

Abstract
The light absorption in light-harvesting complexes is performed by molecules such as chlorophyll, carotenoid, or bilin. Recent experimental findings in some of these complexes suggest the existence of long-lived coherences between the individual pigments at low temperatures. In this context the question arises if the bath-induced fluctuations at different chromophores are spatially correlated or not. Here we investigate this question for the Fenna-Matthews-Olson (FMO) complex of Chlorobaculum tepidum by a combination of atomistic theories, i.e., classical molecular dynamics simulations and semi-empirical quantum chemistry calculations. In these investigations at ambient temperatures, only weak correlations between the movements of the chromophores can be detected at the atomic level and none at the more coarse-grained level of site energies. The often employed uncorrelated bath approximations indeed seems to be valid. Nevertheless, correlations between fluctuations in the electronic couplings between the pigments can be found. Depending on the level of theory employed, also correlations between the fluctuations of site energies and the fluctuations in electronic couplings are discernable.

Introduction
Photosynthesis certainly is one of the key processes of energy transformations on earth. Light is absorbed by individual pigments and its energy is converted into chemical energy. Most photosynthetic systems contain so-called light-harvesting (LH) complexes which collect light and funnel it to the reaction centers.1 Over the past years and even decades, the dynamics of light harvesting systems has been elucidated in detail.2-4 Already in 1985 high-resolution structures of a photosynthetic reaction center became available.5 Examples of further available crystal structures include LH26,7 and LH18 complexes of purple bacteria, LHCII of higher plants9 and several others (see, e.g., Ref. 4). All these structures show that the chromophores are held fixed at their positions by a protein scaffold and reveal many details underlying the absorption and transfer processes in these aggregates. Nevertheless, how and to what extent the very efficient energy transfer is characterised by the structures of the LH complexes and their motions is still an open discussion.
Recent experiments suggest that excitonic coherence is protected by the protein environment3,10-13 and enhance interest in these systems even further.4,14 The surprising feature of the quantum coherent energy transfer is that coherence survives for several hundreds of femtoseconds in a complex biological system. By now several studies have investigated in detail the effect of the environment on quantum coherence in terms of quantum efficiency,15 noise-assisted transport,16,17 entanglement18 and other non-classical effects.19 It has been suggested that long-lived coherence is due to correlations of the fluctuations of the site energies.20 Recently, Womick et al. suggested that, e.g., for allophycocyanin, long-lived electronic coherence requires tuning of the protein environment. 21,22
In DNA, spatial site correlation has been shown to have a drastic effect on transport properties. 23,24 The effect of such correlation on 2D spectra has been studied earlier already, e.g., in Refs. 25 and 26. In multichromophore complexes such as the Fenna-Matthews-Olson (FMO) light-harvesting complex, strong correlation between protein-induced fluctuations in the site energies has been suggested as the source of the experimentally observed coherence beatings.3,10,11,13,20-22 Alternative explanations, namely that the long-lived coherence originates from interference of different quantum pathways, has been put forward recently.27 Concerning correlated fluctuations, Nazir28 investigated the influence of correlations on a donor-acceptor system for strong system-bath coupling. Using a Lindblad approach the influence of correlation on the trapping probability in ring systems was investigated by Fassioli et al.29 It was found that correlation might play a role in tuning the trapping probability. Surprising effects of correlation in the site energy fluctuations were also shown in a study by Nalbach et al30 on a toy model. Furthermore, it has been shown that in a light harvesting complex of purple bacteria, LH2, spatial correlations would modify the optical and transport properties significantly.31 Because of all these possible implications of spatial correlation, we investigate in the present study if such correlation can be observed in simulations at physiological, i.e. ambient, temperatures. This might offer insight into whether spatial correlations are actually important for the biological function of light-harvesting systems.
The specific system under investigation here is the FMO complex of the green sulfur bacterium Chlorobaculum tepidum.32 The optical active entities are bacteriochlorophyll a (BChl a) molecules. In green sulfur bacteria the chlorosomes are the main light-harvesting antennae. The excitation transfer between these chlorosomes and the membrane-embedded reaction center is mediated by the FMO trimer, i.e., the system of interest here. Milder et al.33 have recently reviewed the optical properties of FMO complexes together with the experimental and theoretical approaches comprehensively.33
Arrangements, conformational motions and electronic ground state properties of whole light-harvesting systems or even complexes thereof can be simulated using classical molecular dynamics (MD).34 To obtain optical properties these classical simulation have to be coupled to electronic structure calculations.35-40 In a subsystem-based approach the individual chromophores are usually treated separately. The electronic coupling between the subsystems is determined subsequently. Because of the size of the BChl molecules and the large number of vertical transition energy calculations required along an MD trajectory to capture the quantum mechanical behavior, one often employs the semi-empirical Zerner’s Intermediate Neglect of Differential Orbital method with parameters for spectroscopic properties (ZINDO/S).38-43 We recently tested this method for a LH2 system39 together with the TrEsp approach for the electronic couplings. TrEsp is the abbreviation for the method of transition charges from electrostatic potentials44,45 which was applied to different light-harvesting systems46 before.
Molecular Dynamics
As preparation for the MD simulations, both the monomer and trimer FMO systems, consisting of the pigment-protein complexes, ions and water, were constructed from the crystal structure of Chlorobaculum tedium (PDB code: 3ENI).32 After adding hydrogens to the crystal structures, each pigment-protein complex was embedded in a TIP3P48 water box. The water boxes were sized such that there was a 15 Å distance from the boundaries of each pigment-protein complex to the edge of the water box. Sodium and chloride ions were added to each water box to bring the total ionic concentrations for each system to 0.1 mol/L. System size, atom counts and number of ions are summarized in Table 1. In the monomer calculations the eighth BChl was not associated well and started separating from the structure. Therefore these simulations were only performed with seven BChls.
Table 1.
Summary of simulated systems
| FMO Monomer | |
|---|---|
| Box size: | 100.7×72.5 × 84.1 Å3 |
| # Atoms: | 57 377 |
| # Na+: | 15 |
| # Cl−: | 16 |
|
| |
| FMO Trimer | |
|
| |
| Box size: | 119.7×115.4 × 102.4 Å3 |
| # Atoms: | 133 604 |
| # Na+: | 34 |
| # Cl−: | 37 |
MD simulations were carried out using NAMD249 with the CHARMM27 force field.50,51 BChl parameters used were those reported in Ref. 35. TIP3P water hydrogens were constrained using the SHAKE algorithm.52 Periodic boundary conditions were employed in the simulation together with the particle mesh Ewald (PME) method53,54 for electrostatic summations. Subsequent to an energy minimization, each system was equilibrated for 10 ns at 300 K and 1 atm pressure as an NPT ensemble using 1 fs time steps. Thereafter a production run was carried out with 1 fs time steps for 300 ps. The atomic coordinates from every 5 fs of the 300 ps production run were used for the subsequent QM calculations.
Site energy and electronic coupling calculations
In subsystem-based quantum approaches one calculates the ground and excited state energies of the individual subsystems. Together with electronic couplings (see below) these lead to a time-dependent Hamiltonian. The energy differences which are referred to here as site energies were calculated as reported in Ref. 39 for LH2 of Rhodospirillum (Rs.) molischianum. The ORCA code (University of Bonn, Germany)55 was employed in order to calculate the energy gap between ground and first excited, i.e. the Qy, state for all BChls in the complex at each snapshots. To make the calculations efficient and because the optical properties of BChls are determined by a cyclic conjugated π-electron system, we restricted the quantum system to a truncated structure of the BChl molecule. To this end, each terminal CH3 and CH2CH3 group as well as the pythyl tail were replaced by H atoms.38,56 As a compromise between accuracy and computational efficiency the semiempirical ZINDO/S-CIS(10,10) method was employed using the ten highest occupied and the ten lowest unoccupied states in the configuration interaction description. For a similar system, this approach was employed before.37,38 Point charges stemming from the MD simulations within a cutoff radius of 20 Å around the truncated BChl molecule were included in the ZINDO/S-CIS calculations. In this way one can account for the main effects of surrounding environments on orbital energies.
In addition to describing the electronic excitations of the individual BChls, one needs to determine the coupling among excitations. In this regard, we followed Ref. 39. In the TrEsp approach44,45 the transition density of pigment m is described using atomic transition charges that are localized at the respective pigment, i.e., it is described through where denotes the coordinates of the Ith atom of BChl m. The coupling between two pigment molecules is then given by
| (1) |
The atomic partial charges can be calculated using different electronic structure methods. In Ref. 44 these charges are given for BChl a and Chl a molecules in their planar structures calculated using HF-CIS and TDDFT/B3LYP. The charges calculated by the latter method are used in the present study with a scaling factor of 0.732 to match the size of the experimental dipole moment of 6.3 Debye.57 Furthermore, it is assumed that the charges do not change with varying molecular geometry and, therefore, we employ them for the calculation along the MD trajectory. This approximation is quite plausible since the molecular geometries in an equilibrium MD simulation should fluctuate around the equilibrium positions. To account for solvent effects on the electronic couplings a distance-dependent screening factor f is introduced.39,58
Correlations of atomic motions
In the present study we aim at analyzing the role of correlated atomic fluctuations. The established method to quantify the respective correlations from MD simulation is an extension of the Pearson coefficient to the multidimensional case.59-61 For simplicity we will denote, in the following, this extension as simply the Pearson coefficient. In this approach one considers the positional fluctuations, i.e., the deviation from the respective mean values, x = r − ‹r›. The multidimensional variant of the Pearson coefficient is then defined as the normalized covariance matrix of the fluctuations . This coefficient varies between ± 1, i.e., between maximally positive and negative correlated motions. Though often employed, this correlation measure has several drawbacks.61 The Pearson coefficient is based on the assumption of collinear motion with unit variance and an underlying Gaussian distribution. Furthermore it only detects linear correlation since it is a measure for the quality of the best linear fit.
In contrast to the Pearson coefficient, the generalized correlation coefficient CMI, developed by Lange and Grubmüller,61 is based on the mutual information between atomic fluctuations. This measure has, for example, been applied to analyze correlations for the transporter BtuB.62 The generalized correlation is able to detect correlated motion regardless of the relative orientation and includes nonlinear contributions. CMI captures also the correlation between two atoms fluctuating sinusoidally, but out of phase. For the calculation of the generalized correlation coefficient CMI the g_correlation software developed by Lange and Grubmüller with a density estimator nearest neighbor parameter k = 6 was employed.61 Furthermore, the linearized version of CMI is computed below which is restricted to linear correlations like the Pearson coefficient, but is not based on collinear motions.
Correlation analysis using the different measures was performed on 300 ps equilibrium trajectories for the FMO monomer and 200 ps trajectories for the trimer. Saving frames every 5 fs, 60 000 and 40 000 states were taken into account for the FMO monomer and trimer, respectively. To remove all rotational and translational motions, the conformation of the protein in each frame was aligned to that of the initial frame. Furthermore, to ensure that correlation values are converged and do not show spurious effects,63 calculations were performed for trajectory lengths of 100 ps, 200 ps, and for the monomer also for 300 ps. No significant differences for the correlations were observed for the different trajectory lengths. All figures shown below were obtained using the longest available trajectory length. Hydrogen atoms were not included in this analysis which was restricted to the cores of the BChls as discussed above.
Figure 2 shows the absolute value of the Pearson coefficients for the FMO monomer. The seven blocks on the diagonal show the atomic correlations within each BChl. As mentioned above only a core BChl has been treated including 27 non-hydrogen atoms. Within each BChl strong correlations are visible. Between the different BChls the atomic correlation is maximally 0.4 with the apparently largest values for correlations between atoms in BChls 1 and 2. Fig. S1 shows the same property for the simulation of the trimer with very similar results.
Figure 2.
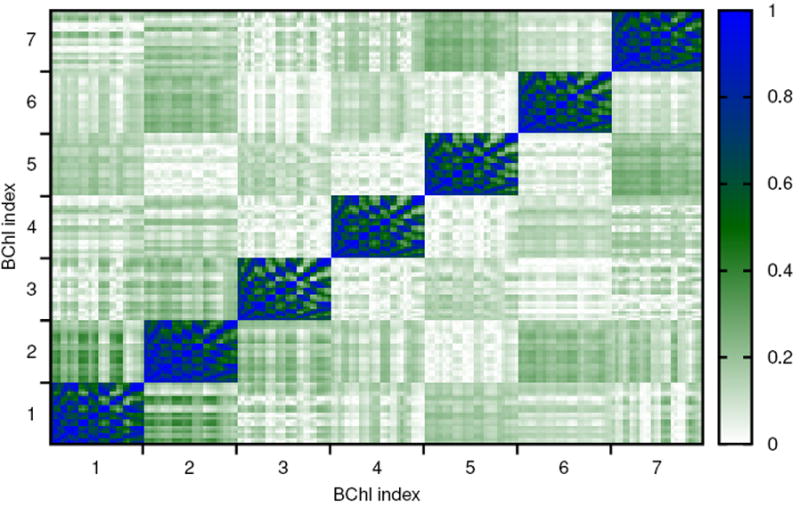
Correlation of atomic motion between the different BChls with 27 atoms per BChl determined using the absolute value of the Pearson coefficient.
As discussed above, the multidimensional Pearson coefficient has several deficiencies. Therefore we also use the generalized correlation coefficient to analyze atomic correlations in FMO. In Figure 3 the generalized correlation coefficient is shown together with its linear approximation. Naturally strong correlations within the individual BChls can be seen. At the same time the minimal values of the correlations, both for the generalized coefficient and its linearized version, are much higher than for the absolute value of the Pearson coefficient. Again somewhat higher correlations are seen for atoms in BChls 1 and 2. The trimer version of these results are shown in Fig. S2, being very similar to the monomer case.
Figure 3.
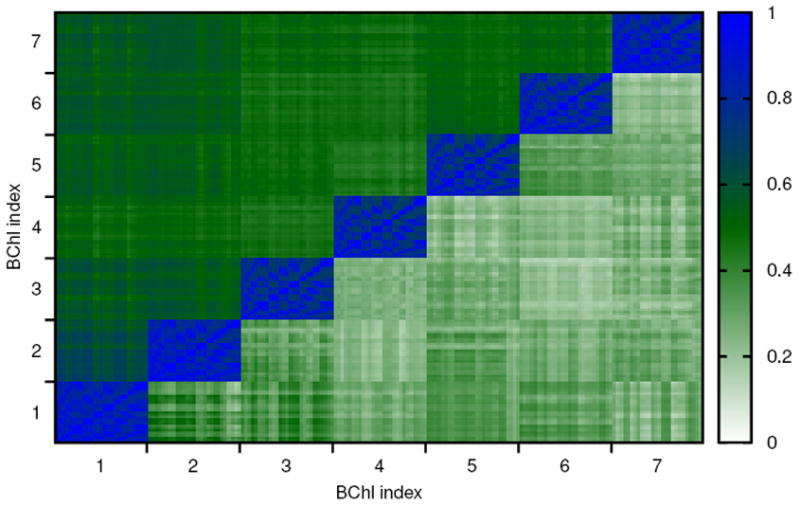
Correlation of atomic motion between different sites with 27 atoms per BChl using the generalized correlation coefficient rMI (upper triangle) as well as its linear approximation (lower triangle).
To understand why the generalized correlation CMI coefficients seem to show larger correlations than the CP even for distant atoms, we plot in Figure 4 the same data as before, not using atom indices, but atom pair distances. This time the Pearson coefficient is shown including its sign. For distances above 10 Å mainly fluctuations around zero arise. For the generalized correlation coefficient the situation is slightly different. This coefficient is larger or equal to zero by definition, i.e., fluctuations around zero are impossible. Furthermore, this measure has been defined by rescaling an information theoretical measure, which varies between zero and infinity, to vary between zero and one; the rescaling is achieved by using the highly nonlinear exponential function which in this case very much overemphasizes small correlations. The data shown in Figure 4 belong to the trimer simulations. The results for the monomer are shown in Fig. S3. Neither the generalized coefficient nor its linearization show much change in the range from 10 Å to 60 Å. At these latter distances it can safely be assumed that no important correlations are present. From this we conclude that there is also no correlation at 10 Å as well. Though the non-linear correlation coefficient shows values of about 0.5, these values do not indicate large correlations between atoms, but are merely artifacts of the positive-definiteness and the highly non-linear scaling in the definition of these generalized coefficients. For an improved generalized coefficient the “background” correlation would have to be taken into account. Nevertheless, the generalized coefficients also show enhanced correlations between atoms in BChl 1 and 2.
Figure 4.
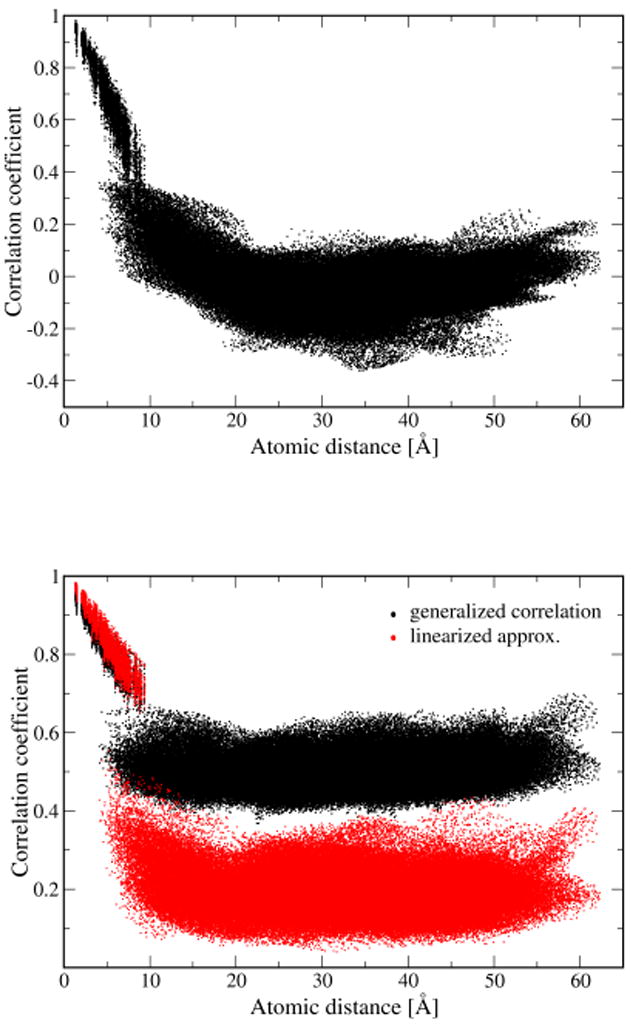
Correlation of atomic motions between different BChls determined using the Pearson approach (top), the generalized correlation coefficient and its linearized approximation (bottom).
Correlations of site energies and couplings
To see if correlations might explain the observed long-lived coherence in the 2D spectra,20 we need to investigate the electronic system, namely site energies and couplings. In direct analogy to the formulas used for atomic correlations, correlations between site energies, between electronic couplings and between site energies and couplings can be determined using the Pearson coefficient, the generalized correlation coefficient and its linearized variant. Since the latter two do not yield new information in the present case, we only show below the results for the one-dimensional Pearson coefficient. As one example of site energy correlations, in the case of individual nucleobases in DNA, a correlation of 0.7 between neighbors and 0.4 between second neighbors was found.23 In Figure 5 the results for the present monomer system are shown in the lower left corner. The values on the diagonal are by definition equal to one. Significant correlation cannot be discerned for in the correlation matrix for BChls 1 and 2, though some of the atom pairs between these BChls showed enlarged correlation coefficients. Apparently the observed atomic correlation is not strong enough to show correlation between the electronic properties. The results for the trimer in Fig. S4 look similar; naturally, the number of combinations is much larger in this case.
Figure 5.
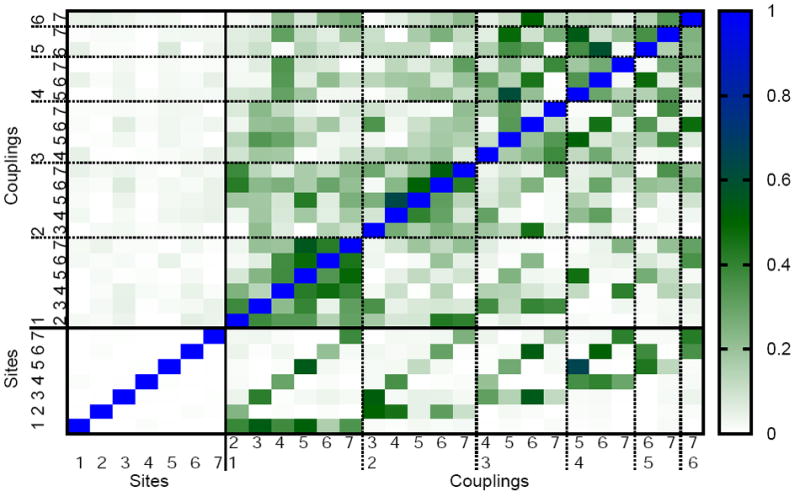
Absolute value of the Pearson coefficient for correlations between site energies and couplings of the monomer system. The lower left corner shows correlations between site energies. The lower triangle of the whole matrix corresponds to couplings determined using the point dipole approximation while the upper triangle is based on TrEsp calculations. The numbers 1 to 7 correspond to the BChls in the FMO monomer.
Despite the observed lack of correlation between site energies, there is still the possibility of correlation between the corresponding electronic couplings. As given in Eq. (1) the electronic coupling depends on positions of individual atoms and, therefore, atomic correlations can lead to spatial correlation in the electronic couplings. Corresponding results are given in Figure 5 for the monomer and in Fig. S4 for the trimer. The couplings have either been determined using the point dipole approximation or the TrEsp approach. In either approach correlations are discernable. The actual degree of correlation depends on the method applied.
It was recently proposed that mixed correlations between fluctuations in site energies and fluctuations in couplings may enhance electronic coherences as seen, for example, in 2D spectra.64 Such mixed correlations are seen in Figure 5. While couplings from the point dipole approximation are substantial for some pairs, couplings based on TrEsp are negligble. The largest correlations between couplings appear for cases in which the two BChl pairs share a common partner. For example, there is a rather large correlation between couplings 4-5 and 5-7 for the TrEsp results, i.e., if pigment 5 is moving, this imposes a change in the coupling between pigments 4 and 7 and, therefore, causes a correlation between the two couplings. Though there appear to be areas of larger correlations in the correlation matrix, these represent rather local correlations. Large correlations appear for movements of BChl 5 since there are, in addition to the just mentioned pair, correlations between the coupling pair 3-5 and 4-5. Also the movement of pigment 2 leads to correlations in the coupling pairs 2-4 and 2-5, 2-5 and 2-6, 2-6 and 2-7.
One has to keep in mind that the correlation matrix does not reveal the absolute values of the couplings involved, i.e., many of the correlations might be unimportant since one of the two couplings might be very small. Furthermore, the size of fluctuations might be rather small and unimportant for dynamical and spectroscopic properties. For example, there is a rather large correlation between the couplings 4-5 and 5-6 for the TrEsp results. This correlation is also very prominent in the weighted correlation matrix Fig. S5. If pigment 5 is moving, this imposes a change in the couplings to pigments 4 and 6 at the same time. Therefore a correlation between both couplings is present. Fig. S5 shows the correlation of the couplings as in Figure 5 but weighted by the widths of the two corresponding coupling distributions. This weighting factor is indicative of their importance. A more detailed analysis is needed in order to clarify if the correlations described here can explain long-lived coherences as observed in experiment. Work in this direction is in progress.
Conclusions
This study focused on spatial correlations in geometrical and electronic properties of the FMO complex based on atomistic simulations. It was motivated by experimental evidence for excitonic coherences in the system.3,10-12 It had been suggested that the observed long-lived coherence is due to correlation between fluctuations of the site energies.20 However, we found only weak atomic correlations in our present study. Only BChls 1 and 2 exhibit somewhat significant correlation in their atomic motion, but not in the fluctuation of their site energies.
The present study was performed at room temperature while the initial experiments finding of long-lived coherence were performed at 77 K.10 Recently experiments have been extended to temperatures of 125 K, 150 K, and 277 K;13 beating signals were seen at all the temperatures.13 The comparison between present results and the reported experimental findings is difficult. It seems to be clear, though, that site correlations do not play a role at physiological conditions and that the biological function of the FMO complex is not affected by spatial site energy correlations. A similar conclusion has already been drawn for the light-harvesting II complex of Rhodospirillum molischianum in a similar study.39 Whether correlations in the couplings or whether an alternative mechanisms27 are responsible for the long-lived coherences is unknown so far.
In our approach we first performed a classical equilibrium MD simulation and then carried out electronic structure calculations employing the trajectory data, i.e., we never really do simulations involving excited states of BChls. The excitation process leads to a redistribution of charges and to dipole moment changes of the excited BChls which has influence the movements of nuclei, i.e., the MD part. However, in contrast to the case of charge transport, e.g., in DNA,23,24 the charge state of the BChls stays the same. Therefore the present results should not be significantly influenced by the missing back-reaction of the electronic onto the nuclear part of our description.
The present combination of MD and quantum chemistry can also be used to derive the so-called spectral density.35,39 The latter quantity is a crucial input parameter for theories of dissipative quantum dynamics. Many such calculations have been performed for light-harvesting systems, see e.g., Refs. 12,15-17,31,65-68. Calculations such as the one presented here help to make a direct connection between atomistic simulations and models of dissipative excitation dynamics.
Supplementary Material
Figure 1.
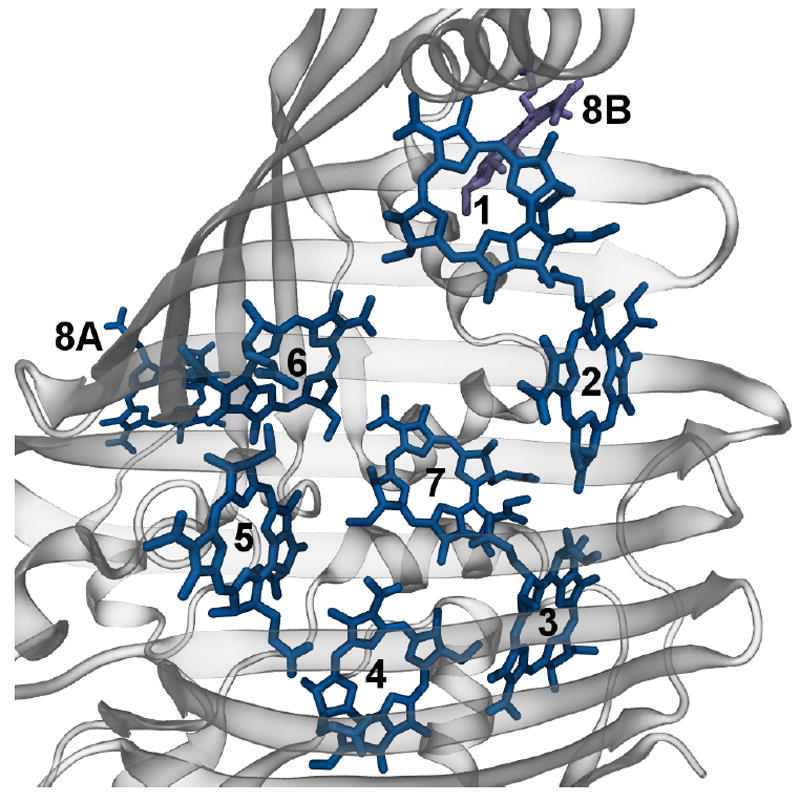
Monomer of the FMO trimer. Shown are the eight BChls of one monomer together with nearby BChl 8B of a neighbouring monomer. The protein structure is shown in cartoon representation. Figure drawn using VMD.47
Acknowledgments
This work has been supported by the Deutsche Forschungsgemeinschaft (DFG), the National Institute of Health (NIH) and the National Science Foundation (NSF). Funding for J.S. and K.S. was provided by NSF grants MCB-0744057, PHY0822613 and NIH grant P41-RR05969.
Footnotes
Supporting Information Available: Atomic and site-energy/coupling correlation matrices of the trimer simulation and weighted correlation matrix of couplings for FMO monomer. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hu X, Ritz T, Damjanović A, Autenrieth F, Schulten K. Photosynthetic apparatus of purple bacteria. Q Rev Biophys. 2002;35:1–62. doi: 10.1017/s0033583501003754. [DOI] [PubMed] [Google Scholar]
- 2.Cogdell RJ, Gall A, Köhler J. The architecture and function of the light-harvesting apparatus of purple bacteria: from single molecules to in vivo membranes. Q Rev Biophys. 2006;39:227–324. doi: 10.1017/S0033583506004434. [DOI] [PubMed] [Google Scholar]
- 3.Cheng YC, Fleming GR. Dynamics of light harvesting in photosynthesis. Annu Rev Phys Chem. 2009;60:241–242. doi: 10.1146/annurev.physchem.040808.090259. [DOI] [PubMed] [Google Scholar]
- 4.Novoderezhkin VI, van Grondelle R. Physical origins and models of energy transfer in photosynthetic light-harvesting. Phys Chem Chem Phys. 2010;12:7352–7365. doi: 10.1039/c003025b. [DOI] [PubMed] [Google Scholar]
- 5.Deisenhofer J, Epp O, Miki K, Huber R, Michel H. Structure of the protein subunits in the photosynthetic reaction centre of Rhodopseudomonas viridis at 3 Å resolution. Nature. 1985;318:618–624. doi: 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- 6.McDermott G, Prince SM, Freer AA, Hawthornthwaite-Lawless AM, Papiz MZ, Cogdell RJ, Isaacs NW. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature. 1995;374:517–521. [Google Scholar]
- 7.Koepke J, Hu X, Muenke C, Schulten K, Michel H. The crystal structure of the light harvesting complex II (B800-850) from Rhodospirillum molischianum. Structure. 1996;4:581–597. doi: 10.1016/s0969-2126(96)00063-9. [DOI] [PubMed] [Google Scholar]
- 8.Roszak AW, Howard TD, Southall J, Gardiner AT, Law CJ, Isaacs NW, Cogdell RJ. Crystal structure of the RC-LH1 core complex from Rhodopseudomonas palustris. Science. 2003;302:1969–1972. doi: 10.1126/science.1088892. [DOI] [PubMed] [Google Scholar]
- 9.Kuhlbrandt W, Wang DN, Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 10.Engel GS, Calhoun TR, Read EL, Ahn TK, Mancal T, Cheng YC, Blankenship RE, Fleming GR. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature. 2007;446:782–786. doi: 10.1038/nature05678. [DOI] [PubMed] [Google Scholar]
- 11.Collini E, Wong CY, Wilk KE, Curmi PM, Brumer P, Scholes GD. Coherently wired light-harvesting in photosynthetic marine algae at ambient temperature. Nature. 2010;463:644–647. doi: 10.1038/nature08811. [DOI] [PubMed] [Google Scholar]
- 12.Ishizaki A, Calhoun TR, Schlau-Cohen GS, Fleming GR. Quantum coherence and its interplay with protein environments in photosynthetic electronic energy transfer. Phys Chem Chem Phys. 2010;12:7319–7337. doi: 10.1039/c003389h. [DOI] [PubMed] [Google Scholar]
- 13.Panitchayangkoon G, Hayes D, Fransted KA, Caram JR, Harel E, Wen J, Blankenship RE, Engel GS. Long-lived quantum coherence in photosynthetic complexes at physiological temperature. Proc Natl Acad Sci USA. 2010;107:12766–12770. doi: 10.1073/pnas.1005484107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholes GD. Quantum-Coherent Electronic Energy Transfer: Did Nature Think of It First? J Phys Chem Lett. 2010;1:2–8. [Google Scholar]
- 15.Olaya-Castro A, Lee CF, Olsen FF, Johnson NF. Efficiency of energy transfer in a light-harvesting system under quantum coherence. Phys Rev B. 2008;78 085115. [Google Scholar]
- 16.Caruso F, Chin AW, Datta A, Huelga SF, Plenio MB. Highly efficient energy excitation transfer in light-harvesting complexes: The fundamental role of noise-assisted transport. J Chem Phys. 2009;131 105106. [Google Scholar]
- 17.Rebentrost P, Mohseni M, Aspuru-Guzik A. Role of Quantum Coherence and Environmental Fluctuations in Chromophoric Energy Transport. J Phys Chem B. 2009;113:9942–9947. doi: 10.1021/jp901724d. [DOI] [PubMed] [Google Scholar]
- 18.Sarovar M, Ishizaki A, Fleming GR, Whaley KB. Quantum entanglement in photosynthetic light-harvesting complexes. Nature Physics. 2010;6:462–467. [Google Scholar]
- 19.Fleming GR, Huelga S, Plenio M. Focus on Quantum Effects and Noise in Biomolecules. New J Phys. 2010;12 065002. [Google Scholar]
- 20.Wolynes PG. Some quantum weirdness in physiology. Proc Natl Acad Sci USA. 2009;106:17247–17248. doi: 10.1073/pnas.0909421106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Womick JM, Moran AM. Exciton Coherence and Energy Transport in the Light-Harvesting Dimers of Allophycocyanin. J Phys Chem B. 2009;113:15747–15759. doi: 10.1021/jp907644h. [DOI] [PubMed] [Google Scholar]
- 22.Womick JM, Miller SA, Moran AM. Toward the origin of exciton electronic structure in phycobiliproteins. J Chem Phys. 2010;133 doi: 10.1063/1.3457378. 024507. [DOI] [PubMed] [Google Scholar]
- 23.Kubař T, Kleinekathöfer U, Elstner M. Solvent Fluctuations Drive the Hole Transfer in DNA: a Mixed Quantum-Classical Study. J Phys Chem B. 2009;113:13107–13117. doi: 10.1021/jp9073587. [DOI] [PubMed] [Google Scholar]
- 24.Dijkstra AG, Tanimura Y. Correlated fluctuations in the exciton dynamics and spectroscopy of DNA. New J Phys. 2010;12 055005. [Google Scholar]
- 25.Venkatramani R, Mukamel S. Correlated line broadening in multidimensional vibrational spectroscopy. J Chem Phys. 2002;117:11089–11101. [Google Scholar]
- 26.Ishizaki A, Tanimura Y. Dynamics of a multimode system coupled to multiple heat baths probed by two-dimensional infrared spectroscopy. J Phys Chem A. 2007;111:9269–9276. doi: 10.1021/jp072880a. [DOI] [PubMed] [Google Scholar]
- 27.Abramavicius D, Mukamel S. Quantum oscillatory exciton migration in photosynthetic reaction centers. J Chem Phys. 2010;133 doi: 10.1063/1.3458824. 064510. [DOI] [PubMed] [Google Scholar]
- 28.Nazir A. Correlation-Dependent Coherent to Incoherent Transitions in Resonant Energy Transfer Dynamics. Phys Rev Lett. 2009;103 doi: 10.1103/PhysRevLett.103.146404. 146404. [DOI] [PubMed] [Google Scholar]
- 29.Fassioli F, Nazir A, Olaya-Castro A. Quantum State Tuning of Energy Transfer in a Correlated Environment. J Phys Chem Lett. 2010;1:2139–2143. [Google Scholar]
- 30.Nalbach P, Eckel J, Thorwart M. Quantum coherent biomolecular energy transfer with spatially correlated fluctuations. New J Phys. 2010;12 065043. [Google Scholar]
- 31.Strümpfer J, Schulten K. The Effect of Correlated Bath Fluctuations on Exciton Transfer. (submitted) 2010 doi: 10.1063/1.3557042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tronrud DE, Wen J, Gay L, Blankenship RE. The structural basis for the difference in absorbance spectra for the FMO antenna protein from various green sulfur bacteria. Photosynth Res. 2009;100:79–87. doi: 10.1007/s11120-009-9430-6. [DOI] [PubMed] [Google Scholar]
- 33.Milder MT, Brüggemann B, van Grondelle R, Herek JL. Revisiting the optical properties of the FMO protein. Photosynth Res. 2010;104:257–264. doi: 10.1007/s11120-010-9540-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sener MK, Strümpfer J, Timney JA, Freiberg A, Hunter CN, Schulten K. Photosynthetic Vesicle Architecture and Constraints on Efficient Energy Harvesting. Biophys J. 2010;99:67–75. doi: 10.1016/j.bpj.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Damjanović A, Kosztin I, Kleinekathöfer U, Schulten K. Excitons in a Photosynthetic Light-Harvesting System: A Combined Molecular Dynamics, Quantum Chemistry and Polaron Model Study. Phys Rev E. 2002;65 doi: 10.1103/PhysRevE.65.031919. 031919. [DOI] [PubMed] [Google Scholar]
- 36.Walker RC, Mercer IP, Gould IR, Klug DR. Comparison of basis set effects and the performance of ab initio and DFT methods for probing equilibrium fluctuations. J Comput Chem. 2007;28:478–480. doi: 10.1002/jcc.20559. [DOI] [PubMed] [Google Scholar]
- 37.Zwier MC, Shorb JM, Krueger BP. Hybrid molecular dynamics-quantum mechanics simulations of solute spectral properties in the condensed phase: evaluation of simulation parameters. J Comput Chem. 2007;28:1572–1581. doi: 10.1002/jcc.20662. [DOI] [PubMed] [Google Scholar]
- 38.Janosi L, Kosztin I, Damjanović A. Theoretical prediction of spectral and optical properties of bacteriochlorophylls in thermally disordered LH2 antenna complexes. J Chem Phys. 2006;125 doi: 10.1063/1.2210481. 014903. [DOI] [PubMed] [Google Scholar]
- 39.Olbrich C, Kleinekathöfer U. Time-dependent atomistic view on the electronic relaxation in light-harvesting system II. J Phys Chem B. 2010;114:12427–12437. doi: 10.1021/jp106542v. [DOI] [PubMed] [Google Scholar]
- 40.Olbrich C, Liebers J, Kleinekathöfer U. Modeling of light-harvesting in purple bacteria using a time-dependent Hamiltonian approach. phys stat sol (b) 2010 in press. [Google Scholar]
- 41.Ridley J, Zerner MC. An intermediate neglect of differential overlap technique for spectroscopy: Pyrrole and the azines. Theor Chim Acta. 1973;32:111–134. [Google Scholar]
- 42.Damjanović A, Vaswani HM, Fromme P, Fleming GR. Chlorophyll Excitations in Photosystem I of Synechococcus elongatus. J Phys Chem B. 2002;106:10251–10262. [Google Scholar]
- 43.Linnanto J, Korppi-Tommola J. Quantum chemical simulation of excited states of chlorophylls, bacteriochlorophylls and their complexes. Phys Chem Chem Phys. 2006;8:663–667. doi: 10.1039/b513086g. [DOI] [PubMed] [Google Scholar]
- 44.Madjet ME, Abdurahman A, Renger T. Intermolecular coulomb couplings from ab initio electrostatic potentials: application to optical transitions of strongly coupled pigments in photosynthetic antennae and reaction centers. J Phys Chem B. 2006;110:17268–81. doi: 10.1021/jp0615398. [DOI] [PubMed] [Google Scholar]
- 45.Renger T. Theory of excitation energy transfer: from structure to function. Photosynth Res. 2009;102:471–485. doi: 10.1007/s11120-009-9472-9. [DOI] [PubMed] [Google Scholar]
- 46.Madjet ME, Müh F, Renger T. Deciphering the influence of short-range electronic couplings on optical properties of molecular dimers: application to “special pairs” in photosynthesis. J Phys Chem B. 2009;113:12603–14. doi: 10.1021/jp906009j. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey WF, Dalke A, Schulten K. VMD – Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 48.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 49.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foloppe N, MacKerell AD., Jr All-Atom Empirical Force Field for Nucleic Acids: 2) Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J Comp Chem. 2000;21:86–104. [Google Scholar]
- 51.MacKerell A, et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 52.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 53.Ewald PP. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann Phys (Berlin) 1921;369:253–287. [Google Scholar]
- 54.Darden T, Perera L, Li L, Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999:55–60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- 55.Petrenko T, Neese F. Analysis and prediction of absorption band shapes, fluorescence band shapes, resonance Raman intensities, and excitation profiles using the time-dependent theory of electronic spectroscopy. J Chem Phys. 2007;127 doi: 10.1063/1.2770706. 164319. [DOI] [PubMed] [Google Scholar]
- 56.Cory MG, Zerner MC, Hu X, Schulten K. Electronic excitations in aggregates of bacteriochlorophylls. J Phys Chem B. 1998;102:7640–7650. [Google Scholar]
- 57.Alden RG, Johnson E, Nagarajan V, Law WWPJ, Cogdell RG. Calculations of Spectroscopic Properties of the LH2 Bacteriochlorophyll: Protein Antenna Complex from Rhodopseudomonas acidophila. J Phys Chem B. 1997;101:4667–4680. [Google Scholar]
- 58.Scholes GD, Curutchet C, Mennucci B, Cammi R, Tomasi J. How Solvent Controls Electronic Energy Transfer and Light Harvesting. J Phys Chem B. 2007;111:6978–6982. doi: 10.1021/jp072540p. [DOI] [PubMed] [Google Scholar]
- 59.Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11:205–207. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 60.Hünenberger PH, Mark AE, van Gunsteren WF. Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J Mol Biol. 1995;252:492–503. doi: 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]
- 61.Lange OF, Grubmüller H. Generalized correlation for biomolecular dynamics. Proteins. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 62.Luan B, Carr R, Caffrey M, Aksimentiev A. The effect of calcium on the conformation of cobalamin transporter BtuB. Proteins. 2010;78:1153–1162. doi: 10.1002/prot.22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meinhold L, Smith JC. Fluctuations and correlations in crystalline protein dynamics: a simulation analysis of staphylococcal nuclease. Biophys J. 2005;88:2554–2563. doi: 10.1529/biophysj.104.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen X, Silbey RJ. Effect of correlation of local fluctuations on exciton coherence. J Chem Phys. 2010;132 doi: 10.1063/1.3435211. 204503. [DOI] [PubMed] [Google Scholar]
- 65.Heřman P, Kleinekathöfer U, Barvík I, Schreiber M. Exciton scattering in light-harvesting systems of purple bacteria. J Lumin. 2001;94&95:447–450. [Google Scholar]
- 66.Heřman P, Kleinekathöfer U, Barvík I, Schreiber M. Influence of Static and Dynamic Disorder on the Anisotropy of Emission in the Ring Antenna Subunits of Purple Bacteria Photosynthetic Systems. Chem Phys. 2002;275:1–13. [Google Scholar]
- 67.Kleinekathöfer U, Barvík I, Heřman P, Kondov I, Schreiber M. Memory effects in the fluorescence depolarization dynamics studied within the B850 ring of purple bacteria. J Phys Chem B. 2003;107:14094–14102. [Google Scholar]
- 68.Strümpfer J, Schulten K. Light harvesting complex II B850 excitation dynamics. J Chem Phys. 2009;131 doi: 10.1063/1.3271348. 225101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.