

Abstract
Background
The ionic mechanisms of electrical heterogeneity in the ischemic ventricular epicardium remain poorly understood.
Objective
To test the hypothesis that the ATP-activated K+ current (IKATP) plays an important role in mediating repolarization differences between the right (RV) and left ventricle (LV) during global ischemia.
Methods
Electrical activity in Langendorff-perfused guinea pig hearts was recorded optically during control, ischemia, and reperfusion. Patch-clamp experiments were used to quantify IKATP density in isolated myocytes. Molecular correlates of IKATP (Kir6/SUR) were probed via RT-PCR. The role of IKATP in modulating repolarization was studied using computer simulations.
Results
Action potential duration (APD) was similar between LV and RV in controls, but significantly different in global ischemia. Pre-treatment of hearts with 10 μM glibenclamide (IKATP blocker) abolished the APD gradient during ischemia. In the absence of ischemia, pinacidil (IKATP opener) tended to shorten the APD more in the LV, and caused a small but significant increase in APD dispersion. In voltage clamp experiments, the density of the whole-cell current activated by pinacidil at depolarized potentials was significantly larger in LV, compared with RV epicardial myocytes. The mRNA levels of Kir6.1/Kir6.2 were significantly higher in LV, compared to RV. Simulations showed that IKATP is the main determinant of LV-RV APD gradient, whereas cell-to-cell coupling modified the spatial distribution of this APD gradient.
Conclusion
IKATP is an important determinant of the epicardial LV-RV APD gradient during global ischemia, in part due to a higher current density and molecular expression in the LV.
Keywords: IKATP, APD gradient, ischemia
INTRODUCTION
Electrical heterogeneities have been reported to exist in the ventricular myocardium, and the ionic mechanisms underlying these differences in various species have been studied extensively in the past two decades.1–3 The transmural disparity in repolarization has been shown to be enhanced in acquired or inherited disease conditions, and hypothesized to increase the vulnerability to reentrant arrhythmias.1 In contrast, although electrophysiological differences between LV and RV have been studied under normal conditions,4,5 little information is available regarding their features in abnormal conditions such as ischemia. Kurz et al.,6,7 found that the action potential duration (APD) shortened more prominently in the LV than RV in rabbits during low flow, global ischemia. However, the underlying ionic determinants were not elucidated.
It is important to study these ionic mechanisms because an enhanced dispersion for repolarization is now well known to act as a substrate for arrhythmogenesis.8,9 Furthermore, recent studies have suggested that such differences may have important implications for maintenance of arrhythmias. For example, LV-RV differences in the inward rectifier K+ current (IK1) have been postulated to affect the spatio-temporal organization of ventricular fibrillation (VF) in the structurally normal heart.10,11 Similarly, a higher density of the acetylcholine-activated K+ current (IKACh) was shown to underlie a faster frequency of activation in the left atrium (than the right) during acute atrial fibrillation in sheep hearts.12
It is well known that activation of ATP-sensitive K+ current (IKATP) shortens the cardiac APD in response to metabolic stress such as ischemia.13,14 We therefore hypothesized that the LV-RV gradient of APD in the globally ischemic heart could be partly attributed to inhomogeneous activation of IKATP across the ventricular chambers (LV versus RV). We tested this hypothesis via optical mapping of isolated Langendorff-perfused hearts, patch-clamp experiments, measuring mRNA levels of the molecular correlates of IKATP, and via computer simulations using a detailed ionic model of the guinea pig ventricular myocyte.15
METHODS
This investigation conformed to US NIH Guidelines for the Care and Use of Laboratory Animals (NIH publication No. 85–23, revised 1996), and approved by local UCUCA protocols at the University of Michigan, Ann Arbor (#10017). The detailed methods are provided in the Online Supplement (In the manuscript, “N” stands for no. of animals used, and “n” stands for cells).
Briefly, hearts were obtained from anesthetized guinea pigs or rabbits. Male guinea pigs (300–600 g) were heparinized and then anesthetized with sodium pentobarbital (35 mg/kg, IP). Rabbits (2–3 Kg) were heparinized and then anesthetized by sodium pentobarbital (50–100 mg/Kg IV). After making sure that the animals were in deep anesthesia, which was monitored via pain reflex, hearts were quickly removed.
We performed optical mapping, patch clamp, molecular biology experiments on isolated hearts, and computer simulations, each of which are briefly described below, and in more details in the online section.
Optical mapping
Isolated hearts were Langendorff-perfused and superfused with Tyrode’s solution at 35.5 ± 1.5°C and bubbled with 95%O2–5%CO2 (pH 7.4). In the guinea pig hearts (N=12), ischemia was induced by stopping perfusion, and bubbling the superfusate with 95%N2–5%CO2. Movies of RH-237 fluorescence were obtained using a Dalsa CCD camera in presence of Blebbistatin (5 μM) to reduce motion artifact, alongwith volume-conducted pseudo-ECGs. Optical mapping experiments were also performed on isolated rabbit hearts, details of which are in the Online supplement.
Patch Clamp
Guinea pig hearts (N=12) were enzymatically digested, and epicardial myocytes from LV and RV were patched in the whole-cell configuration at 35 ± 1.0° C. IKATP was recorded between – 100 and +40 mV during superfusion with pinacidil (100 μM).
Molecular Biology
RNA from epicardial LV and RV tissue from guinea pig hearts (N=6) was also isolated and subjected to RT-PCR to measure and compare levels of Kir6.1/Kir6.2/Sur2A/Sur2B (molecular correlates of IKATP).13,14
Computer Simulations
For simulations, we used a guinea pig ventricular ionic model15 to investigate the changes occurring in the APD and study the role of IKATP versus coupling in inscribing APD heterogeneity.
Statistical Analyses
All data are shown as mean ± SEM. Analysis of variance with Bonferroni’s post-test was used for comparisons among more than 2 groups (2-way or 1-way ANOVA; see individual results); paired or unpaired student’s t-test was used for comparisons between 2 groups. p < 0.05 was considered statistically significant. Graphpad Prism 5 software was used for analyses.
RESULTS
Fig. 1A displays the distribution of APD70 on the anterior epicardial surfaces of the guinea pig ventricles in a representative experiment during control, after 20 minutes of global, no-flow ischemia, and after washout. The underlying volume-conducted ECGs are also shown. These APD maps were recorded at the intrinsic heart rates (i.e. without external pacing). During ischemia the heart rate slowed down, and was always accompanied by AV dissociation. In control, the intrinsic sinus cycle length was 335.00±22.28 ms; it slowed down after 20 minutes of global ischemia to 987.16±64.4 ms (N=6, P<0.05). APD was homogeneous throughout both ventricles in both control and washout. In contrast, as the heart size was reduced during ischemia, the APD70 distribution was inhomogeneous, with a shorter APD in the LV than RV. (Similar differences were also seen in the absence of Blebbistatin during ischemia; see Online Figure 1). As summarized in Fig. 1B, LV-RV APD differences were apparent in all experiments. The mean APD70 shortened by ≈35% in the LV during ischemia, compared to ≈19% in the RV after 20 minutes of ischemia. Thus, the LV-RV APDs were significantly different only during ischemia (N=6, P<0.05; repeated measures 2-way ANOVA; Bonferroni post-test). We also calculated the dispersion of APD70 as the absolute value difference between the mean LV and RV APD values (Fig. 1C). Dispersion was minimal in control and washout (3.65±1.02 ms; 3.71±3.27 ms respectively). Enhanced dispersion was seen after 20 minutes of ischemia (28.44±4.44 ms) (N=6, P<0.05; repeated measures, 1-way ANOVA). The activation pattern was also different in ischemia versus control (see Online Figure 2). We never observed spontaneous arrhythmias during global ischemia; this is possibly related to an absence of an appropriate trigger, despite the presence of an arrhythmogenic substrate.9 However, we did see reperfusion arrhythmias (See Online Figure 3 and Online supplement for details). We also observed similar differences in LV-RV when hearts were paced at a constant cycle length (see Online Figure 5) Since IKATP is activated during ischemia,13,14 we hypothesized that blocking this current would abolish the LV-RV APD gradient. Accordingly, a set of experiments were conducted in the continuous presence of 10μM of the relatively selective IKATP blocker glibenclamide.13,14 In control hearts, treatment with glibenclamide did not significantly affect APD70, which was 136.137±3.68 ms and 141.34±2.94 ms before and during glibenclamide treatment, respectively (N=5, p=NS, paired t-test). Fig. 2A displays representative examples of APD70 distribution and ECG in a glibenclamide treated heart during control, after 20 minutes of global ischemia, and after washout. In the control, the intrinsic cycle length in the presence of glibenclamide was 340.86±32.33 ms. The intrinsic ventricular cycle length slowed down significantly after 20 minutes of global ischemia (1422.2±118.34 ms) (N=5, P<0.05, paired t-test), and AV dissociation was observed. In addition, the APD was significantly prolonged in both LV and RV, compared to controls. Similar results were obtained in 5 experiments, as summarized in Fig. 2B. The APD was increased by ≈36% in both LV and RV during global ischemia compared to controls; the change was not significantly different between ventricles (repeated measures 2-way ANOVA, Bonferroni post-test). The dispersion of APD70 is plotted in Fig. 2C. It is small and similar during control, ischemia and washout (2.10±0.65 ms, 4.79 ± 1.08 ms, and 2.16±0.73 ms respectively; repeated measures, 1-way ANOVA). Thus glibenclamide prolonged the APD and abolished the enhanced dispersion of repolarization seen during ischemia.
Figure 1.
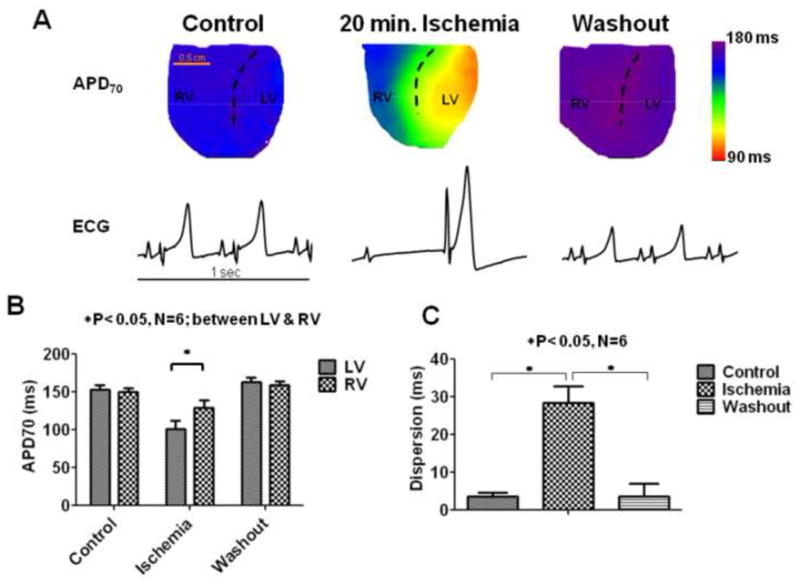
APD distribution during ischemia. (A). APD70 maps on the anterior surface of the guinea pig ventricle and the underlying pseudo-ECGS tracings (each of 1 second duration) during control, after 20 minutes of ischemia, and after washout are shown. The dashed lines are approximately along the left anterior descending (LAD) coronary, which separates the right and left ventricles respectively (RV and LV). The heart can be seen to shrink in size during ischemia. (B) Quantification of the APD70 in LV and RV, and (C) the dispersion of APD70 defined as the absolute difference of the mean values between LV and RV are shown for control (C), 20 minutes of ischemia (I), and washout conditions (W).
Figure 2.
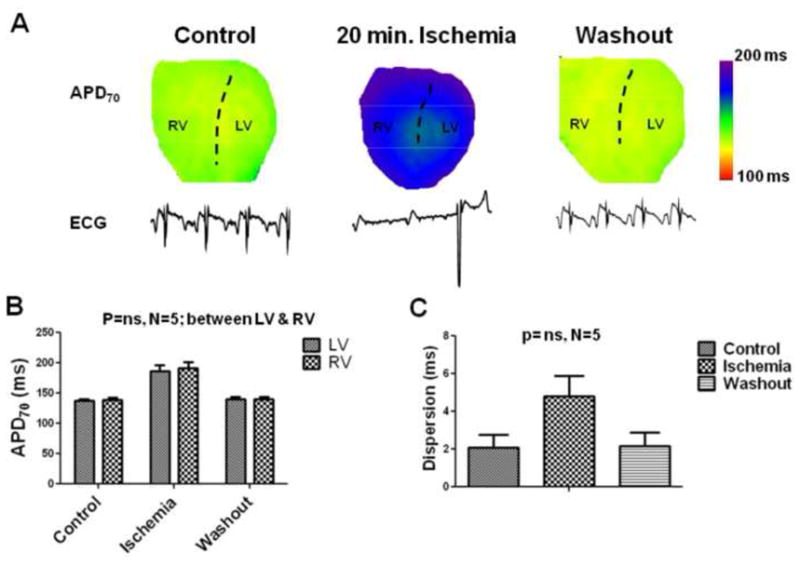
APD distribution during ischemia in the presence of IKATP blocker, glibenclamide. (A). APD70 maps on the anterior surface of the guinea pig ventricle and the underlying volume-conducted-ECGS tracings (1 second duration) during control, after 20 minutes of ischemia, and after washout are shown in hearts pre-treated with 10 μM of glibenclamide. The dashed lines are approximately along the LAD. The heart can be seen to shrink in size during ischemia. (B) Quantification of the APD70 in LV and RV, and (C) the dispersion of APD70 defined as the absolute difference of the mean values between LV and RV are shown for control (C), 20 minutes of ischemia (I), and washout conditions (W).
To further test whether the enhanced dispersion of repolarization during ischemia could be partly attributable to differences in IKATP density between LV and RV, we perfused normal hearts with 90 μM pinacidil, an IKATP activator.13,14 A representative example is shown in Fig. 3A (note the different scales of APD in the middle panel for pinacidil, compared to control and washout). The ECG shows AV block. The effects of pinacidil on the AV node were variable. We observed AV dissociation in 2 experiments, AV block having 2:1 and 4:3 conduction patterns in 2 experiments, and in 1 experiment no AV block. Thus the intrinsic sinus rhythm cycle length calculated from 4 experiments (except for the 4:3 conduction pattern) was regular in 4 experiments, but showed a large standard error. The sinus cycle length during control conditions was 274.58±12.2 ms, and slowed down after 15 minutes pinacidil (616.38±154.01 ms) (N=4). In the experiment shown in Fig. 3, where atrio-ventricular conduction occurred in a 4:3 pattern, the mean cycle length between conducted beats was 297.25 ms, very similar to controls in this experiment (300.8 ms). The data from 4 experiments are summarized in Fig. 3B. On average (and in each individual heart), APD tended to shorten more in LV than RV compared to controls, although the difference between LV and RV in the presence of pinacidil was not statistically significant (repeated measures 2-way ANOVA, Bonferroni post-test). However, pinacidil caused a small, yet statistically significant enhanced dispersion of repolarization compared to control and washout, as shown in Fig. 3C (N=4, P<0.05; repeated measures, 1-way ANOVA) (8.26 ± 1.04 ms, 2.56 ± 0.72 ms, and 4.28 ± 1.25 respectively).
Figure 3.
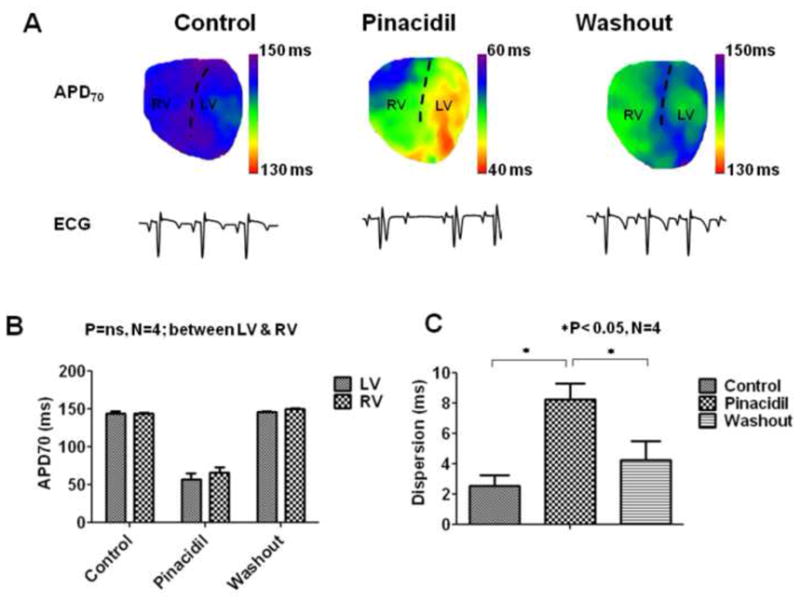
APD distribution during exposure to IKATP opener, pinacidil. (A) APD70 maps on the anterior surface of the guinea pig ventricle and the underlying volume-conducted-ECG tracings (1 second duration) during control, after 15 minutes of 90 μM of pinacidil, and after washout are shown. The dashed lines are approximately along the left anterior descending (LAD) coronary, which separates the right and left ventricles respectively (RV and LV). (B) Quantification of the APD70 in LV and RV, and (C) the dispersion of APD70 defined as the absolute difference of the mean values between LV and RV are shown for control (C), 15 minutes of pinacidil (P), and washout conditions (W).
We next conducted whole-cell patch clamp experiments in isolated single cells obtained from the LV and RV epicardium in guinea pig hearts. Representative traces of the whole-cell, pinacidil activated, and washout currents (in presence of Ca2+ current blockade) at voltages from −100 mV to +40 mV (in steps of 10 mV) are shown in Fig. 4A (LV epicardial) and Fig. 4B (RV epicardial). Cell capacitance in LV epicardium was 106.6±8.4 pF (n=9 cells; N=9 animals), and in RV epicardium was 106.6±7.5 pF (n=12 cells; N=10 animals; ns). The whole-cell current-voltage (I-V) relationships resulting from voltage clamp pulses from −100 to +40 mV in control and in the presence of 100 μM pinacidil (in red) are shown for LV epicardial (Fig. 4C) and RV epicardial cells (Fig. 4D). The whole-cell current was similar in LV and RV epicardial cells, but the pinacidil activated current was significantly greater in LV than RV epicardial cells at voltages >= +10 mV. For example, at +40 mV, the density of the whole-cell current activated by pinacidil was 14.44±1.98 pA/pF in LV epicardial cells (n=9), compared with 7.29±0.89 pA/pF in RV epicardial cells (n=12) (p<0.05). Outward current returned to control values upon pinacidil washout (n=4 for both types of cells).
Figure 4.
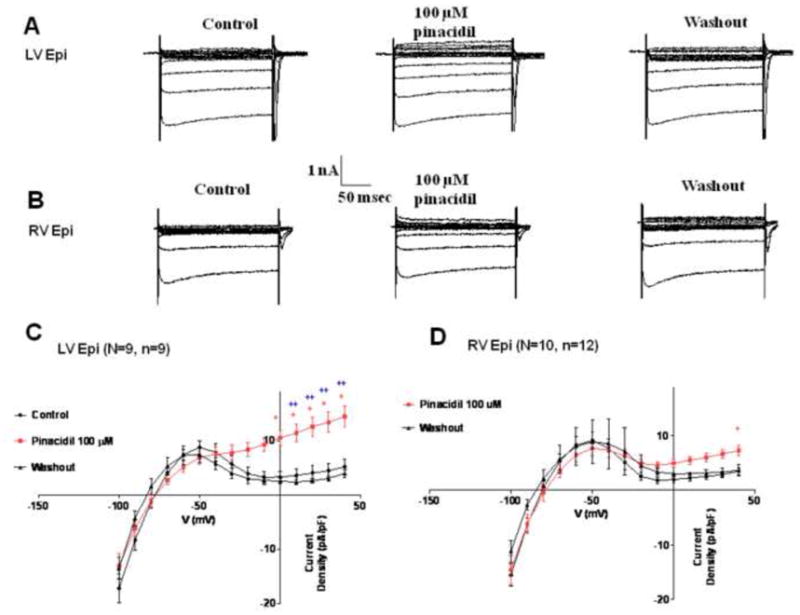
Patch clamp experiments. Representative traces of whole-cell currents (with block of Ca2+ current) at voltage clamp pulses at voltages from −100 to +40 mV in control, in presence of 100 μM pinacidil, and during washout are shown for guinea pig LV epicardial (A) and RV epicardial cells (B). Current-Voltage (I-V) relationship for whole-cell current in the absence and presence of 100 μM pinacidil (red) is shown for guinea pig LV epicardial (C) and RV epicardial cells (D). (+ P< 0.05 RV epi pinacidil versus control; * P< 0.05 LV epi pinacidil versus control; ++ P< 0.05 LV epi pinacidil versus RV epi pinacidil). (N represents no. of hearts used, and n = no. of myocytes).
Expression of Kir6/SUR
Analysis of mRNA levels showed that the LV epicardium had a significantly higher expression of Kir6.1 and Kir6.2 (Figure 5, p<0.05) compared to the RV epicardium. In the LV epicardium, Kir6.1 was expressed at 136% of its expression in the RV. Similarly Kir6.2 gene was 44% more abundant in LV than RV. Analysis of mRNA expression for Sur2A and Sur2B genes in LV and RV epicardial tissue showed that although there was a trend toward larger Sur2A in LV (approx. 40%), this increase was not statistically significant (Figure 5). Expression of Sur2B was similar in both ventricles (Figure 5).
Figure 5.
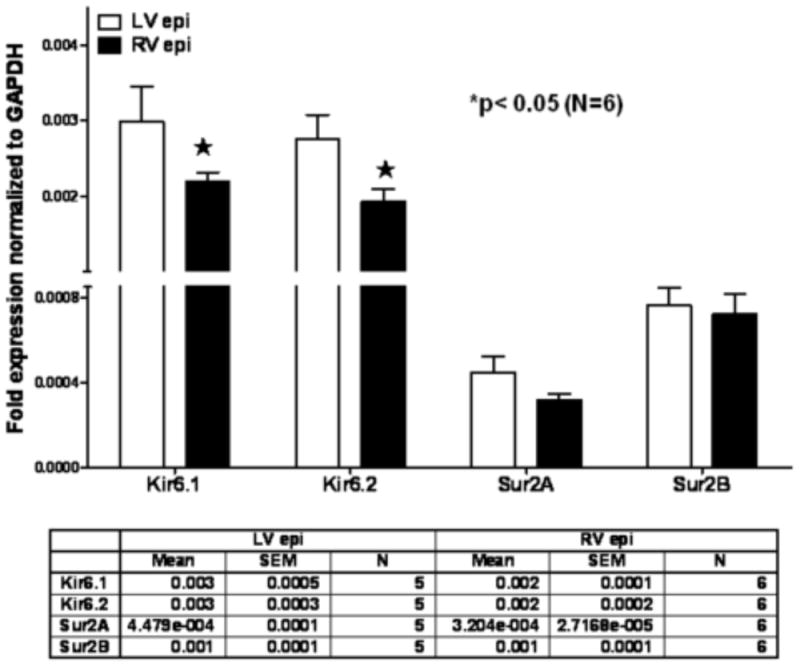
Difference between LV epicardial and RV epicardial expression for Kir6.1, Kir6.2, Sur2A and Sur2B. Real time RT-PCR was performed on RNA of ventricular tissue from adult guinea pigs. RNA expression normalized to expression of GAPDH was determined. Values are mean ± standard error of the mean, n=6 in each group. *, p< 0.05 indicate a significant effect as compared to the respective LV tissue expression. Differences were determined by two-way ANOVA with Bonferroni post-test comparison.
Ischemia leads to altered intercellular coupling which might contribute to the observed LV-RV APD differences. Therefore, we conducted computer simulations to gain insight into the role of cell to cell communication in the mechanism of the APD gradient associated with chamber specific differences in IKATP activation. First, the guinea-pig ventricular action potential was simulated in steady state conditions (after 5 minutes at 1Hz pacing) (Fig. 6A; APD70= 159 ms). Fig. 6B shows the simulated, instantaneous pinacidil-activated IKATP for LV and RV cells that were used to replace the original inward rectifier formulation in the model (see Online Supplement for details). Based on our experimental data (Fig. 4), this current was greater in LV cells at voltages positive to −40 mV, compared to RV cells. Fig. 6C depicts the action potentials in LV and RV cells (APD70= 43ms and 79 ms respectively) when the respective IKATP currents were active, and the cell model was paced at 1 Hz for 30 seconds. Next, we created a 2D rectangular sheet of electrically coupled cells (200 by 50 cells; 20 mm by 5 mm). LV and RV cells were incorporated into the 2D sheet divided equally as shown in Fig 6D, and a wave was allowed to propagate at a speed of 90 cm/s. In presence of an active IKATP, APD70 in the LV and RV regions was 43 and 77 ms, respectively (Fig. 6E). In the pinacidil experiments (Fig 3), APD70 in LV ranged from 42 to 71 ms, and in RV from 51 to 82 ms. To test for a role of an altered intercellular communication in the establishment of LV-RV APD gradient in the presence of IKATP, we changed the diffusion coefficient from 0.2 to 0.05, which reduced the conduction velocity by almost half (from 90 to 43 cm/s), but did not change the absolute value of APD70 in either LV or RV. However, as shown by the increased steepness in the APD profile, the reduced diffusion coefficient did affect the rate of APD change at the LV-RV border (Fig. 6F). The simulated single-cell action potentials in ischemia (See Online Supplement for exact conditions of simulation) are shown in Fig. 6G. APD70 was 123 and 92 ms for RV and LV, respectively. When IK-ATP was blocked (i.e. ḠKATP = 0), the APD70 increased to 164 ms, in part due to the incorporation of a late sodium current in the model. Incorporating the simulated ischemic LV and RV action potentials in a 2D sheet (Fig. 6H) again demonstrated that the electrotonic coupling had a significant effect on the slope of the spatial transition between the right and left steady state APDs, and was sharper with decreasing diffusion coefficients.
Figure 6.
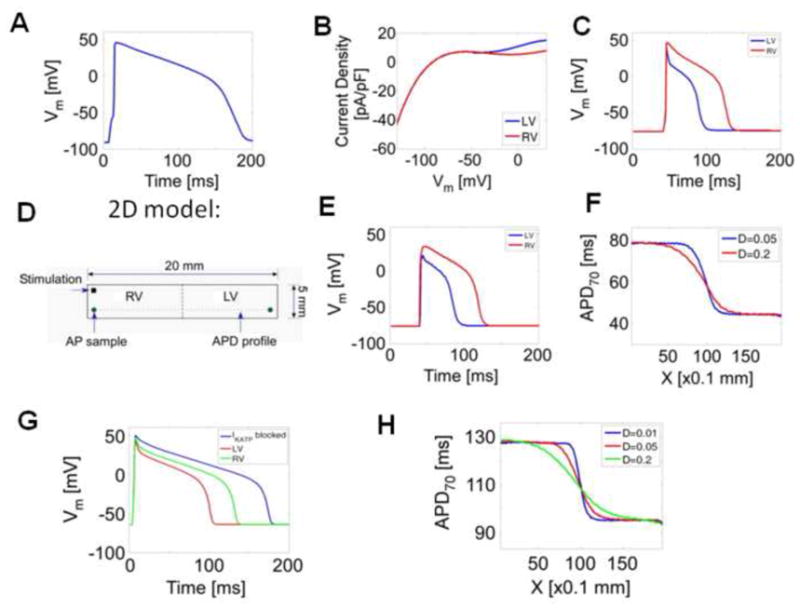
Computer Simulations. (A) Original guinea pig simulated action potential, at extracellular K+ concentration of 4 mM. (B) Simulated I-V curve of IKATP current density difference in LV and RV cells, based on experimental data in Fig. 5. (C) Simulated action potentials in LV and RV when the respective IKATP was incorporated into the model. (D) Schematic of the narrow strand of multicellular model used. (E) LV and RV action potentials in the narrow strand of cells, after the incorporation of IKATP currents. (F) Spatial distribution of APD in the narrow strand of cells at two different coupling coefficients in presence of active IKATP in normal cells. (G) Simulated ischemic action potentials in LV and RV, and when IKATP was blocked are shown. (H) The APD distribution profile between LV and RV in 2D cells during ischemia is shown at different coefficients of coupling.
DISCUSSION
The main new finding from our study is that IKATP contributes to the LV-RV heterogeneity in the anterior epicardial APD during global ischemia in guinea pig hearts. This is related in part to the higher density of IKATP, as well as higher Kir6.1/Kir6.2 mRNA levels in the LV compared to RV.
Heterogeneity of APD in LV and RV
We did not observe any dispersion of APD between LV and RV under normal conditions. This is similar to previous reports in different species including the guinea pig,16,17 rabbits6 and cats.18 In rodents, a larger density of the Ca2+-independent transient outward K+ current Ito has been reported in RV, with a corresponding shorter APD.19,20 In canine hearts, epicardial LV and RV APDs are similar; however a larger Ito in RV leads to a more prominent phase 1 notch in action potentials recorded in RV myocytes.4 Thus, except for rodents, most species do not display LV-RV APD differences in ventricular epicardial cells under normal conditions. Interestingly, recordings from deeper, intramural layers of the ventricle suggest that LV-RV differences may exist under normal conditions; cells from the RV midmyocardium displayed a shorter APD than corresponding LV cells in canine hearts.5 This was attributed to differences in the densities of Ito and the slow delayed rectifier K+ current, IKs. Similarly, longer APDs have been reported in LV than in RV in endocardial and septal regions of guinea pig hearts.16 However the ionic mechanisms remain unknown. In terms of global ischemia, Kurz et al6,7 studied global ischemia in rabbit hearts with MAP recordings, and found that the LV APD shortened more than in the RV. Our study confirmed this finding in the guinea pig, and also explored its putative ionic bases. In our study, the APDs were recorded from the epicardial layer of cells. In contrast, a recent study found that during acute global ischemia in canine hearts, the electrical conduction was slower/blocked in the right ventricular septum, compared to the left ventricular septum, and that this differential effect was abolished in the presence of glibenclamide.21 Thus in this canine model, the RV septum was rendered more inexcitable than the LV. These differences could be related to either the species studied (canine versus guinea pigs/rabbits), or the different layers (epicardium versus septum).
Role of IKATP in electrical heterogeneity
Previous studies have shown that IKATP current is responsible in part for the differential response of the epicardium and the endocardium during acute ischemia.22 It was found that IKATP density was similar, but its sensitivity to ATP was different, which resulted in the epicardium becoming more inexcitable and displaying a shorter APD during ischemia compared to the endocardium.22 The results from our glibenclamide experiments indicate that a relatively selective blockade of IKATP abolishes the LV-RV APD heterogeneity during ischemia. Interestingly, APD was also prolonged in ischemia, when IKATP channels were blocked. The ionic mechanism that underlies this APD prolongation is unknown, but could be related to the presence of a persistent sodium current, that is known to be activated in response to hypoxia in guinea pig cardiac cells.23 However, when IKATP current was activated via pinacidil, the APD tended to shorten more in LV in guinea pig (and rabbit hearts: see Online Figure 4). Our findings from patch clamp experiments suggest that the density of IKATP is responsible in part for the LV-RV heterogeneity. RT-PCR experiments show a higher density of Kir6.1/6.2 mRNA levels in LV compared to RV, and the Sur2A mRNA levels tended to be higher in the LV as well. In addition, it is possible that LV and RV may also exhibit different intrinsic responses to ischemia such as ATP/ADP ratios, catecholamines, pH, and extracellular K+, all of which may modulate IKATP further.13,14 This is borne out by the fact that the normal sequence of activation during sinus rhythm in LV was disrupted (delayed/blocked by ischemia), and not restored by blockade of IKATP by glibenclamide. Thus, this abnormal activation/block in LV may be indicative of the LV being rendered more “ischemic” than RV. Lastly, computer simulations show that although the coupling between cells does affect the steepness of APD gradient distribution, IKATP is the main determinant of the maximum APD shortening, and its absolute value is not altered. In sum, a larger functional activation of IKATP seems to contribute to the shorter APD observed in LV compared to RV during ischemia.
Limitations
The use of Blebbistatin (motion uncoupler) prevents contraction; thus the energy consumption and ATP depletion rate in ischemia is likely altered. However, in the context of our study, we were able to record LV-RV APD differences in ischemia even in the absence of blebbistatin (see Online Figure 1). Blebbistatin has been shown not to affect the APD in normal hearts;24 however whether it alters APD during ischemia, including IKATP remains unknown. Glibenclamide is only a relatively selective blocker of IKATP, and thus the contribution of other ionic mechanisms to the LV-RV heterogeneity, like chloride channels, cannot be ruled out. However at 10 μM concentration used in our experiments, only 10% of cAMP activated chloride channels are blocked in guinea pig cardiomyocytes,25 and the EC50 for blocking volume-activated Cl− channels is 60–200 μM.26 It has also been reported that in addition to partly affecting other transmembrane ionic currents besides the sarcolemmal ATP-activated K+ channel, glibenclamide and pinacidil may also affect the mitochondrial ATP K+ channels.27 We have not tested for the ATP-sensitivity of IKATP in LV versus RV, as was done for epicardium versus the endocardium.22 We have not studied arrhythmogenesis per se in this model of global ischemia. This will require more systematic and extensive studies, with properly timed and spatially appropriate (with respect to the APD gradient) trigger for initiating arrhythmias.9 An additional and important consideration is that epi-endocardial gradients are prominently observed in ischemia and inscribed in part due to K-ATP channel differences.1,22,28 These transmural differences in K-ATP channels may influence arrhythmogenesis not only in the ischemic myocardium,1 but also other substrates such as in Brugada syndrome/J waves,29,30 where genetic defects may cause the activation of the K-ATP channels.31 The relative importance of LV-RV versus epi-endo gradients in arrhythymogenesis thus requires further systematic investigation. Furthermore, we have not studied ionic/molecular bases of K-ATP channels differences that might exist between other regions such epi- versus endocardium, and the anterior versus the posterior regions at this time. In our experiments, pinacidil exposes difference between LV-RV in single cell currents, and although the APD tends to be shorter in LV than RV, it is not statistically significant in the whole heart. This could be related to differences in pinacidil activation properties in cells/versus tissue, concentration of pinacidil, and/or electrotonic effects of the 3D myocardium, or other unknown causes. Because of species-specific differences between ionic currents, caution must be exercised before extrapolating these results to other species and humans. Lastly, the limitations of the computer simulations have been discussed elsewhere.15 Despite these limitations, our study uncovers interesting differences in IKATP between LV and RV in the guinea pig ventricle for the first time, and suggests that these underlie the disparate APD shortening seen during global ischemia.
Supplementary Material
Acknowledgments
We would like to thank Drs. Omer Berenfeld, Colin Nichols and Alena Tolkacheva for useful discussions.
FUNDING
Supported by NHLBI Grants (PO1-HL039707, PO1-HL070074 and RO1-HL080159 to J.J.); and AHA Scientist Development Grant (S.V.P).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293:H2024–H2038. doi: 10.1152/ajpheart.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002;90:939–950. doi: 10.1161/01.res.0000018627.89528.6f. [DOI] [PubMed] [Google Scholar]
- 3.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 4.Di Diego JM, Sun ZQ, Antzelevitch C. I(to) and action potential notch are smaller in left vs. right canine ventricular epicardium. Am J Physiol. 1996;271:H548–561. doi: 10.1152/ajpheart.1996.271.2.H548. [DOI] [PubMed] [Google Scholar]
- 5.Volders PG, Sipido KR, Carmeliet E, Spätjens RL, Wellens HJ, Vos MA. Repolarizing K+ currents ITO1 and IKs are larger in right than left canine ventricular midmyocardium. Circulation. 1999;99:206–210. doi: 10.1161/01.cir.99.2.206. [DOI] [PubMed] [Google Scholar]
- 6.Kurz RW, Xiao-Lin R, Franz MR. Increased dispersion of ventricular repolarization and ventricular tachyarrhythmias in the globally ischaemic rabbit heart. Eur Heart J. 1993;14:1561–1571. doi: 10.1093/eurheartj/14.11.1561. [DOI] [PubMed] [Google Scholar]
- 7.Kurz RW, Ren XL, Franz MR. Dispersion and delay of electrical restitution in the globally ischaemic heart. Eur Heart J. 1994;15:547–554. doi: 10.1093/oxfordjournals.eurheartj.a060541. [DOI] [PubMed] [Google Scholar]
- 8.Han J, Moe GK. Non uniform recovery of excitability in ventricular muscle. Circ Rs. 1964;14:44–60. doi: 10.1161/01.res.14.1.44. [DOI] [PubMed] [Google Scholar]
- 9.Coronel R, Wilms-Schopman FJ, Opthof T, Janse MJ. Dispersion of repolarization and arrhythmogenesis. Heart Rhythm. 2009;6:537–543. doi: 10.1016/j.hrthm.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 10.Samie FH, Berenfeld O, Anumonwo J, et al. Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res. 2001;89:1216–1223. doi: 10.1161/hh2401.100818. [DOI] [PubMed] [Google Scholar]
- 11.Warren M, Guha PK, Berenfeld O, et al. Blockade of the inward rectifying potassium current terminates ventricular fibrillation in the guinea pig heart. J Cardiovasc Electrophysiol. 2003;14:621–631. doi: 10.1046/j.1540-8167.2003.03006.x. [DOI] [PubMed] [Google Scholar]
- 12.Sarmast F, Kolli A, Zaitsev A, et al. Cholinergic atrial fibrillation: I(K,ACh) gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc Res. 2003;59:863–873. doi: 10.1016/s0008-6363(03)00540-6. [DOI] [PubMed] [Google Scholar]
- 13.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 14.Billman GE. The cardiac sarcolemmal ATP-sensitive potassium channel as a novel target for anti-arrhythmic therapy. Pharmacol Ther. 2008;120:54–70. doi: 10.1016/j.pharmthera.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Faber GM, Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watanabe T, Rautaharju PM, McDonald TF. Ventricular action potentials, ventricular extracellular potentials, and the ECG of guinea pig. Circ Res. 1985;57:362–373. doi: 10.1161/01.res.57.3.362. [DOI] [PubMed] [Google Scholar]
- 17.Wan X, Bryant SM, Hart G. A topographical study of mechanical and electrical properties of single myocytes isolated from normal guinea-pig ventricular muscle. J Anat. 2003;202:525–536. doi: 10.1046/j.1469-7580.2003.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleiman RB, Houser SR. Electrophysiologic and mechanical properties of single feline RV and LV myocytes. J Mol Cell Cardiol. 1988;20:973–982. doi: 10.1016/0022-2828(88)90575-5. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe T, Delbridge LM, Bustamante JO, McDonald TF. Heterogeneity of the action potential in isolated rat ventricular myocytes and tissue. Circ Res. 1983;52:280–290. doi: 10.1161/01.res.52.3.280. [DOI] [PubMed] [Google Scholar]
- 20.Knollmann BC, Katchman AN, Franz MR. Monophasic action potential recordings from intact mouse heart: validation, regional heterogeneity, and relation to refractoriness. J Cardiovasc Electrophysiol. 2001;12:1286–1294. doi: 10.1046/j.1540-8167.2001.01286.x. [DOI] [PubMed] [Google Scholar]
- 21.Morita ST, Morita H, Zipes DP, Wu J. Acute ischemia of canine interventricular septum produces asymmetric suppression of conduction. Heart Rhythm. 2008;5:1057–1062. doi: 10.1016/j.hrthm.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa T, Kimura S, Furukawa N, Bassett AL, Myerburg RJ. Role of cardiac ATP-regulated potassium channels in differential responses of endocardial and epicardial cells to ischemia. Circ Res. 1991;68:1693–1702. doi: 10.1161/01.res.68.6.1693. [DOI] [PubMed] [Google Scholar]
- 23.Wang W, Ma J, Zhang P, Luo A. Redox reaction modulates transient and persistent sodium current during hypoxia in guinea pig ventricular myocytes. Pflugers Arch. 2007;454:461–475. doi: 10.1007/s00424-007-0219-1. [DOI] [PubMed] [Google Scholar]
- 24.Fedorov VV, Lozinsky IT, Sosunov EA, et al. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–26. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 25.Tominaga M, Horie M, Sasayama S, Okada Y. Glibenclamide, an ATP-sensitive K+ channel blocker, inhibits cardiac cAMP-activated Cl− conductance. Circ Res. 1995;77:417–423. doi: 10.1161/01.res.77.2.417. [DOI] [PubMed] [Google Scholar]
- 26.Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiol Rev. 2000;80:31–81. doi: 10.1152/physrev.2000.80.1.31. [DOI] [PubMed] [Google Scholar]
- 27.Murata M, Akao M, O’Rourke B, Marbán E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89:891–898. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
- 28.Miyoshi S, Miyazaki T, Moritani K, Ogawa S. Different responses of epicardium and endocardium to KATP modulators during regional ischemia. Am J Physiol. 1996;271:H140–H147. doi: 10.1152/ajpheart.1996.271.1.H140. [DOI] [PubMed] [Google Scholar]
- 29.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 30.Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7:549–558. doi: 10.1016/j.hrthm.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medeiros-Domingo A, Tan BH, Crotti L, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010 Oct;7(10):1466–71. doi: 10.1016/j.hrthm.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.