

Docetaxel is a taxane antineoplasic agent that acts by inducing microtubular stability and disrupting the dynamics of the microtubular network. It is approved for the adjuvant treatment of patients with breast cancer, non-small cell lung cancer (NSCLC), hormone refractory prostate cancer and gastric cancer (http://www.taxotere.com). Moreover, docetaxel is active against different types of solid tumours, including oesophageal squamous cell carcinoma and advanced squamous cell carcinoma of the head and neck [1]. Its dose limiting toxicity is neutropenia, peripheral neurotoxicity and oedema [2].
The pharmacokinetics of docetaxel are complex mainly due to its distribution and metabolism. Docetaxel plasma protein binding is higher than 95% and it is mainly bound to α1-acid glycoprotein (AAG), lipoproteins and albumin. AAG, an acute phase protein, is often elevated during advanced cancer and there are high interindividual differences in the AAG concentrations, which might influence the pharmacokinetics of docetaxel and thereby its toxicity [3, 4]. With respect to its metabolism, in vitro studies indicated docetaxel is primarily eliminated by cytochrome P450 (CYP) 3A4-mediated metabolism. In vivo, co-administration of ketoconazole, a potent CYP 3A4 inhibitor, resulted in a 49% decrease in docetaxel plasma clearance in cancer patients [5]. Moreover, docetaxel oral bioavailability is limited by the effect of P-glycoprotein (PgP) [6] and CYP 3A4 [7–9]. Consequently, the amount of docetaxel absorbed is increased from 19% to 30% when co-administered with ritonavir, a potent PgP and CYP 3A4 inhibitor [6]. The docetaxel label includes a warning related to the concomitant administration of drugs that induce, inhibit or are metabolized by CYP 3A4, such as ciclosporin, terfenadine, ketoconazole or erythromycin, as well as protease inhibitors, particularly ritonavir [1, 10]. However, the docetaxel label does not refer to dietary compounds that can be strong CYP3A4 inhibitors such as grapefruit juice (GFJ).
We report a case of a pharmacokinetic interaction in a 52-year-old Caucasian female with an oesophageal squamous cell carcinoma, who was treated with docetaxel while drinking daily GFJ.
The patient was diagnosed in October 2008 with a locally advanced unresectable middle third oesophageal squamous cell carcinoma. Initially, she was treated with a combination of weekly 1 h intravenous (i.v.) infusion of paclitaxel (40 mg m−2) plus 1.5 h i.v. infusion of cisplatin (20 mg m−2) and radical radiotherapy (total dose of 60 Gy). Therapy was well tolerated except for a transient grade 3 oesophagitis (secondary to radical radiotherapy) and was ended in June 2009. Positron emission tomography-computed tomography evaluation, 1 month later, showed complete disappearance of the tumour. In September 2009, a solitary hepatic lesion, without any other evidence of metastatic disease, was found and resected. She received a combination chemotherapy of 1 h i.v. infusion of paclitaxel (80 mg m−2), 1.5 h i.v. infusion of cisplatin (40 mg m−2) and 24 h i.v. infusion of 5-fluorouracil (2.6 g m−2) biweekly without major toxicity. In March 2010, progressive disease in liver and bones was found, so the chemotherapy regimen was modified to administer 10 min i.v. infusion of pemetrexed (500 mg m−2), 30 min i.v. infusion of doxorubicin (30 mg m−2) and 1 h i.v. infusion of irinotecan (150 mg m−2) on a biweekly basis. This regimen was well tolerated but without clinical activity after 6 weeks of treatment. In April 2010, new combination chemotherapy of 1 h i.v. infusion of docetaxel (40 mg m−2), 30 min i.v. infusion of gemcitabine (800 mg m−2) and 30 min i.v. infusion of doxorubicin (30 mg m−2) was administered biweekly with pharmacokinetically-guided docetaxel dose adjustments, i.e. with therapeutic docetaxel monitoring, to support routine clinical care. She had only moderate asthenia and the tumour had a minimal response.
A total of 74 mg (40 mg m−2) of docetaxel were intravenously administered as a 1 h infusion every 2 weeks. Venous blood samples were collected in heparinized tubes at 1, 1.5, 4 and 6 h after the start of the infusion. All samples were collected in S-monovette® tubes, centrifuged at 3500 rev min−1 for 10 min. and were stored at −80°C until analysis. Docetaxel concentration in plasma samples was determined by high performance liquid chromatography with ultraviolet detection [11]. The lower limit of quantification was 0.01 mg l−1. Over the validated range of the assay (0.01 to 18 mg l−1), the mean intra- and inter-assay coefficients of variation were lower than 6% and 7%, respectively. Additionally, AAG concentrations were measured immunonephelometrically on a routine laboratory technique. The patient was informed of the risk and benefits of repeated blood determination tests for pharmacokinetic studies and provided written informed consent before the chemotherapeutic treatment.
The individual pharmacokinetic parameters were calculated using the POSTHOC option (maximum a posteriori method) in the NONMEM V level 1.1 software package (GloboMax, Hanover, MD, USA) [12]. A three-compartment pharmacokinetic model with first-order elimination was used as a priori information [13]. Baille et al. [14] showed that two points (end of infusion and 6 h after the start of infusion) can be selected for an adequate docetaxel clearance estimation. As docetaxel pharmacokinetics are linear, AUC(0,∞) could be estimated as the ratio of dose to clearance, without significant bias. Furthermore, the application of a limited sampling strategy in the clinical routine is more convenient for patients because it minimizes the number of blood samples and hospital stay, and in conjunction with Bayesian estimation methods provides an adequate estimate of pharmacokinetic parameters. Graphical analyses were performed using S-Plus 6.1 Professional Edition (Insightful, Seattle, WA, USA).
The time course of the plasma concentrations of docetaxel is indicated in Figure 1. Slow docetaxel elimination was evident after the first course of treatment. The estimated docetaxel plasma clearance was 13.2 l h−1 while the typical plasma clearance of docetaxel was 36.7 l h−1. With concomitant GFJ intake, the corresponding 4 h blood sample was not collected. Although only three points were available for the clearance estimation and subsequent AUC calculation, critical points for unbiased and precise clearance estimation were available [14]. Without concomitant GFJ intake, the last point (6 h) had a concentration value below the limit of quantification (BLQ). In this case, an imputation value of BLQ/2 was used as the 6 h docetaxel concentration for clearance estimation [15]. After reviewing the patient's medical history and interviewing the patient, the only factor that could be affecting the elimination of docetaxel was GFJ. The patient reported taking one glass (≈ 250 ml) of GFJ daily for more than 3 months and did not take any concomitant medications that could potentially inhibit CYP 3A4. The patient was asked to stop drinking GFJ. Two weeks later, docetaxel (74 mg) was administered again and docetaxel elimination rate markedly increased relative to the first cycle (Figure 1). An increase of 36% in the plasma clearance value was determined. Docetaxel plasma concentrations in the first cycle were above the median while in the second cycle were below the median plasma concentrations. The estimate of terminal half-life was 10 h which represents a 30% of decrease with respect to the first course of treatment. Finally, the area under the docetaxel plasma concentration–time curve (AUC) form zero to infinity was reduced by 60% (5.61 mg h−1vs. 2.17 mg h−1). In the first cycle, values of albumin, aspartate aminotransferase (AST), alanine aminotransferase (ALT) and alkaline phosphatase (ALPK) were determined to be 5.25 g dl−1, 26 UI l−1, 33 UI l−1 and 159 UI l−1, respectively. AAG was not determined in the first cycle. In the second cycle, the value of AAG was 81.2 mg dl−1, the albumin was not determined, and values of AST, ALT and ALPK were 50 IU l−1, 52 IU l−1 and 157 IU l−1, respectively.
Figure 1.
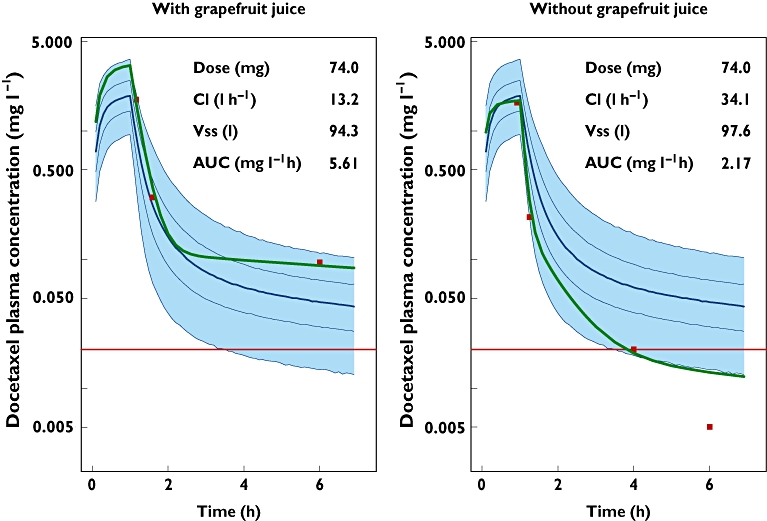
Effect of GFJ on docetaxel PK. Bayesian prediction (solid green line); time course of the 2.5th, 25th, 50th, 75th and 97.5th percentiles of the plasma concentrations (solid blue lines) and their associated model-based prediction of the 95% confidence interval (blue shaded area); observed docetaxel plasma concentrations (red points) and the lower limit of quantification, 0.01 mg l−1 (solid red line)
Docetaxel pharmacokinetics are linear and independent of the dose and the schedule administered [16, 17]. Docetaxel treatment is associated with substantial interpatient variability in exposure that results in a significant risk of under- or overdosing of patients [18] and can directly affect the response to treatment, as the relationship between exposure and response has been previously reported. Bruno et al. demonstrated that exposure to docetaxel was a significant predictor of time to progression and death in NSCLC patients [3]. It is nowadays established that the drug metabolizing enzyme system CYP 3A4 is a major determinant of the variability in docetaxel exposure [12]. PK studies with docetaxel have shown that after i.v. administration, the plasma concentration vs. time profiles follow a three compartment pharmacokinetic model with half-lives of 4 min, 38 min and 12 h in the α, β and γ phase, respectively [2]. Plasma clearance (and its between subject variability) was determined to be 36.7 l h−1 (33.5%) [11]. The interindividual variability in both toxicity and efficacy of docetaxel is thought to be partly related to large interindividual PK variability, mainly in plasma clearance (coefficient of variation around of 30–40%) [3, 19, 20] and, consequently in drug exposure, i.e. AUC.
To date, a well-established AUC value for which the balance between docetaxel efficacy and toxicity is optimal is still lacking. Recently, Engels et al. [21], have chosen a target AUC of 4.90 mg l−1 h that was based on several representative docetaxel pharmacokinetic studies, including a total of 806 patients treated with docetaxel 100 mg m−2. As docetaxel pharmacokinetics are linear, the target docetaxel AUC for a dose of docetaxel of 40 mg m−2 should be 1.96 mg l−1 h.
Traditionally, GFJ has been considered an inhibitor of intestinal CYP 3A4 but with a little effect on hepatic CYP 3A4 activity. However, Veronese et al. [22] have demonstrated that consumption of large amounts of GFJ inhibits both intestinal and hepatic CYP 3A4 activity. As can be seen in Figure 1, this case report showed that the co-administration of 250 ml of GFJ daily reduced docetaxel plasma clearance by 63% (13.2 l h−1vs. 34.1 l h−1). The degree of hepatic metabolism inhibition caused by the GFJ is comparable with ketoconazole, a specific CYP 3A4 inhibitor. Ketoconazole-docetaxel co-administration resulted in a 40% decrease in plasma clearance of docetaxel (22.0 l h−1vs. 36.5 l h−1) [23]. In the presence of GFJ, the AUC is a 65% higher compared with the AUC target (5.61 mg l−1 h vs. 1.96 mg l−1 h). This high exposure had an impact on the haematological toxicity, particularly in the neuthophil counts which were reduced from 2214 × 106 l−1 to 642 × 106 l−1 (a 71% reduction). In the absence of GFJ, the AUC was closer to the AUC target value and the neuthophil count reduction was less than 35%. Previous knowledge of intra-individual variability in docetaxel clearance after i.v. administration suggests that the intrapatient clearance variation is around 15% [24]. Concomitant use of GFJ and docetaxel resulted in a reduction of clearance greater than 15% so, probably, other factors such as the intake of grapefruit juice are involved in the variation of docetaxel clearance in patients.
The interaction between docetaxel and GFJ has been formulated from a theoretical point of view and, to the best of our knowledge, a case report of a pharmacokinetic interaction between i.v. administration of docetaxel and GFJ has not been reported in the literature before. It is the intention of this report to raise the awareness of this food and drug interaction, that is similar to ketoconazole-docetaxel co-administration and which may have significant clinical implications for cancer care. Caution should be taken and patients should stop drinking GFJ while on docetaxel treatment. Alternatively, substantial dose reductions are required if docetaxel has to be administered together with large amounts of GFJ. In this situation docetaxel therapeutic drug monitoring could be useful to deliver the target drug exposure [5].
Acknowledgments
The authors wish to express their gratitude to the anonymous reviewers who gave their comments during the review process to improve the final quality of this manuscript.
Competing Interests
There are no competing interests to declare.
REFERENCES
- 1.Docetaxel summary of product characteristics. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001107/WC500073419.pdf (last accessed 30 November 2010)
- 2.Bruno R, Sanderink GJ. Pharmacokinetics and metabolism of Taxotere (docetaxel) Cancer Surv. 1993;17:305–13. [PubMed] [Google Scholar]
- 3.Brunsvig PF, Andersen A, Aamdal S, Kristensen V, Olsen H. Pharmacokinetic analysis of two different docetaxel dose levels in patients with non-small cell lung cancer treated with docetaxel as monotherapy or with concurrent radiotherapy. BMC Cancer. 2007;7:197. doi: 10.1186/1471-2407-7-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruno R, Hille D, Riva A, Vivier N, ten Bokkel H, van Oosterom AT, Kaye SB, Verweij J, Fossella FV, Valero V, Rigas JR, Seidman AD, Chevallier B, Fumoleau P, Burris HA, Ravdin PM, Sheiner LB. Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J Clin Oncol. 1998;16:187–96. doi: 10.1200/JCO.1998.16.1.187. [DOI] [PubMed] [Google Scholar]
- 5.Engels FK, Ten Tije AJ, Baker SD, Lee CK, Loos WJ, Vulto AG, Verweij J, Sparreboom A. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin Pharmacol Ther. 2004;75:448–54. doi: 10.1016/j.clpt.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Malingré MM, Richel DJ, Beijnen JH, Rosing H, Koopman FJ, Ten Bokkel Huinink WW, Schot ME, Schellens JH. Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J Clin Oncol. 2001;19:1160–6. doi: 10.1200/JCO.2001.19.4.1160. [DOI] [PubMed] [Google Scholar]
- 7.Koolen SL, Oostendorp RL, Beijnen JH, Schellens JH, Huitema AD. Population pharmacokinetics of intravenously and orally administered docetaxel with or without co-administration of ritonavir in patients with advanced cancer. Br J Clin Pharmacol. 2010;69:465–74. doi: 10.1111/j.1365-2125.2010.03621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koolen SL, Beijnen JH, Schellens JH. Intravenous-to-oral switch in anticancer chemotherapy: a focus on docetaxel and paclitaxel. Clin Pharmacol Ther. 2010;87:126–9. doi: 10.1038/clpt.2009.233. [DOI] [PubMed] [Google Scholar]
- 9.van Waterschoot RA, Lagas JS, Wagenaar E, van der Kruijssen CM, van Herwaarden AE, Song JY, Rooswinkel RW, van Tellingen O, Rosing H, Beijnen JH, Schinkel AH. Absence of both cytochrome P450 3A and P-glycoprotein dramatically increases docetaxel oral bioavailability and risk of intestinal toxicity. Cancer Res. 2009;69:8996–9002. doi: 10.1158/0008-5472.CAN-09-2915. [DOI] [PubMed] [Google Scholar]
- 10.Docetaxel label information. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020449s059lbl.pdf (last accessed 10 January 2011)
- 11.Zufia López L, Aldaz Pastor A, Aramendia Beitia JM, Arrobas Velilla J, Giraldez Deiró J. Determination of docetaxel and paclitaxel in human plasma by high performance liquid chromatography. Validation and application to clinical pharmacokinetic studies. Ther Drug Monit. 2006;28:199–205. doi: 10.1097/01.ftd.0000189903.46802.1f. [DOI] [PubMed] [Google Scholar]
- 12.Beal SL, Sheiner LB. NONMEM Users Guides. Hanover, MD: GloboMax, LLC; 1992. [Google Scholar]
- 13.Bruno R, Vivier N, Vergniol JC, De Phillips SL, Montay G, Sheiner LB. A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm. 1996;24:153–72. doi: 10.1007/BF02353487. [DOI] [PubMed] [Google Scholar]
- 14.Baille P, Bruno R, Schellens JHM, Webster LK, Millward M, Verweij J, Montay G. Optimal sampling strategies for Bayesian estimation of docetaxel clearance. Clin Cancer Res. 1997;3:1535–8. [PubMed] [Google Scholar]
- 15.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28:481–504. doi: 10.1023/a:1012299115260. [DOI] [PubMed] [Google Scholar]
- 16.Baker SD, Zhao M, Ck L, Verweij J, Zabelina Y, Brahmer JR, Wolff AC, Sparreboom A, Carducci MA. Comparative pharmacokinetics of weekly and every-three-weeks docetaxel. Clin Cancer Res. 2004;10:1976–83. doi: 10.1158/1078-0432.ccr-0842-03. [DOI] [PubMed] [Google Scholar]
- 17.McLeod HL, Kearns CM, Kuhn JG, Bruno R. Evaluation of the linearity of docetaxel pharmacokinetics. Cancer Chemother Pharmacol. 1998;42:155–9. doi: 10.1007/s002800050799. [DOI] [PubMed] [Google Scholar]
- 18.Baker SD, Sparreboom A, Verweij J. Clinical pharmacokinetics of docetaxel: recent developments. Clin Pharmacokinet. 2006;45:235–52. doi: 10.2165/00003088-200645030-00002. [DOI] [PubMed] [Google Scholar]
- 19.Hirth J, Watkins PB, Strawderman M, Schott A, Bruno R, Baker LH. The effect of an individual's cytochrome CYP3A4 activity on docetaxel clearance. Clin Cancer Res. 2000;6:1255–8. [PubMed] [Google Scholar]
- 20.Veyrat-Follet C, Bruno R, Olivares R, Rhodes GR, Chaikin P. Clinical trial simulation of docetaxel in patients with cancer as a tool for dosage optimization. Clin Pharmacol Ther. 2000;68:677–87. doi: 10.1067/mcp.2000.111948. [DOI] [PubMed] [Google Scholar]
- 21.Engels FK, Loos WJ, van der Bol JM, de Brujin P, Mathijssen RHJ, Verweij J, Mathot RAA. Therapeutic drug monitoring for the individualization of docetaxel dosing: a randomized pharmacokinetic study. Clin Cancer Res. 2011;17:353–62. doi: 10.1158/1078-0432.CCR-10-1636. [DOI] [PubMed] [Google Scholar]
- 22.Veronese ML, Gillen LP, Burke JP, Dorval EP, Hauck WW, Pequignot E, Waldman SA, Greenberg HE. Exposure-dependent inhibition of intestinal and hepatic CYP3A4 in vivo by grapefruit juice. J Clin Pharmacol. 2003;43:831–9. doi: 10.1177/0091270003256059. [DOI] [PubMed] [Google Scholar]
- 23.Lim YW, Goh BC, Wang LZ, Tan SH, Chuah BY, Lim SE, Iau P, Buhari SA, Chan CW, Sukri NB, Cordero MT, Soo R, Lee SC. Pharmacokinetics and pharmacodynamics of docetaxel with or without ketoconazole modulation in chemonaive breast cancer patients. Ann Oncol. 2010;21:2175–82. doi: 10.1093/annonc/mdq230. [DOI] [PubMed] [Google Scholar]
- 24.Sandström M, Lindman H, Nygren P, Lidbrink E, Bergh J, Karlsson MO. Model describing the relationship between pharmacokinetics and hematologic toxicity of the epirubicin-docetaxel regimen in breast cancer patients. J Clin Oncol. 2005;23:413–21. doi: 10.1200/JCO.2005.09.161. [DOI] [PubMed] [Google Scholar]