

Abstract
Diabetic retinopathy is a leading cause of acquired blindness. Visual function disorders have been observed in diabetic patients with very early retinopathy or even before the onset of retinopathy. The aim of the present work was to analyze the visual pathway in an early stage of experimental diabetes. Diabetes was induced in Wistar rats by an i.p. injection of streptozotocin. A deficit in anterograde transport from the retina to the superior colliculus was observed 6 weeks after streptozotocin injection. At this time point, morphologic studies did not reveal retinal ganglion cell loss or substantial alterations in the superior colliculus. The optic nerve was morphometrically evaluated at intraorbital (unmyelinated and myelinated) and intracranial sections. In animals that had been diabetic for 6 weeks, a large increase in astrocyte reactivity occurred in the distal (but not the intraorbital) portion, which coincided with significant axon loss. Moreover, profound myelin alterations and altered morphologic features of oligodendrocyte lineage were observed at the distal (but not the proximal) optic nerve portion. The present results suggest that axoglial alterations at the distal portion of the optic nerve could be the first structural change in the diabetic visual pathway.
Diabetic retinopathy (DR) is a leading cause of acquired blindness. DR is rarely detected in the first few years of diabetes, but its incidence increases to 50% by 10 years and to 90% by 25 years of diabetes. Vision loss mainly occurs due to chronic hyperglycemia, vascular damage and leakage, edema formation, capillary basement membrane thickening, neovascularization, hemorrhage, and ischemia.1 Although DR has long been recognized as a vascular disease, it is becoming increasingly clear that retinal cells are also affected by diabetes, resulting in dysfunction and degeneration of neuronal cells (reviewed by Kern and Barber2). However, the progression rate of retinal cell degeneration is still controversial. Barber et al3 showed decreases in retinal ganglion cell (RGC) number and in inner plexiform layer thickness, which occur after 7.5 months of streptozotocin (STZ)-induced diabetes in rats, whereas Kusari et al4 reported a loss of RGCs at 4 weeks of STZ injection. Moreover, Seigel et al5 demonstrated an increase in the number of apoptotic RGCs after 3 months of diabetes.
Visual function disorders have been demonstrated in diabetic patients with very early retinopathy or even before the onset of retinopathy. Psychophysical methods and electrophysiologic measurements have yielded abnormalities in visual pathway function,6 such as reduced contrast sensitivity,7–10 reduced pattern electroretinogram amplitude,11,12 and conduction delays of visual evoked potentials.13–16 In that sense, it was demonstrated that the impaired visual evoked potentials are due to early involvement of nervous conduction in the optic nerve (ON).15 Moreover, in an experimental model of diabetes in rats, a significant reduction in mean myelinated fiber size in the diabetic ON has been reported.17,18 Thus, it seems likely that diabetic neurosensory disorders may be due not only to subclinical retinal vascular changes but also to those in the ON or higher visual pathway.19 Because most of the previously published literature focuses on the effect of diabetes on the retina, and does not correlate this effect with ON and visual pathway structure, we considered it worthwhile to analyze the morphologic appearance of the visual pathway in an early stage of experimental diabetes.
Materials and Methods
Animals
All the animal procedures were conducted in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. Male Wistar rats (mean ± SD weight, 300 ± 50 g) were housed in a standard animal room with food and water ad libitum under controlled conditions of humidity and temperature (mean ± SD, 21°C ± 2°C) and under a 12-hour light/12-hour dark lighting schedule (lights on at 8:00 AM). For diabetes induction, a single dose of STZ (Sigma-Aldrich, St. Louis, MO), 60 mg/kg (in 0.1 mol/L citrate buffer, pH 4.5), was i.p. injected. Control rats received an equal volume of citrate buffer. The animals were examined 72 hours after injection using a glucose meter (Contour TS, Bayer, Buenos Aires, Argentina), and those with blood glucose levels greater than 350 mg/dL were considered diabetic. Body weight and plasma glucose level were determined weekly.
Tissue Processing
Anesthetized rats were intracardially perfused with saline solution, followed by a fixative solution containing 4% formaldehyde in 0.1 mol/L PBS (pH 7.4). Then, the eyeballs with the intraorbital ON portion and the brain with the intracranial ON portion were carefully removed and immersed for 24 hours in the same fixative. ONs behind the eye (the intraorbital portion containing the ON head) and 2 mm before the optic chiasm (intracranial portion) were dissected out and used for semithin sectioning or immunostaining. After several washings, some tissue blocks were postfixed in 2% aqueous osmium tetroxide in sodium phosphate buffer for 1 hour. Dehydration was accomplished by gradual ethanol series, and tissue samples were embedded in epoxy resin. Semithin sections (1 μm) were obtained using an ultramicrotome, stained with toluidine blue, and used for morphometric analysis. Light microscopic images were digitally captured using a Nikon Eclipse E400 microscope via a Nikon Coolpix s10 camera (Nikon, Tokyo, Japan). In some cases, the ONs were dehydrated and embedded in paraffin. Transverse sections (7 μm) were obtained and used for immunostaining. Eyes were embedded in paraffin and were sectioned (5 μm) along the vertical meridian through the ON head. Sections were stained with H&E.
The brains were coronally sectioned (behind the optic chiasm), and the anterior portion was included in paraffin as previously described.20 Horizontal sections (10 μm) were obtained at the level of the optic chiasm (containing the ON and the optic tract portions). The posterior portion of the brain was immersed in a graded series of sucrose solutions (10%, 20%, and 30%), and coronal sections (40 μm) were obtained using a Leica CM1850 freezing microtome (Leica Microsystems Inc., Buffalo Grove, IL).
Cholera Toxin β-Subunit Injection
Rats (five animals per group) were anesthetized, and a drop of proparacaine (0.5%) was topically administered for local anesthesia. Four microliters of a solution of 0.2% cholera toxin β-subunit (CTB) conjugated to Alexa 488 dye (Molecular Probes Inc., Eugene, OR) in 0.1 mol/L PBS (pH 7.4) was injected into the vitreous using a 30-gauge Hamilton syringe (Hamilton, Reno, NV). The injections were applied 1 mm from the limbus, and the needle was left in the eye for 1 minute to prevent volume loss. Three days after injection, the rats were anesthetized and intracardially perfused as described previously herein. Their brains were carefully removed, postfixed overnight at 4°C, and immersed in a graded series of sucrose solutions; coronal sections (40 μm) were obtained using a freezing microtome. Nuclei were stained with the fluorescent dye DAPI, mounted with antifade medium, and viewed with a fluorescence microscope as described.
TUNEL Assay
For DNA fragmentation of cells undergoing apoptosis, the ApopTag fluorescein in situ apoptosis detection kit [S7110, Chemicon (Millipore), Temecula, CA] was used according to the manufacturer's instructions. For each retinal section, the number of TUNEL+ cells along the entire retina was calculated. For each eye, results obtained from four separate sections were averaged, and the mean of five eyes was recorded as the representative value for each group. For each superior colliculus (SC), three sections per animal were evaluated (rostral, medial, and caudal portions), and the number of TUNEL+ cells was counted in the stratum zonale and the stratum griseum superficiale layers. The mean of five SC values was recorded as the representative value for each group.
Immunostaining Protocol
The following antibodies were used: a mouse monoclonal anti–glial fibrillary acidic protein (GFAP) antibody conjugated to Cy3 (1:800; Sigma-Aldrich), a rabbit polyclonal anti–myelin basic protein (MBP) antibody, a mouse polyclonal IgM O1 antibody, a goat polyclonal anti–platelet-derived growth factor receptor-α (PDGFR-α) antibody (1:100; Neuromics, Edina, MN), a mouse monoclonal anti-CD11b antibody (Abd Serotec, Raleigh, NC), a mouse monoclonal anti-ED1 (CD68) antibody (Abcam Inc., Cambridge, MA), a goat anti-rabbit secondary antibody conjugated to Cy2 (1:100; Jackson ImmunoResearch Laboratories Inc., West Grove, PA), a donkey anti-mouse IgM secondary antibody conjugated to Cy3 (1:200; Jackson ImmunoResearch Laboratories Inc.), and a donkey anti-goat secondary antibody conjugated to DyLight 488 (1:100; Jackson ImmunoResearch Laboratories Inc.). The biotinylated lectin Griffonia simplicifolia I (Isolectin B4, 1:200; Vector Laboratories, Burlingame, CA) was used for microglia staining. The anti-MBP and O1 antibodies were a gift from Dr. A.T. Campagnoni (Mental Retardation Research Center, University of California, Los Angeles, CA). Some sections were immersed in 0.1% Triton X-100 (Roche Diagnostics GmbH, Mannheim, Germany) in 0.1 mol/L PBS for 10 minutes. Antigen retrieval was performed in paraffin sections by heating (90°C) slices for 30 minutes in citrate buffer (pH 6.3). Sections were preincubated with 5% normal horse serum for 1 hour and then were incubated overnight at 4°C with primary antibodies. For double labeling, the antibodies were used at the concentrations listed previously herein. After several washings, secondary antibodies were added, and sections were incubated for 2 hours at room temperature. Regularly, some sections were treated without the primary antibodies to confirm specificity. After immunostaining, nuclei were stained with DAPI, mounted using antifade medium (Vectashield; Vector Laboratories), and viewed using a fluorescence microscope (BX50; Olympus, Tokyo, Japan) connected to a video camera (3CCD; Sony, Tokyo, Japan) attached to a computer running image analysis software (Image-Pro Plus; Media Cybernetics Inc., Bethesda, MD). For microglial cell analysis, sections were incubated for 48 hours at 4°C with lectin, and after several washings, the LSAB2 System-HRP (DakoCytomation, Carpinteria, CA) was used according to the manufacturer's instructions.
Electron Microscopy
After anesthesia and thoracotomy, the animals were perfused through the left ventricle with a fixative solution containing 2% glutaraldehyde and 4% formaldehyde in 0.1 mol/L PBS (pH 7.4). Semithin sections were obtained as described previously hererin, and ultrathin sections were stained with uranyl acetate and lead citrate. Finally, sections were viewed and photographed using a Zeiss EM 10C transmission electron microscope (Carl Zeiss Microscopy, Peabody, MA).
Morphometric Analysis
All the images obtained were assembled and processed using Adobe Photoshop SC (Adobe Systems, San Jose, CA) to adjust the brightness and contrast. No other adjustments were made. For all the morphometric image processing and analysis, digitalized captured TIFF images were transferred to ImageJ software version 1.42q (NIH, Bethesda, MD). All the nomenclature used herein follows that of Paxinos and Watson.21
ON Analysis
ON total area was calculated from light microphotographs of transverse semithin sections obtained at a final magnification of ×20. The nerve was divided into four quadrants, and the number and area of axons were calculated in each section (in areas randomly chosen in each quadrant) from micrographs obtained at a magnification of ×100. Images were converted to 8-bit grayscale, and a manual threshold value, first determined by visual examination, was constantly applied. Finally, images were converted to a binary form. Myelinated fiber sizes were digitally delineated by the outer margin of their myelin sheaths, and axon areas were measured. For each ON, results obtained from four separate sections were averaged, and the mean axon number and area of five nerves were recorded as the representative values for each group. Histograms of nerve fiber size distribution were obtained using Origin software (OriginLab Corp., Northampton, MA). Electron microscopic photographs were obtained at a final magnification of ×3000. Fiber number and axon area were calculated from approximately 10 photographs per nerve. The g-ratios were calculated from no fewer than 500 myelinated axons per nerve. Data from four nerves were averaged and recorded as the representative value for each group.
Ganglion Cell Layer Cell Density
Microscopic images with a final magnification of ×40 were obtained as previously described.22 Total retinal, inner plexiform layer, inner nuclear layer, and outer nuclear layer thicknesses were measured for each eye, and the number of cells in the ganglion cell layer (GCL) was counted along 200 μm for each section. No attempt was made to distinguish cell types in the GCL. Measurements were obtained 1 mm dorsal and ventral to the optic disc. For each eye, results obtained from four separate sections were averaged, and the mean of five eyes was recorded as the representative value for each group.
Immunofluorescence Studies
Comparative digital images from different samples were obtained using identical exposure time, brightness, and contrast settings.
Neuronal Density and GFAP+ Area in the SC
Digital images with a final magnification of ×40 (1000 μm2 area) were obtained from the superficial layers (the stratum zonale and the stratum griseum superficiale) of the SC from five animals per group. For nuclei density analysis and GFAP+ area determination (percentage of area occupied by astrocyte processes), images were converted to 8-bit grayscale, and in each case, a manual threshold value was determined to binarize the images. For each section, results obtained from two images (in the medial and lateral areas of the SC) were averaged, and the mean of three sections from the rostral, medial, and caudal regions was recorded as the representative value for each animal.
CTB Studies
CTB staining was quantified as previously described,23 with some modifications. Coronal sections (every other cut, approximately 30 to 35 sections) were used for the SC reconstruction using Matlab (The MathWorks Inc., Natick, MA). For each section, the retinorecipient SC was outlined using DAPI counterstaining, and the total retinotopic area was calculated. Digital images were converted to 8-bit grayscale, and the optic density of CTB staining was calculated. The total length was measured and divided into bins (4 μm), from the medial to lateral region. The CTB density was obtained by dividing the total pixel area by CTB+ pixels. Finally, a colorimetric thermal representation was applied (from 0% = blue to 100% = red). The number of sections and the thickness (2×) were used for a final reconstruction of the retinal projection to the SC.
Statistical Analysis
Statistical analysis of results was performed using two-way analysis of variance, followed by Dunnett's test or Student's t-test.
Results
Table 1 summarizes the mean weight and blood glucose levels after injection of vehicle or STZ. At 3, 6, and 15 weeks postinjection, significant weight loss and increased blood glucose levels were observed in STZ-treated rats compared with vehicle-injected rats.
Table 1.
Body Weight and Blood Glucose Concentration Assessed at Different Time Points
| Time after vehicle or STZ injection | Body weight (g) |
Blood glucose concentration (mg/dL) |
||
|---|---|---|---|---|
| Control rats | Diabetic rats | Control rats | Diabetic rats | |
| 3 days | 326.8 ± 10.0 | 334.8 ± 9.8 | 115.1 ± 5.1 | 467.7 ± 18.8⁎ |
| 3 weeks | 409.0 ± 12.7 | 311.3 ± 7.7⁎ | 104.9 ± 9.7 | 519.6 ± 16.8⁎ |
| 6 weeks | 443.6 ± 7.6 | 312.5 ± 14.1⁎ | 108.2 ± 8.2 | 537.6 ± 13.7⁎ |
| 15 weeks | 485.9 ± 15.8 | 305.7 ± 18.5⁎ | 118.0 ± 10.8 | 565.6 ± 15.4⁎ |
Data are given as mean ± SEM (n = 10 animals per group). Significant decreases in body weight and increases in blood glucose levels were observed after STZ injection.
P < 0.01 versus aged-matched control animals, by Dunnett's test.
The active anterograde transport of RGC projections to the SC at different time points after diabetes induction was analyzed using CTB. In control animals, CTB labeled the entire retinotopic projection to the stratum zonale and the stratum griseum superficiale, the most superficial layers of the SC (Figure 1). No alterations were found in CTB labeling 3 weeks after diabetes induction, whereas 6 weeks after diabetes onset, a clear reduction in CTB staining was observed in the central and lateral regions of the SC, extending in the rostral to caudal direction. In the lateral region, areas with virtually no CTB staining were also observed (Figure 1). A similar staining profile was observed after 15 weeks of diabetes (Figure 1).
Figure 1.

Retinal terminal field in the SC of control rats and of diabetic rats at different time points after STZ injection. Photomicrographs showing the CTB staining patterns in the superficial layers of the SC of control animals and diabetic rats. Three representative sections (rostral, medial, and caudal) are shown. Dorsal views of a retinotopic SC map reconstruction are also shown. Three weeks after diabetes induction, no signs of alterations were observed. In animals that were diabetic for 6 weeks, a clear reduction in the density of retinal terminals and zones of no CTB staining were found, especially in the lateral area. Fifteen weeks after diabetes induction, a similar reduction in the CTB staining pattern density was observed. Shown are images representative of five animals per group. Scale bar = 1 mm.
Figure 2 shows the analysis of GCL cell density. The number of cells in the GCL significantly decreased after 15 (but not 3 or 6) weeks of diabetes induction (Figure 2F). At this time point, a significant increase in the number of TUNEL+ cells in the GCL was observed in diabetic rats (Figure 2G). At all the assessed time points, no other morphologic alterations or differences in total retina and retinal layer thicknesses were observed (see Supplemental Table S1 at http://ajp.amjpathol.org).
Figure 2.

GCL cell loss in diabetic animals. A–D: Representative photomicrographs showing the histologic appearance of control rat retinas and retinas from animals 3, 6, and 15 weeks after diabetes induction. E: TUNEL+ cells in the GCL (arrows) from an animal that was diabetic for 15 weeks. Cell nuclei were counterstained with DAPI. F: Cell count in the GCL evaluated by H&E staining. G: The number of TUNEL+ cells in the GCL was significantly higher in animals that were diabetic for 15 (but not 3 and 6) weeks than in controls. Scale bars = 50 μm (D and E). Data are mean ± SEM (n = 5 animals per group). *P < 0.01 versus age-matched controls, by Dunnett's test. INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; RGL, retinal ganglion cell layer.
Histologic analysis of the SC at different time points after diabetes induction revealed no substantial alterations in its cytoarchitecture (Figure 3). No significant differences in the number of neurons were found in the SC of diabetic animals (Figure 3B), apoptotic cells were absent (data not shown), and no signs of reactive astrocytes were evident (Figure 3, C–G).
Figure 3.

Morphometric analysis of the SC from control rats and from diabetic rats at different time points after STZ injection. A: The squares indicate the locations of the coronal sections [rostral (R), medial, and caudal (C) portions] used for the quantitative analysis. Neuronal density (B) and area occupied by GFAP+ astrocytes (C) were evaluated in the superficial layers of the SC. No signs of neuron loss or glial reactivity were observed in diabetic animals. Representative photomicrographs showing GFAP immunostaining in controls (D) and in rats after 3 (E), 6 (F), and 15 (G) weeks of diabetes. Data are mean ± SEM (n = 5 animals per group), by Dunnett's test. Scale bar = 50 μm (G). SGS, stratum griseum superficiale; SZ, stratum zonale.
The ON was morphometrically evaluated at different regions (Figure 4). Six weeks after diabetes induction, no changes were observed in transverse sections obtained from the intraorbital myelinated ON portions (Figure 4, A and F). In contrast, clear alterations were evident at the distal region (ie, intracranial ON, near the optic chiasm) (Figure 4, B and G). Light microscopic morphometry of semithin sections of the distal ON revealed significant decreases in the total number of axons (Figure 4D) and the mean axon area (Figure 4E) compared with age-matched controls. The myelinated axon area frequency histograms revealed a clear shift to a smaller-sized area (Figure 4, C and H), with a significant decrease in large fiber number (Figure 4I). No significant morphometric differences were observed in the proximal myelinated portion (data not shown). For all the ON regions analyzed, no differences in the total area were found (data not shown). The proximal region (ie, the intraorbital ON, close to the ON head), named the glial lamina, contains unmyelinated axons surrounded by a meshwork of astrocytes (GFAP immunoreactive). Beyond this area, axons become myelinated toward the chiasm (Figure 5, A–D). Immunostaining of GFAP in transverse ON sections was examined at different portions (Figure 5). No signs of glial reactivity were observed in the intraorbital ON portion (at the glial lamina and the myelinated region) in diabetic animals (Figure 5, E, F, I, and J). In the distal portion, diabetic ON astrocytes showed a reactive morphology with an increased number of processes (Figure 5K). Measurement of the GFAP+ area revealed a significant increase in GFAP levels compared with controls (Figure 5, G and H). In all the sections analyzed, the number of nuclei remained unchanged (Figure 5L).
Figure 4.

Morphometric analysis of the ON from control rats and from diabetic rats after 6 weeks of diabetes. Light micrographs of semithin transverse sections at the proximal and distal myelinated portions from a control (A and B) and a diabetic rat (F and G). Size frequency histograms showing the axon area distribution at the distal ON portion from a representative control (C) and a diabetic rat (H). Note the shift to smaller-sized axons found in the diabetic rat compared with an age-matched control. In diabetic rats, significant reductions were found in total axon number (D) and mean axon area (E). I: Axonal diameter distribution in controls and in animals after 6 weeks of diabetes. A significant decrease in the number of middle and large (but not smaller) axons was observed in diabetic animals. Data are mean ± SEM (n = 5 animals per group). *P < 0.01 versus age-matched controls, by Student's t-test. Scale bar = 25 μm (G).
Figure 5.

Astrocyte analysis at different ON portions from control rats and from rats after 6 weeks of diabetes. A: Schematic representation of a sagittal section through the rat ON showing the glial lamina, the transition zone, and the myelinated region. The horizontal lines (C and D) indicate the location where the sections were obtained along the ON. The pointed lines indicate the arterial blood supply of the retina and the ON. B–D: Representative photomicrographs showing the GFAP (red) and MBP (green) immunostaining patterns. The glial lamina (a transverse section is shown in C) was characterized by intense GFAP labeling and the absence of MBP immunoreactivity. In the myelinated regions, most nerve fibers were fully myelinated (a transverse section is shown in D). Representative GFAP immunostaining of transverse sections at the proximal (glial lamina and myelinated level) and distal ON portions from a normal rat (E–G) and a diabetic rat (I–K) are shown. In the distal portion, the area occupied by astrocytes was significant larger in diabetic rats than in controls (H), and the number of nuclei per section remained unchanged (L). Nuclei were counterstained with DAPI [measured as optical density (OD)]. Data are mean ± SEM (n = 5 animals per group). *P < 0.01 versus age-matched controls, by Student's t-test. Scale bars: 100 μm (B–D); 200 μm (K).
Figure 6 shows MBP immunostaining in horizontal sections obtained at the optic chiasm level. Slight disorganization of myelin, with some demyelinated zones, was observed in the diabetic distal ON and optic tract (Figure 6D). Double-labeling analysis revealed that zones of demyelination were occupied by GFAP+ cells (Figure 6F). To analyze oligodendrocyte (OL) lineage, immature OL (O1+ cells) and OL progenitors (PDGFR-α+ cells) were examined. In the diabetic ON, a marked increase in O1 immunoreactivity was found (Figure 7). O1+ cells showed hypertrophic somas and a high number of processes (Figure 7). Similarly, a marked increase in PDGFR-α immunolabeling, with a high number of hypertrophic cells, was observed in the distal ON (Figure 7).
Figure 6.

Myelin alterations in the diabetic visual pathway. Representative photomicrographs of MBP and GFAP immunostaining of the distal ON (horizontal sections) from control rats (A–C) and from rats after 6 weeks of diabetes (D–F). In diabetic rats, myelin disorganization was observed. Note zones of demyelization and glial reactivity. Shown are images representative of five animals per group. Scale bar = 50 μm (F).
Figure 7.

OL linage evaluation. Immature OL (O1+ cells) and OL precursor (PDGFR-α+ cells) were evaluated by immunostaining and measured as optical density (OD) per section. In the distal ON from animals that were diabetic for 6 weeks, significantly increased O1 and PDGFR-α immunostaining was observed, with the presence of disorganized and hypertrophic cells. Data are mean ± SEM (n = 5 animals per group); *P < 0.01 versus age-matched controls, by Student′s t-test. Scale bar = 50 μm.
At the ultrastructural level, alterations and loss of larger axons were observed in the distal ON from animals that were diabetic for 6 weeks. In these fibers, myelin was highly disorganized, and frequent lamellar membranous bodies were observed (Figure 8). Measurements of electron micrographs demonstrated a significant decrease in the number of axons and the mean axon area in the diabetic ON. The mean ± SEM axon area was 1.28 ± 0.02 μm2 in control rats and 1.20 ± 0.04 μm2 in diabetic rats. In contrast, analysis of the g-ratio (the ratio of the inner axonal diameter to the total outer diameter) revealed no clear differences between groups. The mean ± SEM g-ratio was 0.70 ± 0.04 in control rats and 0.72 ± 0.05 in diabetic rats.
Figure 8.
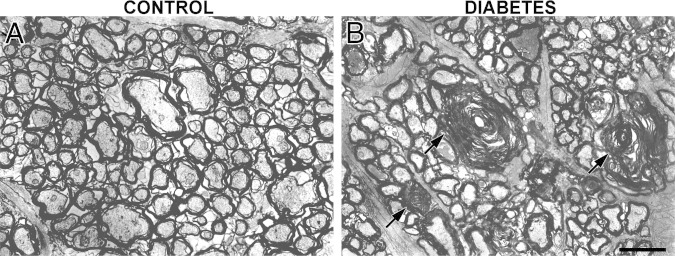
Ultrastructural alterations in the diabetic ON. Transverse sections of the distal ON from a control rat (A) and from a rat after 6 weeks of diabetes (B) are shown. In diabetic animals, clear alterations in the myelin sheaths of larger axons are observed. Note also the presence of lamellar bodies (arrows). Shown are images representative of four animals per group. Scale bar = 2 μm.
No differences in microglial cell morphology assessed by Griffonia simplicifolia I staining in the distal ON were observed between control and diabetic animals. Moreover, no increase in CD11b+ or ED1+ cells (markers of reactive microglia) were observed in the diabetic ON (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Discussion
CTB is a reliable anterograde tracer that binds specifically to surface receptors of neurons and is actively taken up and transported by the axons.24,25 CTB has been extensively used to analyze the communication between the retina and its synaptic targets. The present results demonstrate a deficit in CTB anterograde transport from the retina to the SC, its primary projection site, at an early stage of diabetes (ie, 6 weeks after injection of STZ). At this time point, morphologic studies indicated significant alterations in the distal portion of the ON but no substantial alterations in the retina or SC. After 15 weeks of diabetes, a significant decrease in cell number and an increase in TUNEL+ cell number in the GCL were observed, but no changes in the SC were evident.
The anterograde axonal transport system is responsible for transporting proteins associated with axonal structure and synaptic transmitter function to the axon and its terminals. In the opposite direction is the retrograde axonal transport system, which carries neurotrophic factors that influence steady-state activities in the cell body. It was previously reported as a progressive deficit in the retrograde axonal transport, mainly mediated by large axons, that is evident 1 month after STZ injection and does not involve RGC loss.18 Taken together, these results support that early diabetes provokes a “misconnection” between the retina and the SC, which precedes RGC death.
To identify the locus for the transport disruption along the visual pathway, representative intraorbital (unmyelinated and myelinated) and intracranial ON sections were analyzed. In diabetic rats, the distal (intracranial) portion of the ON degenerated at a time point in which the proximal portions remained unchanged. In animals that had been diabetic for 6 weeks, a large increase in astrocyte reactivity (as shown by GFAP immunoreactivity) occurred in the distal (but not the intraorbital) portion of the ON, which coincided with significant axon loss, making larger axons significantly more vulnerable than smaller axons. In agreement, Zhang et al26 described a higher susceptibility of ON large axons in STZ-treated rats. Furthermore, a preferential loss of ON large axons was demonstrated in experimental models of glaucoma.27,28 Increases in astrocyte reactivity have been observed in many neurologic disorders and acute nervous system injuries and involve increases in gene expression and redistribution of intermediate filaments, and occasionally increases in proliferation (reviewed by Escartin and Bonvento29). Although microglial cell activation was described in the diabetic retina at different stages of the disease,30,31 no changes in the microglia morphologic appearance (as shown by Griffonia simplicifolia I staining) or signs of microglial reactivity were observed in the distal portion of the ON, at least at 6 weeks of diabetes onset.
Structural analyses of peripheral nerves in humans and animals with diabetes have shown that the main lesions involve axonal atrophy and axoglial dysfunction.32 Morphologic studies of ON in autopsy materials demonstrated gliosis, demyelination, focal infarction, and vascular lesions, especially significant in elderly patients with a long history of diabetes.33 Ino-ue et al34 showed a reduction in mean axon size and myelinated fiber atrophy at 3 months of diabetes induction in rats, whereas in the present study, changes in the axoglial structure of the distal ON were observed at an earlier stage of the disease. Notably, these changes were demonstrated by the appearance of a marked decrease in corneal sensitivity, possibly associated with peripheral neuropathy in rats with 5 weeks of diabetes.35
At the ON head, RGC axons pass unmyelinated into the nerve. MBP is an important constituent of peripheral and central nervous system myelin sheaths and plays a major role in myelin membrane formation and structure.36,37 Thus, MBP is a reliable index of OL differentiation and myelin formation. In that sense, myelin disarrangement shown by alterations and disorganization of MBP immunoreactivity and profound myelin alterations at the ultrastructural level were observed in the distal portion of the diabetic ON. In myelin abnormalities and demyelination in the central nervous system, OL remyelination is expected.38 After demyelination, a remyelination process occurs through activation of OL progenitor cells in the lesion. New OLs then are formed, and insulating function recovers.39 However, the remyelination process progressively becomes limited. OL progenitor cells in the lesion, for unknown reasons, gradually lose the ability to form myelinating OLs.40 In animals with 6 weeks of diabetes, progenitor (PDGFR-α+) and immature (O1+) OLs showed an altered morphologic appearance, with hypertrophic somas and multiple processes. The role of these cells in progression of the disease needs to be further examined. It is possible that these reactive cells contribute to a remyelination state but that the axon loss could provoke a fail to complete the differentiation to mature OLs, resulting in aberrant cells that accumulate adjacent to the lesion sites, similar to that seen in chronic models of white matter injury.41 In an experimental model of glaucoma, it was described that OL alteration is not only an early event but also a central mechanism through which the axons are damaged, leading to the subsequent death of RGCs.42 Although DR and glaucoma differ in several aspects (such as causes, risk factors, and retinal cell types involved, among many others), based on the present results, it is tempting to speculate that a similar sequence of events described in glaucoma could occur in experimental diabetes. However, whether changes in GFAP, O1, and PDGFR-α levels are cause or consequence of the decrease in axon number observed in the diabetic ON cannot be determined, and, in that sense, a more detailed temporal course of these alterations will be examined in the near future. The present results suggest a progression of RGC degeneration in experimental diabetes, where axon dysfunction or degeneration preceded neuronal loss. In fact, neuronal compartments often crucially depend on one another for survival, and molecular defects in one compartment can trigger cellular degeneration in distant parts of the neuron.43 Evidence of axon degeneration occurring separate from and before somal degeneration has been observed in many neurodegenerative diseases, such as Alzheimer disease, Parkinson disease, and glaucoma.23,44–46 However, the fact that axoglial alterations at the distal portion of the ON seems to be the first structural alteration in the diabetic visual pathway does not imply that the initial neuronal event is axonal. Instead, a putative sublethal impairment of RGCs could render them unable to support their axons, resulting in progressive dying back of the axon toward the cell body.43 Alternatively, RGC death in experimental diabetes can occur independently from distal axon degeneration. On the other hand, there is a possibility that diabetes causes stress to cells that results in lower efficiency in their mitochondria functioning. Neurons, particularly RGCs (documented by large numbers of their axons in the globe), are more dependent on their mitochondria than are dividing cells. If this were the case, RGC axonal transport (which requires ATP) would be affected before the occurrence of RGC death signs, and, in turn, the interaction between ON axons and glia could be altered. In this scenario, ON glial complications could be secondary rather than being a primary cause for ON alterations. Further studies will clarify whether either compartment dies secondarily to the other or whether both are directly affected by diabetes.
Alterations in the distal ON portion may be involved in the deficit of anterograde transport to the SC and in decreased propagation of nerve impulse and visual evoked potentials,16 contributing to visual disorders observed in early diabetes. In fact, abnormal color vision and contrast sensitivity have been observed before vasculopathy,47 indicating the early presence of neuropathy. In agreement, a growing body of evidence suggests that neural and glial complications may precede vascular changes in DR.3,48,49 In summary, these data indicate, for the first time, that in experimental diabetes, axonal alterations at the distal portion of the ON precede RGC loss. Therefore, early vision loss in diabetes could be abated by interventions that preserve distal axonal function before loss of the neuronal substrate.
Acknowledgment
We thank Juana Pasquini for helpful discussion of the manuscript.
Footnotes
Supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica; the University of Buenos Aires; CONICET, Argentina; and Sigma Xi, The Scientific Research Society.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.09.018.
Supplementary data
Griffonia simplicifolia I–positive cells in normal and diabetic distal ONs. A: In transverse normal sections, weak-stained Griffonia simplicifolia I–positive cells were observed. Note the fine and ramified processes and the rod-shaped soma characteristic from resting microglia (a detail is shown in the inset). B: In diabetic animals, no clear differences were found in the morphologic appearance of microglial cells present in the distal portion of the ON. Shown are images representative of five animals per group. Scale bars: 50 μm (B); 25 μm (inset).
References
- 1.Kowluru R.A., Chan P.S. Oxidative stress and diabetic retinopathy. Exp Diabetes Res. 2007;2007:43603–43614. doi: 10.1155/2007/43603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kern T.S., Barber A.J. Retinal ganglion cells in diabetes. J Physiol. 2008;586:4401–4408. doi: 10.1113/jphysiol.2008.156695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barber A.J., Lieth E., Khin S.A., Antonetti D.A., Buchanan A.G., Gardner T.W. Neural apoptosis in the retina during experimental and human diabetes: early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kusari J., Zhou S., Padillo E., Clarke K.G., Gil D.W. Effect of memantine on neuroretinal function and retinal vascular changes of streptozotocin-induced diabetic rats. Invest Ophthalmol Vis Sci. 2007;48:5152–5159. doi: 10.1167/iovs.07-0427. [DOI] [PubMed] [Google Scholar]
- 5.Seigel G.M., Lupien S.B., Campbell L.M., Ishii D.N. Systemic IGF-I treatment inhibits cell death in diabetic rat retina. J Diabetes Complications. 2006;20:196–204. doi: 10.1016/j.jdiacomp.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Bresnick G.H. Diabetic retinopathy viewed as a neurosensory disorder. Arch Ophthalmol. 1986;104:989–990. doi: 10.1001/archopht.1986.01050190047037. [DOI] [PubMed] [Google Scholar]
- 7.Ghafour I.M., Foulds W.S., Allan D., McClure E. Contrast sensitivity in diabetic subjects with and without retinopathy. Br J Ophthalmol. 1982;66:492–495. doi: 10.1136/bjo.66.8.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Della Sala S., Bertoni G., Somazzi L., Stubbe F., Wilkins A.J. Impaired contrast sensitivity in diabetic patients with and without retinopathy: a new technique for rapid assessment. Br J Ophthalmol. 1985;69:136–142. doi: 10.1136/bjo.69.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sokol S., Moskowitz A., Skarf B., Evans R., Molitch M., Senior B. Contrast sensitivity in diabetics with and without background retinopathy. Arch Ophthalmol. 1985;103:51–54. doi: 10.1001/archopht.1985.01050010055018. [DOI] [PubMed] [Google Scholar]
- 10.Georgakopoulos C.D., Eliopoulou M.I., Exarchou A.M., Tzimis V., Pharmakakis N.M., Spiliotis B.E. Decreased contrast sensitivity in children and adolescents with type 1 diabetes mellitus. J Pediatr Ophthalmol Strabismus. 2011;48:92–97. doi: 10.3928/01913913-20100420-02. [DOI] [PubMed] [Google Scholar]
- 11.Falsini B., Porciatti V., Scalia G., Caputo S., Minnella A., Di Leo M.A., Ghirlanda G. Steady-state pattern electroretinogram in insulin-dependent diabetics with no or minimal retinopathy. Doc Ophthalmol. 1989;73:193–200. doi: 10.1007/BF00155037. [DOI] [PubMed] [Google Scholar]
- 12.Prager T.C., Garcia C.A., Mincher C.A., Mishra J., Chu H.H. The pattern electroretinogram in diabetes. Am J Ophthalmol. 1990;109:279–284. doi: 10.1016/s0002-9394(14)74550-7. [DOI] [PubMed] [Google Scholar]
- 13.Martinelli V., Piatti P.M., Filippi M., Pacchioni M., Pastore M.R., Canal N., Comi G. Effects of hyperglycaemia on visual evoked potentials in insulin-dependent diabetic patients. Acta Diabetol. 1992;29:34–37. doi: 10.1007/BF00572827. [DOI] [PubMed] [Google Scholar]
- 14.Uberall A., Renner C., Edl S., Parzinger E., Wenzel D. VEP and ERP abnormalities in children and adolescents with prepubertal onset of insulin dependent diabetes mellitus. Neuropediatrics. 1996;27:88–93. doi: 10.1055/s-2007-973755. [DOI] [PubMed] [Google Scholar]
- 15.Verrotti A., Lobefalo L., Trotta D., Della Loggia G., Chiarelli F., Luigi C., Morgese G., Gallenga P. Visual evoked potentials in young persons with newly diagnosed diabetes: a long-term follow-up. Dev Med Child Neurol. 2000;42:240–244. doi: 10.1017/s0012162200000414. [DOI] [PubMed] [Google Scholar]
- 16.Wolff B.E., Bearse M.A., Jr, Schneck M.E., Barez S., Adams A.J. Multifocal VEP (mfVEP) reveals abnormal neuronal delays in diabetes. Doc Ophthalmol. 2010;1211:89–96. doi: 10.1007/s10633-010-9245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ino-ue M., Ohgiya N., Yamamoto M. Effect of aminoguanidine on optic nerve involvement in experimental diabetic rats. Brain Res. 1998;800:319–322. doi: 10.1016/s0006-8993(98)00512-5. [DOI] [PubMed] [Google Scholar]
- 18.Ino-Ue M., Zhang L., Naka H., Kuriyama H., Yamamoto M. Polyol metabolism of retrograde axonal transport in diabetic rat large optic nerve fiber. Invest Ophthalmol Vis Sci. 2000;41:4055–4058. [PubMed] [Google Scholar]
- 19.Trick G.L., Burde R.M., Gordon M.D., Kilo C., Santiago J.V. Retinocortical conduction time in diabetics with abnormal pattern reversal electroretinograms and visual evoked potentials. Doc Ophthalmol. 1988;70:19–28. doi: 10.1007/BF00154732. [DOI] [PubMed] [Google Scholar]
- 20.Aldana Marcos H.J., Ferrari C.C., Benitez I., Affanni J.M. Standardization of fixation, processing and staining methods for the central nervous system of vertebrates. Biocell. 1996;20:265–272. [PubMed] [Google Scholar]
- 21.Paxinos G., Watson C. Elsevier; Amsterdam: 1997. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]
- 22.Fernandez D.C., Bordone M.P., Chianelli M.S., Rosenstein R.E. Retinal neuroprotection against ischemia-reperfusion damage induced by postconditioning. Invest Ophthalmol Vis Sci. 2009;50:3922–3930. doi: 10.1167/iovs.08-3344. [DOI] [PubMed] [Google Scholar]
- 23.Crish S.D., Sappington R.M., Inman D.M., Horner P.J., Calkins D.J. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010;107:5196–5201. doi: 10.1073/pnas.0913141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angelucci A., Clascá F., Sur M. Anterograde axonal tracing with the subunit B of cholera toxin: a highly sensitive immunohistochemical protocol for revealing fine axonal morphology in adult and neonatal brains. J Neurosci Methods. 1996;65:101–112. doi: 10.1016/0165-0270(95)00155-7. [DOI] [PubMed] [Google Scholar]
- 25.Prichard J.R., Armacanqui H.S., Benca R.M., Behan M. Light-dependent retinal innervation of the rat superior colliculus. Anat Rec (Hoboken) 2007;290:341–348. doi: 10.1002/ar.20424. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L., Ino-ue M., Dong K., Yamamoto M. Retrograde axonal transport impairment of large- and medium-sized retinal ganglion cells in diabetic rat. Curr Eye Res. 2000;20:131–136. [PubMed] [Google Scholar]
- 27.Moreno M.C., Marcos H.J., Oscar Croxatto J., Sande P.H., Campanelli J., Jaliffa C.O., Benozzi J., Rosenstein R.E. A new experimental model of glaucoma in rats through intracameral injections of hyaluronic acid. Exp Eye Res. 2005;81:71–80. doi: 10.1016/j.exer.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Belforte N., Sande P., de Zavalía N., Knepper P.A., Rosenstein R.E. Effect of chondroitin sulfate on intraocular pressure in rats. Invest Ophthalmol Vis Sci. 2010;51:5768–5775. doi: 10.1167/iovs.10-5660. [DOI] [PubMed] [Google Scholar]
- 29.Escartin C., Bonvento G. Targeted activation of astrocytes: a potential neuroprotective strategy. Mol Neurobiol. 2008;38:231–241. doi: 10.1007/s12035-008-8043-y. [DOI] [PubMed] [Google Scholar]
- 30.Rungger-Brändle E., Dosso A.A., Leuenberger P.M. Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2000;41:1971–1980. [PubMed] [Google Scholar]
- 31.Ibrahim A.S., El-Remessy A.B., Matragoon S., Zhang W., Patel Y., Khan S., Al-Gayyar M.M., El-Shishtawy M.M., Liou G.I. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes. 2011;60:1122–1133. doi: 10.2337/db10-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greene D.A., Sima A.A., Stevens M.J., Feldman E.L., Lattimer S.A. Complications: neuropathy, pathogenetic considerations. Diabetes Care. 1992;15:1902–1925. doi: 10.2337/diacare.15.12.1902. [DOI] [PubMed] [Google Scholar]
- 33.Giarelli L., Grandi G., Delendi M., Falconieri G. Optic nerve in diabetes mellitus: a case control study. Metab Pediatr Syst Ophthalmol. 1986;9:71–73. [PubMed] [Google Scholar]
- 34.Ino-ue M., Yokogawa H., Yamamoto M., Naka H., Kuriyama H. Structural impairments in optic nerve of diabetic rats ameliorated with the aldose reductase inhibitor. Exp Eye Res. 1998;66:397–401. doi: 10.1006/exer.1997.0426. [DOI] [PubMed] [Google Scholar]
- 35.Zagon I.S., Klocek M.S., Sassani J.W., McLaughlin P.J. Dry eye reversal and corneal sensation restoration with topical naltrexone in diabetes mellitus. Arch Ophthalmol. 2009;127:1468–1473. doi: 10.1001/archophthalmol.2009.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Readhead C., Popko B., Takahashi N., Shine H.D., Saavedra R.A., Sidman R.L., Hood L. Expression of a myelin basic protein gene in transgenic shiverer mice: correction of the demyelinating phenotype. Cell. 1987;48:703–712. doi: 10.1016/0092-8674(87)90248-0. [DOI] [PubMed] [Google Scholar]
- 37.Shine H.D., Readhead C., Popko B., Hood L., Sidman R.L. Morphometric analysis of normal, mutant, and transgenic CNS: correlation of myelin basic protein expression to myelinogenesis. J Neurochem. 1992;58:342–349. doi: 10.1111/j.1471-4159.1992.tb09316.x. [DOI] [PubMed] [Google Scholar]
- 38.Bondan E.F., Lallo M.A., Trigueiro A.H., Ribeiro C.P., Sinhorini I.L., Graça D.L. Delayed Schwann cell and oligodendrocyte remyelination after ethidium bromide injection in the brainstem of Wistar rats submitted to streptozotocin diabetogenic treatment. Braz J Med Biol Res. 2006;39:637–646. doi: 10.1590/s0100-879x2006000500011. [DOI] [PubMed] [Google Scholar]
- 39.Arnett H.A., Fancy S.P.J., Alberta J.A., Zhao C., Plant S.R., Kaing S., Raine C.S., Rowitch D.H., Franklin R.J., Stiles C.D. bHLH transcription factor Olig1 is required to repairedmyelinated lesions in the CNS. Science. 2004;306:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- 40.Chang A., Tourtellotte W.W., Rudick R., Trapp B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- 41.Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakazawa T., Nakazawa C., Matsubara A., Noda K., Hisatomi T., She H., Michaud N., Hafezi-Moghadam A., Miller J.W., Benowitz L.I. Tumor necrosis factor-α mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26:12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conforti L., Adalbert R., Coleman M.P. Neuronal death: where does the end begin. Trends Neurosci. 2007;30:159–166. doi: 10.1016/j.tins.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 44.Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- 45.Stokin G.B., Lillo C., Falzone T.L., Brusch R.G., Rockenstein E., Mount S.L., Raman R., Davies P., Masliah E., Williams D.S., Goldstein L.S. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 46.Buckingham B.P., Inman D.M., Lambert W., Oglesby E., Calkins D.J., Steele M.R., Vetter M.L., Marsh-Armstrong N., Horner P.J. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008;28:2735–2744. doi: 10.1523/JNEUROSCI.4443-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shirao Y., Kawasaki K. Electrical responses from diabetic retina. Prog Retin Eye Res. 1998;17:59–76. doi: 10.1016/s1350-9462(97)00005-0. [DOI] [PubMed] [Google Scholar]
- 48.Lieth E., Gardner T.W., Barber A.J., Antonetti D.A. Retinal neurodegeneration: early pathology in diabetes. Clin Exp Ophthalmol. 2000;28:3–8. doi: 10.1046/j.1442-9071.2000.00222.x. [DOI] [PubMed] [Google Scholar]
- 49.Asnaghi V., Gerhardinger C., Hoehn T., Adeboje A., Lorenzi M. A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes. 2003;52:506–511. doi: 10.2337/diabetes.52.2.506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Griffonia simplicifolia I–positive cells in normal and diabetic distal ONs. A: In transverse normal sections, weak-stained Griffonia simplicifolia I–positive cells were observed. Note the fine and ramified processes and the rod-shaped soma characteristic from resting microglia (a detail is shown in the inset). B: In diabetic animals, no clear differences were found in the morphologic appearance of microglial cells present in the distal portion of the ON. Shown are images representative of five animals per group. Scale bars: 50 μm (B); 25 μm (inset).