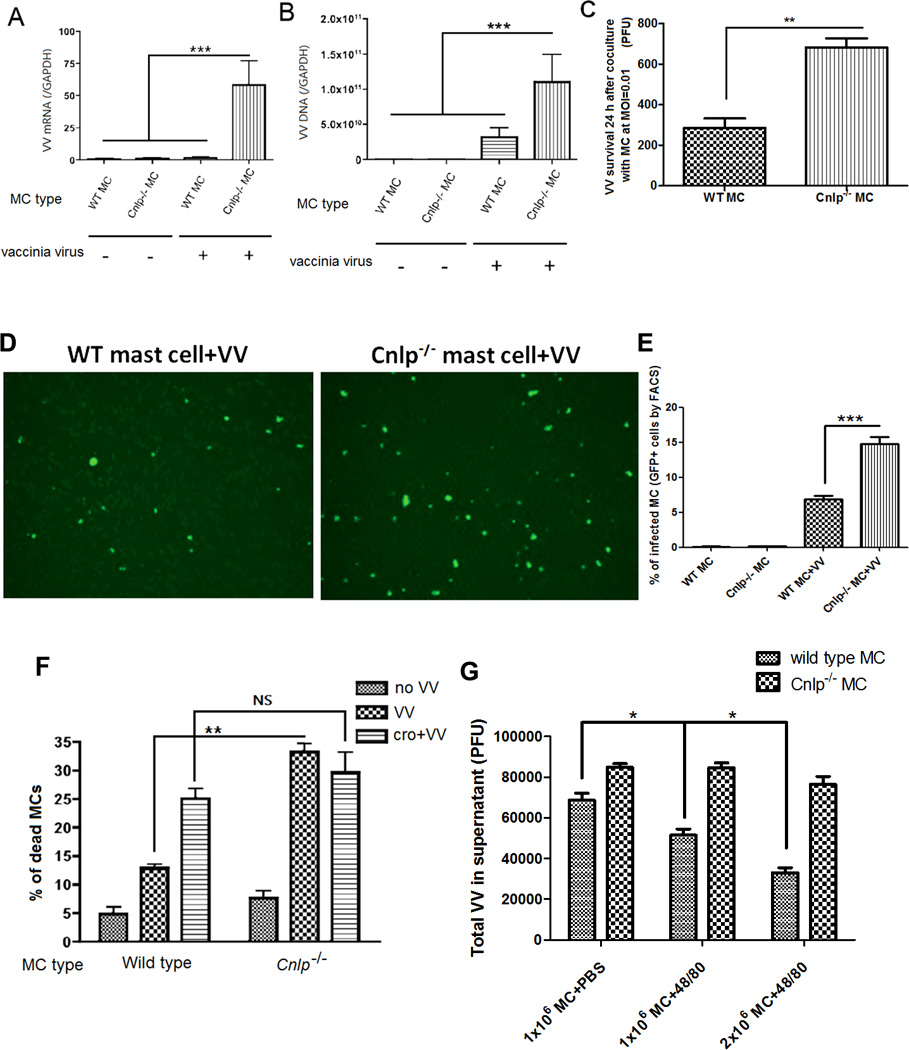
Mast cells generated from Cnlp−/− C57BL/6 mice and their wild type littermate mice inoculated with 106 PFU VV at MOI=1 for 24 h. Quantification of VV early mRNA expression. ***P<0.001 (A) and VV DNA expression. ***P<0.001 (B) by real-time quantitative RT-PCR. (C) VV titer in supernatants from Cnlp−/− and wild-type mast cells with VV at MOI=0.01 for 24 h. **P<0.01. (D) Fluorescence visualization of VV infected Cnlp−/− and wild-type mast cells treated with VV at MOI=1 for 24 h. Infected cells are bright green. The number of infected Cnlp−/− mast cells is significantly higher than infected wild type mast cells. (E) FACS quantification of VV infected Cnlp−/− and wild-type mast cells treated with VV at MOI=1 for 24 h (F) Mast cell death was measured with trypan blue 1 h after contact with VV at MOI=1. One group is pretreated with cromolyn before adding VV (cro+VV). **P<0.01. NS: no significant difference. The total number of cells per well were 111± 12 per sqmm, in the control cells; 98± 8 in the VV treated cells and 99±7 in Cro+VV treated cells for the WT cells; The total number of cells per well were 105± 7 per sqmm, in the control cells; 90± 7 in the VV treated cells and 100±7 in Cro+VV treated cells for the Cnlp−/− mast cells (G) Wild type and Cnlp−/− mast cells purified granule activity against VV. 1×106 mast cells +PBS represents baseline activity, 1×106 mast cells +48/80 represents purified granules from 1×106 mast cells and 2×106 mast cell +48/80 represents purified granules from 2×106 mast cells. With wild type mast cell granules, we observed a dose-dependent effect against VV that was not evident with granules from Cnlp−/− mast cells. *P<0.05. All the in vitro experiments have been performed in triplicate
DISCUSSION
We have demonstrated fundamentally new insights into VV immunity. First, MCs can sense VV and they resist infection proportionally to the viral load. Second, MCs degranulate on VV fusion via interaction with the viral lipid envelope. Third, the response to VV in skin is substantially mediated by degranulation and cathelicidin granule expression in MCs. To our knowledge, this is the first manuscript to describe the direct ability of MCs to elicit an antiviral response through antimicrobial peptide activity.
MCs have been long regarded as essential resident effector cells in the elicitation of the allergic response through the secretion and generation of bioactive mediators. Only in the last few years their nature as defense cells has been exploited. However, their antimicrobial activity has been primarily regarded as secondary to their activation of other host defenses such as neutrophil activation. In the past there have been a few reports that suggested a role for MCs in the initiation of chemokine-dependent host responses to virus infections, and of histamine release in response to viral contact(45, 51, 52); however, the direct capacity of MCs to kill VV through antimicrobial peptides has not been reported before (45–50). Our data provide evidence that MCs are directly involved in host defense against viral infections. Mast cell-deficient mice were significantly more susceptible to viral infections as they developed bigger wounds at the site of infection and had higher viral gene expression. Mast cell reconstitution reverted the phenotype to a normal response even after only 2 weeks from the reconstitution with in vitro matured cells. This in vivo phenomenon could be explained by either MCs directly killing the virus at the port of entry or by MCs releasing chemokines that mediate cell chemotaxis. However, according to our in vitro data, MCs directly kill viruses through degranulation. Many different cytokines have been studied in viral infections. The most representative and best studied for their interaction with VV are TNF-α, IFN-γ, IL-1β, IFN-β, IL-12, IL-6, and IL-10 (92). We therefore chose to study these cytokines and their relationship with MCs during a viral infection. We only reported the cytokines that showed any change in one or more mice groups, which were then selected for further studies. We examined cytokine protein level by ELISA. Mast cell-derived TNF-α plays a crucial role in the influx of neutrophils into lesions. Here, we observed that VV induced IL-6 and TNF-α secretion, especially in mast cell-deficient mice. On the contrary, mast cell presence decreases the viral load in the tissue affected by VV infection. IL-6 and TNF-α levels correlated with the clinical presentation of our infected mice, such that higher levels of IL-6 and TNF-α occur in more severe infections. Therefore, the absence of MCs amplifies the inflammation. IL-6 MC production has been recently implicated in cancer response (11), but levels of IL-6 do not usually increase in VV infections (93). The fact that, in our experiments, IL-6 levels increase in the absence of MCs, indicates a role for MCs in controlling the level of this interleukin in viral infections and inflammation at large. IL-10 is an anti-inflammatory cytokine that has recently been in the center of some mast cell debates (63). The absence of IL-10 when MCs are not present is considered to make lesion worse (57). We demonstrated that IL-10 levels decreased in the presence of VV in both the reconstituted and non-reconstituted Kitwsh−/− mice, indicating that the difference was not specifically due to variations in mast cell presence.
The mast cell degranulation process has been long regarded as secondary to the binding of IgE allergen specific antibodies. Recently, innate immune receptors such as TLRs were identified on the surface of MCs, which could allow MCs to directly recognize pathogens (94). Here, we hypothesized that MCs react to VV on contact and fusion is the probably the mechanism that triggers degranulation. Our in vitro data demonstrated that MCs, which fail to degranulate on viral contact, had higher viral titers and more cell death. This was also observed in vivo where mice with MCs that couldn’t degranulate were more prone to infection. Pretreatment with cromolyn sodium blocked mast cell activation and degranulation creating a phenotype very similar to mast cell-deficient mice during a VV infection. Cromolyn is a chromone medication that has been available for decades to manage allergic disorders. It has been used as an inhaler for asthma, a nasal spray for allergic rhinitis, eye drops for allergic conjunctivitis, an oral liquid for mastocytosis and food allergy, and an ointment for atopic dermatitis. Despite intensive research in many laboratories over the last 40 years, a precise mechanism to explain cromolyn's clinical activity has not been confirmed. However, the primary mode of action of cromolyn was thought for many years to be the stabilization of MCs and thus preventing subsequent release of mediators following appropriate challenge (95–97). We demonstrated that cromolyn inhibited mast cell degranulation in vitro, whereas in vivo, other mechanisms could be present amplifying MCs direct activity against viruses.
The mechanism of VV entering cells is not precisely known. Previous reports indicated VV enters cells through different routes including endocytosis (30, 31) and plasma membrane fusion (32–36, 98). Moreover, VV entry pathways and signaling differed depending on cell types (33, 98–102) as well as viral strains (37, 103). These studies revealed a complex relationship of VV entry processes with host cells. Bisht and co-workers demonstrated that the L1 protein is required for membrane fusion and entry (104) Using a specific monoclonal antibody against the L1 7D11 fusion protein, we demonstrated that viral envelope fusion is required for mast cell degranulation. The study was also supported by UVC treated virions that are able to fuse but not to be infectious. The VV membrane contains a variety of phospholipids (41). In a series of experiments, Ichihashi and collegues (105) showed that the loss of VV infectivity on Nonidet P-40 detergent extraction of lipids could be partially rescued by incubation of the extracted virions with exogenous lipids, including crude mammalian cell membrane preparations. Further studies implicated phosphatidylserine in VV entry (39, 106). We demonstrated a similar mechanism with respect to mast cell viral recognition and degranulation. Because VV envelopes discharge lipids on the mast cell membrane surface, we hypothesized that the sphingomyelin-ceramide-sphingosine-S1P membrane pathway was implicated in the activation of the degranulation process. This pathway ends with the production of S1P to stimulate S1PR2 (44). We demonstrated that blocking this receptor significantly reduced the degranulation response on viral contact. It has been previously recognized that degranulation of MCs derived from bone marrow of S1PR2 knockout mice is impaired (107); we confirmed this observation and we demonstrated that the cells derived from the knockout S1PR2 mice were more susceptible to VV infection.
This is an important mechanism for blocking enveloped viruses because it gives MCs the chance to kill as many viruses as possible before they infect the cells.
In our in vitro model, MCs degranulated when we used Golgi membranes even without the presence of viral proteins; however the quantity of lipids present in the Golgi preparation is very high in sphingomyelin and may generate S1P even without the necessity of fusion according to the hypothesis of the “sphingolipid rheostat" that activates cell membrane (108). Degranulation on envelope contact was not specific to VV as other enveloped viruses, like HSV-1, induced degranulation while adenovirus did not. So we can hypothesize that other enveloped viruses are recognized by mast cells through fusion and lipid discharge.
Other mechanism are certainly in place after the cells have been infected; MCs are equipped with TLR3, TLR7 and TLR9 intracellular receptors and the capacity of further responding to viral infections(9) (2). However, our data suggest that degranulation constitutes the first cellular lines of defense against VV. Thus, the composition of the granules becomes essential to the antiviral capacity of the cell. Antimicrobial peptides are an evolutionarily conserved component of the innate immune response and are found among all classes of life. Two antimicrobial peptides gene families expressed by mammalian mucosal epithelia, Cathelicidin and defensins, have received the most attention (109–111). They are both expressed in MCs. However, mast cell antimicrobial function against bacteria in skin is mediated by cathelicidin activation (58). Cathelicidin has been shown to have an in vitro and in vivo direct effect against viruses (21, 90, 112). Unlike the antibacterial activity of this peptide, the antiviral activity is not fully dependent upon disulfide bond formation. Viral inhibition appears to result, in part, from disruption of the envelope and/or capsid (21).
We demonstrated that the granule-derived antimicrobial peptide cathelicidin is critical for the mast cell response against VV. We first described that in the absence of MCs, mouse cathelicidin was minimally expressed at the site of infection, even 72 hours after the inoculation. The absence of MCs caused a lack of antimicrobial peptide presence either directly due to the absence of mast cell cathelicidin or indirectly due to the capacity of MCs to recruit other cell types producing antimicrobial peptides. In fact, in VV infections neutrophil recruitment begins from day 2 after VV inoculation, and reaches peak on day 10. (113–115). In the case of our experimental model of VV infection, cathelicidin is critical for its direct effect on defense during the initial phase of the infection (mice reconstituted with Cnlp−/− MCs have bigger lesions), but is not critical for the recruitment of other cell types like neutrophils (our data shows no differences in the cell infiltration at 72 hours between mice reconstituted with WT MCs and mice reconstituted with Cnlp−/− cells). However, the presence of VV in the spleen suggests that the initial window phase is critical for the systemic distribution of VV. This led us to confirm these results in vitro and further investigate the mechanism. Our data indicate that Cnlp−/− MCs have a significantly decreased ability to suppress VV proliferation. We demonstrated that Cnlp−/− cells that lack cathelicidin were more susceptible to VV in vitro.
Cathelicidin is reduced in patients with atopic dermatitis and cathelicidin deficiency is linked to the susceptibility to infection with VV and herpes simplex viruses(21, 90, 116). Our data may explain why a disease with normal mast cell number, but low cathelicidin levels has an increased susceptibility to VV infections.
Cathelicidin primarily function as antimicrobials; however, it has been implicated in other immune functions (20); therefore, we had to rule out any indirect antiviral activity that cathelicidin might have on mast cell function. It has recently been described that autophagy is dependent on cathelicidin presence in macrophages (117). As such, we also investigated autophagy formation during a VV infection in Cnlp−/− MCs in order to rule out changes in cellular physiology. As reported in our supplemental data, we did not identify any difference in autophagy formation or in the number of degranulated cells during a VV encounter.
Herein, we provide evidence that MCs intervene in VV infections. Current literature supports the concept that MCs play a relevant role in fighting infections in general, and our data confirm that MCs play an important role in blocking VV pathogens. Our findings suggest that the expression and control of mast cell antimicrobial peptides is key to this transformation. Therefore, any agent or disease that interferes with mast cell’s capacity to release their granules, such as the mast cell stabilizers cromolyn sodium or steroids, will interfere with mast cell’s capacity to block pathogen invasion. This work has the potential to provide significant new insights into the pathogenesis of skin infections and change the way we understand the role of MCs in blocking viral infections in the skin. We provide evidence that MCs interact with VV; however, the mechanisms identified by this study may be applicable to any other enveloped viral infections, and the knowledge obtained from this work will have several therapeutic applications for combating viral diseases.
Supplementary Material
Acknowledgements
We would like to thank Dr. Besmer and the Developmental Biology Program MSKCC at Cornell University, NY for donation of the Kitwsh−/− mice. We would like to acknowledge Dr. Gallo at UCSD for the kind gift of the Cnlp−/− and Tlr2−/− mice. S1PR2−/− mouse bone marrow cells have been a kind donation of Dr. Jerold Chun from The Scripps Research Institute.
This work was supported by the NIH- NIAID grants 1R21A1074766-01A2 and 1R01AI093957 to the corresponding author and by funds to BM by the Division of Intramural Research, NIAID, NIH.
References
- 1.Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol. 2005;6:135–142. doi: 10.1038/ni1158. [DOI] [PubMed] [Google Scholar]
- 2.Matsushima H, Yamada N, Matsue H, Shimada S. TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. J Immunol. 2004;173:531–541. doi: 10.4049/jimmunol.173.1.531. [DOI] [PubMed] [Google Scholar]
- 3.Fehrenbach K, Port F, Grochowy G, Kalis C, Bessler W, Galanos C, Krystal G, Freudenberg M, Huber M. Stimulation of mast cells via FcvarepsilonR1 and TLR2: the type of ligand determines the outcome. Mol Immunol. 2007;44:2087–2094. doi: 10.1016/j.molimm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Malaviya R, Gao Z, Thankavel K, van der Merwe PA, Abraham SN. The mast cell tumor necrosis factor alpha response to FimH-expressing Escherichia coli is mediated by the glycosylphosphatidylinositol-anchored molecule CD48. Proc Natl Acad Sci U S A. 1999;96:8110–8115. doi: 10.1073/pnas.96.14.8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishnaswamy G, Kelley J, Johnson D, Youngberg G, Stone W, Huang SK, Bieber J, Chi DS. The human mast cell: functions in physiology and disease. Front Biosci. 2001;6:D1109–D1127. doi: 10.2741/krishnas. [DOI] [PubMed] [Google Scholar]
- 6.Di Nardo A, Vitiello A, Gallo RL. Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J Immunol. 2003;170:2274–2278. doi: 10.4049/jimmunol.170.5.2274. [DOI] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 8.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulka M, Alexopoulou L, Flavell RA, Metcalfe DD. Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. Journal of Allergy and Clinical Immunology. 2004;114:174–182. doi: 10.1016/j.jaci.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 10.Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood. 2005;106:978–987. doi: 10.1182/blood-2004-07-2656. [DOI] [PubMed] [Google Scholar]
- 11.Oldford SA, Haidl ID, Howatt MA, Leiva CA, Johnston B, Marshall JS. A critical role for mast cells and mast cell-derived IL-6 in TLR2-mediated inhibition of tumor growth. J Immunol. 185:7067–7076. doi: 10.4049/jimmunol.1001137. [DOI] [PubMed] [Google Scholar]
- 12.Varadaradjalou S, Feger F, Thieblemont N, Hamouda NB, Pleau JM, Dy M, Arock M. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human mast cells. Eur J Immunol. 2003;33:899–906. doi: 10.1002/eji.200323830. [DOI] [PubMed] [Google Scholar]
- 13.St John AL, Rathore AP, Yap H, Ng ML, Metcalfe DD, Vasudevan SG, Abraham SN. Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc Natl Acad Sci U S A. 108:9190–9195. doi: 10.1073/pnas.1105079108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prado-Montes de Oca E, Velarde-Felix JS, Rios-Tostado JJ, Picos-Cardenas VJ, Figuera LE. SNP 668C (−44) alters a NF-kappaB1 putative binding site in non-coding strand of human beta-defensin 1 (DEFB1) and is associated with lepromatous leprosy. Infect Genet Evol. 2009;9:617–625. doi: 10.1016/j.meegid.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, Leung DY. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24:341–348. doi: 10.1016/j.immuni.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Bussmann C, Peng WM, Bieber T, Novak N. Molecular pathogenesis and clinical implications of eczema herpeticum. Expert Rev Mol Med. 2008;10:e21. doi: 10.1017/S1462399408000756. [DOI] [PubMed] [Google Scholar]
- 17.Jenssen H. Anti herpes simplex virus activity of lactoferrin/lactoferricin -- an example of antiviral activity of antimicrobial protein/peptide. Cell Mol Life Sci. 2005;62:3002–3013. doi: 10.1007/s00018-005-5228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakashima H, Masuda M, Murakami T, Koyanagi Y, Matsumoto A, Fujii N, Yamamoto N. Anti-human immunodeficiency virus activity of a novel synthetic peptide, T22 ([Tyr-5,12, Lys-7]polyphemusin II): a possible inhibitor of virus-cell fusion. Antimicrob Agents Chemother. 1992;36:1249–1255. doi: 10.1128/aac.36.6.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamamura H, Kuroda M, Masuda M, Otaka A, Funakoshi S, Nakashima H, Yamamoto N, Waki M, Matsumoto A, Lancelin JM, et al. A comparative study of the solution structures of tachyplesin I and a novel anti-HIV synthetic peptide, T22 ([Tyr5,12, Lys7]-polyphemusin II), determined by nuclear magnetic resonance. Biochim Biophys Acta. 1993;1163:209–216. doi: 10.1016/0167-4838(93)90183-r. [DOI] [PubMed] [Google Scholar]
- 20.Gallo RL, Murakami M, Ohtake T, Zaiou M. Biology and clinical relevance of naturally occurring antimicrobial peptides. J Allergy Clin Immunol. 2002;110:823–831. doi: 10.1067/mai.2002.129801. [DOI] [PubMed] [Google Scholar]
- 21.Howell MD, Wollenberg A, Gallo RL, Flaig M, Streib JE, Wong C, Pavicic T, Boguniewicz M, Leung DY. Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol. 2006;117:836–841. doi: 10.1016/j.jaci.2005.12.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moss B. Poxviridae: the viruses and their replication. In: Knipe DM HP, editor. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 2905–2946. [Google Scholar]
- 23.James WD, Berger TG, Elston DM, Odom RB. Andrews' diseases of the skin : clinical dermatology. Philadelphia: Saunders Elsevier; 2006. [Google Scholar]
- 24.Wollenberg A, Engler R. Smallpox, vaccination and adverse reactions to smallpox vaccine. Curr Opin Allergy Clin Immunol. 2004;4:271–275. doi: 10.1097/01.all.0000136758.66442.28. [DOI] [PubMed] [Google Scholar]
- 25.Ryan KJ, Ray CG, Sherris JC. Sherris medical microbiology : an introduction to infectious diseases. New York: McGraw-Hill; 2004. [Google Scholar]
- 26.Johnson L, Gupta AK, Ghafoor A, Akin D, Bashir R. Characterization of vaccinia virus particles using microscale silicon cantilever resonators and atomic force microscopy. Sensors and Actuators B: Chemical. 2006;115:189–197. [Google Scholar]
- 27.Kawakami Y, Tomimori Y, Yumoto K, Hasegawa S, Ando T, Tagaya Y, Crotty S, Kawakami T. Inhibition of NK cell activity by IL-17 allows vaccinia virus to induce severe skin lesions in a mouse model of eczema vaccinatum. J Exp Med. 2009;206:1219–1225. doi: 10.1084/jem.20082835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oyoshi MK, Elkhal A, Kumar L, Scott JE, Koduru S, He R, Leung DY, Howell MD, Oettgen HC, Murphy GF, Geha RS. Vaccinia virus inoculation in sites of allergic skin inflammation elicits a vigorous cutaneous IL-17 response. Proc Natl Acad Sci U S A. 2009;106:14954–14959. doi: 10.1073/pnas.0904021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee MS, Roos JM, McGuigan LC, Smith KA, Cormier N, Cohen LK, Roberts BE, Payne LG. Molecular attenuation of vaccinia virus: mutant generation and animal characterization. J Virol. 1992;66:2617–2630. doi: 10.1128/jvi.66.5.2617-2630.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dales S, Kajioka R. The Cycle of Multiplication of Vaccinia Virus in Earle's Strain L Cells. I. Uptake and Penetration. Virology. 1964;24:278–294. doi: 10.1016/0042-6822(64)90167-9. [DOI] [PubMed] [Google Scholar]
- 31.Payne L. Polypeptide composition of extracellular enveloped vaccinia virus. J Virol. 1978;27:28–37. doi: 10.1128/jvi.27.1.28-37.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armstrong JA, Metz DH, Young MR. The mode of entry of vaccinia virus into L cells. J Gen Virol. 1973;21:533–537. doi: 10.1099/0022-1317-21-3-533. [DOI] [PubMed] [Google Scholar]
- 33.Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol. 2005;86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- 34.Dallo S, Rodriguez JF, Esteban M. A 14K envelope protein of vaccinia virus with an important role in virus-host cell interactions is altered during virus persistence and determines the plaque size phenotype of the virus. Virology. 1987;159:423–432. doi: 10.1016/0042-6822(87)90481-8. [DOI] [PubMed] [Google Scholar]
- 35.Doms RW, Blumenthal R, Moss B. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J Virol. 1990;64:4884–4892. doi: 10.1128/jvi.64.10.4884-4892.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanderplasschen A, Hollinshead M, Smith GL. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J Gen Virol. 1998;79(Pt 4):877–887. doi: 10.1099/0022-1317-79-4-877. [DOI] [PubMed] [Google Scholar]
- 37.Bengali Z, Townsley AC, Moss B. Vaccinia virus strain differences in cell attachment and entry. Virology. 2009;389:132–140. doi: 10.1016/j.virol.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Townsley AC, Weisberg AS, Wagenaar TR, Moss B. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J Virol. 2006;80:8899–8908. doi: 10.1128/JVI.01053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 40.Huang CY, Lu TY, Bair CH, Chang YS, Jwo JK, Chang W. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into HeLa cells through fluid phase endocytosis. J Virol. 2008;82:7988–7999. doi: 10.1128/JVI.00894-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cluett EB, Machamer CE. The envelope of vaccinia virus reveals an unusual phospholipid in Golgi complex membranes. J Cell Sci. 1996;109(Pt 8):2121–2131. doi: 10.1242/jcs.109.8.2121. [DOI] [PubMed] [Google Scholar]
- 42.Olivera A, Mizugishi K, Tikhonova A, Ciaccia L, Odom S, Proia RL, Rivera J. The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity. 2007;26:287–297. doi: 10.1016/j.immuni.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 43.Oskeritzian CA, Price MM, Hait NC, Kapitonov D, Falanga YT, Morales JK, Ryan JJ, Milstien S, Spiegel S. Essential roles of sphingosine-1-phosphate receptor 2 in human mast cell activation, anaphylaxis, and pulmonary edema. J Exp Med. 207:465–474. doi: 10.1084/jem.20091513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugiyama K. Histamine release from rat mast cells induced by Sendai virus. Nature. 1977;270:614–615. doi: 10.1038/270614a0. [DOI] [PubMed] [Google Scholar]
- 46.Mokhtarian F, Griffin DE. The role of mast cells in virus-induced inflammation in the murine central nervous system. Cell Immunol. 1984;86:491–500. doi: 10.1016/0008-8749(84)90404-0. [DOI] [PubMed] [Google Scholar]
- 47.Sorden SD, Castleman WL. Virus-induced increases in airway mast cells in brown Norway rats are associated with enhanced pulmonary viral replication and persisting lymphocytic infiltration. Exp Lung Res. 1995;21:197–213. doi: 10.3109/01902149509068827. [DOI] [PubMed] [Google Scholar]
- 48.King CA, Marshall JS, Alshurafa H, Anderson R. Release of vasoactive cytokines by antibody-enhanced dengue virus infection of a human mast cell/basophil line. J Virol. 2000;74:7146–7150. doi: 10.1128/jvi.74.15.7146-7150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown MG, Huang YY, Marshall JS, King CA, Hoskin DW, Anderson R. Dramatic caspase-dependent apoptosis in antibody-enhanced dengue virus infection of human mast cells. J Leukoc Biol. 2009;85:71–80. doi: 10.1189/jlb.0308167. [DOI] [PubMed] [Google Scholar]
- 50.Burke SM, Issekutz TB, Mohan K, Lee PW, Shmulevitz M, Marshall JS. Human mast cell activation with virus-associated stimuli leads to the selective chemotaxis of natural killer cells by a CXCL8-dependent mechanism. Blood. 2008;111:5467–5476. doi: 10.1182/blood-2007-10-118547. [DOI] [PubMed] [Google Scholar]
- 51.Sun Q, Li W, She R, Wang D, Han D, Li R, Ding Y, Yue Z. Evidence for a role of mast cells in the mucosal injury induced by Newcastle disease virus. Poult Sci. 2009;88:554–561. doi: 10.3382/ps.2008-00468. [DOI] [PubMed] [Google Scholar]
- 52.Ogunbiyi PO, Black WD, Eyre P. Parainfluenza-3 virus-induced enhancement of histamine release from calf lung mast cells--effect of levamisole. J Vet Pharmacol Ther. 1988;11:338–344. doi: 10.1111/j.1365-2885.1988.tb00193.x. [DOI] [PubMed] [Google Scholar]
- 53.Besmer P. The kit ligand encoded at the murine Steel locus: a pleiotropic growth and differentiation factor. Curr Opin Cell Biol. 1991;3:939–946. doi: 10.1016/0955-0674(91)90111-b. [DOI] [PubMed] [Google Scholar]
- 54.Besmer P, Manova K, Duttlinger R, Huang EJ, Packer A, Gyssler C, Bachvarova RF. The kit-ligand (steel factor) and its receptor c-kit/W: pleiotropic roles in gametogenesis and melanogenesis. Dev Suppl. 1993:125–137. [PubMed] [Google Scholar]
- 55.Zhou JS, Xing W, Friend DS, Austen KF, Katz HR. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J Exp Med. 2007;204:2797–2802. doi: 10.1084/jem.20071391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nigrovic PA, Gray DH, Jones T, Hallgren J, Kuo FC, Chaletzky B, Gurish M, Mathis D, Benoist C, Lee DM. Genetic inversion in mast cell-deficient (W(sh)) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am J Pathol. 2008;173:1693–1701. doi: 10.2353/ajpath.2008.080407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grimbaldeston MA, Nakae S, Kalesnikoff J, Tsai M, Galli SJ. Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat Immunol. 2007;8:1095–1104. doi: 10.1038/ni1503. [DOI] [PubMed] [Google Scholar]
- 58.Di Nardo A, Yamasaki K, Dorschner RA, Lai Y, Gallo RL. Mast cell cathelicidin antimicrobial peptide prevents invasive group A Streptococcus infection of the skin. J Immunol. 2008;180:7565–7573. doi: 10.4049/jimmunol.180.11.7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duttlinger R, Manova K, Chu TY, Gyssler C, Zelenetz AD, Bachvarova RF, Besmer P. W-sash affects positive and negative elements controlling c-kit expression: ectopic c-kit expression at sites of kit-ligand expression affects melanogenesis. Development. 1993;118:705–717. doi: 10.1242/dev.118.3.705. [DOI] [PubMed] [Google Scholar]
- 60.Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167:835–848. doi: 10.1016/S0002-9440(10)62055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernstein A, Chabot B, Dubreuil P, Reith A, Nocka K, Majumder S, Ray P, Besmer P. The mouse W/c-kit locus. Ciba Found Symp. 1990;148:158–166. discussion 166–172. [PubMed] [Google Scholar]
- 62.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, Pestonjamasp V, Piraino J, Huttner K, Gallo RL. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature. 2001;414:454–457. doi: 10.1038/35106587. [DOI] [PubMed] [Google Scholar]
- 63.Kalesnikoff J, Galli SJ. Antiinflammatory and immunosuppressive functions of mast cells. Methods Mol Biol. 677:207–220. doi: 10.1007/978-1-60761-869-0_15. [DOI] [PubMed] [Google Scholar]
- 64.Freshney R. Culture of Animal Cells: A Manual of Basic Technique. 1987 [Google Scholar]
- 65.Ward BM, Moss B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J Virol. 2001;75:4802–4813. doi: 10.1128/JVI.75.10.4802-4813.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wolffe EJ, Vijaya S, Moss B. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology. 1995;211:53–63. doi: 10.1006/viro.1995.1378. [DOI] [PubMed] [Google Scholar]
- 67.Earl PL, Moss B, Wyatt LS, Carroll MW. Generation of recombinant vaccinia viruses. Curr Protoc Mol Biol. 2001;Chapter 16(Unit16):17. doi: 10.1002/0471142727.mb1617s43. [DOI] [PubMed] [Google Scholar]
- 68.Earl PL, Cooper N, Wyatt LS, Moss B, Carroll MW. Preparation of cell cultures and vaccinia virus stocks. Curr Protoc Mol Biol. 2001;Chapter 16(Unit16):16. doi: 10.1002/0471142727.mb1616s43. [DOI] [PubMed] [Google Scholar]
- 69.Kowalski WJ, Bahnfleth WP, Witham DL, Severin BF, Whittam TS. Mathematical Modeling of Ultraviolet Germicidal Irradiation for Air Disinfection. Quantitative Microbiology. 2000;2:249–270. [Google Scholar]
- 70.Lytle CD, Aaronson SA, Harvey E. Host-cell reactivation in mammalian cells. II. Survival of herpes simplex virus and vaccinia virus in normal human and xeroderma pigmentosum cells. Int J Radiat Biol Relat Stud Phys Chem Med. 1972;22:159–165. [PubMed] [Google Scholar]
- 71.Collins TJ. ImageJ for microscopy. Biotechniques. 2007;43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 72.Nautiyal KM, Ribeiro AC, Pfaff DW, Silver R. Brain mast cells link the immune system to anxiety-like behavior. Proc Natl Acad Sci U S A. 2008;105:18053–18057. doi: 10.1073/pnas.0809479105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paus R, Heinzelmann T, Robicsek S, Czarnetzki BM, Maurer M. Substance P stimulates murine epidermal keratinocyte proliferation and dermal mast cell degranulation in situ. Arch Dermatol Res. 1995;287:500–502. doi: 10.1007/BF00373436. [DOI] [PubMed] [Google Scholar]
- 74.Luu-The V, Paquet N, Calvo E, Cumps J. Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques. 2005;38:287–293. doi: 10.2144/05382RR05. [DOI] [PubMed] [Google Scholar]
- 75.Amegadzie BY, Ahn BY, Moss B. Identification, sequence, and expression of the gene encoding a Mr 35,000 subunit of the vaccinia virus DNA-dependent RNA polymerase. J Biol Chem. 1991;266:13712–13718. [PubMed] [Google Scholar]
- 76.Schick B, Austen KF. Modulation of chymase-mediated rat serosal mast cell degranulation by trypsin or diisopropyl fluorophosphate. Immunology. 1989;66:434–438. [PMC free article] [PubMed] [Google Scholar]
- 77.Suzuki K, Verma IM. Phosphorylation of SNAP-23 by IkappaB kinase 2 regulates mast cell degranulation. Cell. 2008;134:485–495. doi: 10.1016/j.cell.2008.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwartz LB, Austen KF, Wasserman SI. Immunologic release of beta-hexosaminidase and beta-glucuronidase from purified rat serosal mast cells. J Immunol. 1979;123:1445–1450. [PubMed] [Google Scholar]
- 79.Lindstedt KA, Kovanen PT. Isolation of mast cell granules. Curr Protoc Cell Biol. 2006;Chapter 3(Unit 3):16. doi: 10.1002/0471143030.cb0316s29. [DOI] [PubMed] [Google Scholar]
- 80.Lira FS, Rosa JC, Yamashita AS, Koyama CH, Batista ML, Jr, Seelaender M. Endurance training induces depot-specific changes in IL-10/TNF-alpha ratio in rat adipose tissue. Cytokine. 2009;45:80–85. doi: 10.1016/j.cyto.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 81.Graham JM. Isolation of Golgi membranes from tissues and cells by differential and density gradient centrifugation. Curr Protoc Cell Biol. 2001;Chapter 3(Unit 3):9. doi: 10.1002/0471143030.cb0309s10. [DOI] [PubMed] [Google Scholar]
- 82.Trifilieff A, Baur F, Fozard JR. Role of sphingosine-1-phosphate (S1P) and the S1P(2) receptor in allergen-induced, mast cell-dependent contraction of rat lung parenchymal strips. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:303–309. doi: 10.1007/s00210-009-0438-4. [DOI] [PubMed] [Google Scholar]
- 83.Weller K, Foitzik K, Paus R, Syska W, Maurer M. Mast cells are required for normal healing of skin wounds in mice. FASEB J. 2006;20:2366–2368. doi: 10.1096/fj.06-5837fje. [DOI] [PubMed] [Google Scholar]
- 84.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 207:1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sagripanti JL, Lytle CD. Sensitivity to ultraviolet radiation of Lassa, vaccinia, and Ebola viruses dried on surfaces. Arch Virol. 156:489–494. doi: 10.1007/s00705-010-0847-1. [DOI] [PubMed] [Google Scholar]
- 86.Tsung K, Yim JH, Marti W, Buller RM, Norton JA. Gene expression and cytopathic effect of vaccinia virus inactivated by psoralen and long-wave UV light. J Virol. 1996;70:165–171. doi: 10.1128/jvi.70.1.165-171.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kono M, Belyantseva IA, Skoura A, Frolenkov GI, Starost MF, Dreier JL, Lidington D, Bolz SS, Friedman TB, Hla T, Proia RL. Deafness and stria vascularis defects in S1P2 receptor-null mice. J Biol Chem. 2007;282:10690–10696. doi: 10.1074/jbc.M700370200. [DOI] [PubMed] [Google Scholar]
- 88.Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bauernfeind F, Hornung V. TLR2 joins the interferon gang. Nat Immunol. 2009;10:1139–1141. doi: 10.1038/ni1109-1139. [DOI] [PubMed] [Google Scholar]
- 90.Howell MD, Jones JF, Kisich KO, Streib JE, Gallo RL, Leung DY. Selective killing of vaccinia virus by LL-37: implications for eczema vaccinatum. J Immunol. 2004;172:1763–1767. doi: 10.4049/jimmunol.172.3.1763. [DOI] [PubMed] [Google Scholar]
- 91.Dean RE, O'Brien LM, Thwaite JE, Fox MA, Atkins H, Ulaeto DO. A carpet-based mechanism for direct antimicrobial peptide activity against vaccinia virus membranes. Peptides. 2010;31:1966–1972. doi: 10.1016/j.peptides.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 92.Smith SA, Kotwa GJ. Immune Response to Poxvirus Infections in Various Animals. Critical Reviews in Microbiology. 2002;28:149–185. doi: 10.1080/1040-840291046722. [DOI] [PubMed] [Google Scholar]
- 93.Knorr CW, Allen SD, Torres AR, Smee DF. Effects of cidofovir treatment on cytokine induction in murine models of cowpox and vaccinia virus infection. Antiviral Research. 2006;72:125–133. doi: 10.1016/j.antiviral.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 94.Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 10:440–452. doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cox JS. Disodium cromoglycate (FPL 670) ('Intal'): a specific inhibitor of reaginic antibody-antigen mechanisms. Nature. 1967;216:1328–1329. doi: 10.1038/2161328a0. [DOI] [PubMed] [Google Scholar]
- 96.Church MK, Hiroi J. Inhibition of IgE-dependent histamine release from human dispersed lung mast cells by anti-allergic drugs and salbutamol. Br J Pharmacol. 1987;90:421–429. doi: 10.1111/j.1476-5381.1987.tb08972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Flint KC, Leung KB, Pearce FL, Hudspith BN, Brostoff J, Johnson NM. Human mast cells recovered by bronchoalveolar lavage: their morphology, histamine release and the effects of sodium cromoglycate. Clin Sci (Lond) 1985;68:427–432. doi: 10.1042/cs0680427. [DOI] [PubMed] [Google Scholar]
- 98.Locker JK, Kuehn A, Schleich S, Rutter G, Hohenberg H, Wepf R, Griffiths G. Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol Biol Cell. 2000;11:2497–2511. doi: 10.1091/mbc.11.7.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.de Magalhaes JC, Andrade AA, Silva PN, Sousa LP, Ropert C, Ferreira PC, Kroon EG, Gazzinelli RT, Bonjardim CA. A mitogenic signal triggered at an early stage of vaccinia virus infection: implication of MEK/ERK and protein kinase A in virus multiplication. J Biol Chem. 2001;276:38353–38360. doi: 10.1074/jbc.M100183200. [DOI] [PubMed] [Google Scholar]
- 100.Rahbar R, Murooka TT, Hinek AA, Galligan CL, Sassano A, Yu C, Srivastava K, Platanias LC, Fish EN. Vaccinia virus activation of CCR5 invokes tyrosine phosphorylation signaling events that support virus replication. J Virol. 2006;80:7245–7259. doi: 10.1128/JVI.00463-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sandgren KJ, Wilkinson J, Miranda-Saksena M, McInerney GM, Byth-Wilson K, Robinson PJ, Cunningham AL. A differential role for macropinocytosis in mediating entry of the two forms of vaccinia virus into dendritic cells. PLoS Pathog. 6:e1000866. doi: 10.1371/journal.ppat.1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Whitbeck JC, Foo CH, Ponce de Leon M, Eisenberg RJ, Cohen GH. Vaccinia virus exhibits cell-type-dependent entry characteristics. Virology. 2009;385:383–391. doi: 10.1016/j.virol.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mercer J, Knebel S, Schmidt FI, Crouse J, Burkard C, Helenius A. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc Natl Acad Sci U S A. 107:9346–9351. doi: 10.1073/pnas.1004618107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bisht H, Weisberg AS, Moss B. Vaccinia virus l1 protein is required for cell entry and membrane fusion. J Virol. 2008;82:8687–8694. doi: 10.1128/JVI.00852-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ichihashi Y, Oie M. The activation of vaccinia virus infectivity by the transfer of phosphatidylserine from the plasma membrane. Virology. 1983;130:306–317. doi: 10.1016/0042-6822(83)90085-5. [DOI] [PubMed] [Google Scholar]
- 106.Laliberte JP, Moss B. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc Natl Acad Sci U S A. 2009;106:17517–17521. doi: 10.1073/pnas.0909376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, Milstien S, Spiegel S. Transactivation of sphingosine-1-phosphate receptors by FcepsilonRI triggering is required for normal mast cell degranulation and chemotaxis. J Exp Med. 2004;199:959–970. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Olivera A, Rivera J. Sphingolipids and the balancing of immune cell function: lessons from the mast cell. J Immunol. 2005;174:1153–1158. doi: 10.4049/jimmunol.174.3.1153. [DOI] [PubMed] [Google Scholar]
- 109.Weinberg A, Krisanaprakornkit S, Dale BA. Epithelial antimicrobial peptides: review and significance for oral applications. Crit Rev Oral Biol Med. 1998;9:399–414. doi: 10.1177/10454411980090040201. [DOI] [PubMed] [Google Scholar]
- 110.Yang D, Chertov O, Oppenheim JJ. The role of mammalian antimicrobial peptides and proteins in awakening of innate host defenses and adaptive immunity. Cell Mol Life Sci. 2001;58:978–989. doi: 10.1007/PL00000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yoshio H, Lagercrantz H, Gudmundsson GH, Agerberth B. First line of defense in early human life. Semin Perinatol. 2004;28:304–311. doi: 10.1053/j.semperi.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 112.Gordon YJ, Romanowski EG, Shanks RMQ, Yates KA, Hinsley H, Pereira HA. CAP37-Derived Antimicrobial Peptides Have "In Vitro" Antiviral Activity against Adenovirus and Herpes Simplex Virus Type 1. Current Eye Research. 2009;34:241–249. doi: 10.1080/02713680802714066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Reading PC, Smith GL. A kinetic analysis of immune mediators in the lungs of mice infected with vaccinia virus and comparison with intradermal infection. J Gen Virol. 2003;84:1973–1983. doi: 10.1099/vir.0.19285-0. [DOI] [PubMed] [Google Scholar]
- 114.Jacobs N, Chen RA, Gubser C, Najarro P, Smith GL. Intradermal immune response after infection with Vaccinia virus. J Gen Virol. 2006;87:1157–1161. doi: 10.1099/vir.0.81556-0. [DOI] [PubMed] [Google Scholar]
- 115.Chen RA, Jacobs N, Smith GL. Vaccinia virus strain Western Reserve protein B14 is an intracellular virulence factor. J Gen Virol. 2006;87:1451–1458. doi: 10.1099/vir.0.81736-0. [DOI] [PubMed] [Google Scholar]
- 116.Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 117.Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, Lee ZW, Lee SH, Kim JM, Jo EK. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009;6:231–243. doi: 10.1016/j.chom.2009.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.