

Abstract
Motivation: RNA sequencing (RNA-Seq) is a powerful new technology for mapping and quantifying transcriptomes using ultra high-throughput next-generation sequencing technologies. Using deep sequencing, gene expression levels of all transcripts including novel ones can be quantified digitally. Although extremely promising, the massive amounts of data generated by RNA-Seq, substantial biases and uncertainty in short read alignment pose challenges for data analysis. In particular, large base-specific variation and between-base dependence make simple approaches, such as those that use averaging to normalize RNA-Seq data and quantify gene expressions, ineffective.
Results: In this study, we propose a Poisson mixed-effects (POME) model to characterize base-level read coverage within each transcript. The underlying expression level is included as a key parameter in this model. Since the proposed model is capable of incorporating base-specific variation as well as between-base dependence that affect read coverage profile throughout the transcript, it can lead to improved quantification of the true underlying expression level.
Availability and implementation: POME can be freely downloaded at http://www.stat.purdue.edu/~yuzhu/pome.html.
Contact: yuzhu@purdue.edu; zhaohui.qin@emory.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The transcriptome is the complete set of transcripts in a cell under any given developmental stage or physiological condition. Comprehensively cataloging all the components in the transcriptome is a grand challenge in the post-genome era. In the past decade, microarray technology has played a prominent role in advancing our understanding of transcriptome complexity by allowing scientists to simultaneously monitor the expression of almost all the genes in the genome (Lockhart et al., 1996; Schena et al., 1995). Despite its overwhelming success, microarray technology has its limitations. For example, designing probes on the chip requires knowledge of the genome sequence and annotation; hence, novel transcripts will be missed. Additionally, cross-hybridization, background signal and saturation result in a reduction of microarray's dynamic range and accuracy.
A recently developed sequencing-based technology for measuring gene expression, termed RNA-Seq, has the potential to overcome these limitations (Mortazavi et al., 2008; Wang et al., 2009). The ultra-high-throughput next-generation sequencing technologies capable of producing millions of sequence reads dramatically increase the throughput in DNA sequencing compared with conventional Sanger technology and at a much lower cost. An array of studies has been published that successfully apply these new sequencing technologies to measure mRNA expression levels in cells from various species (Cloonan et al., 2008; Lister et al., 2008; Maher et al., 2009; Marioni et al., 2008; Morin et al., 2008; Mortazavi et al., 2008; Nagalakshmi et al., 2008; Prensner et al., 2011; Trapnell et al., 2010; Wilhelm et al., 2008). Since Illumina's platforms have been widely adopted in RNA-Seq experiments, we focused on short read RNA-Seq data generated from the Illumina platform in this study.
In RNA-Seq experiments using the aforementioned sequencing technologies, RNA molecules are first converted to a library of cDNA fragments with adaptors attached to both ends. Each molecule, often after amplification, is then sequenced using one of the next-generation sequencing technologies. Following sequencing, the resulting reads are aligned to known transcripts or de novo assembled together to produce a genome-scale transcriptional profile.
A fundamental question in RNA-Seq data analysis is to derive expression level from raw sequencing output data. This is the basis of almost all further investigation such as detecting differentially expressed genes, alternative splicing events, etc. Sophisticated and tailor-made data analysis methods are needed to fully realize the power of the new sequencing technologies.
A natural idea is to use the total number of reads mapped inside each transcript, or region of interest (ROI), to represent the expression level. Mortazavi et al. (2008) proposed to use the number of reads per kilobase of a transcript per million mapped reads (RPKM) as the transcript's expression level. The RPKM method is easy to implement and takes into consideration the transcript length and the total number of uniquely mapped reads. However, the RPKM method is oversimplified, as it ignores the variability of read coverage within an ROI demonstrated by real RNA-Seq data. More sophisticated methods are required to account for the complexity and uncertainties associated with read mapping and read depth within an ROI. In that regard, model-based methods can potentially improve upon the RPKM method by explicitly modeling the varying sequencing read coverage within an ROI. A number of models have been proposed in the literature, such as Poisson model (Marioni et al., 2008), generalized Poisson model (Srivastava and Chen, 2010) and negative binomial model (Bullard et al., 2010). However, these models are not sophisticated enough to capture all the variability demonstrated in the observed RNA-Seq data. For example, they do not consider variation in sequencing read coverage within an ROI resulted from local genomic feature and fluctuation in the base-level PCR amplification rate. To accommodate this type of variation, Li et al. (2010) proposed to use Poisson distribution with varying intensity rate to model read counts covering different positions in an ROI and developed a method called mseq to quantify its expression level. The mseq method utilizes neighborhood sequence information and can better explain base-level read counts variation in RNA-Seq data.
Despite improvement over other existing methods, the mseq method assumes that the observed base-level read counts are stochastically independent with each other in an ROI. Analyzing real data indicated that this assumption is not valid in a substantial proportion of the ROIs. We found that in highly expressed transcripts, between-base or spatial dependence is not negligible (see Section 3). Ignoring this dependence may lead to less accurate estimation of the true expression levels.
The presence of location-specific variation along with between-location correlation is an outstanding characteristic of many spatial data generated in geostatistics, spatial epidemiology and image processing, and has been studied in the literature of spatial statistics (Best et al., 2005; Diggle et al., 1998; Wakefield, 2007). Typically, Poisson mixed-effects (POME) models are used to analyze such spatial count data. In this study, in order to model base-specific read coverage while accounting for their dependency simultaneously, we apply the spatial POME model to characterize transcript level RNA-Seq data.
2 METHODS
2.1 POME model
Let Yjk represent the number of reads whose mapping starts at the j-th base of a specified transcript in the k-th sample. Here j=1,…,n, n is the length of the transcript, and k=1,…,m, m is the number of samples. The definition of Yjk is the same as in mseq (Li et al., 2010). Further let θk represent the expression index of the transcript in the k-th sample, which is of primary interest. Our goal is to build a statistical model to capture the base-specific variation and between-base correlation in Yjk. This idea is motivated by the model-based expression index (MBEI) model proposed by Li and Wong (2001) to model probe-level microarray gene expression data.
In order to avoid over-fitting using a complex model, we propose the following POME model for Yjk :
| (1) |
In addition to the fixed effect θk , which is the expression index that is of primary interest, there are two random effect terms in this model: Ujk 's and Vjk 's. As in spatial statistics, Vjk 's are assumed to be independent and identically distributed as N(0, σv2), and used to account for unstructured variability, which may be attributed to some latent factors for over-dispersion; Ujk 's are used to represent the correlation between the read counts of different base pairs. Like in spatial statistics, there are various ways to specify this correlation structure. In POME, we chose the intrinsic conditional autoregressive (ICAR) structure (Besag, 1974). The ICAR structure specifies between-base correlation using a Gaussian Markov random field. Originally, the ICAR structure was proposed for image processing and was later used for disease mapping in spatial epidemiology (Clayton and Kaldor, 1987). For a fixed base j, first we define its neighborhood, denoted by ∂j as the collection of the two bases j−1 and j+1 that are adjacent to base j. Other definitions of neighborhood are possible, for example, bases that are not immediately adjacent to base j can be included in ∂j (Cressie and Chan, 1989). Second, we define a weight matrix W=(wij) as follows. For 1≤i, j≤n, wii=0; wij=1 if i∈∂j; and wij=0 otherwise. Let U(−j)k denote the collection of Uik 's with i≠j. The conditional distribution of Ujk given U(−j)k is assumed to be:
| (2) |
The ICAR structure induces correlation between different bases.
Another option for the correlation structure is the joint structure (Wakefield, 2007). The joint structure assumes that in the k-th sample, Ujk 's follow the multivariate Gaussian distribution N(0, σu2Σ), where Σ=(σij ) is the correlation matrix of Ujk 's and σij=ρdij, 0<ρ<1, dij is the distance or the number of base pairs between the i-th base and the j-th base, i.e. dij=| i−j|. When the ICAR structure in POME is replaced by the joint structure, the resulting model is referred to as the joint model.
The discussion about the pros and cons of the joint versus ICAR structure can be found in the disease mapping literature (Best et al., 2005; Wakefield, 2007). We choose the ICAR structure in POME, because read coverage for some transcripts may be sparse and the excessive zero counts pose a challenge for the joint model. POME is flexible in that covariates or deterministic patterns that affect θk can be incorporated into the POME model in a straightforward fashion.
In the literature on disease mapping, Markov chain Monte Carlo (MCMC) techniques (Gilks et al., 1998; Liu, 2001) are the predominant methods used for fitting the POME model and performing subsequent statistical inference, following Besag et al. (1991) and Diggle et al. (1998). In the literature on image processing, however, maximum likelihood methods are also used for model fitting and inference; see Zhu et al. (2009) for example. The preference for Bayesian computational methods is due to the fact that the random effects Ujk 's and Vjk 's are not directly observable; and it takes high-dimensional integration to integrate them out, which can be computationally challenging. In this study, following the tradition in disease mapping, we use Bayesian computational methods when applying the POME model for transcript level RNA-Seq data analysis.
2.2 Model implementation
The MCMC methods are employed to carry out the fitting of the POME model and subsequent statistical inference. We start from assigning appropriate priors for the model parameters. The marginal distributions of the random effects Ujk and Vjk are N(0, σu2) and N(0, σv2), respectively. Following the approach by Wakefield (2007), we define the total precision as τ=(σu2+σv2)−1, and specify a Gamma prior for it, which is τ~Γ(a, b). Let p=σu2/(σu2+σv2) represent the proportion of base-specific variation in the total variation. We assign a Beta prior Beta(c, d) to p.
If the joint structure is assumed for Ujk 's, there is another parameter ρ determining the extent of between-bases correlation. We assign another Beta prior Beta(e,f) to ρ. A non-informative prior I(0, +∞ ) is assigned for θk.
As the default, we specify c=d=e=f=1 so that the priors for p and ρ are uniform distributions on [0, 1]. The total precision τ=(σu2+σv2)−1 plays a crucial role in the model and the final result is sensitive to the prior assigned to τ. We adopt an empirical Bayesian method to further specify a and b in the prior Γ(a, b) for τ by setting a=1 and 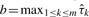 (Section 1 in Supplementary Material).
(Section 1 in Supplementary Material).
It is straightforward to derive the joint posterior distribution involving all the fixed and random effects or parameters (Section 2 in Supplementary Material), and we use Gibbs sampler to iteratively sample parameters from the conditional posterior distributions. Since we use the conjugate priors, the conditional posterior distributions for θk and τ are Gamma distributions, which are easy to sample from. The conditional posterior distributions for random effects Ujk 's and Vjk 's are rather complicated and not in closed forms. Since both are log-concave functions, we use the adaptive rejection sampling (ARS) method (Gilks and Wild, 1992) to draw samples from them. For the other parameters, p and ρ, their conditional posterior distributions are nearly log-concave. We use the adaptive rejection Metropolis-Hasting sampling (ARMS) technique (Gilks et al., 1995) to draw samples from these complicated distributions. One important issue in using ARS and ARMS is to assign appropriate ranges for the parameters. In ARS, we use the interval [-5, 5] for random effects Ujk 's and Vjk 's. For the parameters p and ρ, we use their natural range [0, 1] in ARMS. In addition, to make the fixed parameter θk identifiable, we impose the following two constrains on the random effects in each iteration of the Gibbs sampler:
| (3) |
When fitting the POME model, we ran 10 000 MCMC iterations for each transcript. The first 9000 samplers were dropped as the burn-in stage, and then every 10th sample in the last 1000 samplers were used to calculate the posterior means.
3 RESULTS
3.1 Data description
To evaluate the performance of POME, we choose a published real RNA-Seq data, obtained from 12 prostate cancer cell lines and tissues (Sam et al., 2011). More details about the samples can be found in the Supplementary Table S1. A brief description of the reads mapping procedure used by Sam et al. (2011) is provided in Section 3 in the Supplementary Material. We choose this dataset because these samples were profiled using two types of sequencing instruments with different technologies: Illumina Genome Analyzer using sequencing by synthesis technology and Helicos HeliScope using single-molecule sequencing technology. The Helicos technology represents a new wave of next-generation sequencing technologies in which samples were profiled directly without the polymerase chain reaction (PCR) step, thus eliminating the overrepresentation or underrepresentation biases introduced by the copying process of PCR, a necessary step in Illumina sequencing technology. Given this, we use the expression measure obtained from Helicos as the gold standard when evaluating expression measures reported by different algorithms analyzing RNA-Seq data generated by the Illumina platform.
Transcript-level expression measures from Helicos and mapped Illumina reads information are kindly provided by the Chinnaiyan Lab. The details of the involved data processing procedures can be found in Sam et al. (2011). We plotted log 2 mean versus log 2 variance of the Illumina read coverage of these transcripts in all 12 cell lines (Supplementary Figure S1). We observed that the variance of read count is much larger than the mean, indicating the presence of substantial over-dispersion, especially for highly expressed transcripts. This is consistent with the observation of Li et al. (2010). Furthermore, we explored the top 500 most highly expressed transcripts according to the gold standard Helicos measures, of which 439 also satisfied the minimum read coverage criterion in the Illumina platform (>0.3 RPKM noise level used in the Sam et al. 2011 study) and minimum effective length criterion (with more than 100 non-zero yjk's). When examining these transcripts in the LnCaP_0 sample (Supplementary Table S1), we found that the median of the lag one autocorrelations between base-level read counts of these transcripts is 0.11, the third quartile is 0.18 and the maximum correlation is 0.57. We checked on other samples and observed similar patterns. These findings indicate that dependence between read counts of adjacent base pairs is real and needs to be considered.
3.2 Simulation study
3.2.1 Simulated data from the joint POME model
We conducted a simulation study to compare the performance of the POME method against the commonly used methods in quantifying transcript-level gene expression: RPKM (Mortazavi et al., 2008), mseq (Li et al., 2010) and GPseq (Srivastava and Chen, 2010).
We simulated the read coverage profiles from the spatial POME model with the joint correlation structure, i.e. the joint model which is different from POME. To make our simulation study more realistic, we first used the joint model to fit observed read count data in the 439 transcripts of high expression levels. We then sampled putative sequencing coverage profiles for these transcripts from the joint model. The proportion of base-specific variation in the total variation p was drawn randomly from a uniform distribution defined on the interval (0.1, 0.9).
The simulation was repeated 100 times. For each dataset, we applied RPKM, mseq, GPseq and POME, and calculated the mean square errors (MSEs) of the four resulting estimates of expression index θ.
For all 439 transcripts, the POME method achieved the smallest MSE (1.2024, standard error 0.0379). The MSEs reported by RPKM, mseq and GPseq were much larger: 65.4605 (standard error 4.2217), 87.1559 (standard error 23.3511) and 16.0718 (standard error 0.0692), respectively. The RPKM method overestimated the true expression level when data showed strong over-dispersion. On the other hand, mseq used 40 bp in the neighborhood of each nucleotide as local sequence features that affect the base-level read coverage rate, which may have caused it to over fit the data. Although GPseq is capable of modeling both over-dispersion and under-dispersion patterns in the data, it does not take into account the spatial dependence between adjacent base pairs that may result in less accurate estimates.
3.2.2 Simulated data from the generalized Poisson model
Next we conducted another simulation study with a different simulation strategy. We simulated the read coverage data from the generalized Poisson distribution GP(θ, λ). We again used the 439 highly expressed transcripts in the previous simulation study, and fitted a generalized Poisson model using GPseq (Srivastava and Chen, 2010) to obtain the empirical estimates  and
and 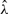 in each transcript.
in each transcript.
We then simulated the read coverage profiles from GP( ,
, 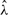 for all 439 transcripts. We applied the RPKM method, mseq and the POME method to estimate θ, separately. We did not apply GPseq in this simulation study since the data were simulated from the generalized Poisson distribution. The simulation was repeated 100 times, and the MSEs of the three resulting estimates of θ were calculated.
for all 439 transcripts. We applied the RPKM method, mseq and the POME method to estimate θ, separately. We did not apply GPseq in this simulation study since the data were simulated from the generalized Poisson distribution. The simulation was repeated 100 times, and the MSEs of the three resulting estimates of θ were calculated.
For all 439 transcripts, the MSEs achieved by POME, mseq and RPKM were 20.6736 (standard error 0.4633), 160.7138 (standard error 33.5309) and 166.1414 (standard error 7.9313), respectively. The results were similar to what we have obtained in the previous simulation study. Although the data were simulated from generalized Poisson distribution with over-dispersion, POME nevertheless was able to provide more accurate estimate of θ through explicitly modeling the position-specific variation.
3.3 Real data analysis
We next analyzed real RNA-Seq data in the 12 prostate cancer samples. Since highly expressed transcripts often display high level of over-dispersion, we selected the 5000 most highly expressed transcripts according to Helicos measures in each sample and removed those that show extreme low read coverage in Illumina [<0.3 RPKM noise level as in the Sam et al. 2011 study]. The numbers of corresponding transcripts measured by Illumina sequencing were listed in Table 1.
Table 1.
Comparison of Spearman's correlation coefficients between four different expression measures (RPKM, mseq, GPseq and POME) in 12 real datasets
| Sample | Na | RPKM | mseq | GPseq | POME |
|---|---|---|---|---|---|
| LnCaP_0 | 4964 | 0.6246 | 0.6155 | 0.5265 | 0.6887 |
| LnCaP_24 | 4956 | 0.6186 | 0.6035 | 0.5466 | 0.6728 |
| LnCaP_48 | 4946 | 0.6001 | 0.5955 | 0.4964 | 0.6407 |
| VCaP_0 | 4939 | 0.5801 | 0.5607 | 0.5822 | 0.6046 |
| VCaP_24 | 4948 | 0.5988 | 0.5474 | 0.5936 | 0.6234 |
| VCaP_48 | 4941 | 0.6222 | 0.4577 | 0.6129 | 0.6569 |
| aT34 | 4869 | 0.5789 | 0.5668 | 0.4351 | 0.5958 |
| aT34N | 4747 | 0.4281 | 0.4245 | 0.4624 | 0.3944 |
| DU145F | 4947 | 0.5945 | 0.5793 | 0.4575 | 0.6350 |
| DU145F2 | 4943 | 0.5939 | 0.5783 | 0.4608 | 0.6263 |
| VCaP | 4944 | 0.5204 | 0.4024 | 0.4398 | 0.5175 |
| RWPE | 4969 | 0.5600 | 0.5514 | 0.4762 | 0.6013 |
In each dataset, the method with the highest Spearman's correlation coefficient is highlighted in bold.
aThe number of transcripts measured in Illumina sequencing.
We applied RPKM (Mortazavi et al., 2008), mseq (Li et al., 2010), GPseq (Srivastava and Chen, 2010) and POME as before. For mseq, we used 40 bp in the neighborhood of each nucleotide, and used the top 500 highly expressed transcripts as the training dataset. To avoid complication of missing data due to unmappable regions or dubious annotation, we removed all positions with zero coverage from each transcript in the data pre-processing step.
We used Helicos measure as the gold standard and compared the Spearman's rank correlation coefficients between the Helicos measure and the estimates of transcript-level gene expression generated by the four tested methods, respectively.
Table 1 shows the overall performances of the four tested methods. POME achieved the highest Spearman's correlation coefficients in 10 of the 12 samples except for sample ‘aT34N’ and sample ‘VCaP’, where GPseq and RPKM were the best, respectively.
To further investigate the differences between POME and the other three competing methods, we focused on a subset of transcripts with high over-dispersion and high spatial dependence. The magnitudes of over-dispersion and spatial dependence were measured by variation-to-mean ratio (also called ‘Fano factor’) and lag one autocorrelation between base-level reads count, respectively. In each sample, around 1500 transcripts were selected with both above-median over-dispersion and above-median spatial dependence. The numbers of selected transcripts in each sample were listed in Table 2.
Table 2.
Comparison of Spearman's correlation coefficients between four different expression measures (RPKM, mseq, GPseq and POME) on transcripts with high over-dispersion and high spatial dependence in 12 real datasets
| Sample | Na | RPKM | mseq | GPseq | POME |
|---|---|---|---|---|---|
| LnCaP_0 | 1495 | 0.6454 | 0.6232 | 0.6071 | 0.7362 |
| LnCaP_24 | 1534 | 0.6248 | 0.5990 | 0.5828 | 0.7118 |
| LnCaP_48 | 1518 | 0.6059 | 0.5963 | 0.5152 | 0.6795 |
| VCaP_0 | 1525 | 0.6641 | 0.6452 | 0.6496 | 0.7106 |
| VCaP_24 | 1523 | 0.6259 | 0.5783 | 0.6691 | 0.6841 |
| VCaP_48 | 1500 | 0.6354 | 0.5094 | 0.6820 | 0.7022 |
| aT34 | 1563 | 0.6705 | 0.6470 | 0.5214 | 0.6914 |
| aT34N | 1476 | 0.6322 | 0.6268 | 0.6203 | 0.6523 |
| DU145F | 1525 | 0.6254 | 0.6017 | 0.5326 | 0.6749 |
| DU145F2 | 1529 | 0.6239 | 0.5984 | 0.5375 | 0.6809 |
| VCaP | 1689 | 0.6211 | 0.5534 | 0.4853 | 0.6301 |
| RWPE | 1548 | 0.6250 | 0.6079 | 0.5173 | 0.6730 |
In each dataset, the method with the highest Spearman's correlation coefficient is highlighted in bold.
aThe number of transcripts measured in Illumina sequencing with high over-dispersion and high spatial dependence.
We again compared the Spearman's rank correlation coefficients between the Helicos measure and the estimates of transcript-level gene expression generated by the four tested methods, respectively. Table 2 shows the overall performances of the four tested methods in the selected subset of transcripts with high over-dispersion and high spatial dependence. The POME method achieved the highest Spearman's correlation coefficients in all 12 samples. Based on the real data results above, we believe that POME provides more accurate quantification of transcript-level expression than the other three competing methods, especially for transcripts with high over-dispersion level and high spatial dependence.
We next repeated the above analyses using the Pearson's correlation coefficients and obtained similar results. The POME method achieved the highest Pearson's correlation coefficients in 7 of 12 samples using highly expressed transcripts (Supplementary Table S2). Using a subset of transcripts with high over-dispersion and high spatial dependence, the POME method achieved the highest Pearson's correlation coefficients in 9 of 12 samples (Supplementary Table S3).
To understand the differences between POME and the other three competing methods (RPKM, mseq and GPseq), we zoomed in on those transcripts for which the tested methods gave dramatically different expression measures. In the discussion below, sample LnCaP_0 was used as an illustrative example. To be specific, we first transferred the estimated transcript-level expression values and the Helicos measures (gold standard) into ranks, since we used Spearman's rank correlation coefficient to measure the performance of a tested method. Next we calculated the rank differences between POME and other three competing methods, and selected the top 10 transcripts with the largest rank differences. We used the ranks provided by the Helicos measure as the gold standard and assumed that the better method provides a closer rank to the Helicos rank. The selected transcripts were listed in Supplementary Table S4 in which we also reported the variance-to-mean ratio and the lag one autocorrelation for each transcript.
First we looked at the 10 transcripts with the largest rank differences between POME and RPKM. We found that POME outperformed RPKM in all 10 transcripts. The RPKM method overestimated the expression levels of these 10 highly over-dispersed transcripts. In contrast, the two random effect terms in the POME model were able to account for extra variability, which contributed to POME's improved accuracy.
For the 10 transcripts with the largest rank differences between POME and GPseq, POME outperformed GPseq in 8 of the 10 transcripts. All these 10 transcripts are extremely highly over-dispersed (variance-to-mean ratios are >10). GPseq was not able to accommodate such huge variability by the two parameters of the assumed generalized Poisson model and in most cases underestimated the expression level.
For the 10 transcripts with the largest rank differences between POME and mseq, POME outperformed mseq in 8 of the 10 transcripts. POME produced more accurate estimates than mseq when the data showed high spatial dependence, since POME explicitly incorporated the spatial dependence while mseq assumed spatial independence between reads counts covering different base pairs.
4 DISCUSSION
In microarray data analysis, it is now widely accepted that estimates of the expression levels based on parametric models such as the model-based expression index (MBEI) (Li and Wong, 2001) are more accurate in reflecting the underlying expression levels than summary statistics of raw intensity values. Inspired by the success of model-based methods in microarray data analysis, in this study, we strived to develop a model-based method for analyzing RNA-Seq data.
When exploring the properties of base-level sequencing depth in RNA-Seq data, we found substantial variation in sequencing depth within most transcripts, especially for those that are highly expressed. We also found that a large proportion of transcripts (~25%) shows over-dispersion in base-level sequencing depth (Supplementary Figure S1), which is consistent with what has been reported in the literature (Li et al., 2010). Additionally, we found that base-level sequencing depth displays substantial dependence between base pairs that are close to each other. This type of dependence resembles spatial correlation between neighboring areas considered in spatial statistics research (Waller and Gotway, 2004), and to the best of our knowledge, has not yet been reported in the literature on RNA-Seq data analysis. Based on these observations, we believe that spatial models that can incorporate correlations such as POME have advantages over existing models in characterizing base-level RNA-Seq data.
In this study, we used POME to estimate the expression level of an individual transcript. Two types of random effects are introduced to characterize two types of variation in read counts, which are between adjacent base pairs (i.e. spatial correlation) and specific to each base pair (i.e. non-spatial variation), respectively. Analyses of both simulated and real RNA-Seq data demonstrate that the expression indices estimated by POME reflect the underlying expression levels more accurately compared with existing methods. We believe that the improvement of estimation accuracy of POME comes from modeling the dependence between the read counts of base pairs adjacent to each other. The POME model can also be used for differential gene expression analysis by incorporating biological or experimental conditions or other covariates. We will pursue research in these directions in the future.
POME model assumes over-dispersion in the observed count data which is the case for a large proportion of transcripts in RNA-Seq data, especially those highly expressed. For this reason, we focused on highly expressed transcripts in this study. From our experience, about one-quarter of all transcripts belong to this category. For these transcripts, our analysis on simulated as well as real data demonstrated that POME offers more accurate expression measure than other methods that we compared. We have also performed small-scale tests on transcripts with medium level expression, and found POME again outperforms RPKM, mseq and GPseq methods (data not shown). For lowly expressed transcripts, we do not recommend to use POME for inference because observed data may be under-dispersed.
Like many other model-based methods, the POME method is more computationally intensive than read enumeration methods. For a 100 bp transcript without replicate, it takes 23 s for POME to complete 10 000 MCMC iterations on a Dell PowerEdge 1950 computing node (2.83 GHz CPU processors and 8 GB RAM). The computation time increases almost linearly with the length of transcripts. However, since model fitting of individual transcript is independent of each other and thus can be performed simultaneously, the computational intensity of the POME method can be much mitigated by parallel computing. The power of modern cluster computers will also help in this regard. After all, we believe that accuracy in statistical inference outweighs computation cost as long as the latter is affordable.
Although we only applied the POME method to RNA-Seq data obtained from the Illumina platform in this study, we believe it can also be applied to RNA-Seq data collected from other sequencing platforms such as SOLiD with little or no modification, because these data possess similar data structures.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the associate editor and three anonymous reviewers for their constructive and helpful suggestions, which substantially improved our manuscript. We thank members of the Arul Chinnaiyan lab, especially Mr Lee Sam for sharing the prostate cancer RNA-Seq data and helpful discussion. We thank Dr Hao Wu for helpful discussion. We thank Mr Han Wu for his help during the revision of this manuscript.
Funding: National Science Foundation grants (DMS 1000617 and DMS 1000443); National Institutes of Health grant (5R01GM080625).
Conflict of Interest: none declared.
REFERENCES
- Besag J. Spatial interaction and the statistical analysis of lattice systems (with Discussion) J. R. Stat. Soc. Ser. B. 1974;36:192–236. [Google Scholar]
- Besag J., et al. Bayesian image restoration, with two applications in spatial statistics. Ann. Inst. Stat. Math. 1991;43:1–20. [Google Scholar]
- Best N., et al. A comparison of Bayesian spatial models for disease mapping. Stat. Methods Med. Res. 2005;14:35–59. doi: 10.1191/0962280205sm388oa. [DOI] [PubMed] [Google Scholar]
- Bullard J.H., et al. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D., Kaldor J. Empirical Bayes estimates of age-standardized relative risks for use in disease mapping. Biometrics. 1987;43:671–681. [PubMed] [Google Scholar]
- Cloonan N., et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. [DOI] [PubMed] [Google Scholar]
- Cressie N., Chan N. Spatial modeling of regional variables. J. Am. Stat. Assoc. 1989;84:393–401. [Google Scholar]
- Diggle P., et al. Model-based geostatistics. J. R. Stat. Soc. Ser. C Appl. Stat. 1998;47:299–350. [Google Scholar]
- Gilks W., Wild P. Adaptive rejection sampling for Gibbs sampling. Appl. Stat. 1992;41:337–348. [Google Scholar]
- Gilks W.R., et al. Adaptive rejection metropolis sampling within Gibbs sampling. Appl. Stat. J. R. Stat. Soc. Ser. C. 1995;44:455–472. [Google Scholar]
- Gilks W.R., et al. Markov Chain Monte Carlo in Practice. Boca Raton, FL: Chapman & Hall; 1998. [Google Scholar]
- Li C., Wong W.H. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl Acad. Sci. USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., et al. Modeling non-uniformity in short-read rates in RNA-Seq data. Genome Biol. 2010;11:R50. doi: 10.1186/gb-2010-11-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.S. Monte Carlo Strategies in Scientific Computing. Berlin: Verlag; 2001. [Google Scholar]
- Lockhart D.J., et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Maher C.A., et al. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni J.C., et al. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin R.D., et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner J.R., et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011;29:742–749. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam L.T., et al. A comparison of single molecule and amplification based sequencing of cancer transcriptomes. PLoS One. 2011;6:e17305. doi: 10.1371/journal.pone.0017305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M., et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Srivastava S., Chen L. A two-parameter generalized Poisson model to improve the analysis of RNA-seq data. Nucleic Acids Res. 2010;38:e170. doi: 10.1093/nar/gkq670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J. Disease mapping and spatial regression with count data. Biostatistics. 2007;8:158–183. doi: 10.1093/biostatistics/kxl008. [DOI] [PubMed] [Google Scholar]
- Waller L.A., Gotway C.A. Applied Spatial Statistics for Public Health Data. Hoboken, NJ: John Wiley & Sons; 2004. [Google Scholar]
- Wang Z., et al. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm B.T., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- Zhu H., et al. Stochastic approximation algorithms for estimation of spatial mixed models. 2009 Manuscript. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.