

Abstract
This review aims to present an overview of the application of stable isotope technology in clinical pharmacology. Three main categories of stable isotope technology can be distinguished in clinical pharmacology. Firstly, it is applied in the assessment of drug pharmacology to determine the pharmacokinetic profile or mode of action of a drug substance. Secondly, stable isotopes may be used for the assessment of drug products or drug delivery systems by determination of parameters such as the bioavailability or the release profile. Thirdly, patients may be assessed in relation to patient-specific drug treatment; this concept is often called personalized medicine. In this article, the application of stable isotope technology in the aforementioned three areas is reviewed, with emphasis on developments over the past 25 years. The applications are illustrated with examples from clinical studies in humans.
Keywords: bioavailability, breath test, clinical pharmacology, pharmacokinetics, phenotyping, stable isotope
Introduction
The use of stable isotopes combined with mass spectrometry is widely accepted as a valuable research methodology. This is illustrated by the more than 33 000 hits obtained with the query ‘stable isotope’ in the Scopus database (January 2011). Stable isotope technology is mainly applied in scientific areas such as agriculture, biochemistry, earth and planetary sciences, environmental science and medicine. In pharmacology, however, its use is relatively limited, as evidenced by about 4500 hits counted with the query ‘stable isotope’ and ‘drug’ (Scopus database, January 2011). The first paper describing the application of stable isotopes in this particular area dates from 1972 [1]. Knapp et al. [1] investigated nortryptiline metabolism using a combination of a well-known experimental set-up from radioactive tracer pharmacokinetic studies and stable isotope technology from metabolic studies. Since then, several groups have used stable isotope technology in their clinical pharmacological studies, especially those led by M. Eichelbaum and T. R. Browne. Their work has stimulated stable isotope technology to obtain a distinct place in clinical pharmacology in the 1980s; however, the most recent review paper on the all applications of stable isotopes in (clinical) pharmacology is over 20 years old [2]. Three main categories of stable isotope technology can be distinguished in clinical pharmacology (Table 1). Firstly, it is applied in the assessment of drug pharmacology to determine the pharmacokinetic profile or mode of action of a drug substance. Secondly, stable isotopes may be used for the assessment of drug products or drug delivery systems by determination of parameters such as the bioavailability or the release profile. Thirdly, patients may be assessed in relation to patient-specific drug treatment; this concept is often called personalized medicine. In this article, the application of stable isotope technology in the aforementioned three areas is reviewed, with emphasis on developments over the past 25 years. Applications are illustrated with examples from clinical studies in humans.
Table 1.
Summary of the applications of stable isotopes in clinical pharmacology
| Assessment of drug pharmacology | Pharmacokinetics |
| Mechanism of action | |
| Mechanism of toxicity and adverse effects | |
| Assessment of drug products or drug delivery systems | Bioavailability |
| Release profile of drug delivery systems | |
| Assessment of patients | Phenotyping |
| Monitoring drug treatment effect | |
| Clinical toxicology |
Sources of information
We selected articles published in international peer-reviewed journals from the Scopus database using the query ‘stable isotope’ combined with ‘drug’, and from our experience in the fields of metabolic research and drug delivery system assessment using stable isotope technology. The main inclusion criterion was that the article covers application of stable isotope technology in humans in relation to drug substances, drug products or drug treatments.
Principles of stable isotope technology and analysis
Physicochemical aspects
Stable isotopes are naturally occurring isotopes that differ from their parent atom (the most abundant form of the element) by a difference of one or more neutrons in the nucleus. Stable isotopes are not radioactive and may be incorporated in molecules that can be traced by analytical techniques discriminating on molecular weights (Table 2). An example of a stable isotopically labelled molecule is 13C-urea, in comparison to the naturally occurring 12C-urea, having molecular weights of 61.06 and 60.06 g mol–1, respectively. In this case, the mass of the labelled molecule is only modestly increased (1.7%). An extreme situation is 2H2O (heavy water, D2O) in which 100% of the 1H atoms are replaced by 2H. The molecular weights of regular water and heavy water are therefore 18 and 20 g mol–1, respectively; an increase of 11%. Physicochemical properties of heavy water are very different from regular water; for example, the freezing points (3.8 vs. 0.0°C) and the boiling points (101.4 vs. 100.0°C) [3]. Furthermore, when substituting hydrogen for deuterium at a chemically reactive site, enzyme-mediated reaction rates are decreased [4]. Apparently, carbon–hydrogen bonds have lower activation energy than carbon–deuterium bonds.
Table 2.
Naturally occurring stable isotopes used in clinical studies in men
| Element | Atomic number | Parent atom | Stable isotope | Abundance (in nature) |
|---|---|---|---|---|
| Hydrogen | 1 | 1H | 2H | 0.015% |
| Carbon | 6 | 12C | 13C | 1.1% |
| Nitrogen | 7 | 14N | 15N | 0.4% |
| Oxygen | 8 | 16O | 17O | 0.04% |
| 18O | 0.20% |
As biological properties relate to physicochemical properties, the different pharmacological and toxicological aspects of the labelled substance always need to be considered before starting studies in humans.
Safety
Safety concerns of stable isotope-labelled drug substances are only of a toxicological nature, because the isotopes are not radioactive. Toxicity aspects were investigated many years after their discovery and first application in animals and humans because of the limited availability of stable isotope-labelled substances. The toxicological properties of stable isotope substitution can be divided into two categories: those that involve deuterium (2H) and those that involve isotopes of higher elements [5].
As the mass of 2H is twice that of 1H, labelling may lead to large differences in metabolic handling, depending on the reactivity of the position of 2H in the molecule. Lewis investigated the effect of 2H2O on the development of seedlings from tobacco seed. Seeds in pure heavy water did not germinate, in contrast to seeds in regular water [6]. Also, Lewis investigated the effect of heavy water when administered to a mouse in a human dose equivalent of 4–5 l heavy water. The mouse showed signs of intoxication but did not die. He concluded that ‘heavy water is never toxic to any high degree and that it is tolerated in high concentrations by lower organisms. In such cases the rate of the vital processes seems to be roughly proportional to the fraction of the total hydrogen which is 1H’. The mode of toxicity that may be related to an isotope effect is the reduction of the rate of biochemical reactions due to the enhanced mass of a labelled atom or molecule. Owing to the great difference in mass between deuterium and hydrogen, a significant isotope effect occurs with deuterium. However, toxicity related to the isotope effect of deuterium can only be produced by very high levels of deuteration (>15% of body water), which greatly exceeds the amount of deuterium given in a typical tracer dose of drug [7]. We only expect deuterium toxicity when it is administered as 2H2O, because hydrogen of body water is an important source for hydrogen incorporation into organic molecules synthesized by a biological system.
No toxicological studies on stable isotopes of higher elements could be retrieved in the peer-reviewed literature additional to those reviewed by Pons et al. in 1999 [7]. Still, to date there is no report on whole-organism responses to 15N-enrichment levels. Mayevsky et al. [8] and Samuel & Steckel [9] administered 18O by inhalation and orally (incorporated in drinking water) to mice and found no biological effect. Administration of 13C-labelled endogenous substances, such as carbohydrates and lipids, has been widely applied in biochemical studies but to our knowledge never induced intoxication, either when administered as a single oral dose or as a continuous intravenous infusion. In practice, when designing an experiment, the dose of a labelled endogenous substance will be chosen between the limit of detection and 10% of the pool size. This upper level is far below toxicological levels. From the information above, it can be concluded that toxicological studies with stable isotopes are incomplete and limited. The body of evidence consists of weakly designed preclinical studies supplemented with a large amount of clinical experience with administrations within the context of metabolic studies in animals and humans. The evidence for the paradigm of stable isotopes having a margin of safety described as virtually limitless [10] is in our opinion not extensive. However, application of stable isotope technology in humans at historically applied enrichment levels has proved not to pose any clinically relevant risk.
Analysis
In almost all pharmaceutical applications of stable isotopes, isotope analysis is to be performed on drugs, drug-derived metabolites or endogenous substances in complex biological matrices, such as plasma or urine. This requires mass spectrometry (MS), if desired in combination with chromatographic separation techniques, such as gas chromatography (GC) or high-pressure liquid chromatography (HPLC), and requires sample preparation (extraction, column separation, hydrolysis) and often also derivatization.
Conventional scanning MS is able to perform measurements at low micromolar concentration levels with sufficient precision when the molar isotope enrichment levels are above 0.1%. When molar isotope enrichments are below 0.1%, isotope ratio mass spectrometry (IRMS) must be applied [11]. This type of MS determines isotope ratios at the atomic level (13C/12C, 2H/1H, 18O/16O or 15N/14N). Isotope ratio mass spectrometry originates from geochemistry, where minor differences of isotopes at the natural abundance level are to be detected to determine the origin of, for instance, fossils. An advantage of IRMS is the high precision for the isotope ratio measurement. The major disadvantage is the requirement of higher metabolite concentrations or larger sample volumes compared with scanning MS. For tracer studies as applied in the clinical situation, two types of IRMS may be distinguished: compound-specific and noncompound-specific systems.
Firstly, compound-specific IRMS is characterized by the introduction of compound-specific atoms into the IRMS. The particular compound is first separated from often complex mixtures by GC or HPLC. Gas chromatography may be coupled directly to the IRMS for breath analysis (e.g. 13C/12C in breath CO2). For this purpose, different options are available. Breath may be introduced directly into the IRMS after GC separation of the breath gases (O2, N2 and CO2). Recently, a nondispersive infrared technology was developed and applied routinely to measure 13C abundance in breath CO2[12, 13]. When nongaseous molecules are to be measured, GC can be coupled to the IRMS by a capillary reaction interface. The type of interface depends on the atom of interest; for 13C/12C, a combustion interface; for 15N/14N analysis, a combustion/reduction interface; and for 2H/1H and 18O/16O analysis, a pyrolysis interface is needed. High-pressure liquid chromatography may be coupled to IRMS by a chemical oxidation interface. So far, HPLC-IRMS can only be applied for 13C/12C measurements. A drawback of this technique is the limitation to aqueous solvent systems. Organic solvents introduce enormous background signals.
Secondly, noncompound-specific systems are available, which are characterized by limited sample preparation requirements. One special technique is the combination of elemental analyser and IRMS interfaced by a combustion/reduction or pyrolysis unit. In this way, 13C/12C, 15N/14N and 2H/1H isotope ratios can be measured in crude biological samples, such as urine.
New developments are in progress to use Fourier transform infrared and laser infrared technology for 13C analysis in CO2 gas [14, 15] In special cases, isotope tracer studies require the measurement of 2H or 18O in body water. In addition to IRMS, laser infrared technology has proved to be very sensitive and accurate for this goal [16].
Availability and quality of labelled substances
Drugs or endogenous substances labelled with various isotopes at specific positions in the molecule can often be purchased from commercial sources. In general, these labelled substances are manufactured by a limited number of companies (Isotec, Cambridge Isotope Laboratories, C/D/N-isotopes). The labelled substances are generally produced by chemical synthesis and sometimes by biological production methods. The concentration of a given isotope in biologically produced labelled substances may depend on the plant species or on the availability of isotope-labelled precursors. An example of the first type is the C4-plant corn. The C4-plants are characterized by a carbon fixation biochemical pathway in which more metabolic products enriched in 13C (percentage of 13C ∼1.097%) are generated compared with those from C3-plants (percentage of 13C ∼1.082%). An example of the second type is to grow plants in a greenhouse with 13C-enriched CO2. As a consequence, typical plant products will also be enriched, as 13C enters plant metabolic pathways following 13CO2 fixation in the photosynthesis process. Potato starch with 99% 13C enrichment is produced in this way (http://www.isolife.nl).
Labelled substances are produced in different quality grades. Most companies offer two grades. Firstly, there is the medicinal grade of labelled substances. This grade is produced according to the Good Manufacturing Practice (GMP) regulations for active pharmaceutical ingredients. Substances of this grade are sold to be applied in humans. The pharmacist is allowed to use these substances in drug products, based on a risk assessment in which the characteristics of the substance and its application are considered. Quality control consists of verification of documents (status of production site, status of manufacturing process, certificate of analysis) and verification of identity by chemical analysis [17]. Unfortunately, a very limited number of substances is commonly available in the GMP grade (i.e. 13C-urea and 13C-octanoic acid). Other substances may be produced according to GMP grade on request but are very expensive. Secondly, there are the laboratory grade labelled substances. They are produced under general laboratory standards, with limited quality control. No stability testing is performed to prove the assigned shelf-life. Cleaning procedures of production equipment are not validated. Sometimes, additional microbiological tests are offered on request (e.g. testing for sterility or bacterial endotoxins). These substances are sold with the restriction ‘not for human use’. The pharmacist is allowed to use these substances in drug products, based on a risk assessment in which the characteristics and lower quality assurance of the substance and its application are considered. Quality control consists of verification of documents (status of production site, status of manufacturing process, certificate of analysis), assessment of manufacture-related contaminants and related substances (based on route of synthesis), verification of chemical, physical and microbiological properties per packaged unit and documented support for shelf-life and eventually stability testing [17].
Applications in clinical pharmacology
Assessment of drug pharmacology
Pharmacokinetics
Pharmacokinetics comprises the absorption, distribution, metabolism and elimination characteristics of drugs. Pharmacokinetic profiling is conventionally executed with an unlabelled drug by administration, sampling tissues or body fluids of interest and subsequent determination of the drug concentration. From the data obtained, drug-specific pharmacokinetic characteristics can be calculated, such as bioavailability, volume of distribution, elimination half-life, clearance and protein binding. Kinetic studies with stable isotope-labelled drug substances are generally not performed. It is only the preferred option when conventional pharmacokinetic profiling of a drug is not feasible. This is the case in the following three situations, which are discussed in more detail below:
studies on pharmacokinetics under chronic administration of the same drug;
studies of the pharmacokinetics of high-clearance drugs; and
studies on the metabolism of drugs yielding metabolites that also occur naturally as endogenous substances.
Firstly, when medical treatment demands chronic administration of a drug, a change in drug pharmacokinetics may occur because of time- (carbamazepine, acetaminophen) or dose-dependent kinetics (phenytoin). When discontinuation of treatment is not acceptable, administration of a single dose of stable isotope-labelled drug may be the method of choice to determine the actual pharmacokinetic parameters in a patient. A typical example is the study by Malik et al. [18], in which the pharmacokinetic profiles of phenobarbital and phenytoin were determined in five three neonates, respectively, who were treated with those drugs at that time because of their history of seizures. A single dose of 2-15N-13C-phenobarbital or 2-13C-1,3-15N-phenytoin was administered orally together with a reduced intravenous dose of unlabelled drug product. Pharmacokinetic parameters such as clearance, half-life and volume of distribution could be calculated from drug plasma concentrations as a function of time. A large variation in pharmacokinetic parameters was observed (coefficient of variation ∼50%), which underlines the necessity of therapeutic drug monitoring for phenobarbital and phenytoin.
Secondly, stable isotopes may be used to correct for the influence of day-to-day variation in clinical studies with multiple-day administration and sampling. This strategy is especially of added value for high-clearance drugs. A nice example is the study of Bode et al. [19], who investigated nifedipine absorption in different regions of the gastrointestinal tract. On separate occasions, nifedipine solution was administered locally into the stomach, the small intestine and two sites in the colon in four healthy male volunteers using a remote-controlled drug delivery device (high-frequency-triggered capsule). In order to assess absolute and relative bioavailabilities, an intravenous infusion of nifedipine was given on a separate occasion, and all treatments were accompanied by a simultaneous oral dose of a 13C4-nifedipine solution to mathematically correct for day-to-day pharmacokinetic variation. The stable isotope-labelled substance was used in this study as an internal standard. A comparable approach was used by Fromm et al. [20] to investigate the presystemic gut wall elimination of verapamil.
Thirdly, studying drug metabolism is conventionally performed by measuring the parent substance and its metabolites. This approach works well as long as the metabolites are not naturally present. This problem of drug metabolites being endogenous substances as well was encountered by Durso et al. [10]. They investigated how carbidopa, a dopa-decarboxylase inhibitor, affects peripheral levodopa pharmacokinetics. They used 1,2,3,4,5,6-13C-levodopa as the drug substance, in which all 12C atoms of the phenyl group were replaced by 13C. Levodopa is metabolized by dopa-decarboxylase to yield dopamine, which in turn is converted into homovanillic acid (HVA) by monoamine oxidase and catechol-O-methyl transferase. Both dopamine and HVA occur naturally in the human body, where dopamine acts as a neurotransmitter. Dopamine and HVA derived from the labelled levodopa still contained the labelled phenyl group and could thus be discriminated from naturally occurring (unlabelled) counterparts. This approach of metabolic tracing may also be well suited to study the metabolism of biological drugs, such as peptides and proteins. The only clinical study in humans available on this subject is the work by Ligthart-Melis et al. [21], who investigated the influence of the route of administration (enteral or intravenous) of l-(2-15N)-glutamine on its metabolic fate; an important topic, because glutamine and its metabolite, arginine, are indicated to be an essential part of the immunonutrition of seriously ill patients [22]. It was shown that enteral compared with intravenous l-(2-15N)-glutamine results in lower arterial plasma 15N-glutamine, higher plasma 15N-citrulline and equal 15N-arginine concentrations. In a follow-up study [23], the authors investigated the same metabolic pathway, but now for oral or intravenous alanyl-(2-15N)-glutamine (Dipeptiven®). Again, enteral administration was shown to contribute more to de novo synthesis of arginine, which stimulates the immune response of patients with critical illnesses (immunonutrition).
In the two earlier reviews from more than two decades ago, racemate kinetics, tissue distribution and studying deep pool effect have been suggested as additional applications for stable isotope technology, but these have not gained extensive follow-up. For these applications, reference is made to earlier reviews [2, 24].
In conclusion, pharmacokinetic profiling is generally performed by studying the unlabelled drug substances in the body. This approach has less complexity, general availability of facilities and lower cost compared with application of stable isotope-labelled drug substances. Stable isotope-labelled drug substance profiling is only the preferred option when the conventional approach is not feasible.
Mechanism of action
Pharmacodynamics often involves binding of drug substances to targets such as receptors, enzymes, ion channels, transporters or nucleic acids, followed by signal transduction leading to a response. A great challenge is to understand the role of these drug targets in relation to physiology and pathology and their interaction with other drug targets and cofactors. Drugs are meant to interfere in pathological pathways by blocking or activating specific targets. In clinical pharmacology, these pathways are confirmed, and the real functionality of a drug in these pathways needs to be tested. Pharmacodynamics research approaches show a wide variety, mainly depending on the class of drugs being investigated. Apart from conventional biochemical testing, stable isotope-labelled substances are widely applied to investigate metabolic processes dynamically. Stable isotope technology can be an excellent tool to investigate the mechanism of action of drug substances.
A nice example is given by the efforts to reduce cholesterol levels in hypercholesterolaemic patients. Drug targets are enzymes in cholesterol metabolism, such as HMG-CoA reductase in cholesterol synthesis (inhibited by statins) or the sterol transporter Niemann-PickC1-Like1 in cholesterol absorption (inhibited by ezetemibe) or cholesterol catabolism (increase via enhanced bile salt formation by bile salt sequestrants such as cholestyramine, colestipol and colesevelam). Functional tests to measure cholesterol synthesis, cholesterol absorption and bile salt synthesis are based on isotope incorporation (cholesterol synthesis) and isotope dilution (cholesterol absorption and bile salt synthesis) [25]. Originally, tests were developed with radioactive isotopes (14C and 3H). Later on, they were modified into methods applying stable isotopes (13C and 2H). Cholesterol synthesis is measured by a constant intravenous infusion of 13C-acetate, which is in the form of acetyl-CoA, the building block of cholesterol. Cholesterol absorption is measured via oral administration of 13C- or 2H-cholesterol.
Two techniques have been developed. One relates the faecal excretion of labelled cholesterol to that of labelled sitostanol. Sitostanol is a non-absorbable sterole, and the malabsorbed fraction is considered 100%. The ratio of labelled cholesterol to labelled sitostanol in faeces reflects the malabsorbed fraction of the orally administered labelled cholesterol. The second technique applies an additional, differently labelled, cholesterol that is administered intravenously at the same time as the first one is administered orally. The ratio of the two labelled versions of cholesterol is determined in plasma after certain time intervals. This ratio represents the absolute bioavailability of orally administered (labelled) cholesterol. To measure bile acid synthesis, 13C- or 2H-labelled bile acids were administered orally, and plasma samples were collected over 4 days. The decay of label represents the fractional turnover rate (day−1), whereas from the intercept at t = 0 the pool size can be calculated (in micromoles). Multiplying pool size and fractional turnover rate results in the daily turnover rate, which equals the synthesis rate. It is of interest to realize that treatment with bile acid sequestrates does not only increase bile acid synthesis. As a consequence of treatment, the hepatic cholesterol pool becomes reduced, leading to an increased cholesterol synthesis. Therefore, most often bile acid sequestrant therapy is combined with statin treatment in order to maximize the clearance of cholesterol from the plasma.
Atorvastatin is known to reduce total cholesterol, triglycerides and low-density lipoprotein-cholesterol. Chan et al. [26] hypothesized that atorvastatin may decrease chylomicron remnant concentrations by increasing their metabolism. The rate of metabolism of a remnant-like intravenous emulsion labelled with cholesteryl-13C-oleate was determined by measuring breath 13CO2 and compared with placebo. Atorvastatin was shown to increase the rate of metabolism of this remnant-like emulsion.
Fibrates have been introduced for the treatment of hypertriglyceridaemia. The lipid-reducing mechanism of action is the interaction in lipoprotein metabolism by affecting apolipoprotein B synthesis and very low-density lipoprotein secretion. To show the mechanism of action in vivo in humans, apoB metabolism has been studied using stable isotopes. 13C-Valine or another labelled amino acid was infused, and the incorporation of 13C-valine into very low-density lipoprotein particles isolated from plasma samples was measured [25, 27].
The use of pre- and probiotics is promoted commercially in order to improve intestinal function. One positive effect is reduction of nitrogen uptake from the colon [28]. The mechanism of this effect has been studied by the oral administration of lactose-15N-ureide. This substance is not absorbed from the small intestine but fermented in the colon, leading to production and subsequent absorption of 15N-ammonia. The results demonstrate the suppression of the generation and accumulation of 15N-ammonia and other toxic fermentation products during treatment with pre- and probiotics [28].
The drug product acarbose interacts with the digestion of starch by inhibiting the intraluminal enzyme α-glycosidase. It is recommended in the treatment of type 2 diabetes and acts by partial inhibition of starch digestion, leading to a slower influx of glucose from the intestine into the circulation. The mechanism of action has been investigated by applying naturally 13C-enriched corn pasta with and without acarbose treatment [29] and 13C-sucrose [30]. Starch digestion, sucrose digestion and availability of glucose were shown to be reduced.
Premature infants have diminished synthesis of the surfactant, phosphatidylcholine (PC). In vitro studies showed that (prenatal) corticosteroids and exogenous surfactants stimulate the synthesis of PC. Bunt et al. showed that intramuscular betamethasone (12 mg) [31] and exogenous surfactants [32] stimulate PC synthesis in premature infants with respiratory distress syndrome. Infants received a 24 h infusion with stable isotope 13C6-glucose, starting around 5 h after birth. The incorporation of 13C into palmitic acid in surfactant PC was measured. Per dose of prenatally administered corticosteroids, the synthesis rate increased by 40% [31]. Surfactant production has also been studied using intravenous infusion of labelled fatty acids [33].
Insulin resistance is a situation where the hormone insulin becomes less effective in lowering blood glucose. It is related to physiological conditions (e.g. puberty), pathological conditions (metabolic syndrome) or drug use. Patients taking antiretroviral drugs, especially certain protease inhibitors and nucleoside reverse transcriptase inhibitors, appear to have an increased risk of hyperglycaemia and diabetes mellitus by induction of insulin resistance [34]. Treatment with insulin-like growth factor-1 has been suggested to decrease insulin resistance. Rao et al. [35] investigated the effect of a 3 month insulin-like growth factor-1 regime in men infected with human immunodeficiency virus who had excess abdominal adiposity and insulin resistance. Stable isotope technology was used to assess glucose and fat metabolism. They found that whole-body glucose uptake and glucose tolerance improved, while hepatic glucose production increased. Fasting triglycerides improved and visceral adiposity remained unchanged.
In conclusion, biochemical testing, in combination with stable isotope technology, is a valuable tool to study pharmacodynamics of a drug product.
Mechanism of toxicity and adverse effects
Metabolism-mediated adverse effects are often discovered clinically and explained by changed plasma levels of endogenous substances or drug-derived metabolites. However, knowledge about the mechanism of toxicity and advrse effects on a molecular level is limited [36]. For example, while troglitazone was withdrawn from the market in 2000 because of serious hepatotoxicity and replaced by chemical analogues, the mechanism of toxicity has to date not been elucidated [37].
The link between drug-derived metabolites and organ toxicity may be studied by modulating drug metabolism. Enzyme-mediated drug metabolism can be modulated by deuteration of drug substances. The advent of organ toxicity may be compared between labelled and unlabelled drug substances to investigate a causal relation. In fact, use is made in these studies of the isotope effect of changed reaction rates (see ‘Safety’). Further detailed insight may be generated by selective deceleration of certain metabolic pathways by selective replacement of hydrogen with deuterium. The relative amounts of different metabolites generated may be changed and related to different toxicity profiles [36].
When the drug target is known to be involved in other metabolic pathways, toxicity and type A adverse effects may be predicted to a certain extent. The mechanisms causing changes in plasma levels of endogenous substances can only be unravelled with biochemical tests, either alone or in combination with stable isotope technology. As an example, we discuss the adverse effects of parenteral nutrition. It is generally known that long-term parenteral nutrition may lead to intrahepatic cholestasis [38]. Using stable isotopes (13C-methionine), it was shown that hepatic mitochondrial function became reduced [39]. Other metabolic effects of parenteral nutrition related to hepatic function included increased cholesterol synthesis and reduced bile acid synthesis. The latter was proved by stable isotope-facilitated measurement of the plasma concentration of 7α-hydroxy-4-cholesten-3-one, an intermediate in bile acid synthesis indicative for rate of synthesis [40]. The changes in cholesterol and bile acid metabolism were explained by the fact that the gall bladder remains unstimulated when food is not administered enterally. This leads to reduced cholesterol absorption and a feedback regulation of cholesterol and bile acid synthesis. In studies using 2H5-phenylalanine, it was demonstrated that albumin production in the liver is increased during parenteral nutrition, whereas at a whole-body level protein catabolism is reduced [41]. This implies a direct effect on hepatic protein metabolism. Interestingly, similar effects on protein metabolism have been documented during treatment with infliximab [41].
It has been shown with stable isotope-labelled amino acids that ciclosporin A treatment after liver transplantation leads to hyperlipidaemia due to interference with lipoprotein metabolism. Hepatic bile acid synthesis is reduced due to ciclosporin A treatment, as has been shown with stable isotope bile acid kinetic studies in rats [42] and humans [43].
In conclusion, stable isotope technology is highly suitable for investigation of the mechanism of toxicity and adverse effects of drug substances. Labelled drug substances may be used to study drug metabolism-mediated toxicity. Labelled endogenous substances may be used to study the mechanism underlying changed plasma levels of endogenous substances. An increased application of stable isotope technology in these types of studies is envisaged, because drug safety is still gaining interest and toxicity is a major cause of market withdrawal of drugs [44].
Assessment of drug products and drug delivery systems
Bioavailability
A key characteristic of a drug product intended to treat a systemic condition is the ability to deliver the drug substance to the systemic circulation in an amount sufficient to elicit the desired response. This characteristic is called bioavailability and captures two features, namely rate and extent of absorption. Absolute and relative bioavailability denote the amount of drug that enters the systemic circulation from an intravenously administered amount of drug and from an orally administered amount relative to an intravenously administered amount of drug, respectively. Most bioavailability studies are of a comparative nature. This means that they compare the bioavailability of the test product to that of a reference. The reference product may be administered intravenously or non-intravenously. The fractions reaching the systemic circulation relative to the reference are called the absolute or relative bioavailability, respectively. Conventionally, a clinical study has a randomized two-period, two-sequence crossover design (Figure 1a). An adequate wash-out period between subsequent periods is needed to avoid drug carry-over effects. The validity of this study design is based on the assumption that distribution, metabolism and clearance of the particular drug remain constant over time. However, for drugs that are subject to extensive and highly variable metabolism, this assumption may be invalid. An alternative to the conventional approach is the application of the stable isotope technology, in which the test and the reference doses are administered concomitantly, thereby minimizing the influence of intra-individual variability of kinetics. Consequently, the clinical study design can be shortened to a randomized one-period, one-sequence crossover design (Figure 1b). Our literature search revealed 53 peer-reviewed papers describing investigations into the bioavailability or release profile of drug products by applying stable isotope technology (Table 3). As can be seen, the determination of the absolute bioavailability is an important application of stable isotope technology in this category of studies (25 of 52 papers). Strong et al. [45] were the first to describe this approach, in 1975. They determined the absolute bioavailability of oral N-acetylprocainamide by comparing its kinetics with that of co-administered 13C-N-acetylprocainamide by intravenous injection. Since then, the absolute bioavailability has been determined for formulations containing a wide variety of drug substances. Special applications are the determination of the absolute bioavailability of separate enantiomers forming a racemate (R- and S-forms of verapamil and ibuprofen) and the studies on intestinal absorption windows (nifedine and verapamil).
Figure 1.
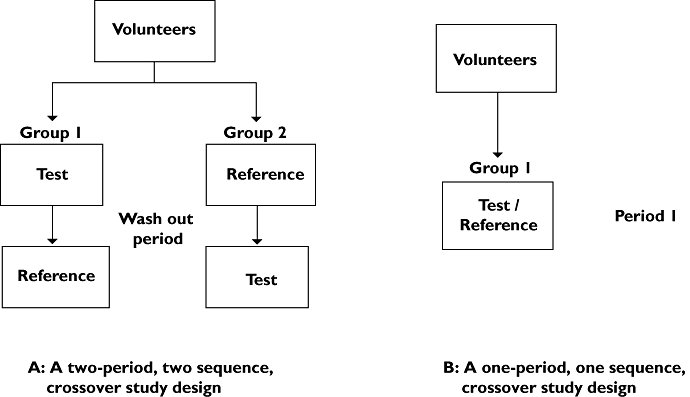
Schematic representation of the study design of a comparative bioavailability study with (a) or without stable isotope technology (b)
Table 3.
Papers describing the application of stable isotope technology to assess the bioavailability or release profile of drug products and delivery systems
| Author | Year | System (reference) | Labelled substance | System (test) | Study objective | Reference |
|---|---|---|---|---|---|---|
| Strong et al. | 1975 | Intravenous injection | 13C-N-Acetylprocainamide | Capsule (immediate release) | ABA | [45] |
| Heck et al. | 1979 | Oral solution | 2H2-Imipramine | Tablet (immediate release) | RBA | [79] |
| Eichelbaum et al. | 1981 | Intravenous infusion | 2H3-Verapamil | Oral solution | ABA | [80] |
| Eichelbaum et al. | 1981 | Oral solution | 2H3-Verapamil | Tablet (prolonged release) | RBA | [81] |
| Meresaar et al. | 1981 | Intravenous injection | 2H2-Methadone | Tablets | ABA | [82] |
| Rigby et al. | 1983 | Pro-banthine, 15 mg | Propantheline bromide | Tablet (new formulations) | RBA | [83] |
| Gammans et al. | 1984 | Oral solution | 2H4-Trazodone | Tablet (immediate release) | RBA | [47] |
| Vogelgesang et al. | 1984 | Intravenous injection | 2H7-Verapamil | ABA | [84] | |
| Marvola et al. | 1985 | Tablet (extended release) | 2H7-Verapamil | Tablet (extended release) | RBA | [85] |
| Shibuya et al. | 1985 | Tablet (excipient lactose) | 15N3-Nitroglycerin | Tablet (excipient methylcellulose) | RBA | [46] |
| Hatch et al. | 1986 | Oral solution | 2H3-Verapamil | Tablet (immediate release) | RBA | [86] |
| Fujioka | 1987 | Endogenous testosterone | 2H3-17α-Testosterone | Intramuscular injection | RP | [87] |
| Looareesuwan et al. | 1987 | Oral solution | 2H-Mefloquine | Tablet (immediate release) | RBA | [88] |
| Mikus et al. | 1987 | Intravenous injection | 13C4-Nitrendipine | Oral solution and tablet (immediate release) | ABA | [89] |
| Hallen et al. | 1988 | Tablet | 2H2-Terodiline | Tablet | RP | [90] |
| Atkinson et al. | 1989 | Intravenous injection | 13C-N-Acetylprocainamide | Capsule (immediate release) | ABA | [91] |
| Fujioka | 1989 | Endogenous testosterone | 2H3-17α-Testosterone proprionate | Intramuscular injection | RP | [92] |
| Shinohara & Baba | 1990 | Solution | 2H3-17α-Testosterone | Tablet | RBA | [93] |
| Aronoff et al. | 1991 | Intraveous infusion | 15N2-Cibenzoline | Capsule (immediate release) | ABA | [94] |
| Benowitz et al. | 1991 | Intravenous infusion | 2H2-Nicotine | Capsule (immediate release) | ABA | [95] |
| Benowitz et al. | 1991 | NA | 2H2-Nicotine | Transdermal delivery system | RP | [96] |
| Wilding et al. | 1991 | Intravenous infusion | 15N-Carbamazepine | Tablet (extended release) | ABA | [49] |
| Oral suspension | Tablet (extended release) | RP | ||||
| Bredberg et al. | 1992 | Intravenous infusion | 2H6-Terbutaline | Tablet | ABA | [97] |
| Oral solution | Tablet | RBA | ||||
| Browne et al. | 1993 | Intravenous infusion (sodium salt of phenytoin) | 15N2-13C3-Phenytoin (sodium salt) and 13C3-phenytoin phosphate | Intravenous infusion (phosphate-ester of phenytoin) | ABA | [98] |
| Hall et al. | 1993 | Intravenous injection | S-2H4-Ibuprofen | Tablet (immediate release) and intravenous injection | ABA | [99] |
| Le Houezec et al. | 1993 | NA | 2H2-Nicotine | Subcutaneous injection | RP | [100] |
| Richard et al. | 1994 | Tablet (immediate and slow release) | H2-Metoprolol | Tablet (extended release) | ABA | [101] |
| Hage et al. | 1995 | Oral solution | 2H3-Flecainide | Tablet (controlled release) | ABA | [102] |
| Sun et al. | 1995 | Intravenous infusion | 15N-Nitroglycerin | Transdermal delivery system | ABA | [103] |
| Voortman & Paanakker | 1995 | Intravenous infusion | 13C-Mirtazapine | Tablet (immediate release) | ABA | [104] |
| Bode et al. | 1996 | Intravenous infusion | 13C4-Nifedipine | Intrajenunal and intracolonic delivered solution | ABA | [105] |
| Cummings et al. | 1996 | NA | 13C-Glucose | Capsule (delayed release) | RP | [50] |
| Gross et al. | 1997 | Capsule (immediate release) | S-2H2-Gallopamil | Capsule (immediate release) | RBA | [106] |
| Benech et al. | 1998 | Intravenous injection | 25Mg and 26Mg | Tablets | ABA | [107] |
| Fromm et al. | 1998 | Intravenous injection | 2H7-Verapamil | Tablet (prolonged release) | ABA | [108] |
| Dilger et al. | 1999 | Intravenous infusion | 2H7-Verapamil | Tablet (prolonged release) | ABA | [109] |
| Pieniaszek et al. | 1999 | Oral solution | 13C6-Moricizine | Tablet (immediate release) | RBA | [110] |
| Yeh et al. | 1999 | Intravenous injection | 2H6-Indinavir | Capsule (immediate release) | ABA | [111] |
| Gross et al. | 2000 | Capsule (immediate release) | S-2H2-Gallopamil | Capsule (immediate release) | RBA | [108] |
| Heikkinen et al. | 2001 | Intravenous injection | 13C-Entacapone | Oral solution | ABA | [112] |
| Glaeser et al. | 2004 | Intravenous injection | 2H7-Verapamil | Intrajenunal delivered solution | ABA | [113] |
| Kasuya et al. | 1985 | Intravenous injection | 2H10-Phenytoin | Tablet | ABA | [114] |
| Verbeke et al. | 2005 | NA | 13C-Urea/15N-urea | Capsule (delayed release) | RP | [52] |
| Foster et al. | 2006 | Intravenous injection | 2H6-Methadone | Oral solution | ABA | [115] |
| Majumdar et al. | 2006 | Intravenous infusion | 13C2-15N3-Aprepitant | Capsule (immediate release) | ABA | [116] |
| Schultz et al. | 2006 | Intravenous injection | 13C-Dichloroacetate | Oral solution | ABA | [117] |
| Schellekens et al. | 2008 | Capsule (immediate release) | 13C-Glucose | Capsule (delayed release) | RP | [51] |
| Schellekens et al. | 2009 | Capsule (immediate release) | 13C-Urea | Capsule (delayed release) | RP | [54] |
| Schellekens et al. | 2010 | Capsule (immediate release) | 13C-Urea | Capsule (delayed release) | RP | [53] |
Abbreviations: ABA, absolute bioavailability; NA, not applicable; RBA, relative bioavailability; and RP, release profile.
The stable isotope technology has also been used to determine the relative bioavailability of a dosage form (13 of 49 papers) A typical example is the study by Shibuya et al. [46], in which the bioavailability of a stabilized formulation of a nitroglycerin sublingual tablet (excipient methylcellulose) was compared with the conventional formulation (excipient lactose). A conventional tablet containing 15N3-nitroglycerin was administered at the same time as the stabilized tablet containing regular nitroglycerin. In a 1 day experiment, it could be shown that both formulations exhibited equal bioavailability.
An interesting study design has been applied by Gammans et al. [47]. The bioavailability of trazodone from a new tablet formulation (dividose) was compared with the marketed tablet formulation (film-sealed tablet). An open three-period crossover design was applied, in which the dividose, a film-sealed tablet or a solution of trazodone was given. In addition, each subject received 5 ml of a 1% solution of 2H4-trazodone concomitantly with each formulation. The addition of the stable isotope-labelled trazodone revealed additional information. Firstly, it became possible to determine intra-individual variability, which turned out to be 6–38%, being less than interindividual variability (26–55%). Secondly, the ability to demonstrate dosage form equivalence could be enhanced by normalizing the data using the results obtained with the concomitantly administered 2H4-trazodone. This was demonstrated by the relatively smaller 95% confidence intervals and increased power for the relative area under the curve data compared with the trazodone area under the curve data alone. Thirdly, the equal plasma concentrations at all time points of trazodone and 2H4-trazodone administered as a solution indicated that the introduction of a stable isotope label does not alter trazodone pharmacokinetics. This result proved that an isotope effect is absent.
In conclusion, stable isotope technology may be used to determine absolute and relative bioavailability of drugs, their racemates and enantiomers. While acknowledging publication bias, the place for this technology in bioavailability studies seems limited, as is concluded from the 49 peer-reviewed papers that could be retrieved. When bioavailability has to be measured in steady-state conditions or when drug substance pharmacokinetics is known to exhibit large inter- and intrasubject variations, stable isotope technology seems to be the appropriate method. In other situations, the bioavailability study should be performed in straightforward manner with unlabelled drug substances.
Release profile of drug delivery systems
To achieve an optimal therapeutic effect, some drug substances can be administered by a drug delivery system with modified-release properties. Objectives that are aimed for by the modified-release profile encompass a modification of the rate and extent to which the drug substance becomes available at the site of action, the maintenance of therapeutic blood levels for a prolonged period of time or the prevention of toxic blood concentrations. For many oral modified-release products, drug delivery occurs in intestinal segments distal to the stomach (e.g. the duodenum, jejunum, ileum or colon). In vivo determination of the precise segment of release is the ultimate proof of a valid drug delivery system. The gold standard for these investigations is imaging. In the international literature, a myriad of approaches are described to image the release profile of modified-release systems; for example, endoscopy, radiology, gamma scintigraphy and magnetic resonance imaging. These technologies are in most cases combined with pharmacokinetic data. Currently, gamma scintigraphy is considered to be the gold standard. To perform a clinical study, a radiolabel is incorporated into the dosage form. For example, cold samarium oxide (152Sm2O3) is incorporated. After completion of manufacturing, the drug product is exposed to a neutron flux that transforms stable 152Sm into radioactive 153Sm with a half-life of 46 h. The 153Sm emits γ-radiation, which can be detected by a gamma camera [48]. Localization and monitoring of the radiolabelled drug product inside the body can thus be performed. Other radiolabels might also be used, such as 99mTc and 111In, but these require hot drug product manufacturing.
A more recent application of stable isotope technology is to determine the release profile of these modified-release drug delivery systems in vivo. When the release profile of a formulated drug product with modified-release technology incorporated has to be assessed, regular bioavailability studies may be performed with or without stable isotope technology. Wilding et al. [49] determined the release profile of carbamazepine formulated in an osmotically controlled-release oral delivery system (OROS). The study design consisted of conventional pharmacokinetics with gamma scintigraphy to localize the drug delivery system. Carbamazepine OROS was administered concomitantly with 15N-carbamazepine oral suspension. A 1 day study design was chosen because of the long elimination half-life (up to 36 h) of carbamazepine that would necessitate a wash-out period of 7 days. Carbamazepine OROS showed a reduced bioavailability (69%) compared with carbamazepine as a suspension. The fraction of the dose recovered was 16.8%. Systemic absorption from the colon proved to be reduced compared with absorption from the upper gastrointestinal segments (stomach and small intestine).
If a drug delivery system needs to be assessed in vivo, a stable isotopically labelled substance may be used to provide information regarding the intestinal segment of release. Stable isotope technology has been used to assess oral colon drug delivery systems in a few cases. To this end, 13C-glucose [50, 51] and 13C-urea [52, 53] have so far been used as marker substances. Both marker substances signal their arrival in bacteria-rich intestinal segments. Bacterial ureases transform 13C-urea into ammonia and 13CO2 by urease, while 13C-glucose is fermented to yield 13CO2, 13C-short-chain fatty acids and H2O. In both cases, 13CO2 is partly (about 55%) absorbed into the systemic circulation, transferred to the lungs and exhaled via breath [54]. The very short transfer time of CO2 from the lumen of the colon to the lung (<5 min [54]) permits the use of 13C appearance in breath as an almost real-time indicator of drug release in the colon. The marker substance of first choice for assessing oral colon delivery systems is 13C-urea. It has good physicochemical characteristics and an excellent safety profile. Urea is freely soluble in water (1 g ml–1) and permeates rapidly through the intestinal wall into the systemic circulation with virtually no presystemic degradation. The distribution of 13C-urea is favourable. The volume of distribution was found to be ∼0.6 l kg–1, indicating uniform distribution throughout the water compartment in the human body [55]. The 13C-urea may be absorbed from the gastrointestinal tract in intact form or fermented, depending on the presence and activity of ureases (generated by bacteria in the colonic flora). As urea is the end-product of nitrogen physiology, no relevant amount of metabolite is formed after intact absorption. Urea is eliminated mainly by renal excretion. This enables easy (non-invasive) sampling to quantify the amount of absorbed intact 13C-urea. Finally, urea exerts no pharmacological effects, making it a very attractive marker for clinical trials, especially in children or healthy volunteers.
In conclusion, stable isotope technology may be a valuable tool in the assessment of modified-release oral drug delivery systems. The labelled substance may be the drug substance itself (formulated as an intravenous injection or oral solution serving as a reference) or a labelled tracer that signals the site of release. Compared with imaging by gamma scintigraphy, stable isotope technology has two distinct advantages. Firstly, trial subjects are not exposed to radioactive radiation. Secondly, exposing the drug delivery device to a neutron flux to activate 152Sm elicits unknown and unpredictable effects on the release profile. This compromises the external validity of the test. On the other hand, there is only one stable isotopically labelled tracer available that has been validated for a specific intestinal segment. For oral colon delivery devices, 13C-urea may be used.
Assessment of patients
Phenotyping
It is generally accepted that certain drug treatments, such as thiopurines (azathioprine and 6-mercoptopurine) [56] and cytostatic drugs, should be pre-adjusted to the characteristics of the individual patient in order to improve drug efficacy or to reduce toxicity and adverse effects. The concept of personalized medicine is nowadays mainly practised by dosing on the basis of weight or body surface or by monitoring drug concentrations in plasma and subsequent dose adjustments [55, 57]. Personalized medicine has increasing momentum because of an increasing number of geno- and phenotyping tests aimed at predicting drug metabolism. These tests would enable physicians to rapidly identify responders or nonresponders to various drugs prior to initiation of therapy. The CYP enzymes are an important subset of phase I drug-metabolizing enzymes. With information on CYP enzyme activity, physicians and clinical pharmacists are able to ensure administration of the right drug, at the right dose, at the right time, to the right individual to obtain the right clinical outcome. Stable isotope technology can be used to provide rapid in vivo phenotype assessment of CYP enzymes. The CYP1A2 activity may be assessed by 13C-methacetin breath test and is relevant for drug and xenobiotic metabolism. The CYP2D6 activity may be assessed by 13C-dextromethorphan breath tests and is relevant for the metabolism of tamoxifen, β-blockers, antiarrhythmics, antidepressants, antipsychotics and opiates. The CYP2C19 activity may be assessed by 13C-pantoprazole breath test and is relevant for the metabolism of proton pump inhibitors, clopidogrel, cyclophosphamide and thalidomide [4]. Currently, application of these breath tests in clinical practice is limited. An interesting upcoming application is the development of a breath test using 2-13C-uracil to determine dihydropyrimidine dehydrogenase (DPD) enzyme activity. Dihydropyrimidine dehydrogenase metabolizes 5-fluorouracil (5-FU), an anticancer agent with a narrow therapeutic window. Dihydropyrimidine dehydrogenase deficiency is an inborn genetic disorder that may lead to severe toxicity or even death [58] in patients treated with 5-FU. Screening for DPD deficiency is therefore recommended before 5-FU-based treatment is started. The American Food and Drug Administration (FDA) has issued a contraindication for the use of 5-FU topical cream for patients with known DPD deficiency. The European Medicines Agency (EMA) demands intensive monitoring when this cream is prescribed to DPD-deficient patients. Currently, genotyping is hampered by insufficient knowledge of identity and clinical relevance of mutations and their interactions. The remaining alternative is phenotyping, i.e. determination of DPD activity [58]. Van Staveren et al. [59] showed that a plasma-based uracil screening test can differentiate between patients with deficient or normal DPD activity based on the uracil/dihydrouracil ratio in plasma. Mattison et al. [60, 61] showed that breath 13CO2 appearance after administration of 2-13C-uracil parallels plasma 2-13C-uracil and 2-13C-dihydrouracil pharmacokinetics and is an accurate measure of interindividual variation in DPD activity. Currently, none of the approaches is preferred. Information on the sensitivity, specificity, positive predictive value and cost of all methods is incomplete. In our opinion, phenotyping may become preferable, especially when a validated genotype–phenotype correlation is unclear. Additional and unresolved problems are phenotype changes during the course of life (e.g. concurrent medication use, environment, age) that are not taken into account by the genetic profile.
Monitoring of drug treatment effects
Monitoring of drug treatment effects is an essential part of medical treatment. Efficacy or toxicity of a drug may be indicated by the presence or amount of biochemical metabolites. As described in sections ‘Mechanism of action’ and ‘Mechanism of toxicity and adverse effects’, biochemical testing by stable isotope technology enables the dynamic assessment of metabolic pathways. As such, the test may be used in medical diagnosis. The same test can also be used to determine the relation between drug treatment, changes in pathological processes and, ultimately, the efficacy of a drug product. In most cases, approaches without application of stable isotope technology may also be suitable. A choice is made based on test-specific characteristics. In general, stable isotope biochemical testing is an approach that scores very well on such aspects as patient safety, comfort (i.e. non-invasive sampling) and costs, while performing equally on specificity and sensitivity compared with plasma- or biopsy-based testing.
An important example of a test applied in medical diagnosis and monitoring drug treatment effect is the 13C-urea breath test. It allows the detection of Helicobacter pylori infection in the stomach [62]. It shows the presence (or absence) of H. pylori urease, which converts orally administered 13C-urea to 13CO2 and ammonia. As 13CO2 is partly expired in the breath, 13C in breath signals presence of H. pylori in the stomach. Upon detection of H. pylori, antibiotic eradication therapy is initiated. The same 13C-urea breath test is then applied to monitor drug treatment efficacy, because absence of 13C in breath indicates eradication of H. pylori and successful therapy. There are many ways of testing: serology, 13C-urea breath test and faecal antigen test. Compared with histology, it is estimated that the 13C-urea breath test is 95% sensitive and 95% specific when diagnosing H. pylori infections [63]. Elwyn et al. [64] performed a cost effectiveness analysis and concluded that serology is less accurate than the 13C-urea breath test and the faecal antigen test, because it is not able to distinguish between present and past infections. According to Elwyn et al., the faecal antigen test should be preferred over the 13C-urea breath test because the breath test is ‘cumbersome to perform’.
Gastric emptying may be investigated in the clinical setting in the case of weight loss, suspicion of mechanical or anatomical obstructions, altered function due to underlying disease (e.g. amyloidosis), nausea or vomiting. Gastric emptying tests are applied for medical diagnosis, as well as for monitoring the effect of drug treatment. Gastroparesis is treated by acceleration of gastric emptying with cisapride. The symptoms of dumping syndrome are treated with octreotide (inhibition of release of insulin) or acarbose (inhibition of α-glycoside hydrolase) to moderate postprandial hypoglycaemia and early hyperglycaemia, respectively. Diagnosis of impaired gastric emptying and monitoring of drug treatment effects are traditionally performed by gamma scintigraphy. Recently, however, two stable isotope breath tests have been introduced for this purpose, one with sodium 13C-acetate [65] to measure gastric emptying of liquids and one with 13C-octanoic acid [66] to measure gastric emptying of solids. Braden et al. [65] investigated the effect of cisapride drug treatment on gastroparesis and found that the half-emptying times in diabetic gastroparesis were improved from 4 to 3 h. Dyspepsia also improved, whereas control of glycaemia did not.
Pancreatic enzyme replacement therapy is prescribed in the case of exocrine pancreatic insufficiency to avoid malnutrition [67]. Dysfunction of the digestive enzymes lipase, protease and α-amylase is difficult to assess. The ideal test to assess pancreatic enzyme function does not exist. Breath tests have been developed [65] and are based on oral intake of 13C-labelled mixed triglyceride (to test lipase activity), 13C-labelled cornstarch (to test α-amylase activity) or 13C-labelled egg protein (to test activity of proteases). An alternative method to detect fat malabsorption is the performance of a fat balance. For this purpose, the intake of fat and the faecal excretion of fat are measured. This requires faecal collection for 3 days. The faecal collection and faecal fat analysis are unpleasant tasks for the patient and analytical personnel.
Lactase enzyme supplements are prescribed in the case of lactose intolerance as a more attractive option than dietary restrictions that have nutritional disadvantages [68]. Lactase activity and effect of supplementation are generally assessed via the combined 13C-lactose/hydrogen breath test [69]. This test has limited accuracy due to false-positive outcomes. The plasma 13C-lactose/2H2-glucose test, however, showed excellent accuracy (no false negatives), but unfortunately is too complex for routine patient care [69].
In conclusion, stable isotope-based tests have a limited but distinct place in monitoring the effect of drug treatment, mainly in a number of gastroenterological diseases.
Clinical toxicology
Drug substances such as acetaminophen, methotrexate or amiodarone and also mushroom intoxication (e.g. death cap poisoning) may cause liver damage (cirrhosis), which, in extreme situations, may progress to acute liver failure shortly after intoxication. The extent of liver damage may be assessed by liver biopsy, biochemical tests, clinical assessments or stable isotope technology. Owing to the many functions of the liver, no single test is able to draw the complete picture. As liver biopsy merely tests inflammation and biochemical monitoring is correlated to metabolic liver capacity to a limited extent, the 13C-breath test is considered to be the only test to allow direct measurement of liver detoxification capacity. The greatest experience exists for the 13C-aminopyrine breath test [70, 71], by which the capacity to carry out hydroxylation and N- and O-methylation is measured. Breath tests are dynamic tests that are able to detect liver damage at an early stage. Therefore, these tests will lead to an earlier start of supportive treatment of liver damage than at the onset clinical symptoms, such as encephalopathy, when the prognosis has worsened. The use of aminopyrine as an analgesic drug has led to serious adverse events (agranulocytosis); therefore, it has been withdrawn from the market as a drug product. As pharmacological doses are prescribed (2 mg kg–1) in the breath test, aminopyrine-induced toxicity cannot be excluded and thus, this test is not preferred by most scientists. Alternatives are 13C-methacetin, 13C-caffeine or 13C-galactose breath tests.
Regulatory aspects of stable isotopes
Finished products containing stable isotopically labelled substances for medical purposes can generally not be procured. If such products are to be used in a clinical trial or in medical diagnostic procedures, the pharmacist is responsible for product design and clinical production. Stable isotope products may be classified as food products, medicinal products or medical devices. The product class determines the regulatory demands that apply for investigational or medical use. There is, to the best of our knowledge, no guidance document available for classifying these products. As it is difficult to classify a stable isotope product, there is a risk of misclassification. In particular, for investigators there is the risk that their product may unfortunately be classified in the category ‘medicinal product’, with a heavy regulatory burden as a consequence. In 2001, a new and more stringent set of rules was adopted by the European Union (EU) for clinical trials with medicinal products. This directive contains rules too strict for noncommercial research with drug products, by increasing the administrative and financial burdens [72]. Based on the definitions in the EU council directives for food products [73] (2002/178/EC), medicinal products [74] (65/65/EEC) and medical devices [75] (93/42/EEC), a decision tree may be drafted to support classification of a product containing a stable isotopically labelled substance (Figure 2). Based on the product class, the requirements for quality assurance can be determined (Table 4). The scope and format of product design documentation as part of the clinical trial application, as well as the GMP requirements for clinical production, can be determined. However, whatever product class applies, the product quality should always be assured, in order not to compromise on the quality of the trial and to protect the subject from unsafe experimentation.
Figure 2.
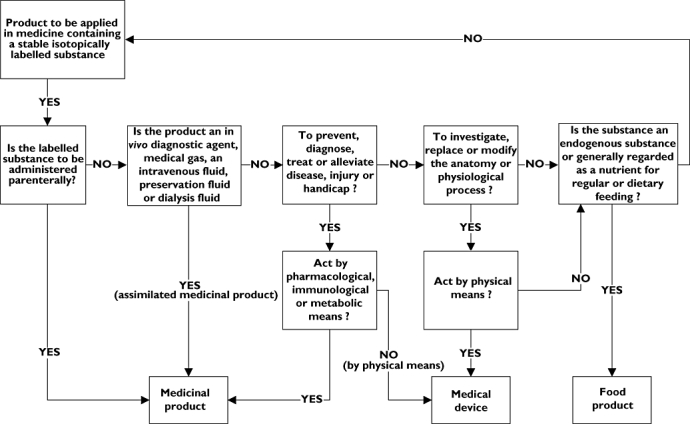
Decision tree to support classification of a product containing a stable isotopically labelled substance
Table 4.
Products containing stable isotopically labelled substances to be used in clinical trials or medical practice; regulatory requirements to assure quality of design and clinical production
| Quality assurance for clinical production | ||||
|---|---|---|---|---|
| Product class | Quality assurance for product design (to be included in clinical trial application) | Manufacturing license | Pharmacy | HACCP |
| Medicinal product | ||||
| IMP | Describe design, clinical production and (pre)clinical pharmacology and toxicology of IMP, conforming to the format of an IMP dossier | + | − | − |
| Magistral or officinal preparation, NIMP | Describe design, clinical production and (pre)clinical pharmacology and toxicology of IMP | − | + | − |
| Medical device | Describe design and clinical production of the medical device | − | + | − |
| Food product | Describe design and clinical production of the food product | − | − | + |
Abbreviations: HACCP, hazard analysis and critical control points; IMP, investigational medicinal product; and NIMP, non-investigational medicinal product.
A stable isotope product is classified as a medicinal product when it is administered parenterally or when applied to restore, correct or modify physiological functions by exerting a pharmacological, immunological or metabolic action or to enable a medical diagnosis [75]. Stable isotope products that are designated as medicinal products are generally unlicensed drug products. When used in clinical investigations, they may be classified as investigational medicinal product (IMP) or non-investigational medicinal product (NIMP). Investigational medicinal products are the subject of investigation in a clinical trial [76], whereas NIMPs perform a supportive role or are used to assess a relevant clinical trial end-point [77]. When the stable isotope product is regarded as an IMP, as such it falls within the scope of the clinical trial directive [78] (2001/20/EC). The clinical trial application needs documented product design in the form of an IMP dossier (IMPD). Clinical production may be executed by a pharmaceutical manufacturer under the provisions of an IMP manufacturer's authorisation for the particular process concerned. The regulations for NIMPs are currently under discussion in the EU [77]. The clinical trial application needs documented product design but not necessarily in the form of an IMPD. On the one hand, the stable isotope product may be produced as an NIMP by a pharmaceutical manufacturer under the provisions of an IMP manufacturer's authorisation for the particular process concerned. On the other hand, it may be produced under national provisions to the principles of GMP and released for use by an appropriately experienced individual [77]. For example, a NIMP can be produced as magistral or officinal preparation in a pharmacy according to Good Preparation Practices regulations under the supervision of a pharmacist. When used in a medical diagnostic procedure, the stable isotope product is generally prepared specifically for one patient and is considered a magistral or officinal preparation [75]. It may be produced in a licensed (hospital) pharmacy under the same conditions as a NIMP.
Products that are not classified as a medicinal product but are used to investigate a physiological process may be classified as a medical device when they act by physical means. Part of the definition of a medical device is that it does not achieve its principal action by pharmacological, immunological or metabolic means [74]. A stable isotope product will therefore seldom be classified as a medical device.
In clinical trials, a stable isotope-labelled substance may be incorporated in a medical device. For example, when a colon-specific drug delivery device is investigated by incorporating a stable isotopically labelled substance as a marker, this combination is probably classified as a medical device.
Food products and nutrients cover all substances not regarded as drug substances or medical devices, i.e. not applied for a medicinal purpose or a medical indication. We also classify nonparenteral products containing stable isotopically labelled substances.
We present two examples from our practice in a university medical centre to illustrate the classification system. As a first example, we present a clinical investigation into bile acid metabolism and gall bladder function. In the study protocol, stable isotope technology is used to measure bile acid kinetics dynamically. Stable isotopically labelled bile salts (e.g. 2,2,4,4-2H2-chenodeoxycholic acid) are administered orally as part of a metabolic study. As the product is administered orally, is not an assimilated medicinal product, is not administered to prevent, diagnose, treat or alleviate disease, injury or handicap, but is used to investigate a physiological process not by physical means and is an endogenous substance, it should be classified as a food product and treated accordingly. As a second example, we present a clinical investigation into the optimal composition of an infant formula as clinical nutrition. In the study protocol, stable isotope technology is applied to assess fatty acid metabolism. Stable isotopically labelled fatty acids are mixed into a conventional infant formula (e.g. 1,1,1-13C3-tripalmitin). Tripalmitin is a natural constituent of palm oil and as such considered to be a food product. Infant formula with the addition of 1,1,1-13C3-tripalmitin is therefore classified as a food product and should be treated accordingly.
Concluding remarks and future perspectives
Stable isotope technology has been applied in clinical pharmacology since 1972. It may be considered as a technology with proven and promising value, depending on the area of application in the field of clinical pharmacology. Firstly, in drug pharmacology it has obtained a distinctive place when conventional methods are not able to deliver the required information. In the near future, the possibility of studying the metabolism of biotechnology-derived drug substances (that may also exist endogenously) is of special interest considering the rapid developments in this field over recent years. With the advent of peptides and proteins, the study of their metabolism is important to understand and improve drug treatment. Until now, this subject has gained very limited interest. Secondly, in the assessment of drug products, stable isotope technology is well established and recognized as a method to shorten bioavailability trials and to reduce the number of subjects to be included. In the performance assessment of drug delivery systems (e.g. the release profile of modified-release drug products), stable isotope tracers have not been extensively developed and investigated. Applications in this area can be further developed to improve the quality of acquired data from these studies. Only 13C-urea is considered as a useful alternative to gamma scintigraphy in the assessment of colonic delivery systems. Thirdly, in the assessment of patients, stable isotope technology is able to phenotype patients and thus to support personalized medicine. As this field is currently making strong progress, now is the time to investigate the utilization of stable isotope-facilitated monitoring of metabolic processes. Geno- and phenotyping are different approaches to determine the patient's sensitivity to a drug prior to treatment. It remains to be determined which approach the medical community should favour. We envisage a combination of geno- and phenotyping, because both approaches give different but necessary and complementary information.
Translation of stable isotope technologies into commercially viable products is a dismal story. As described in this review, researchers have investigated several stable isotope tracers, but only one product (13C-urea breath test) has been registered on the market for clinical diagnostic purposes. For investigational purposes, commercial test kits that include analytical facilities and calculations can be purchased. Failure to commercialize more stable isotope tracers is related to the following three factors: the unfamiliarity with the methodology due to the limited number of applications; the complex regulatory system; and the small market potential. As these roadblocks are likely not to be eliminated in the near future, application of stable isotope technologies will most probably be confined to academic centres or commercial research organizations. In specific cases, investigators, clinical pharmacologists and medical doctors will require access to this technology as a tool in clinical trials, medical diagnosis or clinical pharmacology.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Knapp DR, Gaffney TE, McMahon RE. Use of stable isotope mixtures as a labeling technique in drug metabolism studies. Biochem Pharmacol. 1972;21:425–9. doi: 10.1016/0006-2952(72)90357-7. [DOI] [PubMed] [Google Scholar]
- 2.Browne TR. Stable isotopes in pharmacology studies: present and future. J Clin Pharmacol. 1986;26:485–9. doi: 10.1002/j.1552-4604.1986.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 3.Lewis GN, MacDonald RT. Some properties of pure H2H2O [8] J Am Chem Soc. 1933;55:3057–9. [Google Scholar]
- 4.Nelson SD, Trager WF. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab Dispos. 2003;31:1481–98. doi: 10.1124/dmd.31.12.1481. [DOI] [PubMed] [Google Scholar]
- 5.Klein PD, Roseland Klein E. Stable isotopes: origins and safety. J Clin Pharmacol. 1986;26:378–82. doi: 10.1002/j.1552-4604.1986.tb03544.x. [DOI] [PubMed] [Google Scholar]
- 6.Lewis GN. The biology of heavy water. Science. 1934;79:151–3. doi: 10.1126/science.79.2042.151. [DOI] [PubMed] [Google Scholar]
- 7.Pons G, Rey E. Stable isotopes labeling of drugs in pediatric clinical pharmacology. Pediatric. 1999;104:633–9. [PubMed] [Google Scholar]
- 8.Mayevsky A, Samuel D, Ortega C, Amsel G. In vivo measurement of variations in the oxygen 18 content of water in rat brain. J Appl Physiol. 1975;39:300–4. doi: 10.1152/jappl.1975.39.2.300. [DOI] [PubMed] [Google Scholar]
- 9.Samuel D, Steckel F. Research with the stable isotopes of oxygen (O17 and O18) during 1958–1960. Int J Appl Radiat Isot. 1961;11:190–229. doi: 10.1016/0020-708x(61)90021-7. [DOI] [PubMed] [Google Scholar]
- 10.Durso R, Evans JE, Josephs E, Szabo G, Evans B, Fernandez HH, Browne TR. Variable absorption of carbidopa affects both peripheral and central levodopa metabolism. J Clin Pharmacol. 2000;40:854–60. doi: 10.1177/00912700022009585. [DOI] [PubMed] [Google Scholar]
- 11.Brenna JT, Corso TN, Tobias HJ, Caimi RJ. High-precision continuous-flow isotope, ratio mass spectrometry. Mass Spectrom Rev. 1997;16:227–58. doi: 10.1002/(SICI)1098-2787(1997)16:5<227::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 12.Braden B, Haisch M, Duan LP, Lembcke B, Caspary WF, Hering P. Clinically feasible stable isotope technique at a reasonable price: analysis of 13CO2/12CO2-abundance in breath samples with a new isotope selective-nondispersive infrared spectrometer. Z Gastroenterol. 1994;32:675–8. [PubMed] [Google Scholar]
- 13.Barth E, Tugtekin I, Weidenbach H, Wachter U, Vogt J, Radermacher P, Adler G, Georgieff M. Determination of 13CO2/12CO2 ratio by IRMS and NDIRS. Isotopes Environ Health Stud. 1998;34:209–13. [PubMed] [Google Scholar]
- 14.Esler MB, Griffith DW, Wilson SR, Steele LP. Precision trace gas analysis by FT-IR spectroscopy. 2. The 13C/12C isotope ratio of CO2. Anal Chem. 2000;72:216–21. doi: 10.1021/ac990563x. [DOI] [PubMed] [Google Scholar]
- 15.Castrillo A, Casa G, Palmieri A, Gianfrani L. Measuring the 13C/12C isotope ratio in atmospheric CO2 by means of laser absorption spectrometry: a new perspective based on a 2.05-microm diode laser. Isotopes Environ Health Stud. 2006;42:47–56. doi: 10.1080/10256010500503361. [DOI] [PubMed] [Google Scholar]
- 16.Stellaard F, Henk E. Analytical techniques in biomedical stable isotope applications: (isotope ratio) mass spectrometry or infrared spectrometry? Isotopes Environ Health Stud. 2005;41:345–61. doi: 10.1080/10256010500384333. [DOI] [PubMed] [Google Scholar]
- 17.Ververs FFT, Smeets OSNM, Schobben AFAM. Assessment of not for human use chemicals for human use [Beoordeling voor humane toepassing van chemicaliën ‘not for human use’] Pharm Weekbl. 2006;141:1469–71. [Google Scholar]
- 18.Malik SI, Painter MJ, Venkataramanan R, Alvin JD. Phenytoin and phenobarbital stable isotope studies in neonates. Pediatr Neurol. 2003;29:376–80. doi: 10.1016/s0887-8994(03)00304-7. [DOI] [PubMed] [Google Scholar]
- 19.Bode H, Brendel E, Ahr G, Fuhr U, Harder S, Staib AH. Investigation of nifedipine absorption in different regions of the human gastrointestinal (GI) tract after simultaneous administration of 13C- and 12C-nifedipine. Eur J Clin Pharmacol. 1996;50:195–201. doi: 10.1007/s002280050092. [DOI] [PubMed] [Google Scholar]
- 20.Fromm MF, Dilger K, Busse D, Kroemer HK, Eichelbaum M, Klotz U. Gut wall metabolism of verapamil in older people: effects of rifampicin-mediated enzyme induction. Br J Clin Pharmacol. 1998;45:247–55. doi: 10.1046/j.1365-2125.1998.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ligthart-Melis GC, Van De Poll MCG, Boelens PG, Dejong CHC, Deutz NEP, Van Leeuwen PAM. Glutamine is an important precursor for de novo synthesis of arginine in humans. Am J Clin Nutr. 2008;87:1282–9. doi: 10.1093/ajcn/87.5.1282. [DOI] [PubMed] [Google Scholar]
- 22.Andrews FJ, Griffiths RD. Glutamine: essential for immune nutrition in the critically ill. Br J Nutr. 2002;87(Suppl. 1):S3–8. doi: 10.1079/bjn2001451. [DOI] [PubMed] [Google Scholar]
- 23.Ligthart-Melis GC, Van De Poll MCG, Vermeulen MAR, Boelens PG, Van Den Tol MP, Van Schaik C, De Bandt J-P, Deutz NEP, Dejong CHC, Van Leeuwen PAM. Enteral administration of alanyl-[2-15N]glutamine contributes more to the de novo synthesis of arginine than does intravenous infusion of the dipeptide in humans. Am J Clin Nutr. 2009;90:95–105. doi: 10.3945/ajcn.2008.26399. [DOI] [PubMed] [Google Scholar]
- 24.Browne TR, editor. Stable Isotopes in Pharmaceutical Research. Amsterdam: Elsevier; 1997. [Google Scholar]
- 25.Stellaard F, Kuipers F. Assessment of modes of action and efficacy of plasma cholesterol-lowering drugs: measurement of cholesterol absorption, cholesterol synthesis and bile acid synthesis and turnover using novel stable isotope techniques. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:209–18. doi: 10.2174/1568008054064913. [DOI] [PubMed] [Google Scholar]
- 26.Chan DC, Watts GF, Hugh R, Barrett P, Martins IJ, James AP, Mamo JCL, Mori TA, Redgrave TG. Effect of atorvastatin on chylomicron remnant metabolism in visceral obesity: a study employing a new stable isotope breath test. J Lipid Res. 2002;43:706–12. [PubMed] [Google Scholar]
- 27.De Sain-Van Der Velden MG, Kaysen GA, Barrett HA, Stellaard F, Gadellaa MM, Voorbij HA, Reijngoud D-J, Rabelink TJ. Increased VLDL in nephrotic patients results from a decreased catabolism while increased LDL results from increased synthesis. Kidney Int. 1998;53:994–1001. doi: 10.1111/j.1523-1755.1998.00831.x. [DOI] [PubMed] [Google Scholar]
- 28.De Preter V, Geboes K, Verbrugghe K, De Vuyst L, Vanhoutte T, Huys G, Swings J, Pot B, Verbeke K. The in vivo use of the stable isotope-labelled biomarkers lactose-[15N]ureide and [2H4]tyrosine to assess the effects of pro- and prebiotics on the intestinal flora of healthy human volunteers. J Nutr. 2004;92:439–46. doi: 10.1079/bjn20041228. [DOI] [PubMed] [Google Scholar]
- 29.Wachters-Hagedoorn RE, Priebe MG, Heimweg JAJ, Heiner AM, Elzinga H, Stellaard F, Vonk RJ. Low-dose acarbose does not delay digestion of starch but reduces its bioavailability. Diabetic Med. 2007;24:600–6. doi: 10.1111/j.1464-5491.2007.02115.x. [DOI] [PubMed] [Google Scholar]
- 30.Gerard J, Jandrain B, Pirnay F. Utilization of oral sucrose load during exercise in humans: effect of the α-glucosidase inhibitor Acarbose. Diabetes. 1986;35:1294–301. doi: 10.2337/diab.35.11.1294. [DOI] [PubMed] [Google Scholar]
- 31.Bunt JEH, Carnielli VP, Wattimena JLD, Hop WC, Sauer PJJ, Zimmermann LJI. The effect in premature infants of prenatal corticosteroids on endogenous surfactant synthesis as measured with stable isotopes. Am J Respir Crit Care Med. 2000;162:844–9. doi: 10.1164/ajrccm.162.3.9906139. [DOI] [PubMed] [Google Scholar]
- 32.Bunt JEH, Carnielli VP, Janssen DJ, Wattimena JLD, Hop WC, Sauer PJ, Zimmermann LJI. Treatment with exogenous surfactant stimulates endogenous surfactant synthesis in premature infants with respiratory distress syndrome. Crit Care Med. 2000;28:3383–8. doi: 10.1097/00003246-200010000-00001. [DOI] [PubMed] [Google Scholar]
- 33.Cogo PE, Carnielli VP, Bunt JE, Badon T, Giordano G, Zacchello F, Sauer PJ, Zimmermann LJ. Endogenous surfactant metabolism in critically ill infants measured with stable isotope labelled fatty acids. Pediatr Res. 1999;45:242–6. doi: 10.1203/00006450-199902000-00015. [DOI] [PubMed] [Google Scholar]
- 34.Ismail WIW, King JA, Pillay TS. Insulin resistance induced by antiretroviral drugs: current understanding of molecular mechanisms. J Endocrinol Metab Diabetes S Afr. 2009;14:129–32. [Google Scholar]
- 35.Rao MN, Mulligan K, Tai V, Wen MJ, Dyachenko A, Weinberg M, Li X, Lang T, Grunfeld C, Schwarz J-M, Schambelan M. Effects of Insulin-Like Growth Factor (IGF)-I/IGF-binding protein-3 treatment on glucose metabolism and fat distribution in human immunodeficiency virus-infected patients with abdominal obesity and insulin resistance. J Clin Endocrinol Metab. 2010;95:4361–6. doi: 10.1210/jc.2009-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mutlib AE. Application of stable isotope-labelled compounds in metabolism and in metabolism-mediated toxicity studies. Chem Res Toxicol. 2008;21:1672–89. doi: 10.1021/tx800139z. [DOI] [PubMed] [Google Scholar]
- 37.Park KB, Dalton-Brown E, Hirst C, Williams DP. Selection of new chemical entities with decreased potential for adverse drug reactions. Eur J Pharmacol. 2006;549:1–8. doi: 10.1016/j.ejphar.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 38.Teitelbaum DH. Parenteral nutrition-associated cholestasis. Curr Opin Pediatr. 1997;9:270–5. doi: 10.1097/00008480-199706000-00016. [DOI] [PubMed] [Google Scholar]
- 39.Duro D, Duggan C, Valim C, Bechard L, Fitzgibbons S, Jaksic T, Yu Y-M. Novel intravenous 13C-methionine breath test as a measure of liver function in children with short bowel syndrome. J Pediatr Surg. 2009;44:236–40. doi: 10.1016/j.jpedsurg.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sauter G, Berr F, Beuers U, Fischer S, Paumgartner G. Serum concentrations of 7alpha-hydroxy-4-cholesten-3-one reflect bile acid synthesis in humans. Hepatology. 1996;24:123–6. doi: 10.1053/jhep.1996.v24.pm0008707250. [DOI] [PubMed] [Google Scholar]
- 41.Steiner SJ, Pfefferkorn MD, Fitzgerald JF, Denne SC. Effects of infliximab and parenteral nutrition on albumin and fibrinogen synthesis rates in pediatric crohn disease. J Pediatr Gastroenterol Nutr. 2008;47:579–84. doi: 10.1097/mpg.0b013e3181653a89. [DOI] [PubMed] [Google Scholar]
- 42.Hulzebos CV, Wolters H, Plösch T, Kramer W, Stengelin S, Stellaard F, Sauer PJJ, Verkade HJ, Kuipers F. Cyclosporin A and enterohepatic circulation of bile salts in rats: decreased cholate synthesis but increased intestinal reabsorption. J Pharmacol Exp Ther. 2003;304:356–63. doi: 10.1124/jpet.102.041640. [DOI] [PubMed] [Google Scholar]
- 43.Hulzebos CV, Bijleveld CMA, Stellaard F, Kuipers F, Fidler V, Slooff MJH, Peeters PMJG, Sauer PJJ, Verkade HJ, Cyclosporine A. Induced reduction of bile salt synthesis associated with increased plasma lipids in children after liver transplantation. Liver Transpl. 2004;10:872–80. doi: 10.1002/lt.20168. [DOI] [PubMed] [Google Scholar]
- 44.Schuster D, Laggner C, Langer T. Why drugs fail – A study on side effects in new chemical entities. Curr Pharm Des. 2005;11:3545–59. doi: 10.2174/138161205774414510. [DOI] [PubMed] [Google Scholar]
- 45.Strong JM, Dutcher JS, Lee WK, Atkinson AJ., Jr Absolute bioavailability in man of N acetylprocainamide determined by a novel stable isotope method. Clin Pharmacol Ther. 1975;18:613–22. doi: 10.1002/cpt1975185part1613. [DOI] [PubMed] [Google Scholar]
- 46.Shibuya F, Kuramoto K, Koizumi H. Biopharmaceutical evaluation of methylcellulose as an excipient for nitroglycerin tablets. J Pharmacobiodyn. 1985;8:352–6. doi: 10.1248/bpb1978.8.352. [DOI] [PubMed] [Google Scholar]
- 47.Gammans RE, Mackenthun AV, Russell JW. Comparative bioavailability of trazodone formulations using stable isotope methodology. Br J Clin Pharmacol. 1984;18:431–7. doi: 10.1111/j.1365-2125.1984.tb02485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilding IR, Coupe AJ, Davis SS. The role of γ-scintigraphy in oral drug delivery. Adv Drug Deliv Rev. 2001;46:103–24. doi: 10.1016/s0169-409x(00)00135-6. [DOI] [PubMed] [Google Scholar]
- 49.Wilding IR, Davis SS, Hardy JG, Robertson CS, John VA, Powell ML, Leal M, Lloyd P, Walker SM. Relationship between systemic drug absorption and gastrointestinal transit after the simultaneous oral administration of carbamazepine as a controlled-release system and as a suspension of 15N-labelled drug to healthy volunteers. Br J Clin Pharmacol. 1991;32:573–9. doi: 10.1111/j.1365-2125.1991.tb03954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cummings JH, Milojevic S, Harding M, Coward WA, Gibson GR, Botham RL, Ring SG, Wraight EP, Stockham MA, Allwood MC, Newton JM. In vivo studies of amylose-and ethylcellulose-coated [13C]glucose microspheres as a model for drug delivery to the colon. J Control Release. 1996;40:123–31. [Google Scholar]
- 51.Schellekens RCA, Stellaard F, Mitrovic D, Stuurman FE, Kosterink JGW, Frijlink HW. Pulsatile drug delivery to ileo-colonic segments by structured incorporation of disintegrants in pH-responsive polymer coatings. J Control Release. 2008;132:91–8. doi: 10.1016/j.jconrel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 52.Verbeke K, De Preter V, Geboes K, Daems T, Van Den Mooter G, Evenepoel P, Rutgeerts P. In vivo evaluation of a colonic delivery system using isotope techniques. Aliment Pharmacol Ther. 2005;21:187–94. doi: 10.1111/j.1365-2036.2005.02323.x. [DOI] [PubMed] [Google Scholar]
- 53.Schellekens RCA, Stellaard F, Olsder GG, Woerdenbag HJ, Frijlink HW, Kosterink JGW. Oral ileocolonic drug delivery by the colopulse-system: a bioavailability study in healthy volunteers. J Control Release. 2010;146:334–40. doi: 10.1016/j.jconrel.2010.05.028. [DOI] [PubMed] [Google Scholar]
- 54.Schellekens RCA, Olsder GG, Langenberg SMCH, Boer T, Woerdenbag HJ, Frijlink HW, Kosterink JGW, Stellaard F. Proof-of-concept study on the suitability of 13C-urea as a marker substance for assessment of in vivo behaviour of oral colon-targeted dosage forms. Br J Pharmacol. 2009;158:532–40. doi: 10.1111/j.1476-5381.2009.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alffenaar JW, Wessels AM, van Hateren K, Greijdanus B, Kosterink JG, Uges DR. Method for therapeutic drug monitoring of azole antifungal drugs in human serum using LC/MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:39–44. doi: 10.1016/j.jchromb.2009.11.017. [DOI] [PubMed] [Google Scholar]
- 56.Slanař O, Chalupná P, Novotný A, Bortlík M, Krška Z, Lukáš M. Fatal myelotoxicity after azathioprine treatment. Nucleosides Nucleotides Nucleic Acids. 2008;27:661–5. doi: 10.1080/15257770802143905. Biochemical Pharmacology, 21 (3), pp. 425-429. [DOI] [PubMed] [Google Scholar]
- 57.Press RR, De Fijter JW, Guchelaar H-J. Individualizing calcineurin inhibitor therapy in renal transplantation – Current limitations and perspectives. Curr Pharm Des. 2010;16:176–86. doi: 10.2174/138161210790112782. [DOI] [PubMed] [Google Scholar]
- 58.Eidens M, Prause S, Weise A, Klemm M, Weber MM, Pfützner A. Dihydropyrimidine dehydrogenase genotyping and phenotyping for 5-fluorouracil dysmetabolism: moving towards personalized chemotherapy in patients with cancer. Curr Pharmacogenom Pers Med. 2009;7:275–83. [Google Scholar]
- 59.Van Staveren MC, Van Kuilenburg ABP, Visser T, Maring JG. Sensitivity and specificity of an oral uracil loading test in patients with normal DPD activity and with DPD deficiency. Pharm Weekbl. 2009;144:183–6. [Google Scholar]
- 60.Mattison LK, Fourie J, Hirao Y, Koga T, Desmond RA, King JR, Shimizu T, Diasio RB. The uracil breath test in the assessment of dihydropyrimidine dehydrogenase activity: pharmacokinetic relationship between expired13CO 2 and plasma [2-13C] dihydrouracil. Clin Cancer Res. 2006;12:549–55. doi: 10.1158/1078-0432.CCR-05-2020. [DOI] [PubMed] [Google Scholar]
- 61.Mattison LK, Ezzeldin H, Carpenter M, Modak A, Johnson MR, Diasio RB. Rapid identification of dihydropyrimidine dehydrogenase deficiency by using a novel 2-13C-uracil breath test. Clin Cancer Res. 2004;10:2652–8. doi: 10.1158/1078-0432.ccr-03-0374. [DOI] [PubMed] [Google Scholar]
- 62.Goddard AF, Logan RPH. Diagnostic methods for Helicobacter pylori detection and eradication. Br J Clin Pharmacol. 2003;56:273–83. doi: 10.1046/j.1365-2125.2003.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thijs JC, Van Zwei AA, Thijs WJ, Oey HB, Karrenbeld A, Stellaard F, Luijt DS, Meyer BC, Kleibeuker JH. Diagnostic tests for Helicobacter pylori: a prospective evaluation of their accuracy, without selecting a single test as the gold standard. Am J Gastroenterol. 1996;91:2125–9. [PubMed] [Google Scholar]
- 64.Elwyn G, Taubert M, Davies S, Brown G, Allison M, Phillips C. Which test is best for Helicobacter pylori? A cost-effective model using decision analysis. Br J Gen Pract. 2007;57:401–3. [PMC free article] [PubMed] [Google Scholar]
- 65.Braden B, Adams S, Duan L-P, Orth K-H, Maul F-D, Lembcke B, Hor G, Caspary WF. The [13C]acetate breath test accurately reflects gastric emptying of liquids in both liquid and semisolid test meals. Gastroenterology. 1995;108:1048–55. doi: 10.1016/0016-5085(95)90202-3. [DOI] [PubMed] [Google Scholar]
- 66.Ghoos YF, Maes BD, Geypens BJ, Mys G, Hiele MI, Rutgeerts PJ, Vantrappen G. Measurement of gastric emptying rate of solids by means of a carbon- labelled octanoic acid breath test. Gastroenterology. 1993;104:1640–7. doi: 10.1016/0016-5085(93)90640-x. [DOI] [PubMed] [Google Scholar]
- 67.Sikkens ECM, Cahen DL, Kuipers EJ, Bruno MJ. Pancreatic enzyme replacement therapy in chronic pancreatitis. Best Pract Res Clin Gastroenterol. 2010;24:337–47. doi: 10.1016/j.bpg.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 68.Ojetti V, Gigante G, Gabrielli M, Ainora ME, Mannocci A, Lauritano EC, Gasbarrini G, Gasbarrini A. The effect of oral supplementation with Lactobacillus reuteri or tilactase in lactose intolerant patients: randomized trial. Eur Rev Med Pharmacol Sci. 2010;14:163–70. [PubMed] [Google Scholar]
- 69.Vonk RJ, Stellaard F, Priebe MG, Koetse HA, Hagedoorn RE, De Bruijn S, Elzinga H, Lenoir-Wijnkoop I, Antoine J-M. The 13C/2H-glucose test for determination of small intestinal lactase activity. Eur J Clin Invest. 2001;31:226–33. doi: 10.1046/j.1365-2362.2001.00791.x. [DOI] [PubMed] [Google Scholar]
- 70.Schneider JF, Schoeller DA, Nemchausky B. Validation of 13CO2 breath analysis as a measurement of demethylation of stable isotope labelled aminopyrine in man. Clin Chim Acta. 1978;84:153–62. doi: 10.1016/0009-8981(78)90489-8. [DOI] [PubMed] [Google Scholar]
- 71.Braden B, Lembcke B, Kuker W, Caspary WF. 13C-breath tests: current state of the art and future directions. Dig Liver Dis. 2007;39:795–805. doi: 10.1016/j.dld.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 72.van Veen EB. Implementatie van richtlijn 2011/20/EC, Tijdschrift voor Gezondheidsrecht 6 oktober. 2009.
- 73.European Commision. Directive 2002/178/EC laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. 2002.
- 74.European Commision. Directive 65/65/EEC on the approximation of provisions laid down by law, regulation or administrative action relating to medicinal products. 1965.
- 75.European Commision. Directive 93/42/EEC concerning medical devices. 1993.
- 76.European Commision. The rules governing medicinal products in the European Union, Volume 10 – guidance documents applying to clinical trials, questions & answers. Version 7.0, September 2010.
- 77.European Commision. The rules governing medicinal products in the European Union, Volume 10 – guidance documents applying to clinical trials, Harmonised requirements for Non Investigational Medicinal products in CTA submissions. June 2010.
- 78.European Commision. Directive 2001/20/EC. 4 April 2001.
- 79.Heck d'AH, Buttrill SE, Jr, Flynn NW. Bioavailability of imipramine tablets relative to a stable isotope-labelled internal standard: increasing the power of biovailability tests. J Pharmacokinet Biopharm. 1979;7:233–48. doi: 10.1007/BF01060015. [DOI] [PubMed] [Google Scholar]
- 80.Eichelbaum M, Somogyi A, Von Unruh GE, Dengler HJ. Simultaneous determination of the intravenous and oral pharmacokinetic parameters of D,L-verapamil using stable isotope-labelled verapamil. Eur J Clin Pharmacol. 1981;19:133–7. doi: 10.1007/BF00568400. [DOI] [PubMed] [Google Scholar]
- 81.Eichelbaum M, Dengler HJ, Somogyi A, Von Unruh GE. Superiority of stable isotope techniques in the assessment of the bioavailability of drugs undergoing extensive first pass elimination. Studies of the relative bioavailability of verapamil tablets. Eur J Clin Pharmacol. 1981;19:127–31. doi: 10.1007/BF00568399. [DOI] [PubMed] [Google Scholar]
- 82.Meresaar U, Nilsson M-I, Holmstrand J, Anggard E. Single dose pharmacokinetics and bioavailability of methadone in man studied with a stable isotope method. Eur J Clin Pharmacol. 1981;20:473–8. doi: 10.1007/BF00542102. [DOI] [PubMed] [Google Scholar]
- 83.Rigby GV, Vose CW, Haskins NJ. Propantheline bromide plasma level, urinary excretion and pharmacological data in a comparison of the bioavailability of three oral formulations of Pro-Banthine. Eur J Drug Metab Pharmacokinet. 1983;8:219–24. doi: 10.1007/BF03188751. [DOI] [PubMed] [Google Scholar]
- 84.Vogelgesang B, Echizen H, Schmidt E, Eichelbaum M. Stereoselective first-pass metabolism of highly cleared drugs: studies of the bioavailability of L- and d-verapamil examined with a stable isotope technique. Br J Clin Pharmacol. 1984;18:733–40. doi: 10.1111/j.1365-2125.1984.tb02536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marvola M, Kannikoski A, Taskinen J, Ottoila P. Assessment of bioavailability of experimental single-unit sustained release tablets of verapamil hydrochloride using the stable isotope technique. J Pharmacy Pharmacol. 1985;37:766–70. doi: 10.1111/j.2042-7158.1985.tb04965.x. [DOI] [PubMed] [Google Scholar]
- 86.Hatch F, McKellop K, Hansen G, MacGregor T. Relative bioavailability of metaproterenol in humans utilizing a single dose, stable isotope approach. J Pharm Sci. 1986;75:886–90. doi: 10.1002/jps.2600750913. [DOI] [PubMed] [Google Scholar]
- 87.Fujioka M, Shinohara Y, Baba S. Acute suppression of endogenous testosterone levels by exogenous testosterone in normal men. Life Sci. 1987;41:945–9. doi: 10.1016/0024-3205(87)90681-3. [DOI] [PubMed] [Google Scholar]
- 88.Looareesuwan S, White NJ, Warrell DA. Studies of mefloquine bioavailability and kinetics using a stable isotope technique: a comparison of Thai patients with falciparum malaria and healthy caucasian volunteers. Br J Clin Pharmacol. 1987;24:37–42. doi: 10.1111/j.1365-2125.1987.tb03133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mikus G, Fischer C, Heuer B, Langen C, Eichelbaum M. Application of stable isotope methodology to study the pharmacokinetics, bioavailability and metabolism of nitrendipine after i.v. and p.o. administration. Br J Clin Pharmacol. 1987;24:561–9. doi: 10.1111/j.1365-2125.1987.tb03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hallen B, Guilbaud O, Stromberg S, Lindeke B. Single-dose pharmacokinetics of terodiline, including a stable isotope technique for improvement of statistical evaluations. Biopharm Drug Dispos. 1988;9:229–50. doi: 10.1002/bod.2510090302. [DOI] [PubMed] [Google Scholar]
- 91.Atkinson AJ, Jr, Ruo TI, Piergies AA, Breiter HC, Connelly TJ, Sedek GS, Juan D, Hubler GL, Hsieh A-M. Pharmacokinetics of N-acetylprocainamide in patients profiled with a stable isotope method. Clin Pharmacol Ther. 1989;46:182–9. doi: 10.1038/clpt.1989.124. [DOI] [PubMed] [Google Scholar]
- 92.Fujioka M, Shinohara Y, Baba S, Irie M. Inoue k., Endogenous and exogenous testosterone levels after administration of deuterium-labelled testosterone propionate in hypogonadotropic hypogonadism. Chem Pharm Bull (Tokyo) 1989;37:3100–1. doi: 10.1248/cpb.37.3100. [DOI] [PubMed] [Google Scholar]
- 93.Shinohara Y, Baba S. Stable isotope methodology in the pharmacokinetic studies of androgenic steriods in humans. Steroids. 1990;55:170–6. doi: 10.1016/0039-128x(90)90106-l. [DOI] [PubMed] [Google Scholar]
- 94.Aronoff G, Brier M, Mayer ML, Barbalas M, Aogaichi K, Sloan R, Brazzell R, Massarella J. Bioavailability and kinetics of cibenzoline in patients with normal and impaired renal function. J Clin Pharmacol. 1991;31:38–44. doi: 10.1002/j.1552-4604.1991.tb01884.x. [DOI] [PubMed] [Google Scholar]
- 95.Benowitz NL, Jacob P, III, Denaro C, Jenkins R. Stable isotope studies of nicotine kinetics and bioavailability. Clin Pharmacol Ther. 1991;49:270–7. doi: 10.1038/clpt.1991.28. [DOI] [PubMed] [Google Scholar]
- 96.Benowitz NL, Chan K, Denaro CP, Jacob P., III Stable isotope method for studying transdermal drug absorption: the nicotine patch. Clin Pharmacol Ther. 1991;50:286–93. doi: 10.1038/clpt.1991.138. [DOI] [PubMed] [Google Scholar]
- 97.Bredberg U, Karlsson MO, Borgstrom L. A comparison between the semisimultaneous and the stable isotope techniques for bioavailability estimation of terbutaline in humans. Clin Pharmacol Ther. 1992;52:239–48. doi: 10.1038/clpt.1992.136. [DOI] [PubMed] [Google Scholar]
- 98.Browne TR, Szabo GK, McEntegart C, Evans JE, Evans BA, Miceli JJ, Quon C, Dougherty CL, Kres J, Davoudi H. Bioavailability studies of drugs with nonlinear pharmacokinetics: II. Absolute bioavailability of intravenous phenytoin prodrug at therapeutic phenytoin serum concentrations determined by double-stable isotope technique. J Clin Pharmacol. 1993;33:89–94. doi: 10.1002/j.1552-4604.1993.tb03910.x. [DOI] [PubMed] [Google Scholar]
- 99.Hall SD, Rudy AC, Knight PM, Brater DC. Lack of presystemic inversion of (R)- to (S)-ibuprofen in humans. Clin Pharmacol Ther. 1993;53:393–400. doi: 10.1038/clpt.1993.42. [DOI] [PubMed] [Google Scholar]
- 100.Le Houezec J, Jacob P, III, Benowitz NL. A clinical pharmacological study of subcutaneous nicotine. Eur J Clin Pharmacol. 1993;44:225–30. doi: 10.1007/BF00271362. [DOI] [PubMed] [Google Scholar]
- 101.Richard J, Cardot J-M, Godbillon J. Stable isotope methodology for studying the performance of metoprolol Oros tablets in comparison to conventional and slow release formulations. Eur J Drug Metab Pharmacokinet. 1994;19:375–80. doi: 10.1007/BF03188865. [DOI] [PubMed] [Google Scholar]
- 102.Hage K, Buhl K, Fischer C, Knebel NG. Estimation of the absolute bioavailability of flecainide using stable isotope technique. Eur J Clin Pharmacol. 1995;48:51–5. doi: 10.1007/BF00202172. [DOI] [PubMed] [Google Scholar]
- 103.Sun JX, Piraino AJ, Morgan JM, Joshi JC, Cipriano A, Chan K, Redalieu E. Comparative pharmacokinetics and bioavailability of nitroglycerin and its metabolites from transderm-nitro, nitrodisc, and Nitro-Dur II systems using a stable-isotope technique. J Clin Pharmacol. 1995;35:390–7. doi: 10.1002/j.1552-4604.1995.tb04079.x. [DOI] [PubMed] [Google Scholar]
- 104.Voortman G, Paanakker JE. Bioavailability of mirtazapine from Remeron® tablets after single and multiple oral dosing. Hum Psychopharmacol. 1995;10(Suppl. 2):S83–96. [Google Scholar]
- 105.Bode H, Brendel E, Ahr G, Fuhr U, Harder S, Staib AH. Investigation of nifedipine absorption in different regions of the human gastrointestinal (GI) tract after simultaneous administration of 13C- and 12C-nifedipine. Eur J Clin Pharmacol. 1996;50:195–201. doi: 10.1007/s002280050092. [DOI] [PubMed] [Google Scholar]
- 106.Gross AS, Mikus G, Ratge D, Wisser H, Eichelbaum M. Pharmacokinetics and pharmacodynamics of the enantiomers of gallopamil. J Pharmacol Exp Ther. 1997;281:1102–12. [PubMed] [Google Scholar]
- 107.Benech H, Pruvost A, Batel A, Bourguignon M, Thomas J-L, Grognet J-M. Use of the stable isotopes technique to evaluate the bioavailability of a pharmaceutical form of magnesium in man. Pharm Res. 1998;15:347–51. doi: 10.1023/a:1011947525377. [DOI] [PubMed] [Google Scholar]
- 108.Fromm MF, Dilger K, Busse D, Kroemer HK, Eichelbaum M, Klotz U. Gut wall metabolism of verapamil in older people: effects of rifampicin-mediated enzyme induction. Br J Clin Pharmacol. 1998;45:247–55. doi: 10.1046/j.1365-2125.1998.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dilger K, Eckhardt K, Hofmann U, Kucher K, Mikus G, Eichelbaum M. Chronopharmacology of intravenous and oral modified release verapamil. J Clin Pharmacol. 1999;47:413–9. doi: 10.1046/j.1365-2125.1999.00910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pieniaszek HJ, Jr, Mayersohn M, Adams MP, Reinhart RJ, Barrett JS. Moricizine bioavailability via simultaneous, dual, stable isotope administration: bioequivalence implications. J Clin Pharmacol. 1999;39:817–25. doi: 10.1177/00912709922008489. [DOI] [PubMed] [Google Scholar]
- 111.Yeh KC, Stone JA, Carides AD, Rolan P, Woolf E, Ju WD. Simultaneous investigation of indinavir nonlinear pharmacokinetics and bioavailability in healthy volunteers using stable isotope labeling technique: study design and model-independent data analysis. J Pharm Sci. 1999;88:568–73. doi: 10.1021/js9802392. [DOI] [PubMed] [Google Scholar]
- 112.Heikkinen H, Saraheimo M, Antila S, Ottoila P, Pentikäinen PJ. Pharmacokinetics of entacapone, a peripherally acting catechol-O-methyltransferase inhibitor, in man -A study using a stable isotope technique. Eur J Clin Pharmacol. 2001;56:821–6. doi: 10.1007/s002280000244. [DOI] [PubMed] [Google Scholar]
- 113.Glaeser H, Drescher S, Hofmann U, Heinkele G, Somogyi AA, Eichelbaum M, Fromm MF. Impact of concentration and rate of intraluminal drug delivery on absorption and gut wall metabolism of verapamil in humans. Clin Pharmacol Ther. 2004;76:230–8. doi: 10.1016/j.clpt.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 114.Kasuya Y, Mamada K, Baba S, Matsukura M. Stable-isotope methodology for the bioavailability study of phenytoin during multiple-dosing regimens. J Pharm Sci. 1985;74:503–7. doi: 10.1002/jps.2600740503. [DOI] [PubMed] [Google Scholar]
- 115.Foster DJR, Morton EB, Heinkele G, Mürdter TE, Somogyi AA. Stereoselective quantification of methadone and a d6-labelled isotopomer using high performance liquid chromatography-atmospheric pressure chemical ionization mass-spectrometry: application to a pharmacokinetic study in a methadone maintained subject. Ther Drug Monit. 2006;28:559–67. doi: 10.1097/00007691-200608000-00012. [DOI] [PubMed] [Google Scholar]
- 116.Majumdar AK, Howard L, Goldberg MR, Hickey L, Constanzer M, Rothenberg PL, Crumley TM, Panebianco D, Bradstreet TE, Bergman AJ, Waldman SA, Greenberg HE, Butler K, Knops A, De Lepeleire I, Michiels N, Petty KJ. Pharmacokinetics of aprepitant after single and multiple oral doses in healthy volunteers. J Clin Pharmacol. 2006;46:291–300. doi: 10.1177/0091270005283467. [DOI] [PubMed] [Google Scholar]
- 117.Schultz IR, Shangraw RE. Effect of short-term drinking water exposure to dichloroacetate on its pharmacokinetics nd oral bioavailability in human volunteers: a stable isotope study. Toxicol Sci. 2006;92:42–50. doi: 10.1093/toxsci/kfj193. [DOI] [PubMed] [Google Scholar]