

Abstract
Phosphorylation of the Saccharomyces cerevisiae Snf1 kinase activation loop is determined by the integration of two reaction rates: the rate of phosphorylation by upstream kinases and the rate of dephosphorylation by Glc7. The activities of the Snf1-activating kinases do not appear to be glucose-regulated, since immune complex kinase assays with each of the three Snf1-activating kinases show similar levels of activity when prepared from cells grown in either high or low glucose. In contrast, the dephosphorylation of the Snf1 activation loop was strongly regulated by glucose. When de novo phosphorylation of Snf1 was inhibited, phosphorylation of the Snf1 activation loop was found to be stable in low glucose but rapidly lost upon the addition of glucose. A greater than 10-fold difference in the rates of Snf1 activation loop dephosphorylation was detected. However, the activity of the Glc7-Reg1 phosphatase may not itself be directly regulated by glucose, since the Glc7-Reg1 enzyme was active in low glucose toward another substrate, the transcription factor Mig1. Glucose-mediated regulation of Snf1 activation loop dephosphorylation is controlled by changes in the ability of the Snf1 activation loop to act as a substrate for Glc7.
The Snf1 protein kinase of Saccharomyces cerevisiae is a founding member of a family of protein kinases that includes the mammalian AMP-activated protein kinase (AMPK)2 (1) and is present in all eukaryotes. Interest in this family has been increased by the finding that the medications used to treat type 2 diabetes activate AMPK (2). Indeed, the metabolic consequences of AMPK activation, which include increased glucose uptake and oxidation, increased fatty acid oxidation, inhibition of anabolic reactions, and stimulation of reactions that regenerate ATP (3), are beneficial to patients experiencing hyperglycemia. With the rise in the prevalence of obesity and type 2 diabetes in Western cultures, a more complete understanding of the mechanisms that regulate the activity of the Snf1/AMPK enzymes is needed.
The Snf1/AMPK enzymes function as heterotrimers with a catalytic α subunit associated with regulatory β and γ subunits. The α subunit contains a canonical kinase domain in its N terminus and an autoinhibitory domain in its C terminus (4). The catalytic activities of the Snf1/AMPK enzymes are regulated in a complex manner. First, subunit interactions within the Snf1/AMPK heterotrimer regulate enzymatic activity in response to the cellular energy status. Although not all agree on the mechanism, the γ subunit appears to play an important role in the regulation of Snf1/AMPK catalytic activity. The mammalian γ subunit can bind AMP (5) and can interact with the α subunit autoinhibitory domain to abrogate its inhibitory potential (4, 6). An alternative but not necessarily exclusive model posits the presence of a pseudosubstrate sequence in the γ subunit (7). Second, Snf1/AMPK kinases require phosphorylation of a conserved threonine residue in their activation loops by a distinct upstream kinase (8–10). Concerted effort by several research groups led to the identification of the activating kinases for Snf1 and AMPK (11–15). Yeasts encode three Snf1-activating kinases (SAKs), Sak1, Tos3, and Elm1 (11–13), and all three SAKs must be deleted to block Snf1 signaling. In mammalian cells, LKB1 is the primary activating kinase of AMPK under conditions of energy stress (15, 16), although other kinases may contribute to AMPK activation in specific cell types or in response to other stimuli (17–20).
It was hoped that the identification of the activating kinases for Snf1 and AMPK would rapidly lead to an understanding of the means by which cellular energy status controlled the activity of the Snf1/AMPK pathway. However, several lines of evidence suggest that the Snf1/AMPK activating kinases are not themselves regulated by energy status. First, LKB1, the primary activator of AMPK (14, 15), is the activation loop kinase for at least 12 other kinases that are responsive to different stimuli (16). If LKB1 were in fact regulated by cellular energy status, one would predict increased activity toward all of its substrates under conditions of nutrient limitation. This is not observed. Furthermore, when LKB1 was expressed in yeast in place of the SAKs, Snf1 activation loop phosphorylation responded normally to changes in glucose levels (21). Either LKB1 could sense energy status in both yeast and mammalian cells or the phosphorylation status of the Snf1 activation loop was not regulated at the level of phosphate addition. Third, reactions with purified components showed that the addition of AMP did not stimulate LKB1-mediated phosphorylation of AMPK but rather inhibited its dephosphorylation (22, 23). Fourth, mutations in the γ subunit of AMPK affect the ability of the PP2C α phosphatase to dephosphorylate the AMPK activation loop (23). Finally, one of the three SAKs, Elm1, is required for normal cell morphology (24, 25). Cells lacking Elm1 display the characteristic elongated morphology for which Elm1 was named. Since yeast cells display normal morphology in both glucose-rich and glucose-limited media, the Elm1 kinase must be active in both growth conditions. However, the Snf1 kinase activation loop shows a much greater degree of phosphorylation when cells are grown in limiting glucose conditions (8). The fact that Snf1 is not phosphorylated in glucose-rich media even when Elm1 is active suggests that the phosphorylation status of the Snf1 activation loop might not be regulated by phosphate addition, but rather by dephosphorylation. All of these data taken together suggest that the dephosphorylation reaction may be the regulated step in determining the phosphorylation status of the Snf1/AMPK activation loop.
Genetic studies have long indicated that the dephosphorylation of the Snf1 activation loop was mediated by the yeast PP1 protein phosphatase Glc7 (26). Deletion of the GLC7 gene is lethal, thus complicating study of the PP1 phosphatase in yeast. Glc7 can bind to several alternative regulatory subunits that are thought to direct the phosphatase to different substrates (27). The accessory subunit that directs Glc7 to the Snf1 signaling pathway is Reg1 (28). Deletion of the REG1 gene results in constitutive activation of Snf1 and hyperphosphorylation of its activation loop (8). Models for the Snf1/AMPK pathway in which dephosphorylation is the key to regulation have been lacking, perhaps because the laboratories studying AMPK and Snf1 (ours included) tend to have a kinase-centric world view. The experiments in this report are designed to directly test whether the SAKs, the deactivating phosphatase, or both are regulated by glucose.
EXPERIMENTAL PROCEDURES
Media and Strains
Strains used in this study are presented in Table 1. Yeast were grown at 30 °C in standard media. Glucose was present at 2 or 0.05% (g/100 ml), as indicated in the relevant figure legends. Raffinose medium contained 2% (g/100 ml) raffinose, 0.05% (g/100 ml) glucose, and 1 μg/ml antimycin A. In order to create a complete deletion of the REG1 gene, a reg1Δ::HIS3 allele was created using the HIS3 gene from pRS403 (1622-bp SspI fragment) flanked by the sequences from the REG1 locus. The upstream sequences from REG1 were the 824-bp EcoRI-BsaAI fragment representing nucleotides −953 to −129 relative to the REG1 ATG codon. The downstream sequences from REG1 were the 237-bp BsaAI-XbaI fragment representing sequences 143–380 downstream from the REG1 stop codon. REG1 gene replacement was confirmed by PCR.
TABLE 1.
S. cerevisiae strains
| Strain | Genotype |
|---|---|
| MSY857 | MATα ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 sak1Δ::KAN tos3Δ::KAN elm1Δ::KAN |
| MSY923 | MATα ura3 leu2 his3 sak1Δ::KAN tos3Δ::KAN elm1Δ::KAN snf1Δ10 |
| MSY955 | MATα ura3-52 leu2Δ1 his3Δ200 SNF1-3HA |
| MSY963 | MATα ura3-52 leu2Δ1 his3Δ200 trp1Δ63 SNF1-3HA reg1Δ::HIS3 |
| MSY951 | MATα ura3 leu2 his3 sak1Δ::KAN tos3Δ::KAN elm1Δ::KAN SNF1-3HA |
| MSY966 | MATα ura3 leu2 his3 sak1Δ::KAN tos3Δ::KAN elm1Δ::KAN SNF1-3HA reg1Δ::HIS3 |
| MSY961 | MATα ura3 leu2 his3 tos3Δ::KAN elm1Δ::KAN SNF1-3HA |
| MSY983 | MATα ura3 leu2 his3 tos3Δ::KAN elm1Δ::KAN SNF1-3HA reg1Δ::HIS3 |
| MSY958 | MATa ura3 leu2 trp1Δ63 his3 sak1Δ::KAN elm1Δ::KAN SNF1-3HA |
| MSY965 | MATa ura3 leu2 trp1Δ63 his3 sak1Δ::KAN elm1Δ::KAN SNF1-3HA reg1Δ::HIS3 |
| MSY956 | MATα ura3 leu2 trp1Δ63 his3 lys2Δ0 sak1Δ::KAN tos3Δ::KAN SNF1-3HA |
| MSY964 | MATα ura3 leu2 trp1Δ63 his3 lys2Δ0 sak1Δ::KAN tos3Δ::KAN SNF1-HA reg1Δ::HIS3 |
| MSY987 | MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 snf1Δ10 reg1Δ::HIS3 |
| FY1193 | MATα ura3-52 leu2Δ1 trp1Δ63 his3Δ200 snf1Δ10 |
Plasmids and Mutagenesis
All Snf1-activating kinases were expressed from their own cognate promoters, tagged with five copies of the V5 epitope at the C terminus, and introduced to cells on low copy plasmids based on pRS315 (29). Point mutations in the ATP-binding pockets of Sak1, Tos3, and Elm1 were introduced with the Stratagene QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). TAP-tagged versions of the three SAKs have been described previously (30). A low copy plasmid expressing the Mig1 protein tagged with the HA epitope at the C terminus has been previously described (31).
Screen for Altered Sensitivity Alleles
Overnight saturated yeast cultures (200 μl) grown at 30 °C were suspended in 3 ml of 47 °C top agar (containing 0.07% (g/100 ml) Bacto Agar, synthetic complete media lacking uracil with 2% (g/100 ml) sucrose, 1 μg/ml antimycin A, and 0.01% (g/100 ml) glucose). The mixture was carefully mixed and poured onto agar plates containing synthetic complete media lacking uracil with 2% (g/100 ml) sucrose, 0.01% (g/100 ml) glucose, and 1 μg/ml anti-mycin A. The top agar mixture was allowed to cool and harden. Sterile filter discs were gently placed on the surface of the top agar. 3 μl of 2.5 or 25 μM candidate inhibitor drugs (in Me2SO) were applied to the filter discs. Me2SO alone was used as a negative control. Plates were incubated at 30 °C overnight and photographed. The design and use of the altered sensitivity allele of Snf1 will be described in detail elsewhere.3
Invertase Assay
Invertase activity of midlog cells grown in high and low glucose was determined quantitatively using a colorimetric assay coupled to glucose oxidase (32). Three independent cultures were assayed, and the mean and S.E. values are plotted. The units of invertase activity used were milliunits/OD where one unit equals 1 μmol of glucose released/min.
Western Blotting
Protein extracts were prepared using the NaOH cell lysis method as described previously (8). To detect Snf1 activation loop (Thr210) phosphorylation, 600–1200 μg of protein was immunoprecipitated in radioimmune precipitation buffer as previously described (8). Rabbit polyclonal antibody directed against phosphorylated Snf1 T210 was used as the primary antibody (1:1000) to detect phosphorylated Snf1. Horseradish peroxidase-conjugated goat anti-rabbit antibody was used as secondary antibody (1:15,000) (Santa Cruz Biotechnology, Santa Cruz, CA). For Snf1-HA detection, 20–40 μg of protein extract was used. Snf1-HA was detected by a horseradish peroxidase-conjugated antibody (1:1000) recognizing the HA epitope (Santa Cruz Biotechnology).
Snf1-activating Kinase Assay
Protein extracts were prepared from cells expressing a single SAK as a C-terminal TAP fusion (30) or without any SAK. SAKs were collected from 300 μg of protein extract by immunoprecipitation with IgG beads. Precipitates were washed five times in radioimmune precipitation buffer and two times in kinase assay buffer as described (30). Precipitates were then incubated in 20 μl of kinase assay buffer with 0.2 mM [γ-32P]ATP (1000 cpm/pmol) and purified recombinant Snf1 kinase domain as the substrate. Reactions were incubated for 1 h at 30 °C and then subjected to SDS-PAGE and autoradiography.
RESULTS
Snf1-activating Kinases Are Active in High and Low Glucose
If the phosphorylation status of the Snf1 activation loop were regulated by phosphate addition, then one would predict that the SAKs would be inactive in high glucose and activated by low glucose. We have described above several lines of evidence that suggest the opposite, that the SAKs may be active in both high and low glucose. Three different experiments were conducted to assess whether glucose regulates the activity of one or more of the SAKs. First we measured invertase activity in cells expressing a single SAK with and without the PP1 regulatory subunit Reg1. When Reg1 is absent, Snf1-dependent invertase expression is elevated in both high and low glucose (28), and the Snf1 activation loop becomes phosphorylated in high glucose (8). Consistent with prior observations, strains expressing Reg1 and all three SAKs exhibit normal, glucose-regulated invertase induction (Fig. 1). In the absence of Reg1, cells with all three SAKs display elevated invertase activity independent of glucose, indicating that one or more of the SAKs is active in high glucose. In the absence of all three SAKs, invertase activity is minimal regardless of the glucose concentration or the presence or absence of Reg1, confirming previous work indicating that Sak1, Tos3, and Elm1 represent the complete complement of SAKs (12, 13). In order to determine if any of the SAKs were responsive to changes in glucose levels, Sak1, Tos3, and Elm1 were each expressed as the only SAK in the presence and absence of Reg1. In the absence of Reg1, invertase is elevated in both high and low glucose regardless of which SAK is present (Fig. 1), indicating that each of the three SAKs is capable of activating Snf1 in high glucose. When Reg1 is present, glucose regulation of invertase activity is restored. These results support the idea that the SAKs are active regardless of the glucose concentration.
FIGURE 1. Snf1-activating kinases are active in high and low glucose.

Invertase activity was measured in cells containing all three, none, or only one of the SAKs in the presence or absence of REG1 as indicated. Aliquots were assayed from cells grown in high glucose (2%) or after shifting to low glucose (0.05%) for 2 h. The mean value from three independent cultures is plotted with the error bars indicating one S.E.
Earlier work has suggested that Sak1 is the primary SAK. Of the three SAKs, only Sak1 is found in stable complex with Snf1 (30). Sak1 most efficiently activates all three β subunit-specific isoforms of Snf1 under multiple conditions (33) and is the only SAK that can promote nuclear localization of the Gal83 isoform of Snf1 (34). Consistent with these data, Sak1 most robustly induces invertase activity, particularly in the absence of Reg1 (Fig. 1). The reduced induction of invertase in cells expressing Elm1 as the only SAK may be a reflection of sequestration of Elm1 at the bud-neck for its role in regulating cell cycle and morphogenesis (25).
A second measure of SAK activity is the measurement of activation loop phosphorylation using a phosphopeptide antibody specific for Snf1 phosphorylation on the activation loop threonine 210 (PT210) (8). In this experiment, cells were grown in high glucose in the presence or absence of Reg1, protein extracts were prepared, and total Snf1 and Thr210 phosphorylation was determined by Western blotting with either HA antibodies (Fig. 2, bottom) or PT210 antibodies (top), respectively. In multiple independent experiments, the presence or absence of Reg1 affected the phosphorylation status of the Snf1 activation loop but did not affect the level of total Snf1 protein. Results from a representative experiment are shown in Fig. 2. In wild type cells, phosphorylation of the Snf1 activation loop is regulated in response to changes in glucose levels (lanes 1 and 2). In high glucose, cells expressing all three SAKs show low but detectable Thr210 phosphorylation when Reg1 is present and distinctly elevated phosphorylation when Reg1 is absent (lanes 3 and 4). In the absence of all three SAKs, no phosphorylation of Thr210 is detected, regardless of whether Reg1 is present or not (lanes 5 and 6). When the SAKs are expressed individually, the level of Thr210 phosphorylation is increased by the absence of Reg1, regardless of which SAK is present (lanes 7–12). Some variation is observed between the individual SAKs. Sak1 and Elm1 show higher basal phosphorylation (lanes 7 and 11) compared with Tos3 (lane 9). Nonetheless, the conclusion we reach is that the SAKs are capable of phosphorylating Snf1 in high glucose, supporting the idea that the SAKs are active in both high and low glucose conditions. However, these two experiments do not exclude the possibility that the Glc7-Reg1 complex regulates the SAKs. Therefore, our next experiment was designed to measure SAK activity without disrupting the Glc7-Reg1 complex.
FIGURE 2. Phosphorylation of the Snf1 kinase activation loop in reg1Δ cells.
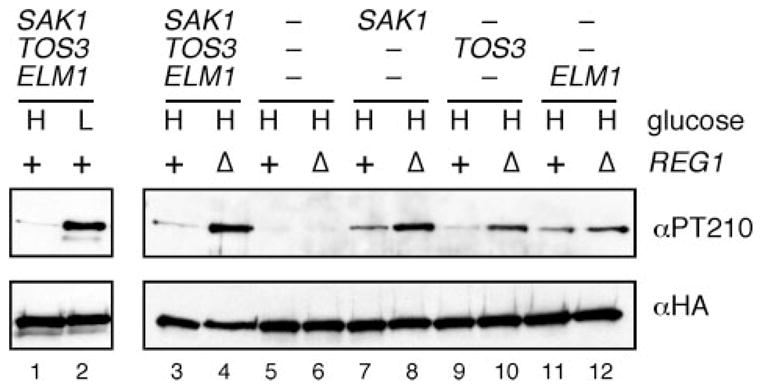
Western blot of Snf1 protein using antibodies directed against the phosphorylated form of the Snf1 activation loop (αPT210) or against the HA epitope. Cells expressing all three, one, or none of the SAKs (as shown) were grown in high (H) or low (L) glucose. Cells contained the wild-type REG1 gene (+) or a complete deletion of the of REG1 (Δ) as indicated.
Previously, we have used the tandem affinity purification protocol to isolate the three SAKs, Sak1, Tos3, and Elm1 (30). In that study, we also showed that the Sak1 complex constituents and activity were not affected by carbon source (glucose versus sucrose). The activity of Tos3 and Elm1 prepared from different carbon sources was not measured. In the third experiment to measure the activity of the SAKs, we conducted in vitro kinase assays with each SAK isolated from cells grown in high glucose or 2 h after a shift to low glucose. The SAKs were collected by immunoprecipitation and assayed for the ability to phosphorylate purified recombinant Snf1 kinase domain (Fig. 3). In earlier experiments, we have shown that the SAKs phosphorylate the Snf1 kinase domain exclusively on threonine 210 (30). In order to be certain that we were in the linear range of the assay, immune complexes were collected from increasing quantities of extract (0–300 μg). The reaction depended on exogenous substrate, since no incorporation was observed when recombinant Snf1 kinase domain was omitted from the reaction. For each SAK, a similar level of activity was observed in reactions using extracts prepared from cells grown in high and low glucose. The first three experiments described here all support the idea that the SAKs are active in both high and low glucose. Since the phosphorylation status of the Snf1 activation loop is regulated by glucose, the simplest model remaining is that the dephosphorylation of Snf1 is regulated by glucose.
FIGURE 3. Activity of Snf1-activating kinases is independent of glucose.

In vitro kinase assays were performed with SAKs immunoprecipitated from protein extracts of cells grown in either high or low glucose. Cells expressed a single, TAP-tagged SAK (as shown). Precipitates were incubated in the presence of [γ-32P]ATP with (+) or without (−) the addition of recombinant Snf1 kinase domain (Snf1-KD) as a substrate. Activity present in a titration from 0 to 300 μg of protein extract is shown.
Identification of an Altered Sensitivity Allele of TOS3
To monitor the rate of Snf1 activation loop dephosphorylation, we sought to develop a means to rapidly and selectively inactivate the SAKs. In the absence of de novo phosphate addition, changes in Snf1 phosphorylation status may be attributed to the phosphatase, enabling us to determine if the activity of Glc7-Reg1 is regulated by changes in glucose concentration. A chemical genetic method has been developed for specifically inactivating protein kinases (35). By changing the “gatekeeper” residue in the ATP binding pocket from a bulky hydrophobic residue to glycine or alanine, kinases can be engineered that are selectively inhibited by adenine analogues. An analogue-sensitive variant of the SAK Elm1 (T200G) has been reported (36). Because the primary role of Elm1 seems not to be Snf1 activation but rather participation in cell morphology signaling, we sought to identify analogue-sensitive alleles of the other SAKs, Sak1 and Tos3, whose only described role is activation of Snf1.
The gatekeeper residues of Sak1 (Leu223) and Tos3 (Leu135) were changed to glycine and alanine. Plasmids encoding these kinase variants were introduced to cells that lacked the genes for all three SAKs. A library of adenine analogues was screened for the ability to block Snf1 signaling in cells expressing analogue-sensitive variants of the SAKs without affecting signaling in cells expressing the wild-type kinase. All attempts to identify a specific inhibitor for the altered sensitivity alleles of Sak1 were unsuccessful. However, we were successful with Tos3, using the Tos3-L135G allele in combination with the adenine analogue CZ22 (Fig. 4). In a screen to identify specific inhibitors for our mutant kinases, cells were grown as a lawn with sucrose as the carbon source and antimycin to prevent aerobic metabolism. Under these conditions, Snf1 signaling is required for growth. Adenine analogues suspended in Me2SO were spotted onto filter discs. Formation of a halo around the disc was indicative of compromised Snf1 signaling, an outcome expected when the only SAK present in cells is inhibited. When cells were exposed to CZ22, a halo of growth inhibition was observed when Tos3-L135G was the only SAK. No halo was observed with cells expressing wild-type Tos3 (Fig. 4A). Therefore, the growth inhibition by CZ22 is specific to the sensitized allele of TOS3.
FIGURE 4. Analogue-sensitive allele of the Snf1-activating kinase Tos3.

A, cells expressing either Tos3 or Tos3-L135G as the only SAK were mixed with top agar and overlaid onto a plate containing sucrose as the carbon source. Sterile discs were spotted with either Me2SO (DMSO) or the adenosine analogue CZ22 dissolved in Me2SO. B, invertase assay. Cells expressing either the wild-type Tos3 or the analogue-sensitive Tos3-L135G as the only SAK were assayed for invertase induction after growth in high glucose (H) or following 2 h of incubation in media containing 0.05% glucose (L). The adenosine analogue CZ22 was added to the final concentration indicated. Mean values of invertase activity from three independent cultures are plotted with the error bar signifying one S.E. C, CZ22 blocks Snf1 activation loop phosphorylation. Cells expressing Tos3-L135G as the only SAK were grown in high glucose (H) or shifted to low glucose (L) in the presence of added Me2SO or 2.5 μM CZ22 dissolved in Me2SO. Total and activated Snf1 protein was assessed by Western blotting with anti-HA antibodies or the phosphopeptide antibody specific for the phosphorylated activation loop of Snf1 (α-PT210). Representative Western blots are shown. D, structure of the adenosine analogue CZ22.
In order to determine the effective concentration of CZ22, induction of the enzyme invertase was measured in cells grown in high glucose and shifted to low glucose as an indicator of Snf1 activation by Tos3-L135G or wild-type Tos3 (Fig. 4B). Cells expressing the wild-type or sensitized Tos3 kinase as the only SAK were initially grown in glucose-rich media. CZ22 was included in the media at the shift to low glucose at concentrations of 0 (Me2SO only), 1, 2.5, or 25 μM. In the absence of CZ22, cells expressing either the wild-type Tos3 or the sensitized Tos3-L135G show robust invertase induction upon shifting to low glucose, indicating that the Tos3-L135G allele is fully functional. When CZ22 is included in the reactions at 1 and 2.5 μM, cells expressing wild-type Tos3 exhibited no defect in the induction of invertase. In contrast, significant inhibition (85%) of invertase induction was observed in cells expressing Tos3-L135G exposed to the same analogue concentration. Increasing the concentration of CZ22 did not produce a significant improvement in the degree of inhibition. All subsequent experiments with Tos3-L135G used 2.5 μM CZ22.
We observed a subtle but reproducible stimulation of invertase activity when Tos3 cells were subjected to glucose limitation in the presence of 1 μM CZ22 (Fig. 4B). The mechanism by which this occurs is not clear. However, the purpose of these experiments was to generate a tool with which we could rapidly and efficiently shut off SAK-to-Snf1 signaling. Here we have generated a variant of Tos3 that functions like the wild-type kinase but can be efficiently inhibited by the addition of low concentrations of CZ22.
We next directly analyzed the capacity of CZ22 to prevent Tos3-L135G-mediated phosphorylation of Snf1 (Fig. 4C). Cells expressing analogue-sensitive Tos3 as the only SAK were grown in glucose-rich media and shifted to low glucose in the presence of Me2SO or 2.5 μM CZ22. The phosphorylation state of the Snf1 activation loop as well as the level of total Snf1 protein were assessed by Western blotting. Glucose-grown cells exhibit low level basal phosphorylation. As previously reported, Snf1 becomes phosphorylated when cells are shifted to low glucose media for 2 h in the absence of CZ22 (8). When CZ22 is included at the shift to low glucose, however, Snf1 never becomes phosphorylated beyond basal levels, clearly demonstrating that CZ22 inhibits the activity of Tos3-L135G toward the Snf1 activation loop. These data establish our analogue-sensitive allele of TOS3 as a legitimate tool to study the regulation of Glc7-Reg1 protein phosphatase activity.
Snf1 Dephosphorylation Is Regulated by Glucose
To test whether dephosphorylation of the Snf1 activation loop is regulated by glucose, the rate of dephosphorylation was measured in cells with active (phosphorylated) Snf1. In one case, de novo phosphorylation of Snf1 was blocked by inhibiting the sole SAK, and the rate of dephosphorylation in low glucose was then measured. In the second case, de novo phosphorylation was blocked, glucose was added back to cultures, and the rate of dephosphorylation upon shifting to high glucose was measured. By comparing these two conditions, we were able to assess whether the dephosphorylation reaction is regulated by glucose. In multiple experiments, we found that the rate of Snf1 dephosphorylation was strongly regulated by glucose. A representative experiment is shown in Fig. 5. Cells expressing the sensitized Tos3-L135G kinase as the only SAK were grown in high glucose (2%) and then shifted for 2 h to media containing only 0.05% glucose. Snf1 activation loop phosphorylation status and total Snf1 protein levels were again assessed by Western blotting. The phosphorylation status of the Snf1 activation loop is greatly increased when cells are shifted to low glucose (lanes 1 and 2). As a negative control, Me2SO was added to the low glucose culture. When aliquots were removed after an additional 1, 3, or 5 min in low glucose plus Me2SO, the activation loop phosphorylation remained stable and high (lanes 3–5). In contrast, when glucose was added to the cultures with or without CZ22, dephosphorylation of Snf1 to basal levels was observed after only 1 min of glucose addition (lanes 13 and 18). Therefore, the rate of dephosphorylation upon glucose addition is extremely rapid (t½ < 0.5 min). Technical limitations prevent analysis of shorter time points. To assess the rate of dephosphorylation occurring in low glucose, de novo phosphorylation was inhibited by the addition of CZ22 to 2.5 μM. In the absence of new phosphorylation events, the activation loop of Snf1 retained its high level of phosphorylation (lanes 8–10); therefore, the half-time for the dephosphorylation reaction in low glucose must be greater than 5 min. This indicates that the rate of Snf1 dephosphorylation is at least 10 times slower under glucose limitation than is observed under glucose abundance (compare t½ of <0.5 min and >5 min). These results provide the first direct evidence that the rate of Snf1 dephosphorylation is regulated by glucose concentration in vivo. These experiments have also been undertaken with the analogue-sensitive Elm1-T200G (36) with similar results (data not shown).
FIGURE 5. Activity of the Glc7-Reg1 phosphatase is regulated by glucose.

Cells expressing the analogue sensitive Tos3-L135G protein as the only SAK were grown in high glucose media (H) and then shifted to low glucose (L) for 30 min. Me2SO (DMSO), CZ22 dissolved in Me2SO, or glucose was added, and aliquots were removed after 1, 3, or 5 min. Extracts were prepared, and the HA-tagged Snf1 protein was collected by immunoprecipitation and analyzed by Western blotting using antibodies directed against the phosphorylated form of the Snf1 activation loop (α-PT210) or against total Snf1 protein (α-HA).
Glc7-Reg1 Phosphatase Complex Is Active in Low Glucose
The glucose-regulated dephosphorylation of Snf1 could occur by one of two mechanisms. First, glucose concentration could control the phosphatase activity of the Glc7-Reg1 complex. Second, glucose levels might regulate the availability of phosphorylated Snf1 as a substrate to the inactivating phosphatase. Supporting the latter model, in vitro experiments with purified mammalian proteins have shown that AMP binding to the γ subunit of AMPK decreases the rate of AMPK activation loop dephosphorylation (22, 23). To test whether the regulation of Snf1 activation loop dephosphorylation is controlled by changes in Glc7-Reg1 activity or accessibility of the phosphatase to the Snf1 activation loop, we analyzed the glucose-regulated activity of Glc7-Reg1 toward another substrate, the transcription factor Mig1.
In order to examine the activity of Glc7-Reg1 phosphatase toward Mig1 in low glucose, we utilized an adenine analogue-sensitive variant of Snf1, Snf1-I132G, to be described elsewhere.3 In a manner similar to the inhibition of Tos3-L135G by CZ22, the I132G mutation in the ATP binding pocket of Snf1 rendered it sensitive to the related compound, 2NM-PP1.
We first tested the capacity of 2NM-PP1 to completely inhibit Snf1-I132G phosphorylation of Mig1 tagged with the HA epitope (31). The phosphorylation status of Mig1-HA can be determined by anti-HA Western blotting, as phosphorylated isoforms exhibit a slower electrophoretic migration (37). Cells expressing Mig1-HA and either wild-type Snf1 or Snf1-I132G were grown in high glucose, where Mig1 is normally not phosphorylated. Aliquots were taken, and the remainder of the cells were divided and treated with either Me2SO (as a negative control) or 25 μM 2NM-PP1 (suspended in Me2SO) for 10 min prior to and throughout the shift to low glucose. In cells expressing either wild-type Snf1 or Snf1-I132G, Mig1-HA was seen as the dephosphorylated faster migrating isoform in high glucose (Fig. 6A, lanes 1 and 4). The addition of 2NM-PP1 completely blocked the ability of Snf1-I132G to phosphorylate Mig1 (lane 6) but had no effect on the wild-type Snf1 (lane 3). The addition of Me2SO did not inhibit either form of Snf1 kinase. Thus, the Snf1-I132G mutant is functional but can be selectively inhibited by 2NM-PP1.
FIGURE 6. Glc7-Reg1 phosphatase is active toward Mig1 in high and low glucose.
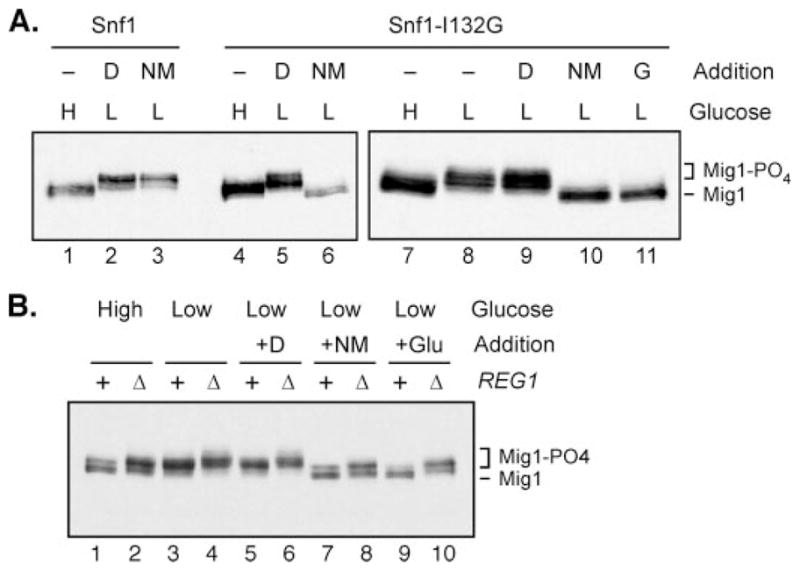
A, phosphorylation of Mig1 was measured by Western blotting of protein extracts prepared from cells grown in high glucose (H) or 1 h after shifting to low glucose (L). Cells expressed either wild-type Snf1 (lanes 1–3) or the analogue-sensitive Snf1-I132G (lanes 4 –11). Cells were treated with Me2SO (D) or the Snf1 inhibitor, 2NM-PP1 (NM), prior to and throughout the shift to low glucose as indicated (lanes 1– 6). Cells were grown in high glucose (H) and shifted to low (L) glucose for 1 h (lanes 7 and 8, respectively). After 1 h in low glucose, cells were treated for 5 min with the addition of Me2SO (D), the Snf1-I132G inhibitor, 2NM-PP1 dissolved in Me2SO (NM), or glucose (G) (lanes 9 –11). B, cells carrying a complete deletion of the REG1 gene were transformed with low copy plasmids expressing wild-type Reg1 protein (+) or empty vector (Δ). Cells also expressed HA-tagged Mig1 and the analogue-sensitive Snf1-L132G. Cells were grown in high glucose and shifted to low glucose to allow for phosphorylation of the Mig1 protein and were subsequently treated for 5 min with either Me2SO (+D), 2NM-PP1 (+NM), or glucose (+Glu), as indicated. Extracts were prepared and phosphorylation of Mig1 determined by Western blotting.
We next wanted to analyze the activity of the Glc7-Reg1 phosphatase toward phosphorylated Mig1-HA in high and low glucose. In multiple experiments, we have found that Mig1 is rapidly dephosphorylated in low glucose when Snf1 kinase is inactivated by 2NM-PP1. A representative experiment is shown in Fig. 6. Cells expressing the analogue-sensitive Snf1-I132G and Mig1-HA were grown in high glucose, shifted to low glucose for 1 h such that Mig1-HA would be in the phosphorylated state. Cells were treated with either Me2SO, 2NM-PP1 suspended in Me2SO, or glucose for 5 min. Mig1-HA was found as the faster, unphosphorylated migrating form in high glucose (Fig. 6A, lane 7) and the slower, phosphorylated migrating form upon shifting to low glucose (lane 8). The addition of Me2SO had no effect on the Mig1-HA phosphorylation status (lane 9). Within 5 min of the addition of glucose to the media, Mig1-HA was quantitatively returned to the faster, unphosphorylated form (lane 11). When the de novo phosphorylation of Mig1 was blocked with 2NM-PP1, Mig1-HA returned to the faster, unphosphorylated isoform within the same 5 min (lane 10). Therefore, the Glc7-Reg1 phosphatase is active toward Mig1 even in low glucose. These experiments demonstrate that Snf1 dephosphorylation by Glc7-Reg1 is regulated by glucose (Fig. 5), whereas Mig1 dephosphorylation is not (Fig. 6). Taken together, these results suggest that the catalytic activity of the Glc7-Reg1 phosphatase is not controlled by glucose availability; rather, it seems the availability of phosphorylated Snf1 as a substrate for dephosphorylation by Glc7-Reg1 is regulated by environmental glucose concentration.
The conclusion that the Glc7-Reg1 phosphatase is active toward one substrate (Mig1) under conditions where it is not active toward another (Snf1 activation loop) is based on the assumption that Mig1 is a direct substrate of the Glc7-Reg1 phosphatase. Although this conclusion is sometimes stated as if it were proven, an alternative interpretation for the effect of mutations in the REG1 and GLC7 genes on Mig1 phosphorylation has been noted (38). Mutations in REG1 and GLC7 cause hyperactivation of Snf1, which may in turn explain the increased phosphorylation state of Mig1. We used the analogue-sensitive allele of Snf1 to examine the effect of deletion of the REG1 gene on the phosphorylation of Mig1. In this way, any changes in the phosphorylation state of Mig1 as a result of REG1 deletion would be independent of the hyperactivation of Snf1. Cells expressing HA-tagged Mig1 and Snf1-I132G were shifted to low glucose for 1 h to allow for phosphorylation of Mig1. Cells were then treated for 5 min with 2NM-PP1 to inhibit the Snf1-I132G kinase activity. For controls, cells were either treated with Me2SO or glucose (Fig. 6B). When Snf1 kinase was inhibited by the addition of glucose, the dephosphorylation of Mig1 was rapid when Reg1 was present (lane 9). Deletion of REG1 caused an increase in the phosphorylation state of Mig1 in both high and low glucose (lanes 2 and 4). When the activity of Snf1 was inhibited by the addition of 2NM-PP1, Mig1 was rapidly dephosphorylated (lane 7). Deletion of REG1 inhibited the dephosphorylation of Mig1 (lane 8). Therefore, the effect of Reg1 on the phosphorylation state of Mig1 was not dependent on the hyperactivation of Snf1 kinase. This experiment is the first to separate the role of Glc7-Reg1 in the dephosphorylation of Mig1 from the hyperactivation of Snf1. These data provide strong evidence that Glc7-Reg1 is indeed active toward Mig1 in low glucose, although it is largely inactive toward the activation loop of Snf1.
DISCUSSION
Glucose abundance regulates the Snf1 signaling pathway. The identity of the glucose signal and the mechanism(s) by which it is transduced to the Snf1 kinase have remained elusive. The phosphorylation status of the Snf1 activation loop is determined by the relative rates of phosphorylation by the SAKs and dephosphorylation by the inactivating phosphatase PP1 (Glc7-Reg1). Until the present report, it has been unclear whether phosphate addition, phosphate removal, or both are regulated by changes in glucose availability.
We first asked if phosphorylation of Snf1 is regulated by glucose concentration. Snf1-dependent invertase activity was elevated in high and low glucose conditions in the absence of the PP1 regulatory subunit Reg1 when Sak1, Tos3, or Elm1 were present as the only SAK (Fig. 1). Thus, each of the SAKs is capable of activating the Snf1 pathway in high glucose. Additionally, the fraction of total Snf1 that was phosphorylated was higher in glucose-rich conditions in reg1Δ strains than in REG1+ strains regardless of which SAK(s) was expressed (Fig. 2). These experiments support the idea that the SAKs are active independent of glucose, but two other possibilities existed. First, it was conceivable that the SAKs actually did exhibit glucose regulation but that the lower level of activity in high glucose was sufficient to activate Snf1 when PP1 function was disrupted. Second, the activity of the SAKs themselves might be regulated by PP1. These two possibilities were addressed by our third experiment; SAKs immunoprecipitated from extracts of cells grown in high and low glucose exhibited similar activity toward exogenous Snf1 kinase domain substrate. These results strongly suggest that the activities of the SAKs are glucose-independent. Additional lines of evidence supporting this idea were presented above.
If the SAKs are not regulated by glucose, then the dephosphorylation of Snf1 by Glc7-Reg1 PP1 must be regulated by glucose. Using an adenine analogue-sensitive mutant of the SAK Tos3, we showed that the rate of Snf1 dephosphorylation is at least 10-fold greater in high glucose than in low glucose (Fig. 5). This experiment did not distinguish between a direct effect of cellular energy status on the catalytic activity of the Glc7-Reg1 complex and an effect on the availability of the Snf1 activation loop to serve as a Glc7-Reg1 substrate. An adenine analogue-sensitive allele of Snf1 allowed us to determine that Glc7-Reg1 was active in low glucose, rapidly dephosphorylating another substrate, Mig1, in conditions under which Snf1 is resistant to dephosphorylation. This result suggests that glucose regulation of Snf1 dephosphorylation is achieved by controlling access of the phosphorylated Snf1 activation loop to PP1.
Our model of the regulation of Snf1 activation loop phosphorylation is presented in Fig. 7. The three SAKs and the PP1 phosphatase are catalytically active regardless of glucose availability. In low glucose, however, the phosphorylated activation loop of Snf1 is protected from dephosphorylation by an unknown factor (X in our schema), allowing the phosphorylated isoform of Snf1 to accumulate.
FIGURE 7. Model for the regulation of Snf1 kinase at the level of dephosphorylation.
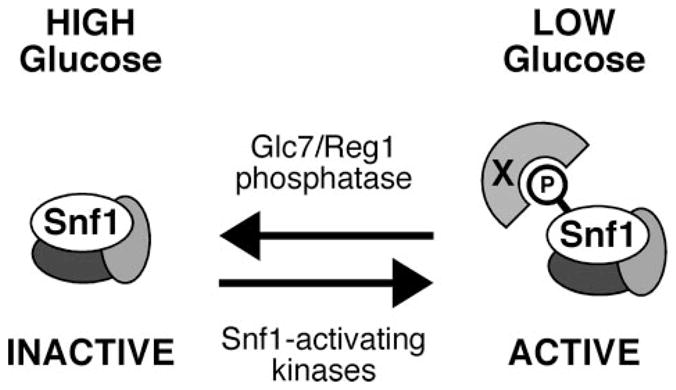
In high glucose media, the Snf1 kinase is largely unphosphorylated and inactive due to the accessibility of the activation loop threonine to the Glc7-Reg1 phosphatase. Under conditions of glucose limitation, the dephosphorylation of the Snf1 activation loop is inhibited by an unknown factor (X), leading to the accumulation of phosphorylated and active Snf1 kinase.
How might the access of phosphorylated Snf1 to Glc7-Reg1 be controlled? We consider here two possibilities. The first is that, in low glucose, phosphorylated Snf1 and Glc7-Reg1 are maintained in distinct subcellular compartments, thus protecting phosphorylated Snf1 from PP1. Dombek et al. (39) have reported that Reg1-GFP fusions display constitutively cytoplasmic localization. Snf1, by contrast, has been shown to exhibit a complex, glucose-regulated subcellular localization pattern. In glucose abundance, all three β subunit-specific isoforms of Snf1 are cytoplasmic (40). Upon a shift to low glucose, however, the Sip1-Snf4-Snf1 complex becomes localized to the vacuolar periphery, the Gal83-Snf4-Snf1 complex becomes enriched in the nucleus, and the Sip2-Snf4-Snf1 complex seems to remain cytoplasmic under all conditions (40, 41). The idea that subcellular localization might control Snf1 dephosphorylation is challenged by the finding that the three isoforms of Snf1, each with a distinct localization pattern, all show similar regulation by glucose (31, 33). For this reason, we do not favor a model in which Snf1 dephosphorylation is controlled by subcellular localization.
A second possibility is that the phosphorylated Thr210 residue is protected from dephosphorylation by Glc7 in low glucose by some protein factor (X in our model; Fig. 7). The identity of this protein factor is unclear; however, two strong candidates come immediately to mind. One candidate is Snf4, the γ subunit of the Snf1 heterotrimer. In the mammalian enzyme, the γ subunit binds AMP and inhibits the dephosphorylation reaction (22, 23). In yeast, the γ subunit does not appear to bind AMP, and whether or not it binds some other small metabolite is a topic of great interest in the field. Two-hybrid interactions between Snf1 and Snf4 are reported to increase in response to glucose limitation (6). It is possible that interactions of Snf1 with the γ subunit Snf4, possibly in conjunction with the β subunits, preclude dephosphorylation by PP1 in low glucose. It is clear, however, that although Snf4 and the beta subunits are required for Snf1 function in vivo, at least some degree of glucose regulation of Snf1 phosphorylation is independent of the β and γ subunits, since cells lacking either all three β subunits, the γ subunit, or both retain glucose-regulated phosphorylation of Thr210 (44). In fact, when Snf1 kinase lacking its C-terminal regulatory domain is expressed in the absence of β and γ subunits, the activation loop still exhibits glucose-regulated phosphorylation (44). Thus, some regulation must lie outside the Snf1 regulatory domain as well as the β and γ subunits.
The second strong candidate for X in our model is Reg1. Although Reg1 is thought to act as the targeting subunit for Glc7 that promotes Snf1 dephosphorylation in high glucose, it is possible that Reg1 maintains dual roles in Snf1 activation loop regulation. Reg1 is an abundant component of the Snf1 kinase complex as determined by two-hybrid analysis (42) and mass spectrometry (30). Furthermore, the interaction of Reg1 with the Snf1 kinase domain requires the activation loop threonine and is stronger in low glucose (42). In low glucose Reg1 could function to protect the phosphorylated Thr210. Some as yet unidentified signal would control whether Reg1 serves as protector of Snf1 activation loop phosphorylation or recruiter for the PP1 phosphatase.
Additional candidates for the identity of the X factor are other proteins found in the Snf1 complex, including chaperones and the phosphoprotein-binding 14-3-3 proteins, Bmh1 and Bmh2 (30, 45). Finally, the Snf1 kinase domain has been reported to dimerize in a manner that could make the activation loop inaccessible (46). Whether or not there is regulatory significance to the proposed Snf1 dimerization is unknown. Clearly, further experimentation will be needed to determine precisely how glucose abundance controls Snf1 availability for dephosphorylation by PP1.
Finally, the glucose signal controlling Snf1 phosphorylation status is not known. Our data do not distinguish between a low glucose signal that protects Snf1 from dephosphorylation and a high glucose signal that promotes its dephosphorylation. Identifying the molecular cue(s) that regulates Snf1 activation loop phosphorylation status is of great interest. The primary consequence of this study will be to redirect the focus of future investigations of Snf1 activation loop phosphorylation away from the regulation of phosphate addition and toward the regulation of phosphate removal.
Acknowledgments
We thank Karin Elbing for valuable insights and suggestions and Annie Bedison for purification of recombinant proteins.
Footnotes
The abbreviations used are: AMPK, AMP-activated protein kinase; SAK, Snf1-activating kinases; HA, hemagglutinin.
M. K. Shirra, R. R. McCartney, C. Zhang, K. M. Shokat, M. C. Schmidt, and K. M. Arndt, manuscript in preparation.
This work was supported by National Institutes of Health Grants GM46443 (to M. C. S.), GM52593 (to K. M. A.), DK74654 (to K. M. A.), and AI44009 (to K. M. S.) and American Heart Association Predoctoral Fellowship 0615379U (to E. M. R.).
References
- 1.Hardie DG, Carling D, Carlson M. Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- 2.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. Diabetes. 2002;51:2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 3.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 4.Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. J Biol Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- 5.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang R, Carlson M. Genes Dev. 1996;10:3105–3115. doi: 10.1101/gad.10.24.3105. [DOI] [PubMed] [Google Scholar]
- 7.Scott JW, Ross FA, Liu JK, Hardie DG. EMBO J. 2007;26:806–815. doi: 10.1038/sj.emboj.7601542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCartney RR, Schmidt MC. J Biol Chem. 2001;276:36460–36466. doi: 10.1074/jbc.M104418200. [DOI] [PubMed] [Google Scholar]
- 9.Estruch F, Treitel MA, Yang X, Carlson M. Genetics. 1992;132:639–650. doi: 10.1093/genetics/132.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 11.Nath N, McCartney RR, Schmidt MC. Mol Cell Biol. 2003;23:3909–3917. doi: 10.1128/MCB.23.11.3909-3917.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, Hardie DG. Curr Biol. 2003;13:1299–1305. doi: 10.1016/s0960-9822(03)00459-7. [DOI] [PubMed] [Google Scholar]
- 13.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Proc Natl Acad Sci U S A. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 16.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. J Biol Chem. 2005;280:29060–29066. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 19.Momcilovic M, Hong SP, Carlson M. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 20.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Hong SP, Momcilovic M, Carlson M. J Biol Chem. 2005;280:21804–21809. doi: 10.1074/jbc.M501887200. [DOI] [PubMed] [Google Scholar]
- 22.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. J Biol Chem. 2006;281:32207–32216. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 23.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Biochem J. 2006;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sreenivasan A, Kellogg D. Mol Cell Biol. 1999;19:7983–7994. doi: 10.1128/mcb.19.12.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouquin N, Barral Y, Courbeyrette R, Blondel M, Snyder M, Mann C. J Cell Sci. 2000;113:1435–1445. doi: 10.1242/jcs.113.8.1435. [DOI] [PubMed] [Google Scholar]
- 26.Tu J, Carlson M. Mol Cell Biol. 1994;14:6789–6796. doi: 10.1128/mcb.14.10.6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Tatchell K. Biochemistry. 2001;40:7410–7420. doi: 10.1021/bi002796k. [DOI] [PubMed] [Google Scholar]
- 28.Tu J, Carlson M. EMBO J. 1995;14:5939–5946. doi: 10.1002/j.1460-2075.1995.tb00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elbing K, McCartney RR, Schmidt MC. Biochem J. 2006;393:797–805. doi: 10.1042/BJ20051213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt MC, McCartney RR. EMBO J. 2000;19:4936–4943. doi: 10.1093/emboj/19.18.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldstein A, Lampen JO. Methods Enzymol. 1975;42:504–511. doi: 10.1016/0076-6879(75)42159-0. [DOI] [PubMed] [Google Scholar]
- 33.McCartney RR, Rubenstein EM, Schmidt MC. Curr Genet. 2005;47:335–344. doi: 10.1007/s00294-005-0576-2. [DOI] [PubMed] [Google Scholar]
- 34.Hedbacker K, Hong SP, Carlson M. Mol Cell Biol. 2004;24:8255–8263. doi: 10.1128/MCB.24.18.8255-8263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bishop AC, Shah K, Liu Y, Witucki L, Kung C, Shokat KM. Curr Biol. 1998;8:257–266. doi: 10.1016/s0960-9822(98)70198-8. [DOI] [PubMed] [Google Scholar]
- 36.Sreenivasan A, Bishop AC, Shokat KM, Kellogg DR. Mol Cell Biol. 2003;23:6327–6337. doi: 10.1128/MCB.23.17.6327-6337.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Treitel MA, Carlson M. Proc Natl Acad Sci U S A. 1995;92:3132–3136. doi: 10.1073/pnas.92.8.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Wever V, Reiter W, Ballarini A, Ammerer G, Brocard C. EMBO J. 2005;24:4115–4123. doi: 10.1038/sj.emboj.7600871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dombek KM, Voronkova V, Raney A, Young ET. Mol Cell Biol. 1999;19:6029–6040. doi: 10.1128/mcb.19.9.6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vincent O, Townley R, Kuchin S, Carlson M. Genes Dev. 2001;15:1104–1114. doi: 10.1101/gad.879301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedbacker K, Carlson M. Eukaryot Cell. 2006;5:1950–1956. doi: 10.1128/EC.00256-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ludin K, Jiang R, Carlson M. Proc Natl Acad Sci U S A. 1998;95:6245–6250. doi: 10.1073/pnas.95.11.6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanz P, Alms GR, Haystead TA, Carlson M. Mol Cell Biol. 2000;20:1321–1328. doi: 10.1128/mcb.20.4.1321-1328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elbing K, Rubenstein EM, McCartney RR, Schmidt MC. J Biol Chem. 2006;281:26170–26180. doi: 10.1074/jbc.M603811200. [DOI] [PubMed] [Google Scholar]
- 45.Dombek KM, Kacherovsky N, Young ET. J Biol Chem. 2004;279:39165–39174. doi: 10.1074/jbc.M400433200. [DOI] [PubMed] [Google Scholar]
- 46.Nayak V, Zhao K, Wyce A, Schwartz MF, Lo WS, Berger SL, Marmorstein R. Structure. 2006;14:477–485. doi: 10.1016/j.str.2005.12.008. [DOI] [PubMed] [Google Scholar]