

Abstract
The World Health Organization estimates that the number of obese and overweight adults has increased to 1.6 billion, with concomitant increases in comorbidity. While genetic factors for obesity have been extensively studied in Caucasians, fewer studies have investigated genetic determinants of body mass index (BMI; weight (kg)/height (m)2) in African Americans. A total of 38 genes and 1,086 single nucleotide polymorphisms (SNPs) in African Americans (n = 1,173) and 897 SNPs in Caucasians (n = 1,165) were examined in the Southern Community Cohort Study (2002–2009) for associations with BMI and gene × environment interactions. A statistically significant association with BMI survived correction for multiple testing at rs4140535 (β = −0.04, 95% confidence interval: −0.06, −0.02; P = 5.76 × 10−5) in African Americans but not in Caucasians. Gene-environment interactions were observed with cigarette smoking and a SNP in ADIPOR1 in African Americans, as well as between a different SNP in ADIPOR1 and physical activity in Caucasians. A SNP in PPARGC1A interacted with alcohol consumption in African Americans, and a different SNP in PPARGC1A was nominally associated in Caucasians. A SNP in CYP19A1 interacted with dietary energy intake in African Americans, and another SNP in CYP191A had an independent association with BMI in Caucasians.
Keywords: African continental ancestry group, body mass index, European continental ancestry group, genetics, molecular epidemiology, obesity
Globally, the number of overweight (body mass index (BMI) ≥25) and obese (BMI ≥30) adults has increased to approximately 1.6 billion, with correspondingly increasing comorbid disease burdens (1). The public health impact and economic burden of obesity is substantial, since obesity is associated with increased risks of type 2 diabetes mellitus, cardiovascular disease, dyslipidemia, hypertension, sleep apnea, and several forms of cancer (2, 3).
Multiple lines of evidence indicate that a large proportion of obesity risk is mediated by genetic factors, with studies estimating that 40%–90% of human BMI variation is due to genetic factors (4–6). Several genome-wide association studies have identified reproducible associations, although these appear to explain only a small proportion of obesity risk. Several BMI genes are now known to function in the central nervous system or in energy homeostasis (7–12).
In the United States, the obesity epidemic disproportionately affects certain racial/ethnic minorities, including African Americans (13). The causes of this disparity are poorly understood, but they may be related to a combination of behavioral, environmental, and genetic risk factors (13). Although understanding the way in which genetic and environmental/behavioral risk factors interact may offer insights into modifiable factors, few studies have been performed to evaluate those phenomena (14).
In this study, we examined single nucleotide polymorphisms (SNPs) in 38 candidate genes selected for a putative function in pathways leading to obesity, as well as genes previously associated with body size in published studies of African Americans and Caucasians. We tested single marker associations and gene × environment interactions with known obesity risk factors, including smoking, physical activity, alcohol consumption, time spent sitting and sleeping, and dietary energy intake.
MATERIALS AND METHODS
Study population and data collection
Participants were enrolled in the Southern Community Cohort Study (SCCS), a prospective epidemiologic cohort study designed to examine racial/ethnic disparities in cancer incidence and mortality (15) in 12 southeastern states from 2002 to 2009. Participants were selected primarily from community health centers, government-funded facilities providing health services to low-income persons in underserved areas. Participants were aged 40–79 years, spoke English, and had not undergone treatment for cancer within the past year. All eligible participants completed a baseline in-person interview on lifestyle, medical, and other personal and familial characteristics and self-reported their height and weight. A copy of the study questionnaire can be found at www.southerncommunitystudy.org. For assessment of the accuracy of self-reported weight, 20% of participants had weight data abstracted from community health center medical records; this information was highly correlated with self-reported weight (r > 0.96). BMI was calculated as weight (kg) divided by the square of height (m2). Participants were asked to provide a biologic specimen; over 90% provided a blood, saliva (mouth rinse), and/or urine specimen, with approximately half donating a 20-mL blood sample.
From SCCS participants enrolled at a community health center between March 2002 and November 2004 who had provided a blood sample at the time of enrollment, 395 women and 396 men were randomly selected for the SCCS Pilot Biomarker Project on the basis of 4 factors, including sex (male/female), race/ethnicity (Caucasian or African-American), smoking status (current, former, or never smoker), and BMI (18–24, 25–29, or 30–45). In 2006, an additional sample of 1,605 women was randomly selected according to criteria similar to those used for the SCCS Pilot Biomarker Project, although 4 BMI strata were used (18–24, 25–29, 30–34, and 35–45). After combining the participants, 2,396 persons were eligible for genotyping. We excluded 10 subjects who reported a previous diagnosis of breast cancer at baseline. This resulted in 2,386 subjects eligible for genotyping. A detailed description of sample inclusions and exclusions is provided in Web Figures 1 and 2, which appear on the Journal’s Web site (http://aje.oxfordjournals.org/).
Participants were classified as current, former, or never smokers, and pack-years of smoking were calculated. The questionnaire also obtained data on physical activity and amounts of time spent sitting and sleeping and included an 89-item food frequency questionnaire (16, 17). We excluded participants who had extreme values for total energy intake (for women, <500 kcal/day or >3,500 kcal/day (156 African Americans, 86 Caucasians); for men, <800 kcal/day or >4,200 kcal/day (48 African Americans, 25 Caucasians)) (18) when analyzing interactions with energy intake. Alcohol consumption was self-reported by participants according to beverage type (beer, white wine, red wine, and liquor) and then totaled as grams of alcohol per day. Participants self-reported diagnoses of medical conditions and treatments, including diabetes, heart attack, coronary bypass surgery, and cancer. Institutional review boards at Vanderbilt University and Meharry Medical College approved the study.
Genotyping and SNP selection
We evaluated 38 candidate genes from relevant obesity and related biologic pathways. Obesity-related genes were selected on the basis of evidence from at least 2 studies indicating an association with obesity-related phenotypes and information available in the Obesity Gene Map (1). SNPs were selected to include functional candidate SNPs and haplotype tag SNPs tagging 10,000-base-pair flanking regions of each gene with an r2 value greater than or equal to 0.8 and a minor allele frequency greater than or equal to 0.05 using the Caucasian (CEU) and Yoruban (YRI) data sets in the HapMap Project (19). A total of 1,244 SNPs for the 38 candidate genes were selected. A complete list of genes and the number of SNPs examined is provided in Web Table 1.
A list of 300 ancestry informative markers (AIMs) was generated by comparing CEU and YRI subjects from HapMap and from a list of 1,508 AIMs from an Illumina-designed panel of ancestry estimation (Illumina, Inc., San Diego, California) (19). To minimize selection of AIMs associated with BMI, we excluded SNPs lying within 5 megabases of the boundaries of a candidate gene. Of the 300 AIMs selected, 292 passed through the Illumina scoring algorithm and were included in the final genotyping assay.
Genomic DNA was extracted from buffy coat using Qiagen’s DNA purification kit (Qiagen, Inc., Valencia, California) according to the manufacturer’s instructions. Of the 2,386 participants genotyped, 2,371 were successfully genotyped using the Illumina GoldenGate genotyping platform (Illumina, Inc.), and 2,369 had available BMI data. There was a 99.9% genotype concordance rate when duplicated samples were compared across plates.
Quality control
A total of 1,444 SNPs were genotyped (1,152 from the candidate genes and 292 AIMs). We included 1,374 total genotyped SNPs in the final analytic data set including the AIMs after removing SNPs with low genotyping efficiency (<95%), low concordance rates (<95%), and X-chromosome heterozygosity inconsistent with reported sex. All samples included in the final analysis had genotyping efficiency greater than 95%. A total of 2,338 participants remained in the final data set. All SNPs were tested for deviation from Hardy-Weinberg equilibrium using PLINK software, stratified by race/ethnicity (20). We excluded SNPs with a Hardy-Weinberg equilibrium P value less than or equal to 1.00 × 10−6, dropping 9 SNPs in African Americans and 6 SNPs in Caucasians. SNPs with a minor allele frequency less than 0.01 were also excluded, which resulted in our dropping 13 SNPs in African Americans and 205 SNPs in Caucasians. A detailed description of SNP inclusion and exclusion criteria is given in Web Figure 2.
We used Structure software (version 2.3.3; http://pritch.bsd.uchicago.edu/structure.html) and EIGENSTRAT (http://genepath.med.harvard.edu/∼reich/Software.htm) to examine AIMs in order to remove racially misclassified persons, as well as to create a term representing ancestry to include in our statistical models (21, 22). We conducted analyses adjusting for ancestry both with STRUCTURE variables and with EIGENSTRAT principal components separately for each racial/ethnic group using the AIMs and determined that the STRUCTURE variables resulted in the lowest genomic inflation factor. As a result, we adjusted for STRUCTURE variables in linear models evaluating associations between SNPs and BMI. EIGENSTRAT was used to examine the first and second principal components plotted according to self-reported race/ethnicity (Web Figure 3). This identified 18 self-reported Caucasians and 13 self-reported African Americans whose genotypes were not consistent with their reported race/ethnicity. The final data set included 1,086 SNPs in African Americans (n = 1,173) and 897 SNPs in Caucasians (n = 1,165), not including AIMs.
Statistical analysis
All analyses were conducted using STATA 11.0 statistical software (StataCorp LP, College Station, Texas). Demographic variables were analyzed with 2-sample rank sum tests to compare racial/ethnic groups when variables were continuous, and chi-square tests were used for binary variables.
The relation between BMI and candidate risk factors for obesity, including smoking, physical activity, time spent sitting, time spent sleeping, alcohol consumption, and dietary energy intake were investigated. Each factor was regressed separately on BMI in a generalized linear model while adjusting for sex and age. BMI values were not normally distributed; therefore, we performed an analysis assuming a Gaussian distribution with a log link function and applied a robust measure of standard error. The normality of regression residuals was verified to evaluate the modeling approach. Histograms of the BMI distributions are provided in Web Figure 4. Similarly, the association between each genetic marker and BMI was assessed using a generalized linear model stratified by race/ethnicity, using a log link function and robust standard errors, while adjusting for percent ancestry. The most significant SNPs in African-American and Caucasian subjects were also evaluated using logistic regression subdividing BMI into strata (<25, 25–29, 30–34, and ≥35) and evaluating each category in comparison with BMI <25 as the outcome to estimate the changes in risk of overweight, obesity, and severe obesity, respectively. Given the known differences in environmental risk factors, we also performed sensitivity analyses for gene-environment interactions. We did not observe significant differences in point estimates or levels of statistical significance from the sex- and age-adjusted analyses.
Obesity risk factors examined for gene-environment interactions included cigarette smoking (current smoking and pack-years analyzed individually), physical activity, alcohol consumption, time spent sitting, time spent sleeping, and total energy intake. For investigation of a BMI gene-environment interaction, we included adjustments for ancestral variation, age at interview, terms for the environmental variable and SNP, and sex. We considered only SNPs with a minor allele frequency greater than or equal to 0.10 to maintain the stability of effect estimates and statistical validity. Age and sex were not included as covariates in single-SNP analyses because they are not confounders for the genetic effects, as assessed by observing effect-size point estimates of SNP regression terms with and without sex and age. Alcohol consumption was analyzed with adjustment for smoking (current smoking vs. never smoking) after we observed that effect estimates for alcohol were attenuated when results were adjusted for smoking but smoking estimates were unchanged when adjusted for alcohol. Additionally, the most significant interactions were stratified by the levels of discrete environmental factors or by strata of continuous factors to evaluate the magnitude and direction of effect modification. Multiple testing was assessed through a false discovery rate (FDR) correction for multiple testing with q* = 0.10 for all single-SNP analyses and gene-environment interactions (23). For Caucasians, the FDR threshold for the most significant of 897 tests was P < 1.1 × 10−4; for African Americans, the analogous threshold for 1,086 tests was 9.2 × 10−5. For the gene-environment interactions, there were 824 tests per environmental variable in African-American subjects, with an FDR threshold for the most significant test of P < 1.2 × 10−4, and in Caucasian subjects there were 699 tests per variable, with an FDR threshold of P < 1.4 × 10−4. Statistically significant associations were those that passed an FDR correction for multiple testing, and nominally significant associations were those that had a P value less than or equal to 0.05 but did not pass an FDR correction for multiple testing. All P values are 2-sided.
RESULTS
Demographic data
This study included 1,173 African-American participants and 1,165 Caucasian participants; approximately 84% of participants were female (Table 1). African Americans reported approximately one-half the cigarette smoking of Caucasians but higher average amounts of time spent sitting (P < 1.00 × 10−4) or sleeping (P = 0.003). Reported alcohol (P < 1.00 × 10−4) and dietary energy (P < 1.00 × 10−4) intakes and rates of diabetes (P = 0.010) were also higher among African Americans. By design, mean BMIs were similar for African Americans and Caucasians (30.06 (standard deviation, 6.27) and 30.04 (standard deviation, 6.53), respectively; P = 0.822).
Table 1.
Demographic, Behavioral, and Health Status Characteristics of Participants, by Race/Ethnicity, Southern Community Cohort Study, 2002–2009
| Variable | African Americans (n = 1,173) |
Caucasians (n = 1,165) |
||||
| No. of Persons | Mean (SD) | % | No. of Persons | Mean (SD) | % | |
| Body mass indexa | 1,173 | 30.06 (6.27) | 1,165 | 30.04 (6.53) | ||
| <25 | 304 | 25.92 | 307 | 26.35 | ||
| 25–29 | 313 | 26.68 | 304 | 26.09 | ||
| 30–34 | 282 | 24.04 | 280 | 24.03 | ||
| ≥35 | 274 | 23.36 | 274 | 23.52 | ||
| Weight, poundsb | 1,173 | 183.31 (40.84) | 1,165 | 181.65 (42.48) | ||
| Age at interview, years | 1,173 | 50.11 (8.72) | 1,165 | 50.31 (8.67) | ||
| Male sex | 191 | 16.28 | 193 | 16.57 | ||
| Heart disease (yes) | 50 | 4.26 | 74 | 6.35 | ||
| Prevalent diabetes (yes) | 251 | 21.40 | 200 | 17.17 | ||
| Cigarette smoking | 1,171 | 1,160 | ||||
| Never smoker | 477 | 40.73 | 379 | 32.67 | ||
| Current smoker | 434 | 37.06 | 503 | 43.36 | ||
| Former smoker | 260 | 22.20 | 278 | 23.97 | ||
| Pack-years of cigarette smoking | 1,168 | 9.85 (14.88) | 1,156 | 20.67 (25.40) | ||
| Physical activity, MET-hours | 1,169 | 22.21 (16.60) | 1,160 | 22.91 (17.47) | ||
| Time spent sitting, hours/day | 1,164 | 9.35 (5.03) | 1,158 | 8.50 (4.41) | ||
| Time spent sleeping, hours/day | 1,167 | 7.30 (1.98) | 1,159 | 6.97 (1.81) | ||
| Alcohol consumption, g/day | 1,118 | 13.25 (38.78) | 1,137 | 7.46 (34.40) | ||
| Total dietary energy intake, kcal/day | 916 | 1,969.12 (751.02) | 1,026 | 1,876 (719.94) | ||
Abbreviations: MET, metabolic equivalent of task; SD, standard deviation.
Weight (kg)/height (m)2.
1 pound = 0.45 kg.
Environmental/behavioral risk factors and BMI
A summary of the association between BMI and smoking status or other candidate environmental/behavioral risk factors is provided in Table 2. We observed statistically significant inverse associations between BMI and current cigarette smoking and alcohol consumption among both African Americans and Caucasians. Among Caucasians, but not among African Americans, we observed an inverse association with energy expenditure and a positive association with amount of time spent sitting.
Table 2.
Associations Between Environmental/Behavioral Risk Factors and Body Mass Indexa (Continuous) After Adjustment for Sex and Age at Interview, by Race/Ethnicity, Southern Community Cohort Study, 2002–2009
| Variable | African Americans (n = 1,173) |
Caucasians (n = 1,165) |
||||
| βb | 95% CI | P Valuec | βb | 95% CI | P Valued | |
| Cigarette smoking | ||||||
| Former smoking | −0.020 | 0.051, 0.011 | 0.205 | 0.012 | −0.019, 0.043 | 0.446 |
| Current smoking | −0.093 | −0.121, −0.066 | <1.00 × 10−10 | −0.068 | −0.097, −0.038 | 7.93 × 10−6 |
| Pack-years of smoking | −0.001 | −0.002, −0.001 | 0.001 | −0.0005 | −0.001, −0.0004 | 0.033 |
| Physical activity | −0.0005 | −0.001, 0.0002 | 0.157 | −0.001 | −0.002, −0.0005 | 0.001 |
| Time spent sitting | 0.001 | −0.002, 0.004 | 0.432 | 0.006 | 0.004, 0.009 | 2.41 × 10−6 |
| Time spent sleeping | −0.002 | −0.008, 0.004 | 0.571 | 0.0005 | −0.007, 0.008 | 0.894 |
| Alcohol consumptione | −0.0007 | −0.001, −0.0003 | 0.0003 | −0.0006 | −0.001, −0.0002 | 0.004 |
| Dietary energy intakef | 0.0002 | −0.002, 0.002 | 0.815 | 0.0003 | −0.002, 0.002 | 0.763 |
Abbreviations: BMI, body mass index; CI, confidence interval.
Weight (kg)/height (m)2.
Beta coefficient from the generalized linear model for the variable indicated, which is the log of the average change in BMI.
Test of the association between the risk factor and BMI in African Americans.
Test of the association between the risk factor and BMI in Caucasians.
Analyses of alcohol consumption also included adjustment for current smoking.
Dietary energy intake was divided by 100 in order to reduce the scale and make the coefficients more interpretable.
Single-locus associations
A summary of the gene information and BMI association results for SNPs with P values less than 0.01 is provided in Table 3. Eight SNPs in 6 genes showed evidence of association among African Americans (P < 0.01); all SNPs were nominally associated, with the exception of 1 SNP that survived correction for multiple testing. Nine SNPs in 7 genes showed evidence of association with BMI among Caucasians, at a nominal significance level.
Table 3.
Associations of Single Genetic Markers With Body Mass Indexa (Continuous) After Adjustment for Ancestry (P < 0.01) and Minor Allele Frequency (Frequency > 0.01), Southern Community Cohort Study, 2002–2009b
| Population and Gene | Chromosome | rs No. | Base Pair Position | Minor Allele | Minor Allele Frequency | βc | 95% Confidence Interval | P Value |
| African Americans | ||||||||
| HTR1B | 6 | rs13212041 | 78171124 | A | 0.43 | −0.02 | −0.04, −0.01 | 0.008 |
| HTR1B | 6 | rs6296 | 78172260 | G | 0.23 | −0.03 | −0.05, −0.01 | 0.006 |
| HTR1B | 6 | rs4140535 | 78175052 | A | 0.38 | −0.04 | −0.06, −0.02 | 0.00006 |
| UCP2 | 11 | rs17132534 | 73691774 | G | 0.19 | 0.03 | 0.01, 0.05 | 0.009 |
| UCP3 | 11 | rs7110607 | 73710478 | C | 0.08 | −0.04 | −0.07, −0.01 | 0.009 |
| VDR | 12 | rs4334089 | 48286015 | G | 0.38 | −0.02 | −0.04, −0.01 | 0.008 |
| IGF1 | 12 | rs6214 | 102793569 | G | 0.46 | 0.02 | 0.01, 0.04 | 0.008 |
| MC4R | 18 | rs17066829 | 58030066 | T | 0.46 | 0.02 | 0.01, 0.04 | 0.007 |
| Caucasians | ||||||||
| IL6R | 1 | rs12083537 | 154381103 | G | 0.22 | −0.03 | −0.05, −0.01 | 0.003 |
| IL6R | 1 | rs1386821 | 154382049 | C | 0.20 | −0.03 | −0.05, −0.01 | 0.004 |
| GHSR | 3 | rs11929140 | 172155437 | G | 0.11 | −0.04 | −0.07, −0.02 | 0.002 |
| GHSR | 3 | rs2948694 | 172165163 | G | 0.11 | −0.04 | −0.07, −0.01 | 0.003 |
| PPARGC1A | 4 | rs6821591 | 23797000 | A | 0.48 | 0.02 | 0.01, 0.04 | 0.008 |
| LEP | 7 | rs2278815 | 127881851 | G | 0.44 | −0.03 | −0.04, −0.01 | 0.006 |
| CYP19A1 | 15 | rs1902584 | 51611654 | A | 0.08 | 0.05 | 0.02, 0.08 | 0.001 |
| GLDN | 15 | rs1961177 | 51625078 | A | 0.11 | 0.04 | 0.01, 0.07 | 0.006 |
| PPARA | 22 | rs135543 | 46555321 | A | 0.30 | −0.03 | −0.05, −0.01 | 0.005 |
Abbreviations: CYP19A1, cytochrome P450, family 19; GHSR, growth hormone secretagogue receptor isoform 1a; GLDN, gliomedin; HTR1B, 5-hydroxytryptamine (serotonin) receptor 1B; IGF1, insulin-like growth factor 1 (somatomedin C); IL6R, interleukin 6 receptor isoform 1 precursor; LEP, leptin precursor; MC4R, melanocortin 4 receptor; PPARA, peroxisome proliferator-activated receptor A; PPARGC1A, peroxisome proliferator-activated receptor G coactivator 1; rs, reference SNP; SNP, single nucleotide polymorphism; UCP2, uncoupling protein 2; UCP3, uncoupling protein 3 isoform UCP3L; VDR, vitamin D (1,25-dihydroxyvitamin D3) receptor.
Weight (kg)/height (m)2.
There were no statistically significant deviations from Hardy-Weinberg equilibrium in these associated SNPs.
Beta coefficient from the generalized linear model.
The most statistically significant single-locus association, which also passed an FDR correction for multiple testing, was seen among African Americans for the 5-hydroxytryptamine receptor 1B (HTR1B) SNP rs4140535 (minor allele frequency = 0.38; β = −0.04, 95% confidence interval (CI): −0.06, −0.02; P = 0.00006). The presence of the minor allele (A) for rs4140435 was associated with lower BMI (Figure 1), such that participants with the African-American genotype had a median BMI 2.4 units lower than participants with the GG genotype. Additional analyses performed after subdividing BMI into strata (<25, 25–29, 30–34, and ≥35) further demonstrated the declining risk of severe obesity per minor allele for rs4140535, with an odds ratio for exposure to the minor allele of 0.58 (95% CI: 0.45, 0.75) among African Americans with BMI ≥35 as compared with African Americans with BMI <25 (Table 4). Two other SNPs within HTR1B also were associated with BMI (rs6296 and rs13212041). These 3 SNPs in HTR1B had minor alleles associated with a lower BMI, and these SNPs were in moderate-to-high linkage disequilibrium (r2 = 0.17–0.47; D′ = 0.48–0.99) (Web Figures 5 and 6). Rs4140535 passed an FDR correction for single-locus multiple testing among African Americans.
Figure 1.
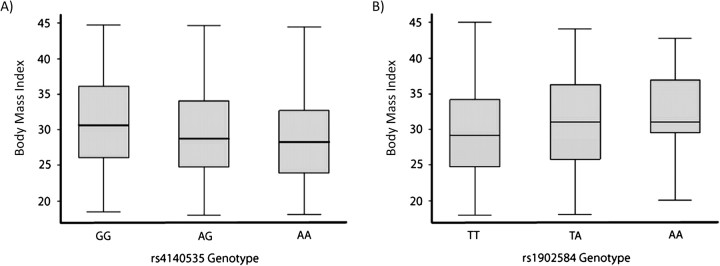
Box plots of the distribution of body mass indices (weight (kg)/height (m)2; continuous) according to genotype in A) African Americans (HTR1B marker rs4140535) and B) Caucasians (CYP19A1 rs1902584), Southern Community Cohort Study, 2002–2009. HTR1B, 5-hydroxytryptamine (serotonin) receptor 1B; CYP19A1, cytochrome P450, family 19. The T-shaped bars represent the 5th and 95th percentiles of the data.
Table 4.
Associations of the Most Statistically Significant Single Nucleotide Polymorphisms With Body Mass Indexa (Categorical), by Race/Ethnicity, Southern Community Cohort Study, 2002–2009
| Population and Gene | rs No. | Body Mass Index Category | Odds Ratio | 95% Confidence Interval | P Value |
| African Americans | |||||
| HTR1B | rs4140535 | <25 | 1.00 | ||
| 25–29 | 0.85 | 0.67, 1.07 | 0.168 | ||
| 30–34 | 0.77 | 0.60, 0.98 | 0.031 | ||
| ≥35 | 0.58 | 0.45, 0.75 | 2.33 × 10−5 | ||
| CYP19A1 | rs1902584 | <25 | 1.00 | ||
| 25–29 | 1.20 | 0.78, 1.83 | 0.409 | ||
| 30–34 | 1.01 | 0.62, 1.63 | 0.977 | ||
| ≥35 | 1.34 | 0.85, 2.10 | 0.207 | ||
| Caucasians | |||||
| HTR1B | rs4140535 | <25 | 1.00 | ||
| 25–29 | 0.75 | 0.59, 0.96 | 0.021 | ||
| 30–34 | 0.93 | 0.73, 1.18 | 0.538 | ||
| ≥35 | 0.87 | 0.68, 1.12 | 0.274 | ||
| CYP19A1 | rs1902584 | <25 | 1.00 | ||
| 25–29 | 0.88 | 0.55, 1.40 | 0.592 | ||
| 30–34 | 1.22 | 0.78, 1.92 | 0.382 | ||
| ≥35 | 1.91 | 1.26, 2.88 | 0.002 |
Abbreviations: CYP19A1, cytochrome P450, family 19; HTR1B, 5-hydroxytryptamine (serotonin) receptor 1B; rs, reference SNP; SNP, single nucleotide polymorphism.
Weight (kg)/height (m)2.
The single-locus association of highest statistical significance among Caucasians, which did not pass a correction for multiple testing, was for the cytochrome p450, family 19, subfamily A, polypeptide 1 (CYP19A1) SNP rs1902584 (minor allele frequency = 0.08; β = 0.05, 95% CI: 0.02, 0.08; P = 0.001) (Table 3). The minor allele (A) for rs1902584 was associated with an increase in BMI, but this SNP did not pass correction for multiple testing (Figure 1). The odds ratio for exposure to the minor allele increased with rising BMI; for BMI ≥35, the odds ratio was 1.91 (95% CI: 1.26, 2.88) (Table 4). Linkage disequilibrium plots for this gene among Caucasians are presented in Web Figures 7 and 8.
No associations were observed at P < 0.01 at specific SNPs or at the gene level in either racial/ethnic group.
Gene-environment interactions
Potential interactions between each genetic marker and candidate obesity risk factors were examined within each racial/ethnic group. This included gene interactions with current cigarette smoking (never smoker reference group, with former smokers deleted from the analysis), pack-years of cigarette smoking (never smoker reference group), physical activity, alcohol consumption, time spent sitting, time spent sleeping, and total energy intake. The most significant results from each set of tests are provided in Table 5, and detailed results are presented in Web Tables 2–15. Tests of association for the SNPs stratified by levels of environmental factors are provided in Web Table 16. Among African Americans, the strongest statistical interaction was between peroxisome proliferator-activated receptor-gamma coactivator 1, alpha (PPARGC1A) rs4619879 and alcohol consumption (β = 0.001, 95% CI: 0.001, 0.001; P = 3.75 × 10−5). The minor allele of rs4619879 in combination with increasing alcohol consumption was associated with increased BMI. The most significant interaction affecting BMI in Caucasians, though it was only nominally significant, was between ghrelin (GHRL) rs1617161 and time spent sleeping, where a combination of the minor allele and increasing amount of time spent sleeping was associated with lower BMI (β = −0.03, 95% CI: −0.047, −0.013; P = 5.08 × 10−4). No significant interactions were observed consistently in both African Americans and Caucasians.
Table 5.
Associations of the Most Statistically Significant Gene × Environment Interactions and Overlapping Interactions With Body Mass Indexa, by Race/Ethnicity, Southern Community Cohort Study, 2002–2009
| Population and Gene | Chromosome | Gene-Environment Interaction | Base Pair Position | Minor Allele | Minor Allele Frequency | βb | 95% Confidence Interval | P Value |
| African Americans | ||||||||
| HTR2C | X | rs17095676 × cigarette smoking (current) | 113776514 | A | 0.18 | −0.073 | −0.119, −0.028 | 0.001 |
| ADIPOR1 | 1 | rs6672643 × cigarette smoking (current) | 201167175 | G | 0.26 | −0.070 | −0.112, −0.028 | 0.001 |
| ADIPOR1 | 1 | rs6672643 × cigarette smoking (pack-years) | 201167175 | G | 0.26 | −0.003 | −0.005, −0.001 | 1.44 × 10−4 |
| PPARGC1A | 4 | rs4619879 × alcohol consumption | 23443974 | C | 0.46 | 0.001 | 0.001, 0.001 | 3.75 × 10−5 |
| IL6 | 7 | rs2069824 × dietary energy intake | 22731757 | G | 0.12 | −0.006 | −0.010, −0.003 | 3.00 × 10−4 |
| PPARA | 22 | rs7284616 × physical activity | 45020136 | A | 0.19 | 0.002 | 0.001, 0.003 | 7.05 × 10−5 |
| INSIG2 | 2 | rs17047728 × time spent sitting | 118555487 | A | 0.14 | 0.008 | 0.004, 0.013 | 4.46 × 10−4 |
| LEPR | 1 | rs6697315 × time spent sleeping | 65766144 | G | 0.38 | 0.016 | 0.008, 0.023 | 9.20 × 10−5 |
| Caucasians | ||||||||
| INSR | 19 | rs11667110 × cigarette smoking (current) | 7087609 | C | 0.32 | 0.074 | 0.031, 0.118 | 7.21 × 10−4 |
| IGFBP3 | 7 | rs6670 × smoking (pack-years) | 45918779 | T | 0.23 | −0.002 | −0.002, −0.001 | 6.52 × 10−4 |
| LEPR | 1 | rs17127677 × alcohol consumption | 65730862 | A | 0.14 | 0.001 | 0.000, 0.001 | 0.001 |
| GHSR | 3 | rs9819506 × dietary energy intake | 173652798 | A | 0.30 | −0.006 | −0.009, −0.003 | 1.87 × 10−4 |
| ADIPOR | 1 | rs12045862 × physical activity | 201183429 | A | 0.26 | 0.002 | 0.001, 0.003 | 0.001 |
| PPARG | 3 | rs709157 × time spent sitting | 12437024 | A | 0.32 | −0.006 | −0.01, −0.002 | 0.001 |
| PPARG | 3 | rs1175540 × time spent sitting | 12440243 | A | 0.36 | −0.006 | −0.009, −0.002 | 0.001 |
| GHRL | 3 | rs1617161 × time spent sleeping | 10311853 | A | 0.11 | −0.030 | −0.047, −0.013 | 5.08 × 10−4 |
Abbreviations: ADIPOR1, adiponectin receptor 1; GHRL, ghrelin precursor; GHSR, growth hormone secretagogue receptor isoform 1a; HTR2C, 5-hydroxytryptamine (serotonin) receptor 2C; IGFBP3, insulin-like growth factor binding protein 3; IL6, interleukin 6 (interferon, beta 2); INSIG2, insulin-induced protein 2; INSR, insulin receptor; LEPR, leptin receptor isoform 3; PPARA, peroxisome proliferator-activated receptor A; PPARGC1A, peroxisome proliferator-activated receptor G coactivator 1; PPARG, peroxisome proliferator-activated receptor G; rs, reference SNP; SNP, single nucleotide polymorphism.
Weight (kg)/height (m)2.
Beta coefficient from the generalized linear model.
DISCUSSION
In this study, most environmental/behavioral factors that were associated with BMI differed between African Americans and Caucasians. Exceptions included a strong inverse association with current smoking status and an inverse association between alcohol consumption and BMI, after adjustment for smoking. The latter association has been previously reported in some observational studies, although results have been inconsistent (24). Physical activity was inversely associated with BMI in both groups, while increasing sitting time was associated with increased BMI only among Caucasians. Total energy intake was significantly inversely associated with BMI in African Americans but not in Caucasians.
Our analysis most strongly implicated HTR1B as a determinant of BMI in African Americans. This SNP was especially associated with a high BMI (≥35) in African Americans but not in Caucasians. The HTR1B protein is a G-protein-linked receptor for serotonin (25, 26) and has been associated with reward pathways and addictive behaviors in mice (27). This gene is also reported to be associated with chemical dependency (28, 29), attention-deficit/hyperactivity disorder (30), and suicide (31), as well as extreme neurologic traits associated with eating behavior, such as BMI in bulimia nervosa patients (32). Response to treatment with selective serotonin reuptake inhibitors for depression, aggression, and obsessive-compulsive disorder has been associated with HTR1B genotype (33–35). Additionally, a nominally associated nonsynonymous HTR1B SNP from our study, rs6296 G861C, has been associated with substance abuse disorder, major depression, attention-deficit/hyperactivity disorder, and antisocial personality in alcohol-dependent patients (36–39). Our finding is thus consistent with observations from previous work in Caucasians in which several neurologic rather than metabolic genes were observed to be associated with BMI (12).
The gene CYP19A1 contained the most statistically significant SNP (it was nominally associated and did not pass a correction for multiple testing) in Caucasians, and there was evidence for an interaction between variation in this gene and dietary energy intake in African Americans. These SNPs (rs2445762 and rs1902584) were not in tight linkage disequilibrium in either racial/ethnic group (Caucasians: r2 = 0.03; African Americans: r2 = 0.05), and both associations persisted when conditioned on the other SNP (data not shown). This SNP was associated with a high BMI (≥35) in Caucasians but not in African Americans. CYP19A1 is expressed in many tissues, including the ovaries, and CYP19A1 activity in fat tissue is a primary estrogen source and sets the androgen/estrogen ratio for men and postmenopausal women. It is feasible that variants in CYP19A1 associated with BMI can extend the testosterone limits of adipose deposition or interact with insulin growth factors to maintain lean body mass or growth or induce tissue differentiation (40). SNPs in CYP19A1 have also been strongly associated with height in a Japanese cohort (41) and a European-ancestry sample (42), as well as with bone mineral density (43) and risk of fracture (44).
In addition, we observed nominally significant evidence for an association with SNPs in the PPARGC1A gene for Caucasian subjects and a statistically significant interaction with alcohol consumption in African Americans. These associated SNPs (rs6821591 and rs4619879) were in moderate linkage disequilibrium in both racial/ethnic groups (Caucasians: r2 = 0.06; African Americans: r2 = 0.20). Peroxisome proliferator-activated receptor gamma (PPARG) is a regulator of adaptive thermogenic activity, insulin action, and energy homeostasis, is probably inhibited by ethanol, and is a regulator of energy intake (45–49). PPARGC1A acts as a key transcriptional cofactor for PPARG function by binding to enhancer elements of key mitochondrial genes involved in adenosine triphosphate synthesis, and it is also expressed in the brain in areas implicated in addiction and in GABAergic neurons (49, 50). Obesity (51, 52) and several metabolic traits have been associated with PPARGC1A variation, such as waist-to-hip ratio hyperinsulinemia (53), birth weight (54), and type 2 diabetes mellitus (55–58). Furthermore, associations between PPARGC1A G482S genotypes and alcohol consumption have been observed in a Mediterranean population (59). It is thereby feasible that variation in this gene influences both the amount of alcohol consumed and measures of adiposity and metabolism, which might vary by population because of the underlying unobserved genetic variation. Stratified analysis of this SNP and alcohol consumption in African-American subjects suggests that this effect is observable only when the term for interaction with alcohol is included in the model.
Nominally significant interactions were observed between variants in the adiponectin receptor 1 gene (ADIPOR1) and smoking among African Americans and between ADIPOR1 variants and physical activity in Caucasians. ADIPOR1 is a receptor for globular and full-length adiponectin and mediates the expression of peroxisome proliferator-activated receptor A (PPARA) ligand activities, as well as fatty acid oxidation and glucose uptake by adiponectin (60, 61). Plasma adiponectin levels are decreased in obesity, insulin resistance, and type 2 diabetes mellitus, and adiponectin-deficient mice exhibit insulin resistance and diabetes (62–64). Suppression of ADIPOR1 expression decreases PPARGC1A expression and DNA deacetylation, decreases mitochondrial content and enzymes, and decreases oxidative stress-detoxifying enzymes in skeletal muscle, which are associated with insulin resistance and decreased exercise endurance (65). In a study of ADIPOR1 SNPs in African-American men, Beebe-Dimmer et al. (66) detected an association with BMI. Stratified analysis of these interactions suggested that the effect of ADIPOR1 variation was stronger in African-American nonsmokers and was greatly reduced in smokers. The stratified analysis of Caucasians suggested that more physical activity was associated with higher BMI levels per allele, while less activity was associated with lower BMI per allele. More research is required to elucidate more precisely the molecular mechanisms underlying the differences observed in the interaction for ADIPOR1.
One limitation of this study is the fact that several SNPs in genes most recently associated with BMI were not included in our analysis. Since all SNPs investigated were previously identified as associated with obesity, we attempted in our study to validate those prior findings in Caucasians and extend the results to African Americans. However, we realize that an independent replication sample for analyses of African Americans is needed prior to drawing conclusions. Additionally, we note that differences in the unobserved underlying genetic variation from the ancestral populations, as well as differences in the demographic histories of these populations, make direct replication of specific SNPs less likely than in 2 cohorts of the same race/ethnicity. Given the candidate gene approach, we chose not to adjust all P values for an increase in type 1 error due to the number of statistical tests. The self-reported environmental/behavioral variables employed in this study, particularly diet and physical activity exposures, are subject to measurement error that effectively reduces the power to detect associations for these variables. An additional limitation of this analysis was the use of self-reported height and weight values to calculate BMI (67); however, self-reported body size was strongly correlated with objective measurements in a sample of SCCS participants, as we noted above.
This study explored relations of SNPs in several candidate genes, environmental factors, and gene-environment interactions with BMI in African-American and Caucasian subjects. Although some but not all of the most statistically significant associations survived FDR correction for multiple testing, they warrant further study. While Caucasian populations have been extensively studied for genetic factors influencing BMI, the African-American population has been studied much less, despite a higher prevalence of obesity. More effort is required to discover whether the genetic determinants of BMI are population-specific or overlap between these populations. While commonalities are likely, this study suggests that there are potential differences in the genetic determinants of obesity by race/ethnicity. These differences may derive from specific associations with environmental factors within each racial/ethnic group and highlight the need to consider the environment and environmental interactions in genetic investigations of obesity.
Acknowledgments
Author affiliations: Department of Medicine, Vanderbilt University Medical Center, Nashville, Tennessee (Todd L. Edwards, Raquel Villegas, Maciej S. Buchowski, Jay H. Fowke, Ji Rong Long, Qiuyin Cai, Wei Zheng, Xiao-Ou Shu, Smith Jeffrey, Lisa B. Signorello, William J. Blot); Vanderbilt Epidemiology Center, Institute of Medicine and Public Health, Vanderbilt University Medical Center, Nashville, Tennessee (Todd L. Edwards, Raquel Villegas, Jay H. Fowke, Ji Rong Long, Qiuyin Cai, Wei Zheng, Xiao-Ou Shu); Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, Tennessee (Digna R. Velez Edwards); International Epidemiology Institute, Rockville, Maryland (Sarah S. Cohen, Lisa B. Signorello, William J. Blot); Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, Tennessee (Maciej S. Buchowski, Jay H. Fowke, David Schlundt, Ji Rong Long, Qiuyin Cai, Wei Zheng, Xiao-Ou Shu, Margaret K. Hargreaves, Smith Jeffrey, Lisa B. Signorello, William J. Blot); Department of Internal Medicine, Meharry Medical College, Nashville, Tennessee (Margaret K. Hargreaves); Medical Research Service, Veterans Affairs Tennessee Valley Healthcare System, Nashville, Tennessee (David Schlundt, Smith Jeffrey); Center for Human Genetics Research, Department of Molecular Physiology and Biophysics, Vanderbilt University Medical Center, Nashville, Tennessee (Scott M. Williams); and Nutritional Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Bethesda, Maryland (Charles E. Matthews).
Drs. Todd L. Edwards and Digna R. Velez Edwards are co-first authors and contributed equally to this work.
This project was funded in part by grant OP05-0927-DR1 from the Susan G. Komen for the Cure Foundation. The Southern Community Cohort Study is funded by grant R01 CA092447 from the National Cancer Institute. Sample preparation was conducted by the Survey and Biospecimen Shared Resource, which is supported in part by the Vanderbilt-Ingram Cancer Center (grant P30 CA68485). Dr. Todd L. Edwards also received support through Vanderbilt Clinical and Translational Research Scholar award 5KL2RR024975-04. Additional funds were provided by the Building Interdisciplinary Research Careers in Women's Health career development program (grant K12HD4383) for Dr. Digna Velez Edwards' salary.
Conflict of interest: none declared.
Glossary
Abbreviations
- AIM
ancestry informative marker
- BMI
body mass index
- CI
confidence interval
- FDR
false discovery rate
- SCCS
Southern Community Cohort Study
- SNP
single nucleotide polymorphism
References
- 1.World Health Organization. Global Database on Body Mass Index (BMI) Geneva, Switzerland: World Health Organization; 2009. ( http://www.who.int/mediacentre/factsheets/fs311/en/index.html). (Accessed June 6, 2010) [Google Scholar]
- 2.Field AE, Coakley EH, Must A, et al. Impact of overweight on the risk of developing common chronic diseases during a 10-year period. Arch Intern Med. 2001;161(13):1581–1586. doi: 10.1001/archinte.161.13.1581. [DOI] [PubMed] [Google Scholar]
- 3.Must A, Spadano J, Coakley EH, et al. The disease burden associated with overweight and obesity. JAMA. 1999;282(16):1523–1529. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]
- 4.Hjelmborg JB, Fagnani C, Silventoinen K, et al. Genetic influences on growth traits of BMI: a longitudinal study of adult twins. Obesity (Silver Spring). 2008;16(4):847–852. doi: 10.1038/oby.2007.135. [DOI] [PubMed] [Google Scholar]
- 5.Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27(4):325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 6.Wardle J, Carnell S, Haworth CM, et al. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87(2):398–404. doi: 10.1093/ajcn/87.2.398. [DOI] [PubMed] [Google Scholar]
- 7.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loos RJ, Lindgren CM, Li S, et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet. 2008;40(6):768–775. doi: 10.1038/ng.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyre D, Delplanque J, Chèvre JC, et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet. 2009;41(2):157–159. doi: 10.1038/ng.301. [DOI] [PubMed] [Google Scholar]
- 10.Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorleifsson G, Walters GB, Gudbjartsson DF, et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet. 2009;41(1):18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 12.Willer CJ, Speliotes EK, Loos RJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Genetic Investigation of Anthropometric Traits Consortium. Nat Genet. 2009;41(1):25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogden CL, Carroll MD, Curtin LR, et al. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295(13):1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 14.Hetherington MM, Cecil JE. Gene-environment interactions in obesity. Forum Nutr. 2010;63:195–203. doi: 10.1159/000264407. [DOI] [PubMed] [Google Scholar]
- 15.Signorello LB, Hargreaves MK, Steinwandel MD, et al. Southern Community Cohort Study: establishing a cohort to investigate health disparities. J Natl Med Assoc. 2005;97(7):972–979. [PMC free article] [PubMed] [Google Scholar]
- 16.Buchowski MS, Schlundt DG, Hargreaves MK, et al. Development of a culturally sensitive food frequency questionnaire for use in the Southern Community Cohort Study. Cell Mol Biol (Noisy-le-Grand). 2003;49(8):1295–1304. [PubMed] [Google Scholar]
- 17.Signorello LB, Munro HM, Buchowski MS, et al. Estimating nutrient intake from a food frequency questionnaire: incorporating the elements of race and geographic region. Am J Epidemiol. 2009;170(1):104–111. doi: 10.1093/aje/kwp098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joshipura KJ, Hu FB, Manson JE, et al. The effect of fruit and vegetable intake on risk for coronary heart disease. Ann Intern Med. 2001;134(12):1106–1114. doi: 10.7326/0003-4819-134-12-200106190-00010. [DOI] [PubMed] [Google Scholar]
- 19.Thorisson GA, Smith AV, Krishnan L, et al. The International HapMap Project Web site. Genome Res. 2005;15(11):1592–1593. doi: 10.1101/gr.4413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 22.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9(7):811–818. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 24.Togo P, Osler M, Sørensen TI, et al. Food intake patterns and body mass index in observational studies. Int J Obes Relat Metab Disord. 2001;25(12):1741–1751. doi: 10.1038/sj.ijo.0801819. [DOI] [PubMed] [Google Scholar]
- 25.Hamblin MW, Metcalf MA, McGuffin RW, et al. Molecular cloning and functional characterization of a human 5-HT1B serotonin receptor: a homologue of the rat 5-HT1B receptor with 5-HT1D-like pharmacological specificity. Biochem Biophys Res Commun. 1992;184(2):752–759. doi: 10.1016/0006-291x(92)90654-4. [DOI] [PubMed] [Google Scholar]
- 26.Jin H, Oksenberg D, Ashkenazi A, et al. Characterization of the human 5-hydroxytryptamine1B receptor. J Biol Chem. 1992;267(9):5735–5738. [PubMed] [Google Scholar]
- 27.Rocha BA, Scearce-Levie K, Lucas JJ, et al. Increased vulnerability to cocaine in mice lacking the serotonin-1B receptor. Nature. 1998;393(6681):175–178. doi: 10.1038/30259. [DOI] [PubMed] [Google Scholar]
- 28.Lerer E, Kanyas K, Karni O, et al. Why do young women smoke? II. Role of traumatic life experience, psychological characteristics and serotonergic genes. Mol Psychiatry. 2006;11(8):771–781. doi: 10.1038/sj.mp.4001855. [DOI] [PubMed] [Google Scholar]
- 29.Sun HF, Chang YT, Fann CS, et al. Association study of novel human serotonin 5-HT(1B) polymorphisms with alcohol dependence in Taiwanese Han. Biol Psychiatry. 2002;51(11):896–901. doi: 10.1016/s0006-3223(01)01366-x. [DOI] [PubMed] [Google Scholar]
- 30.Faraone SV, Perlis RH, Doyle AE, et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57(11):1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 31.Wang S, Zhang K, Xu Y, et al. An association study of the serotonin transporter and receptor genes with the suicidal ideation of major depression in a Chinese Han population. Psychiatry Res. 2009;170(2-3):204–207. doi: 10.1016/j.psychres.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Levitan RD, Kaplan AS, Masellis M, et al. Polymorphism of the serotonin 5-HT1B receptor gene (HTR1B) associated with minimum lifetime body mass index in women with bulimia nervosa. Biol Psychiatry. 2001;50(8):640–643. doi: 10.1016/s0006-3223(01)01201-x. [DOI] [PubMed] [Google Scholar]
- 33.Denys D, Van Nieuwerburgh F, Deforce D, et al. Prediction of response to paroxetine and venlafaxine by serotonin-related genes in obsessive-compulsive disorder in a randomized, double-blind trial. J Clin Psychiatry. 2007;68(5):747–753. doi: 10.4088/jcp.v68n0512. [DOI] [PubMed] [Google Scholar]
- 34.Silva H, Iturra P, Solari A, et al. Fluoxetine response in impulsive-aggressive behavior and serotonin transporter polymorphism in personality disorder. Psychiatr Genet. 2010;20(1):25–30. doi: 10.1097/YPG.0b013e328335125d. [DOI] [PubMed] [Google Scholar]
- 35.Villafuerte SM, Vallabhaneni K, Sliwerska E, et al. SSRI response in depression may be influenced by SNPs in HTR1B and HTR1A. Psychiatr Genet. 2009;19(6):281–291. doi: 10.1097/YPG.0b013e32832a506e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fehr C, Grintschuk N, Szegedi A, et al. The HTR1B 861G>C receptor polymorphism among patients suffering from alcoholism, major depression, anxiety disorders and narcolepsy. Psychiatry Res. 2000;97(1):1–10. doi: 10.1016/s0165-1781(00)00215-8. [DOI] [PubMed] [Google Scholar]
- 37.Hawi Z, Dring M, Kirley A, et al. Serotonergic system and attention deficit hyperactivity disorder (ADHD): a potential susceptibility locus at the 5-HT(1B) receptor gene in 273 nuclear families from a multi-centre sample. Mol Psychiatry. 2002;7(7):718–725. doi: 10.1038/sj.mp.4001048. [DOI] [PubMed] [Google Scholar]
- 38.Huang YY, Oquendo MA, Friedman JM, et al. Substance abuse disorder and major depression are associated with the human 5-HT1B receptor gene (HTR1B) G861C polymorphism. Neuropsychopharmacology. 2003;28(1):163–169. doi: 10.1038/sj.npp.1300000. [DOI] [PubMed] [Google Scholar]
- 39.Soyka M, Preuss UW, Koller G, et al. Association of 5-HT1B receptor gene and antisocial behavior in alcoholism. J Neural Transm. 2004;111(1):101–109. doi: 10.1007/s00702-003-0064-0. [DOI] [PubMed] [Google Scholar]
- 40.Harada N, Ogawa H, Shozu M, et al. Genetic studies to characterize the origin of the mutation in placental aromatase deficiency. Am J Hum Genet. 1992;51(3):666–672. [PMC free article] [PubMed] [Google Scholar]
- 41.Okada Y, Kamatani Y, Takahashi A, et al. A genome-wide association study in 19 633 Japanese subjects identified LHX3-QSOX2 and IGF1 as adult height loci. Hum Mol Genet. 2010;19(11):2303–2312. doi: 10.1093/hmg/ddq091. [DOI] [PubMed] [Google Scholar]
- 42.Ellis JA, Stebbing M, Harrap SB. Significant population variation in adult male height associated with the Y chromosome and the aromatase gene. J Clin Endocrinol Metab. 2001;86(9):4147–4150. doi: 10.1210/jcem.86.9.7875. [DOI] [PubMed] [Google Scholar]
- 43.Somner J, McLellan S, Cheung J, et al. Polymorphisms in the P450 c17 (17-hydroxylase/17,20-lyase) and P450 c19 (aromatase) genes: association with serum sex steroid concentrations and bone mineral density in postmenopausal women. J Clin Endocrinol Metab. 2004;89(1):344–351. doi: 10.1210/jc.2003-030164. [DOI] [PubMed] [Google Scholar]
- 44.Riancho JA, Valero C, Naranjo A, et al. Identification of an aromatase haplotype that is associated with gene expression and postmenopausal osteoporosis. J Clin Endocrinol Metab. 2007;92(2):660–665. doi: 10.1210/jc.2006-1616. [DOI] [PubMed] [Google Scholar]
- 45.Cecil JE, Palmer CN, Fischer B, et al. Variants of the peroxisome proliferator-activated receptor gamma- and beta-adrenergic receptor genes are associated with measures of compensatory eating behaviors in young children. Am J Clin Nutr. 2007;86(1):167–173. doi: 10.1093/ajcn/86.1.167. [DOI] [PubMed] [Google Scholar]
- 46.Ceni E, Crabb DW, Foschi M, et al. Acetaldehyde inhibits PPARγ via H2O2-mediated c-Abl activation in human hepatic stellate cells. Gastroenterology. 2006;131(4):1235–1252. doi: 10.1053/j.gastro.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Festuccia WT, Oztezcan S, Laplante M, et al. Peroxisome proliferator-activated receptor-gamma-mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology. 2008;149(5):2121–2130. doi: 10.1210/en.2007-1553. [DOI] [PubMed] [Google Scholar]
- 48.Hollenberg AN, Susulic VS, Madura JP, et al. Functional antagonism between CCAAT/enhancer binding protein-alpha and peroxisome proliferator-activated receptor-gamma on the leptin promoter. J Biol Chem. 1997;272(8):5283–5290. doi: 10.1074/jbc.272.8.5283. [DOI] [PubMed] [Google Scholar]
- 49.Puigserver P, Wu Z, Park CW, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 50.Cowell RM, Blake KR, Russell JW. Localization of the transcriptional coactivator PGC-1α to GABAergic neurons during maturation of the rat brain. J Comp Neurol. 2007;502(1):1–18. doi: 10.1002/cne.21211. [DOI] [PubMed] [Google Scholar]
- 51.Esterbauer H, Oberkofler H, Linnemayr V, et al. Peroxisome proliferator-activated receptor-gamma coactivator-1 gene locus: associations with obesity indices in middle-aged women. Diabetes. 2002;51(4):1281–1286. doi: 10.2337/diabetes.51.4.1281. [DOI] [PubMed] [Google Scholar]
- 52.Ridderstråle M, Johansson LE, Rastam L, et al. Increased risk of obesity associated with the variant allele of the PPARGC1A Gly482Ser polymorphism in physically inactive elderly men. Diabetologia. 2006;49(3):496–500. doi: 10.1007/s00125-005-0129-8. [DOI] [PubMed] [Google Scholar]
- 53.Weng SW, Lin TK, Wang PW, et al. Gly482Ser polymorphism in the peroxisome proliferator-activated receptor gamma coactivator-1α gene is associated with oxidative stress and abdominal obesity. Metabolism. 2010;59(4):581–586. doi: 10.1016/j.metabol.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 54.Gemma C, Sookoian S, Alvariñas J, et al. Maternal pregestational BMI is associated with methylation of the PPARGC1A promoter in newborns. Obesity (Silver Spring). 2009;17(5):1032–1039. doi: 10.1038/oby.2008.605. [DOI] [PubMed] [Google Scholar]
- 55.Kim JH, Shin HD, Park BL, et al. Peroxisome proliferator-activated receptor gamma coactivator 1 alpha promoter polymorphisms are associated with early-onset type 2 diabetes mellitus in the Korean population. Diabetologia. 2005;48(7):1323–1330. doi: 10.1007/s00125-005-1793-4. [DOI] [PubMed] [Google Scholar]
- 56.Kunej T, Globocnik Petrovic M, Dovc P, et al. A Gly482Ser polymorphism of the peroxisome proliferator-activated receptor-gamma coactivator-1 (PGC-1) gene is associated with type 2 diabetes in Caucasians. Folia Biol (Praha). 2004;50(5):157–158. doi: 10.14712/fb2004050050157. [DOI] [PubMed] [Google Scholar]
- 57.Sun L, Yang Z, Jin F, et al. The Gly482Ser variant of the PPARGC1 gene is associated with type 2 diabetes mellitus in northern Chinese, especially men. Diabet Med. 2006;23(10):1085–1092. doi: 10.1111/j.1464-5491.2006.01949.x. [DOI] [PubMed] [Google Scholar]
- 58.Zhang SL, Lu WS, Yan L, et al. Association between peroxisome proliferator-activated receptor-gamma coactivator-1α gene polymorphisms and type 2 diabetes in southern Chinese population: role of altered interaction with myocyte enhancer factor 2C. Chin Med J (Engl). 2007;120(21):1878–1885. [PubMed] [Google Scholar]
- 59.Francès F, Verdú F, Portolés O, et al. PPAR-alpha L162V and PGC-1 G482S gene polymorphisms, but not PPAR-gamma P12A, are associated with alcohol consumption in a Spanish Mediterranean population. Clin Chim Acta. 2008;398(1-2):70–74. doi: 10.1016/j.cca.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 60.Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423(6941):762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 61.Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13(3):332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 62.Hotta K, Funahashi T, Arita Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol. 2000;20(6):1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 63.Kubota N, Terauchi Y, Yamauchi T, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277(29):25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 64.Maeda N, Shimomura I, Kishida K, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8(7):731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 65.Iwabu M, Yamauchi T, Okada-Iwabu M, et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464(7293):1313–1319. doi: 10.1038/nature08991. [DOI] [PubMed] [Google Scholar]
- 66.Beebe-Dimmer JL, Zuhlke KA, Ray AM, et al. Genetic variation in adiponectin (ADIPOQ) and the type 1 receptor (ADIPOR1), obesity and prostate cancer in African Americans. Prostate Cancer Prostatic Dis. 2010;13(4):362–368. doi: 10.1038/pcan.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stewart AL. The reliability and validity of self-reported weight and height. J Chronic Dis. 1982;35(4):295–309. doi: 10.1016/0021-9681(82)90085-6. [DOI] [PubMed] [Google Scholar]