

Abstract
One of the main regulators of gene expression during embryonegesis and stem cell differentiation is DNA methylation. The recent identification of hydroxymethylcytosine (5hmC) as a novel epigenetic mark sparked an intense effort to characterize its specialized enzymatic machinery and to understand the biological significance of 5hmC. The recent discovery of recurrent deletions and somatic mutations in the TET gene family, which includes proteins that can hydroxylate methylcytosine (5mC), in a large fraction of myeloid malignancies further suggested a key role for dynamic DNA methylation changes in the regulation of stem cell differentiation and transformation.
One of the major epigenetic modifications of the human genome is methylation at the 5-position of cytosine (5mC). Genome-wide reprogramming events during early embryonic development are characterized by the loss and reacquisition of DNA methylation (Hajkova et al., 2008; Hajkova et al., 2010; Surani et al., 2007), a process orchestrated by DNA methyltranferases (DNMTs) that are essential for the establishment and maintenance of 5mC in the genome (Li et al., 1992; Okano et al., 1999). The discovery that hydroxymethylcytosine (5hmC) is also abundant in embryonic stem cells (ESCs) and adult cells (Ito et al., 2010; Koh et al., 2011; Kriaucionis and Heintz, 2009; Song et al., 2011; Szwagierczak et al., 2010; Tahiliani et al., 2009) prompted several studies aimed at identifying enzymes that could generate 5hmC from 5mC in mammalian cells and the role of this epigenetic modification in the regulation of DNA methylation.
In tumor cells, the normal pattern of DNA methylation is often altered, resulting in global hypomethylation of the genome in conjunction with hypermethylation at CpG islands within the promoters of critical genes such as tumor suppressors (Esteller, 2008). Recent studies have identified a novel class of epigenetic mutations in myeloid malignancies. Specifically, deletions and inactivating mutations in the TET2 gene were identified in almost 30% of all myeloid malignancies (Abdel-Wahab et al., 2009; Delhommeau et al., 2009; Jankowska et al., 2009; Langemeijer et al., 2009). With the parallel discovery that the TET family of proteins, TET1, TET2, and TET3, were able to modify DNA by hydroxylating 5mC to 5hmC (Tahiliani et al., 2009) and in so doing promote active DNA demethylation (Cortellino et al., 2011; Guo et al., 2011; He et al., 2011), a new field in epigenetic regulation has emerged aimed at understanding the role of 5hmC and TETs in development and disease pathogenesis.
5hmC and the TET family proteins
The role of 5mC in epigenetic regulation has been extensively studied, however, until recently very little was known about the role of 5hmC despite its identification in the mammalian genome several decades ago (Penn et al., 1972). In 2009, Kriaucionis and Heintz showed that global 5hmC levels in the genome of mouse Purkinje cells and granule neurons constitute approximately 0.6% and 0.2% of all nucleotides, respectively (Kriaucionis and Heintz, 2009). In addition, Tahiliani et al found that 5hmC levels comprised 0.03% of total nucleotides in mouse embryonic stem cells (ESCs) (Tahiliani et al., 2009). While these frequencies seem small, the biological significance of 5hmC levels becomes apparent when compared to 5mC, which is also considered to be a minor base in mammalian DNA, constituting approximately 1% of all DNA bases in the genome, yet plays a crucial role in the regulation of gene expression. In the search for mammalian enzymes able to modify 5mC, using computational analysis, Tahiliani et al. made the discovery that human TET1, TET2, and TET3 all contain in their c-terminus a 2-oxoglutarate- (2-OG) and Fe(II)-dependant dioxygenase domain homologous to the thymidine hydroxylase domain of the trypanosomal proteins JBP1 and JBP2 (Tahiliani et al., 2009). Using WT and catalytic mutants of human and mouse TET1, TET2 and TET3 it was subsequently shown that all the TET proteins are able to convert 5mC to 5hmC in an Fe(II) and α-ketoglutarate-dependent manner in vitro (Ito et al., 2010; Tahiliani et al., 2009). Although all three TET proteins can catalyze 5hmC production from 5mC, only the over-expression of TET1 and TET2 in vivo caused a global decrease in 5mC (Ito et al., 2010; Tahiliani et al., 2009). More recently it has also been shown that the TET proteins are able to further oxidize 5hmC into 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (He et al., 2011; Khachatryan et al., 2011), raising the possibility that loss of 5mC may occur through TET-catalyzed oxidation followed by decarboxylation. These findings and the subsequent identification of 5hmC in many other mammalian tissues and cell types (Globisch et al., 2010; Song et al., 2011) have prompted several studies designed to investigate the role of this DNA modification in epigenetic and transcriptional regulation.
Active DNA demethylation catalyzed by TET, 5hmC, deamination and DNA repair
The presence of 5hmC may contribute to both passive and active DNA demethylation in the genome (Figure 1). Maintenance methylation normally follows replication, and this process may be inhibited due to the inability by DNMT1 to recognize 5hmC (Valinluck and Sowers, 2007), causing 5mC to be lost passively during cell division by replacement with unmethylated cytosine. However, during mammalian development the rapid loss of DNA methylation immediately after fertilization (Hajkova et al., 2010; Mayer et al., 2000; Oswald et al., 2000) and again in primordial germ cells (Hajkova et al., 2002; Lee et al., 2002) within the period of a single cell cycle suggests the presence of enzymes that can actively remove 5mC independently of replication. In addition, modifying methylation patterns in non-replicating cells such as neurons would require either direct enzymatic removal of the methyl group from cytosine, a role once proposed for the methylated CpG DNA-binding protein MBD2b (Bhattacharya et al., 1999) or excision and replacement of cytosine by DNA repair pathways. Plants employ BER to achieve active demethylation by excising 5mC directly (Gehring et al., 2009) (Zhu, 2009) however DNA glycosylases that recognize 5mC have not been identified in mammals.
Figure 1. The role of TET proteins in Passive or Active DNA demethylation.
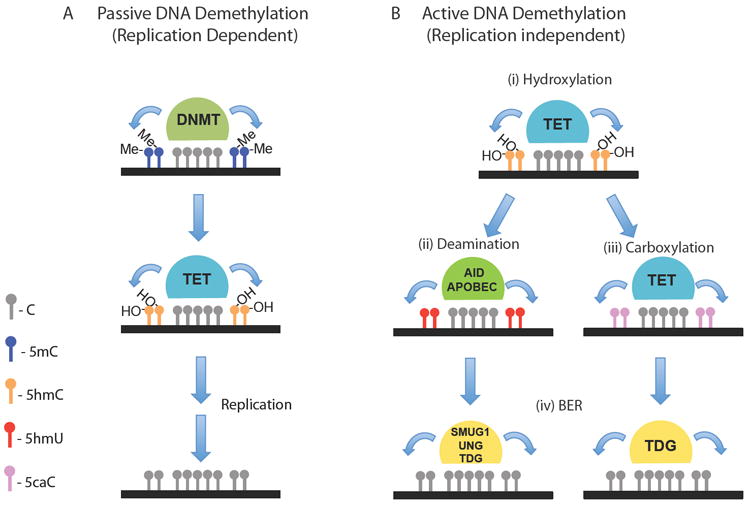
A) The inability of the maintenance methyltransferase, DNMT1, to recognize 5hmC-containing DNA may lead to passive demethylation following replication. B) Active DNA methylation can be catalyzed by TET-mediated (i) hydroxylation of 5mC to 5hmC followed by AID and APOBEC-mediated (ii) deamination of 5hmC to 5hmU or (iii) further carboxylation of 5hmC to 5caC by the TET proteins and the subsequent cleavage of 5hmU or 5caC by DNA glycosylases (e.g. SMUG1, UNG and TDG) and replacement with cytosine by (iv) a base excision repair (BER) enzymatic pathway.
Recent studies have shown that TET hydroxylases may catalyze active DNA demethylation induced by oxidation of 5mC to 5hmC and its replacement with unmethylated cytosine by a sequential deamination and base excision repair (BER) pathway. DNA deaminases of the AID/APOBEC family catalyze the conversion of cytosine to uracil and 5hmC to 5hmU. Activation induced cytidine deaminase (AID) is required for antibody diversification mediated by somatic hypermutation and class switching in germinal center B cells (Muramatsu et al., 2000; Muramatsu et al., 1999). APOBEC1 is the most well characterized of the APOBEC family of enzymes that catalyses the conversion of a C residue to U in apolipoprotein B mRNA resulting in a premature stop codon (Teng et al., 1993). Interestingly, while AID expression was thought to be restricted to lymphoid tissue, it was recently identified as a factor required for DNA demethylation during reprogramming of somatic cells (Bhutani et al., 2010) and AID-null mice have been shown to exhibit significant defects in genome-wide DNA demethylation in primordial germ cells (Popp et al., 2010).
Two recent studies have now shown that both AID and APOBEC can mediate active DNA demethylation in mammalian cells in collaboration with TET hydroxylase activity and the demethylation intermediate 5hmC. In the first study by Guo et al., AID over-expression in HEK 293 cells led to a significant decrease of endogenous 5hmC levels induced by TET1 (Guo et al., 2011) and co-expression of these two enzymes promoted the loss of 5mC. Given the abundance of 5hmC in various regions of the brain such as the hippocampal dentate gyrus (Munzel et al., 2010), the authors used adeno-associated viruses to introduce TET1, or AID into this area of the adult mouse brain to measure effects on 5hmC production and stability. Over-expression of TET1 further increased 5hmC levels whereas over-expression of AID significantly decreased 5hmC, supporting its role for 5hmC-removal in vivo (Guo et al., 2011). Active demethylation was also analyzed at endogenous loci known to exhibit neuronal-activity induced active DNA demethylation in adult dentate granule cells (Fernandez et al., 2009). TET1 or AID over-expression mediated significant decreases in CpG methylation levels at these genomic loci and both TET1 and AID-induced demethylation was accompanied by a significant upregulation in the expression of these genes in vivo (Guo et al., 2011). Repeating this experiment using shRNAs to target both endogenous TET1 and TET2 and the most abundant endogenously expressed deaminase in this tissue, APOBEC1, showed that demethylation was impaired in a locus-specific manner. In a subsequent study by Cortellino et al., it was also shown using TDG-deficient mice that demethylation is an active process that requires TDG catalytic activity immediately downstream of the deaminase-catalyzed conversion of 5mC into thymine and/or 5hmC into 5hmU (Cortellino et al., 2011). Co-immunoprecipitation experiments also revealed that AID and TDG directly interact in a ternary complex with growth arrest and DNA damage-inducible protein 45-α (Gadd45a). Gadd45a is a genome stability protein known to initiate demethylation in mammalian cells (Barreto et al., 2007) (Schmitz et al., 2009), Xenopus (Barreto et al., 2007) and in zebrafish embryos via an AID/APOBEC-mediated deamination reaction followed by removal of the mismatched thymine by the zebrafish thymine glycosylase MBD4a (Rai et al., 2008).
Interestingly, the TET-mediated oxidation of 5hmC to 5caC may also trigger DNA demethylation by base excision repair independently of deamination. Recently it has been shown that mouse ESCs, iPSCs and other mouse organs contain low but detectable levels of 5caC in their genome (He et al., 2011; Ito et al., 2011). Using an in vitro assay, He et al., identified glycosylase activity in mouse ESC nuclear extracts specific only for 5caC- and not 5hmC-containing DNA substrates. Depletion of TDG in mouse ES or iPS cells caused significant 5caC accumulation (He et al., 2011) providing further evidence that TDG may act both on 5hmU and 5caC, downstream of TET activity, to initiate BER in vivo. These combined studies suggest that the role of the TET proteins may be to prevent hypermethylation and promote demethylation by a sequential process involving AID/APOBEC mediated deamination of 5mC to T (Rai et al., 2008) (Cortellino et al., 2011) or 5hmC to 5hmU or 5caC followed by BER (Cortellino et al., 2011; Guo et al., 2011; He et al., 2011). Loss of either AID or TDG alone, however, is not sufficient to completely block DNA demethylation. Therefore, other DNA repair pathways or unknown DNA demethylation mechanisms may also contribute to the regulation of DNA methylation patterns during developmental reprogramming events and differentiation.
Genome-wide mapping of 5hmC and TET1 binding in the mammalian genome
Based on the findings that 5hmC is enriched in mouse ESCs (Tahiliani et al., 2009) and neuronal cells (Kriaucionis and Heintz, 2009) several groups have employed 5hmC-specific immunoprecipitation techniques to map its genomic distribution in mouse ESCs (Ficz et al., 2011; Robertson et al., 2011; Williams et al., 2011; Wu et al., 2011a; Xu et al., 2011b), human ESCs (Stroud et al., 2011) and in brain tissue (Jin et al., 2011; Song et al., 2011).
The distribution of 5hmc in ESCs localizes mainly to TSSs and within gene bodies, and in general is found more highly enriched at gene exons compared to introns (Ficz et al., 2011; Williams et al., 2011; Wu et al., 2011a; Xu et al., 2011b). This distribution is similar to that seen in mouse cerebellum (Song et al., 2011) however in an adult human brain study there was greater enrichment for 5hmC in the promoter region than gene bodies (Jin et al., 2011) suggesting perhaps differences in 5hmC distribution between species or during maturation. The combined studies in ESCs for the most part agree that 5hmC modifications are found predominantly localized within gene bodies with intermediate density CpG promoters of both active and repressed genes.
TET1, unlike TET2 and TET3, possesses a CXXC zinc finger DNA-binding motif and three candidate bipartite nuclear localization signals in its N-terminal domain (Lorsbach et al., 2003). Studies have shown that TET1 has a preference for binding CpG rich DNA (Xu et al., 2011c) and has a high affinity for binding to non-methylated CpG sequences similar to other CXXC-containing chromatin modifiers such as DNMT and MLL (Zhang et al., 2010). It also significantly binds to methylated CpG and 5hmC sequences (Xu et al., 2011b), which would be consistent with its role in catalyzing the conversion of 5mC to 5hmC (Zhang et al., 2010).
Given that TET1 is highly expressed in ESCs, and that loss of TET1 expression causes a global decrease in 5hmC levels (Koh et al., 2011), several groups also performed TET1 ChIP-seq analysis in parallel with genome-wide 5hmC mapping in mouse ESCs that resulted in highly similar TET1 binding profiles (Figure 2A). The consensus from studies thus far is that TET1 binds preferentially at TSSs in promoters compared to gene bodies and with an affinity that positively correlates with CpG density (Williams et al., 2011; Wu et al., 2011b; Wu and Zhang, 2011; Xu et al., 2011b). High CpG containing promoters (HCPs) are bound most strongly by TET1 and display univalent H3K4me3 marks compared to intermediate CpG containing promoters (ICPs), which are poorly bound by TET1 and display bivalent K3K4me3 and H3K27me3, and very little TET1-binding is seen at low CpG promoters (LCPs) that are univalent H3K27me3. Interestingly, knockdown of TET1 causes an increase in 5mC at TSS regions of its target genes and a partial decrease in 5hmC at specific gene loci (Williams et al., 2011) at both promoters and within gene bodies of TET1-target genes (Wu et al., 2011a; Xu et al., 2011b). What remains unclear is how loss of TET1 influences the 5hmC distribution of genes with ICPs and LCPs, which exhibit more 5hmC in their gene bodies, increasing toward the TTS where there is negligible TET1 binding (Williams et al., 2011; Xu et al., 2011b). It is interesting to speculate that lack of TET1-binding indirectly shifts the balance of 5hmC distribution from the TSS toward the TTS, perhaps due to the activity of another enzyme, such as TET2, which is also highly expressed in ESCs but lacks the CXXC-motif that would potentially target it to the same CpG-rich regions in the promoters of genes recognized by TET1.
Figure 2. A dual role for TET1 and 5hmC in transcriptional regulation.
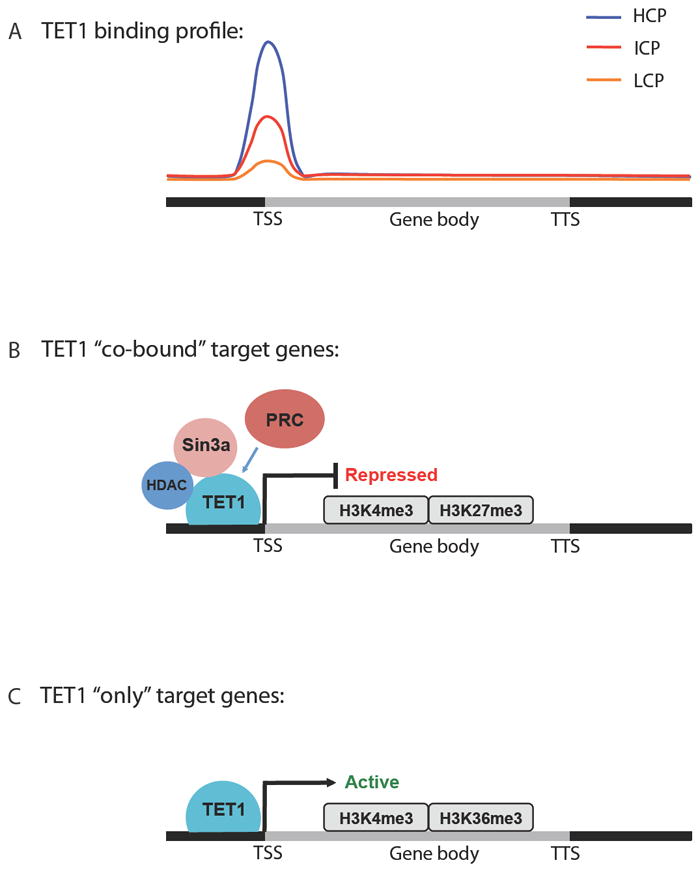
A) Schematic of TET1 binding profile in the genome of mouse ESCs. TET1 is most highly enriched at the transcriptional start site (TSS) of genes with high CpG promoters (HCPs) compared to those with intermediate and low CpG promoters (ICPs and LCPs, respectively). B) TET1 directly binds to the co-repressors Sin3a and HDACs. TET1 target genes co-bound by polycomb repressor complex (PRC) proteins and Sin3a are repressed and display bivalent H3K4me3 and H3K27me3 marks. C) TET1-only target genes are actively transcribed and enriched for H3K4me3 and H3K36me3.
Additional complexity is seen at the transcriptional level, with TET1 playing a dual role in both activation and repression of its target genes (Williams et al., 2011; Wu et al., 2011b; Xu et al., 2011b) (Figure 2B and 2C). In all three studies, TET1 knockdown causes more genes to become upregulated than downregulated and TET1 binds directly to more of the upregulated genes. Interestingly, two groups found extensive overlap between TET1 and polycomb repressor complex (PRC) target genes, and despite a requirement of TET1 for the transcriptional activity of PRC2 (Wu et al., 2011b), there was no direct interaction between these proteins (Williams et al., 2011; Wu et al., 2011b). The co-repressor Sin3a was instead found to directly associate with TET1 and not TET2 in a pull down-assay using HEK293 cells and showed extensive overlap with TET1 in both binding profiles and target genes (Williams et al., 2011). The direct association of Sin3a and recruitment of PRC2 at the TSS of target genes bound by TET1 may explain how TET1 plays a role as a transcriptional repressor (Williams et al., 2011; Wu et al., 2011b). The perplexing conclusion from these studies is that TET1 binding cannot however be used to predict whether a gene will be activated or repressed and how activation could be mediated was not addressed. Subsequent studies will need to ascertain TET2 and TET3 localization in the mammalian genome and to determine if TET2 and TET3 can function as transcriptional repressors in vitro as has been shown for TET1.
Gene expression was also correlated to 5hmC mapping with WT or TET1-deficient ESCs and associated histone modifications. In the study by Wu et al., 5hmC was preferentially enriched at the TSS of lowly expressed genes that exhibit bivalent H3K4me3 and H3K27me3, consistent with TET1-repressed PRC2 co-occupied target genes (Wu et al., 2011a; Wu et al., 2011b). Instead, 5hmC is depleted at the TSS and enriched within gene bodies peaking at the TTS in highly expressed genes that are H3K4me3 and H3K36me3 consistent with TET1-mediated gene activation (Wu et al., 2011a; Wu et al., 2011b). While TET1 is able bind to genes that are expressed at either high or low levels (Wu et al., 2011a; Xu et al., 2011b) Xu et al identified two additional groups of TET1-bound target genes that do not depend on TET1 for transcriptional regulation; genes that are either silenced by univalent H3K27me3 or highly expressed genes with abundant H3K4me3 and H3K36me3 occupancy. That TET1 might influence chromatin accessibility independently of its effect on gene transcription is exciting but requires further validation using catalytically inactive, DNA-binding mutant or truncated proteins and genetic approaches targeting TET proteins and PRC complexes in tandem.
5hmC may play distinct roles in gene transcription in pluripotent stem cells versus specialized cells such as neurons, particularly given that the correlation between gene expression and 5hmC levels at promoters and gene bodies between ESCs (Williams et al., 2011; Xu et al., 2011b) (Wu et al., 2011a) and mouse cerebellum (Song et al., 2011) is different. Global 5hmC and locus-specific 5hmC may be differentially regulated in different cell types or at different stages of development, as is the case during maturation in the brain (Song et al., 2011). From the combined studies performed in ESCs it could be concluded that TET1 and TET2, 5hmC and its precursor 5mC are not needed for ESC pluripotency but are required for normal differentiation during ESC lineage specification (Koh et al., 2011). Further evidence for this proposal is provided by Dnmt TKO ESCs, that lack both 5mC and 5hmC (Williams et al., 2011) and are defective in differentiation (Jackson et al., 2004) yet display only limited alteration in gene expression and can maintain self-renewal and an undifferentiated ESC state (Tsumura et al., 2006). Lack of TET2 in hematopoietic cells may parallel loss of TET1 in mESCs and render myeloid progenitors unable to achieve normal patterns of methylation that are associated with differentiation but instead permit the stem cell self-renewing state, a trait that may then contribute to leukemic transformation in TET2-deficient mice (Moran-Crusio et al., 2011; Quivoron et al., 2011). Genome-wide mapping of 5hmC in the hematopoietic system has not yet been reported, however it will be intriguing to see how this epigenetic marker might change in distribution between HSC and differentiated lineage-specific genomes. Given that all three TET proteins exhibit differential expression in hematopoietic cells (Lorsbach et al., 2003; Moran-Crusio et al., 2011; Quivoron et al., 2011) it remains to be seen what the contribution of each TET will be in similar studies that employ ChIP-seq, gene expression and histone modification profiling in correlation with 5hmC mapping.
Identification of TET2 mutations in myeloid malignancies
Initial reports of alterations involving the TET family of proteins came in 2002 and 2003 when two groups identified TET1 as a fusion partner of MLL in a small subset of patients with acute myeloid leukemia associated with a t(10;11)(q22;q23) translocation (Lorsbach et al., 2003; Ono et al., 2002). Lorsbach, Downing, and colleagues used homology searches to identify TET2 and TET3 as members of the TET family, and demonstrated that all three TET proteins were differentially expressed in different tissue compartments with TET2 being most widely expressed, including within the hematopoietic compartment (Lorsbach et al., 2003). Notably, given that TET1 was fused to MLL in all patients with the t(10;11)(q22;q23) translocation, functional studies of TET1 or of the other TET proteins were not undertaken at that time.
In 2009 two groups independently identified somatic alterations in TET2 in patients with myeloproliferative neoplasms (MPN) and myelodysplastic syndrome (MDS) (Delhommeau et al., 2009; Langemeijer et al., 2009). Delhommeau and colleagues investigated a subset of MPN patients with expansion of the immature, CD34+ hematopoietic progenitor compartment, and using comparative genomic hybridization and single nucleotide polymorphism (SNP) arrays they identified, recurrent, focal deletions and copy neutral loss of heterozygosity (CN-LOH) on chromosome 4q24. In one patient they mapped the deletion to a single gene, TET2, and then performed candidate gene resequencing of TET2 and identified somatic mutations in 10-20% of MPN and MDS patients. Langemeijer and colleagues performed SNP array analysis of 102 patients with MDS, and in five patients they identified focal deletions or CN-LOH involving the region on chromosome 4q24, which includes the TET2 locus. They then performed candidate gene sequencing and identified missense, nonsense, and frameshift mutations in 26% of the patients in their MDS cohort. These two landmark studies demonstrated that deletions, LOH, and mutations are a frequent somatic event in MPN and MDS and the presence of nonsense/frameshift mutations and deletions provides strong genetic evidence that TET2 mutations in leukemia patients result in reduced/absent TET2 function. Importantly, although these studies did not include extensive functional analysis, Delhommeau and colleagues demonstrated improved engraftment of TET2-mutant MPN cells in immunodeficient mice, consistent with enhanced stem cell function in vivo (Delhommeau et al., 2009).
Clinical Spectrum of TET2 mutations in Hematopoietic Malignancies
Subsequent to the discovery of TET2 mutations in MPN and MDS patients, many studies have performed TET2 resequencing in large cohorts of leukemia patients to delineate the frequency of TET2 mutations in different malignancies (Figure 3), and to investigate whether TET2 mutations are associated with specific clinical features or with poor outcome (Table 1). Many groups have investigated TET2 mutations in MPN patients; the largest studies suggest that TET2 mutations can be identified in 2-10% of polycythemia vera (PV) and essential thrombocytosis (ET) patients, and in 10-20% of patients with primary myelofibrosis (PMF) or post PV/ET myelofibrosis (Abdel-Wahab et al., 2009; Tefferi et al., 2009). Importantly, TET2 mutations are observed at a higher frequency in MPN patients who transform to acute myeloid leukemia (AML), suggesting TET2 loss is a common genetic event associated with leukemic transformation (Abdel-Wahab et al., 2010). However, the prognostic relevance of TET2 mutations in MPN has not been assessed in sufficiently large cohorts to date. Similarly, many groups have confirmed that TET2 mutations occur in 25-30% of MDS patients (Witkowski et al., 2009), and while smaller studies suggested TET2 mutations were associated with adverse outcome in MDS more recent investigation of a large MDS cohort failed to demonstrate a relationship between TET2 mutations and poor prognosis (Bejar et al., 2011). TET2 mutations also occur in almost 50% of patients diagnosed with chronic myelomonocytic leukemia (CMML), an overlapping malignancy with features of both MPN and MDS. Given the CMML phenotype of TET2-deficient mice (Moran-Crusio et al., 2011) it is notable that CMML is associated with the highest frequency of TET2 mutations to date.
Figure 3. Schema of somatic TET2 mutations in hematopoietic malignancies.
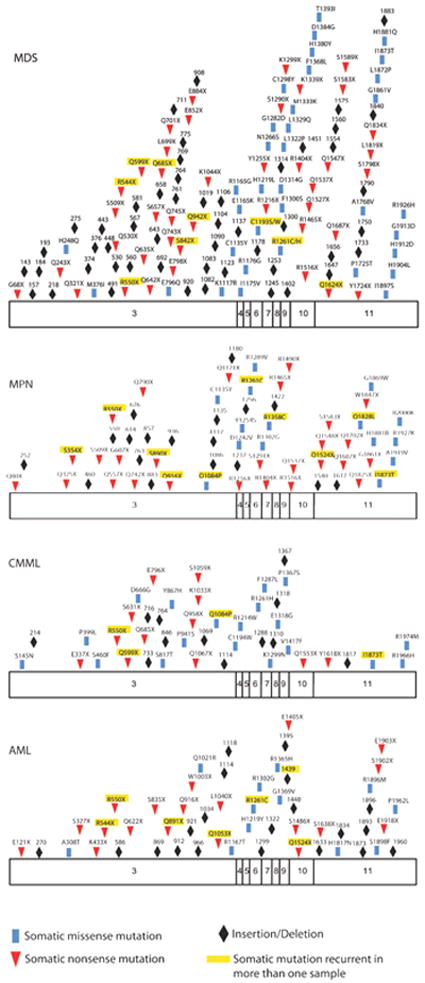
Displayed are the known somatic missense, nonsense, and frameshift mutations throughout the open-reading frame of TET2 in myelodisplastic syndromes (MDS), myeloproliferative neoplasms (MPN), chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML). In addition mutations that were seen in more than one sample amongst studies are highlighted in yellow. Data are compiled from several studies (Abdel-Wahab et al., 2009; Bejar et al., 2011; Delhommeau et al., 2009; Figueroa et al., 2010; Jankowska et al., 2009; Koh et al., 2011).
Table 1.
Studies evaluating the potential prognostic significance of TET2 mutations in patients with myeloid malignancies
| Disease | Reference | Patients studied | Prognostic relevance |
|---|---|---|---|
| AML | (Abdel-Wahab et al., 2009) | 119 AML patients including de novo AML, therapy-related AML, and AML with an antecedent hematologic disorder | TET2 mutations were associated with significantly worsened overall survival. |
| AML | (Nibourel et al., 2010) | 111 patients with de novo AML | TET2 mutations did not impact overall survival. |
| AML | (Metzeler et al., 2011) | 427 patients with normal karyotype AML | TET2 mutations were associated with a lower complete remission rate and shorter disease free and overall survival. |
| Secondary AML | (Kosmider et al., 2011) | 247 patients with AML derived from MDS or therapy-related AML | TET2 mutations did not influence complete remission rates or overall survival. |
| MDS | (Kosmider et al., 2009a) | 89 MDS patients and 7 with MDS transformed to AML | TET2 mutations associated with a significantly improved overall and progression-free survival. |
| MDS | (Bejar et al., 2011) | 439 de novo MDS patients | TET2 mutations did not impact overall survival or effect clinical characteristics in an adverse way. |
| MPN | (Tefferi et al., 2009) | 239 BCR-ABL negative MPN patients | TET2 mutations did not impact overall survival or effect clinical characteristics in an adverse way. |
| CMML | (Kosmider et al., 2009b) | 88 patients with CMML | TET2 mutations strongly associated with monocytosis. In the overall cohort, TET2 mutations did impact survival but TET2 imparted significantly worse overall survival in patients with CMML I specifically. |
In addition, TET2 mutations are observed in a subset of patients with AML. Initial studies identified TET2 mutations in 10% of AML patients, and found that TET2-mutant AML patients had poorer outcome compared to patients without TET2 mutations (Abdel-Wahab et al., 2009). Subsequent studies in larger, homogenously treated cohorts of de novo AML patients have confirmed that TET2 mutations occur in 7-23% of patients with de novo AML, and that the frequency of TET2 mutations increases with increasing age (Figueroa et al., 2010; Metzeler et al., 2011). Importantly, TET2 mutations were found to be associated with adverse outcome in karyotypically normal AML (Metzeler et al., 2011), suggesting that TET2 mutational status represents a novel biomarker that can be used to improve risk stratification in AML and to select patients for stem cell transplantation. By contrast, TET2 mutations occur in a higher proportion of patients who transform from MPN to AML, and studies of paired MPN and AML samples from individual patients demonstrated that TET2 mutations are commonly acquired in transformation to AML from a chronic myeloid neoplasm (Abdel-Wahab et al., 2010), implicating TET2 loss as an important genetic event in conferring self-renewal and acute leukemic transformation to chronic myeloid malignancies.
Most recently, candidate gene resequencing of TET2 in hematologic malignancies led to the identification of recurrent TET2 mutations in B and T-cell lymphoid malignancies, including in 30% of patients with angioimmunoblastic T-cell lymphoma (Quivoron et al., 2011). In addition, they demonstrated that the TET2 mutant clone could be identified in hematopoietic stem/progenitor cells (HSPCs) from patients with T-cell lymphoma, suggesting that TET2 mutations are acquired in the stem cell compartment in lymphoma patients without co-existent myeloid disease. These data suggest that TET2 mutations are acquired in HSPCs but that the disease phenotype associated with TET2 loss is dictated by the presence of known myeloid (JAK2, FLT3) or lymphoid disease alleles.
Insights into Mechanisms of Transformation from Investigation of Primary Patient Samples
As noted above, TET2-mutant MPN samples exhibited improved engraftment (Delhommeau et al., 2009), consistent with improved stem-cell function of TET2-mutant HSPCs, which has subsequently been confirmed in murine genetic studies (see below). Importantly, two additional important insights have emanated from studies of primary patient samples with TET2 mutations. Rao, Maciejewski, and colleagues assessed 5hmC content in CMML patients with TET2 mutations and demonstrated that TET2-mutant CMML is characterized by a marked reduction in global levels of 5hmC (Ko et al., 2010); this has subsequently been confirmed in MPN patients (Pronier et al., 2011). In addition, they demonstrated that introduction of TET2 missense mutations seen in leukemia patients into the TET2 catalytic domain resulted in impaired hydroxymethylation. Taken together, these data show that leukemia-associated TET2 mutations cause loss-of-function with respect to TET2-mediated hydroxymethylation.
More recently, genetic studies in AML patients have provided additional insight into the role of TET2 in leukemic transformation. Detailed genetic and methylation analysis of a large cohort of AML patients led to the observation that TET2-mutant AML is characterized by a hypermethylation phenotype (Figueroa et al., 2010). This is consistent with the biochemical data establishing 5hmC as an intermediate to demethylated DNA, and suggests that impaired hydroxymethylation leads to increased DNA cytosine methylation in TET2-mutant AML patients. However, in a separate study, TET2 loss-of-function was associated with hypomethylation rather than hypermethylation at differentially methylated CpG sites (Ko et al., 2010). However there are important differences between these two studies; notably the study by Figueora and colleagues focused on a large, homogeneous clinical trial cohort of patients with a single disease entity, AML, and only included patient samples obtained at diagnosis before patients received any treatment. By contrast, the work by Koh and colleagues included a smaller, retrospective cohort of heterogeneous primary samples from patients with CMML, AML, MPN, MDS and MPN/MDS overlap syndromes. As such the discrepancies between these two studies likely reflect the differences between the patient cohorts in these two studies, and between the assays used to measure DNA methylation by the two groups. Given aberrant DNA methylation has been shown to be an important pathogenetic mechanism in MDS progressing to AML (Jiang et al., 2009) it is likely the effects of TET2 mutations on cytosine methylation will differ in different oncogenic contexts. Additional DNA methylation profile studies are required that combine TET2 mutational status, 5hmC measurements, and DNA methylation analyses in patients with MPN, MDS and AML.
Recent mutational studies in AML have revealed that TET2 mutations are mutually exclusive with somatic mutations in the isocitrate dehydrogenase genes, IDH1 and IDH2 (Figueroa et al., 2010). The IDH enzymes are NADP-dependent enzymes that normally function as homodimers to catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) with the concomitant production of NADPH (Figure 4). Somatic mutations affecting residue R132 of IDH1 are found in over 70% of secondary glioblastomas (Parsons et al., 2008; Yan et al., 2009); subsequent studies identified the same recurrent somatic mutation in AML patients(Mardis et al., 2009) Subsequent studies identified IDH2 mutations in glioma and AML patients including mutations at codon R172 in glioma/AML patients (Gross et al., 2010; Marcucci et al., 2010; Ward et al., 2010; Yan et al., 2009) and at R140 in patients with AML, notably IDH2R140 mutations are the most common IDH1/2 mutations seen in myeloid malignancies (Marcucci et al., 2010; Ward et al., 2010). Mutation of IDH1 R132 and IDH2 R172 residues along with the novel AML mutation in IDH2, R140, are predicted to interfere with the ability to bind the β-carboxyl of isocitrate, and the initial studies suggested that IDH1/2 mutations were purely loss-of-function disease alleles (Yan et al., 2009). However, subsequent studies demonstrated the loss in the ability of mutant IDH proteins to convert isocitrate to α-KG is replaced by a neomorphic enzymatic function. Specifically, the mutant IDH proteins catalyze the NADPH-dependent reduction of α-KG to 2-hydroxyglutarate (2-HG), a rare metabolite present at very low levels in normal cells. IDH mutant gliomas and AML cells accumulate 10-100-fold higher levels of 2-HG (Dang et al., 2009; Gross et al., 2010; Ward et al., 2010), which can also be detected in elevated quantities in the sera of IDH1/2 mutant AML patients (Gross et al., 2010).
Figure 4. Mutation of TET2 or IDH proteins is associated with myeloid malignancies and aberrant DNA methylation.
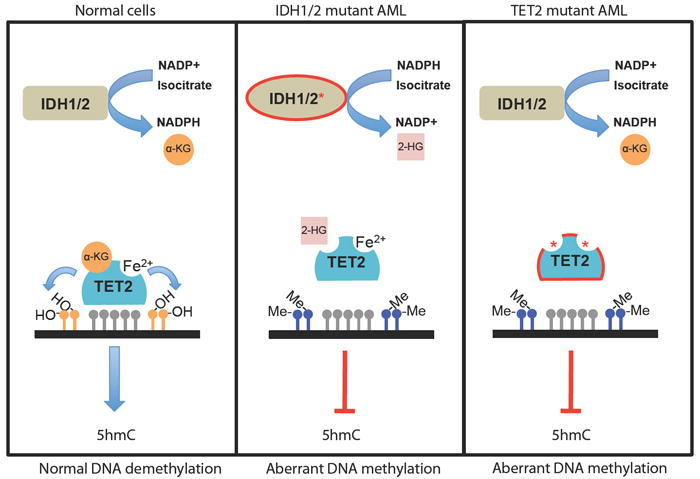
IDH proteins normally catalyze the conversion of NADP+ into NADPH and isocitrate into α-ketoglutarate (α-KG), a substrate required for normal TET2 enzymatic activity. Mutant IDH proteins cause loss of function of TET2 by exhibiting neomorphic enzymatic activity that results in the consumption of NADPH and production of the oncometabolite 2-hydroxyglutarate (2-HG) instead of α-KG. IDH mutations are mutually exclusive with TET2 mutations found in patients that exhibit reduced hydroxylase activity, due in part to mutations in the 2-OG and Fe(II)-dependant dioxygenase domain that cause decreased 5hmC production and aberrant DNA methylation patterns.
While these previous studies had shown that IDH mutations were associated with aberrant production of 2-hydroxyglutarate (2-HG) in AML and glioblastoma multiforme patients (Dang et al., 2009; Gross et al., 2010; Ward et al., 2010), the mechanism by which 2-HG production contributed to oncogenic transformation had not been elucidated. IDH1/2-mutant AML was also characterized by increased cytosine methylation, suggesting a convergent mechanism of transformation by TET2 and IDH1/2 mutant disease alleles in AML. Given the TET family of proteins require α-ketoglutarate for their enzymatic function, Figueora and colleagues then investigated whether IDH1/2-mutants could inhibit TET2 enzymatic activity, and demonstrated that co-expression of IDH-mutant alleles with wild-type TET2 led to impaired DNA hydroxymethylation (Figueroa et al., 2010). Subsequent enzymatic studies demonstrated that direct exposure to 2-HG directly inhibits TET2 enzymatic function (Xu et al., 2011a). In addition, in vitro studies showed that ectopic expression of IDH-mutant alleles or shRNA mediated depletion of TET2 led to similar effects on hematopoietic differentiation (Figueroa et al., 2010). These data demonstrate that a hypermethylation signature characterizes TET2-mutant and IDH-mutant leukemia, and that neomorphic IDH mutations and TET2 loss-of-function mutations converge on a shared mechanism of hematopoietic transformation characterized by impaired hydroxymethylation.
In vitro and in vivo models of Tet2 deficiency: Effects of Tet2 deletion on HSC differentiation and induction of leukemia
The identification of TET2 mutations in myeloid neoplasms suggested that TET2 function could be both an oncogenic trigger and a regulator of physiological hematopoiesis. As mentioned previously, mutational analysis suggested that the majority of these mutations severely affect enzymatic activity by either inhibiting gene expression or the function of essential catalytic domains of the protein. These studies underlined the need for models of Tet2 deficiency in the hematopoietic system, to directly address the function of this gene in both physiological differentiation and leukemic transformation.
Rao and colleagues initially used RNAi-mediated Tet2 silencing to address such questions. They reported that Tet2 knockdown in early hematopoietic progenitors affects myelopoiesis as it promotes expansion of monocyte-macrophage cells in the presence of G-CSF and GM-CSF, which promote granulocyte and granulocyte/monocyte differentiation respectively. No such effects were noted in the presence of M-CSF, a cytokine promoting the development of monocytic progenitors (Ko et al., 2010). More recently similar knock-down studies were performed using human progenitor cells (Pronier et al., 2011). TET2 silencing led to skewed differentiation towards the myelo-monocytic lineage, at the expense of lymphopoiesis and erythropoiesis. Interestingly, monocytic cell differentiation was specifically favored, providing a possible explanation for the high frequency of TET2 mutations in CMML, a disease associated with the expansion of the monocytic compartment.
A different outcome was reported in another recent study in which primary bone marrow cells were infected with viruses expressing either Tet2 shRNAs or mutant IDH2 molecules (thus affecting Tet2 enzymatic activity). After culturing the cells on both methylcellulose and liquid cultures, a higher percentage of more immature c-Kit+ cells were observed (Figueroa et al., 2010). Higher c-kit levels suggested that Tet2 silencing is able to suppress stem/progenitor cell differentiation and retain the cells in a more immature state. In agreement with these studies, Moran-Crusio and colleagues have shown that Tet2 silencing by shRNA and by genetic deletion not only increases the c-Kit+ fraction but increases the replating ability of the cells in vitro, suggesting direct effects on self-renewal (Moran-Crusio et al., 2011).
Although these studies provided valuable information of the role of Tet2, hematopoiesis and leukemogenesis are dynamic processes that can be optimally studied in vivo. Three recent studies have reported the generation and analysis of Tet2 deficient animals (Li, 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011). Interestingly, four different Tet2-/- models were described: a) a “gene-trapped” Tet2 allele targeting a lacZ/GFP cassette at the transcriptional start of Tet2, b) a “gene-trapped” allele targeting a lacZ cassette at the intron between exons 9 and 10, c) a conditional allele targeting the first (and larger) coding exon (exon 3) of the gene and d) a conditional allele targeting Exon 11 which encodes for the catalytic domain of the enzyme. Although one of the “gene-trapped” alleles resulted in a hypomorphic allele and different Cre recombinase-deleter strains were used for the deletion of the gene, all four mice led to highly similar phenotypes, underlining the reproducibility of the findings and mimicking the diverse mutational targets found in human disease.
Tet2 deletion led to the progressive enlargement of the stem/progenitor compartment (Lineage-Sca1+c-Kit+, LSK cells) however no compartment (defined by SLAM marker staining) was specifically enlarged. Strikingly, these mice developed significant extramedullary hematopoiesis, with the appearance of both LSK and myelo-erythroid progenitors in peripheral lymphoid organs. These include bona fide HSCs as shown by transplantation assays. Moreover, in addition to their relative enlargement in absolute numbers, Tet2-/- HSCs have increased self-renewing abilities as proven by both in vitro re-plating assays and in vivo competitive transplantation experiments. Indeed, Tet2-/- total bone marrow and purified LSK cells have the ability to serially replate. These cells express high levels of c-Kit and remain arrested at a common myeloid progenitor-like (CMP) stage. Whole-transcriptome analysis has shown that the replating pre-myeloblasts have downregulated expression of several genes characteristic of myeloid differentiation and upregulated expression of genes indicative of stem cell self-renewal (c-Kit, Evi1, Meis1). Interestingly, previous studies of DNA methylation in TET2WT and TET2MUT patient samples, identified the Evi1 promoter as differentially methylated in these two cell types, suggesting that Tet2-mediated differential methylation of specific gene-targets could lead to restrained stem cell self-renewal in normal HSPCs. Future studies that will map whole-genome Tet2, 5hmC and 5mC enrichment in distinct gene loci during HSC differentiation will identify which TET2-regulated genes are in fact direct Tet2 targets. We should mention here that in all three studies Tet2 deletion led to the loss of 5hmC consistent with the suggested enzymatic activity of the protein.
One of the most intriguing findings was the progressive development of myeloid disease, in all the animal models, starting at 4-6 months after the deletion of the gene. Although there were slight differences in the disease onset and the kinetics of progression, most likely due to different backgrounds and efficiency/mode of gene deletion, Tet2 deficient mice developed myeloid neoplasms and predominantly CMML-like disease in agreement with the high prevalence of TET2 mutations in human CMML (Abdel-Wahab et al., 2009). Indeed, Tet2-/- mice developed progressive leukocytosis, splenomegaly, neutrophilia, blood monocytosis and myeloid dysplasia. Although CMML-like disease was the most predominant, other types of neoplasms, including MDS with erythroid predominance and MPD-like disease were detected in a fraction of the animals (Li et al., 2011). These studies demonstrated that Tet2 functions as a bona fide tumor suppressor and its deletion is sufficient to initiate myeloid transformation. However, the kinetics of disease induction suggests that additional genetic lesions can co-operate with Tet2 loss to induce leukemia. Indeed, Tet2 mutations co-occur with a wide range of genetic lesions in both myeloid leukemia (Klinakis et al., 2011) and lymphocytic neoplasms (Quivoron et al., 2011). It is intriguing to speculate that these additional genetic lesions not only co-operate with Tet2 but also influence the generation of distinct types of blood neoplasms. Further experiments are required to address these issues.
Finally, all three studies demonstrated that Tet2+/- mice also develop similar types of disease albeit with slower kinetics. These findings suggest a haploinsufficient tumor suppressor function for Tet2 and are clinically important as the majority of patients carry heterozygous TET2 mutations or deletions. However, Tet2+/- mice develop disease with longer latency and milder phenotype suggesting gene-dosage effects. Interestingly all three TET family proteins are expressed in hematopoietic progenitors and deletion of Tet2 does not lead to the upregulation of the expression of Tet1 and Tet3. It is thus startling that reduction of the dosage of one of the three genes (Tet2) is sufficient to induce significant 5hmC changes, a skewing in hematopoietic differentiation and development of myeloid neoplasms. Further studies should focus on the relative redundancy between the three proteins and putative interactions between them in developing hematopoietic progenitor cells.
TET protein function and role in the differentiation of other stem cell types
Germline deletion of Tet2 leads exclusively to hematopoietic defects suggesting that Tet2 plays a specific role in HSC differentiation. However, to fully understand the role of Tet2 in other adult stem cell compartments further studies are required, targeting specific stem cell types such as neural stem cells, as substantial amounts of 5hmC have been detected in brain tissue (Guo et al., 2011; Kriaucionis and Heintz, 2009) where Tet2 is also expressed (Lorsbach et al., 2003).
Although the study of TET protein function is at its infancy there is a significant amount of evidence suggesting their importance during embryonic stem (ES) cell differentiation. Tet1 (and Tet2) and are both expressed in ES cells and silencing of Tet1 affects self-renewal and maintenance (Ito et al., 2010). Ito and colleagues reported that Tet1 silencing leads to methylation of the Nanog promoter, resulting in loss of Nanog expression and concomitant effects in ESC differentiation. In the same study, Tet1 was shown to be expressed in pre-implantation embryos and play an essential role in the specification of the cells of the inner cell mass (ICM). A more recent study further supported the idea that Tet1 function is associated with ESC pluripotency, and suggested that Tet1 and Tet2 transcription itself is regulated by the master regulator of ESC self-renewal, Oct4, by directly binding to 5’UTR regulatory regions (Koh et al., 2011). This same study demonstrated an altered developmental potential of Tet1-depleted ESCs, as their differentiation is skewed towards the trophectoderm fate, which is accompanied by the increased expression of Cdx2, Eomes and Elf2 and decreased expression of the Nodal antagonists Lefty1 and Lefty2. These and other studies indicated that 5hmC exists at significant levels in self-renewing ESCs and its levels decline during differentiation (Szwagierczak et al., 2010; Tahiliani et al., 2009; Xu et al., 2011c). Tet1 appears to be an important regulator of 5hmC availability as its depletion leads to a significant reduction of global 5hmC levels in ESCs (Ito et al., 2011; Koh et al., 2011; Xu et al., 2011c). Furthermore, it was shown the Tet1 regulates CpG methylation at promoters of specific genes that could be essential mediators of ESC differentiation. Intriguingly, last month in Cell Stem Cell, Jaenisch and colleagues report the conditional deletion of Tet1 in ESCs and early embryogenesis, and questio the validity of the previous in vitro studies. As expected, Tet1-/- ESCs have reduced levels of global 5hmC. However, the cells do not lose pluripotency upon Tet1 deletion, express near-physiological levels of Nanog, Oct4 and Sox2 and are able to support organism development (Dawlaty et al., 2011). Indeed, Tet1-/- animals are born at normal Mendelian ratios and are fertile. In vitro, Tet1-/- ESCs display a skewing towards trophectoderm, in agreement with Koh et al., however this phenotype is not recapitulated in vivo during embryogenesis. These important data, together with the generation of Tet2-/- germline knockouts (Li, 2011; Moran-Crusio et al., 2011; Quivoron et al., 2011) seriously question in vitro findings suggesting that the “safest” way to study effects on ESC pluripotency is to actually target the gene using homologous-recombination-based approaches. Further studies, including generation of combined Tet gene knockouts are required to assess their role on DNA methylation and early embryogenesis.
Conclusions
How both ESCs and HSCs utilize TET proteins and 5hmC modifications as they differentiate from pluripotent stem cells into adult cells is of great interest and under intense study. One of the key upcoming challenges will be the detailed understanding of TET family function in hematopoietic stem and progenitor cells and the continued identification of TET binding partners and target genes responsible for establishment of leukemia. Blood malignancies have been at the forefront of our understanding of the epigenetic regulation of cell transformation and targeting aberrant DNA methylation using hypomethylating agents as an alternative to conventional chemotherapy is already underway.
Acknowledgments
We would like to thank the members of the Aifantis and Levine labs for critical reading of the manuscript. IA is supported by the National Institutes of Health (RO1CA133379, RO1CA105129, R21CA141399, RO1CA149655, RO1GM088847), the Leukemia & Lymphoma Society (TRP grant), the American Cancer Society (RSG0806801), the V Foundation for Cancer Research, the Irma T. Hirschl Trust, the Chemotherapy Foundation, the Dana Foundation and the Alex’s Lemonade Stand Foundation. LC is supported by a NHMRC biomedical research fellowship. OAW is a Basic Research Fellow of the American Society of Hematology. This work was supported in part by grants from the National Institutes of Health (U54CA143798-01, Physical Sciences Oncology Center) and 1R01CA138234-01 to RLL, and by grants from the Starr Cancer Consortium and Howard Hughes Medical Institute to RLL. RLL is a Geoffrey Beene Junior Faculty Chair at Memorial Sloan Kettering Cancer Center. I.A is a Howard Hughes Medical Institute Early Career Scientist.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Wahab O, Manshouri T, Patel J, Harris K, Yao J, Hedvat C, Heguy A, Bueso-Ramos C, Kantarjian H, Levine RL, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010;70:447–452. doi: 10.1158/0008-5472.CAN-09-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397:579–583. doi: 10.1038/17533. [DOI] [PubMed] [Google Scholar]
- Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty MM, G K, Powell BE, Hu Y, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi S, Page DC, Jaenisch R. Tet1 is dispensible for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, Racevskis J, Dewald GW, Ketterling RP, Bennett JM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361:1249–1259. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M, Reik W, Henikoff S. DNA demethylation by DNA repair. Trends Genet. 2009;25:82–90. doi: 10.1016/j.tig.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, Lee C, Almouzni G, Schneider R, Surani MA. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science. 2011 doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science. 2011 doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, Ramsahoye B. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, O’Keefe CL, Ganetzky R, McDevitt MA, Maciejewski JP. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113:6403–6410. doi: 10.1182/blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C, Sekeres M, Saunthararajah Y, Maciejewski JP. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113:1315–1325. doi: 10.1182/blood-2008-06-163246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39:5015–5024. doi: 10.1093/nar/gkr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachatryan V, Sirunyan AM, Tumasyan A, Adam W, Bergauer T, Dragicevic M, Ero J, Fabjan C, Friedl M, Fruhwirth R, et al. Search for stopped Gluinos in pp collisions at square root s=7 TeV. Phys Rev Lett. 2011;106:011801. doi: 10.1103/PhysRevLett.106.011801. [DOI] [PubMed] [Google Scholar]
- Klinakis A, Lobry C, Abdel-Wahab O, Oh P, Haeno H, Buonamici S, van De Walle I, Cathelin S, Trimarchi T, Araldi E, et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473:230–233. doi: 10.1038/nature09999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmider O, Delabesse E, Mas VM, Cornillet-Lefebvre P, Blanchet O, Delmer A, Recher C, Raynaud S, Bouscary D, Viguie F, et al. TET2 mutations in secondary acute myeloid leukemias: a French retrospective study. Haematologica. 2011;96:1059–1063. doi: 10.3324/haematol.2011.040840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, Viguie F, Quesnel B, Beyne-Rauzy O, Solary E, et al. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs) Blood. 2009a;114:3285–3291. doi: 10.1182/blood-2009-04-215814. [DOI] [PubMed] [Google Scholar]
- Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N, Quesnel B, Fenaux P, Bastie JN, Beyne-Rauzy O, et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica. 2009b;94:1676–1681. doi: 10.3324/haematol.2009.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- Lee J, Inoue K, Ono R, Ogonuki N, Kohda T, Kaneko-Ishino T, Ogura A, Ishino F. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development. 2002;129:1807–1817. doi: 10.1242/dev.129.8.1807. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Li Z, Cai X, Cai C, Wang J, Zhang W, Peterson BE, Yang F, Xu M. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stemm Cells and Subsequent Development of Myeloid Malignancies. Blood. 2011 doi: 10.1182/blood-2010-12-325241. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, Curfman J, Holland KB, Schwind S, Whitman SP, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzel M, Globisch D, Bruckl T, Wagner M, Welzmiller V, Michalakis S, Muller M, Biel M, Carell T. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem Int Ed Engl. 2010;49:5375–5377. doi: 10.1002/anie.201002033. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- Nibourel O, Kosmider O, Cheok M, Boissel N, Renneville A, Philippe N, Dombret H, Dreyfus F, Quesnel B, Geffroy S, et al. Incidence and prognostic value of TET2 alterations in de novo acute myeloid leukemia achieving complete remission. Blood. 2010;116:1132–1135. doi: 10.1182/blood-2009-07-234484. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23) Cancer Res. 2002;62:4075–4080. [PubMed] [Google Scholar]
- Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J. Active demethylation of the paternal genome in the mouse zygote. Curr Biol. 2000;10:475–478. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn NW, Suwalski R, O’Riley C, Bojanowski K, Yura R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem J. 1972;126:781–790. doi: 10.1042/bj1260781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, Jacobsen SE, Reik W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, Masse A, Le Couedic JP, Pendino F, Carbonne B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulo-monocytic differentiation of human hematopoietic progenitors. Blood. 2011 doi: 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivoron C, Couronne L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, Do Cruzeiro M, Delhommeau F, Arnulf B, Stern MH, et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J, Robertson AB, Klungland A. The presence of 5-hydroxymethylcytosine at the gene promoter and not in the gene body negatively regulates gene expression. Biochem Biophys Res Commun. 2011 doi: 10.1016/j.bbrc.2011.06.077. [DOI] [PubMed] [Google Scholar]
- Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schafer A, Grummt I, Mayer C. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell. 2009;33:344–353. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Szwagierczak A, Bultmann S, Schmidt CS, Spada F, Leonhardt H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010;38:e181. doi: 10.1093/nar/gkq684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Pardanani A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Gangat N, Finke CM, Schwager S, Mullally A, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23:905–911. doi: 10.1038/leu.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S, Sakaue M, Matsuoka C, Shimotohno K, Ishikawa F, Li E, Ueda HR, et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006;11:805–814. doi: 10.1111/j.1365-2443.2006.00984.x. [DOI] [PubMed] [Google Scholar]
- Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkowski A, Maciejewski P, Wasek W, Malek LA, Niewada M, Kaminski B, Drzewiecki J, Kosmider M, Kubica J, Ruzyllo W, et al. Influence of different antiplatelet treatment regimens for primary percutaneous coronary intervention on all-cause mortality. Eur Heart J. 2009;30:1736–1743. doi: 10.1093/eurheartj/ehp114. [DOI] [PubMed] [Google Scholar]
- Wu H, D’Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011a;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011b;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Tet1 and 5-hydroxymethylation: A genome-wide view in mouse embryonic stem cells. Cell Cycle. 2011;10 doi: 10.4161/cc.10.15.16930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011a;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011b;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, et al. Genome-wide Regulation of 5hmC, 5mC, and Gene Expression by Tet1 Hydroxylase in Mouse Embryonic Stem Cells. Mol Cell. 2011c doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang X, Clark E, Mulcahey M, Huang S, Shi YG. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010;20:1390–1393. doi: 10.1038/cr.2010.156. [DOI] [PubMed] [Google Scholar]
- Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–166. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]